Изобретение относится к способу . очистки высших алифатических вторич ных и третичных аминов. Известен ряд способов очистки аминов, основанных на химической обработке последних раствором едког натра, хлорангидридом карбоновой ки слоты Cl 1 и 2 . Эти способы не позволяют очистит высшие вторичные от примеси третичных и наоборот. Способ ПО не пригоден пот«5му, что щелочи.не действу ют на вторичные и третичные амины. Спо соб j 2 совершенно неприменим для очи стки вторичных аминов, поскольку они разрушаются хлорангидридом карбоново кисдоты, и практически непригоден для очистки высших третичных аминов ввиду их малой летучести (трудность отгонки амина из реакционной смеси) Известен способ очистки высших первичных алкиламинов, основанный на экстракции примеси из водного раствора аминной соли жидкими углеводородами 3 3 Этот способ также непригоден для очистки высших вторичных и третичных аминов ввиду очень.малой растворимости солей в воде и усиленного их перехода из водной в углеводородную фазу. Наиболее близким к изобретению является способ разделения вторичных и третичных аминов, основанный на экстракции их бутиловым эфиром диэтиленгликоля f 3 Этот способ отличается громоздкостью (требуется шестикратная экстракция вторичного амина эфиром диэтиленгликоля и шестикратная промывка его фазы гексаном с целью доочистки вторичного амина), недоста точно эффективен ( чистота .целевого вторичного амина не превышает 95%); ведет к значительным потерям третич ного амина, поскол ьку в фазе эфира внэтиленгликоля содержатся примерно равные количества вторичного и третичного амина (примерно по 20% от общего количества смеси, что составляет 20/80-100 25% от исходного количества третичного амина). Кроме того, этот способ не позволяет добиться эффективного отделения от высших вторичных и третичных аминов окрашенных примесей. Цель изобретения - улучшение качества целевого продукта. Поставленная цель дЬстигается тем, что согласно способу очистки высших алифатических вторичных (С,- €2,4)-аминов от примеси высших алифатических третичных ( С-)аминов или высших алифатических третичных (С24- С)-аминов от примеси высших алифатических вторичных ( С24)-эминов путем экстракции, экстракцию ведут смесью углеводорода и диметилформамида или углеводорода и водного диметилформамида, со- держащего 5 - 20об.% воды, с одновременным подкислением азотной кисло той до рН 2,0-6,5 при соотношении объемов углеводорода и диметилформамида или углеводорода и водного диметилформамида (I:) - ). Причем в случае содержания в высших алифатических третичных ( аминах 50 - 100 вес. высших алифатических вторичных ( -аминов подкисление азотной кислотой ведут до рН 5,0-6,2 при cooTrioшении объемов углеводорода и водного (диметилформамида от :k до {:1. В качестве углеводородов предпочтительно используют петропейный эфир, гептан, октан, нонан. Процесс осуществляют следующим образом. Примеси,высших алифатических третичных амиЯов в высших алифатических вторичных аминах и окрашенные примеси экстрагируют углеводородами из водио-диметилформамидного раствора нитратной соли высшего вторичного амина, содержащего 5-20об. воды при рН 6,0-6,5 и соотношении объемов углеводород:водный диметилформамид 1:2, а подкисление осуществляется азотной кислотой. Использование соляной и особенно серной кислоты приводит к образованию плохо растворимых в водном диметилформамиде солей и к получению осадков и труднорасслаивающихся эмульсий. Примеси высших алифатических вторичных аминов в высших алифатических третичных аминах экстрагируются из углеводородного раствора высших третичных аминов диметилформамидным или водно-диметилформамидным раствором, содержащим 5 20 об.% воды, при подкислении азотной кислотой C|o рН 5(0-6,0, а окрашенные примеси углеводородами из диметилформамйдного или вод но-диметилформамид но го рас.т3 . 10271514
вора нитратной соли высшего алифати- .ричных и третичных аминов углеводоческого третичного амина при рН 2,0- родной фазой в зависимости от рН 2,5 водного диметилформамида при соотноВ табл. 1 k приведены степени шении объемов углеводород:водный извлечения высших алифатических вто- 5 диметилформамид 1:2.
Таблица







| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения третичных аминов в водных растворах | 1978 |
|
SU792117A1 |
| Способ очистки первичных алифатических аминов с @ -с @ | 1986 |
|
SU1353772A1 |
| Способ очистки алифатическогоТРЕТичНОгО АМиНА | 1979 |
|
SU845772A3 |
| СПОСОБ ВЫДЕЛЕНИЯ НИЗКОМОЛЕКУЛЯРНЫХ АЛИФАТИЧЕСКИХ КИСЛОТ ИЗ ВОДНЫХ РАСТВОРОВ, СОДЕРЖАЩИХ МУРАВЬИНУЮ КИСЛОТУ | 2001 |
|
RU2197471C1 |
| Способ получения третичных алифатических аминов | 1976 |
|
SU745359A3 |
| Способ очистки солей высших четвертичныхАММОНиЕВыХ ОСНОВАНий, СОдЕРжАщиХ OT16 дО 36 ATOMOB углЕРОдА | 1979 |
|
SU833949A1 |
| Способ количественного определения первичных алифатических аминов | 1979 |
|
SU883738A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ ТРЕТИЧНЫХ АЛИФАТИЧЕСКИХ ИЛИ АРИЛАЛИФАТИЧЕСКИХ АМИНОВ | 1972 |
|
SU351843A1 |
| Способ очистки первичных алифатических аминов С @ -С @ | 1989 |
|
SU1643525A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВТОРИЧНЫХ АМИДОВ ПУТЕМ КАРБОНИЛИРОВАНИЯ СООТВЕТСТВУЮЩИХ ТРЕТИЧНЫХ АМИНОВ | 2009 |
|
RU2525400C2 |

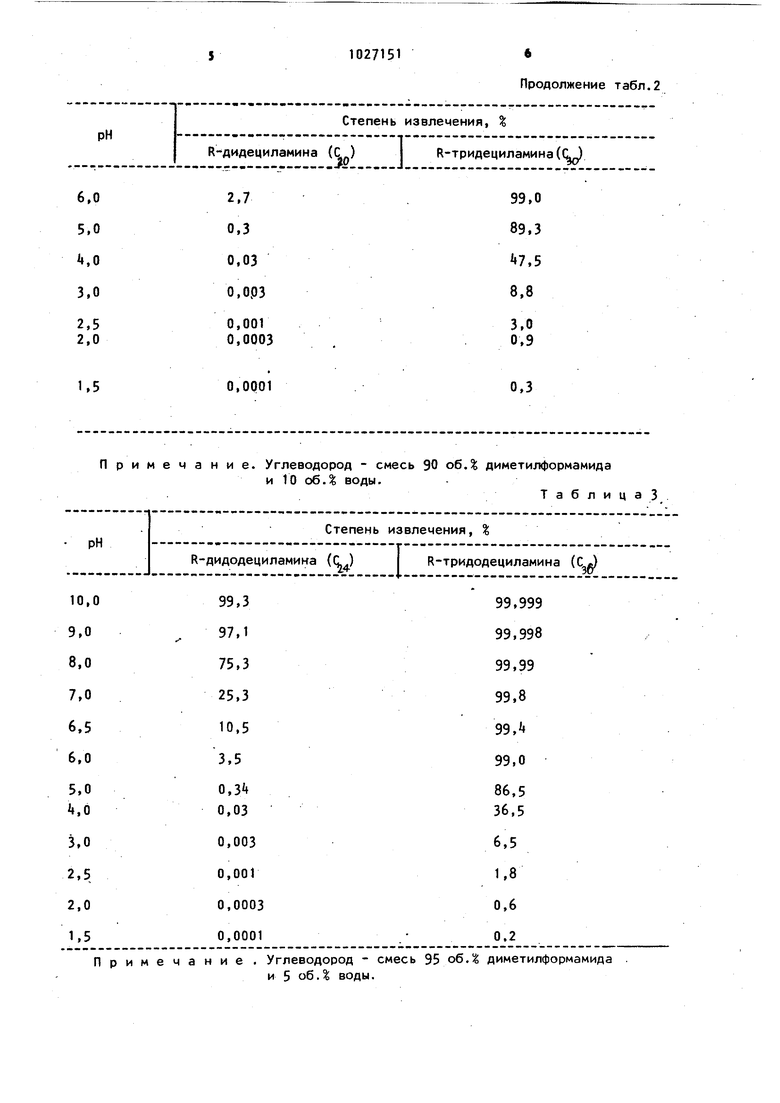
Л ip и м е ч а н и е ..Углеводород смесь и 20 об.% воды. 80 об.% диметилформамида Таблица2 Примечание. Углеводород - смесь и 10 об.% воды. Примечание . Углеводород - смесь и 5 об.% воды.
Продолжение табл.2 90 об. диметилформамида ТаблицаЗ 95 об. диметилформамида Примечание. Углеводор Как видно из табл. 1 - , наиболее удобной при очистке высших алиф тических вторичных аминов от примеси высших алифатических третичных аминов является рН 6,0-6,5, при которой практически весь высший алифа тический вторичный амин (на 90-98) остается в водном диметилформамиде и вместе с тем достаточно велика степень извлечения примеси высшего алифатического третичного амина i. 96-99). При более высоких рН степень извлечения примеси высшего али фатического третичного амина возрастает, но возрастают потери очища мого продукта. Так, при рН 7,0 в углеводородной фазе остается до 25 высшего алифатического вторичного амина, хотя степень извлечения примеси высшего алифатического третичного амина составляет 99i8-99 9%. Поэтому в зависимости от требуемой ЧИСТОТЫ и выхода высшего алифатичес
Таб. лица диметилформамид. кого вторичного амина его очистку проводят при рН 6,0-7,0. При очистке высших алифатических третичных аминов от примеси высших алифатических вторичных наиболее удобен интервал рН 5i0-6,О, при котором практически весь третичный амин находится в углеводородной фазе, а примесь высшего алифатического вторичного - в водном диметилформамиде (табл. 1 - t) . При очистке от окрашенных примесей используют рН 2,0-2,5 с целью уменьшения потерь очищаемого высшего алифатического третичного амина. Вместе с тем, степень извлечения окрашенных примесей углеводородной фазой практически не зависит от рН. С целью экономии диметилформамида очистку высших алифатических третич- . ных аминов проводят в следующем порядке: вначале добавлением азотной кислоты переводят весь высший ал фатический третичный амин и примесь высшего алифатического вторичного амина в водный дйметилформамид (рН 2,0-2,5)- Углеводородом при этом извлекаются окрашенные примеси. Зате добавлением аммиака до рН ,О разрушают соль высшего алифатического третичного амина и экстрагируют выделившийся амин другой порцией углеводорода. Примесь высшего алифатического вторичного амина при этом не экстрагируется {табл. 1 - ). Если извлекать примесь высшего алифати ческого вторичного аминэ и высший алифатический третичный амин отдельными порциями водно-диметилформамидйого раствора, расход диметилформамида увеличится вдвое. В случае, если высшие алифатические третичные (Сг.ф С)-амины содер жат 50 - 100 вес. высших алифатичес ких вторичных 02.4)аминов, процесс проводят следующим образом. . При кислотности, соответствующей рН 5, водного диметилформамида в углеводородной фазе локализуется 98,5 триалкиламина и 1,51 диалкиламина, а в водном диметилформамиде 98,5 нитратной соли вторичного амина и 1,5 нитратной соли третичного амина. При отсутствии подкисления водного дйметилформамида оба амина количественно локализуются 8 углеводородной фазе: высший алифатический вторичный амин - на 99/5%, высший алифатический третичный амин на 99f999 « Недостаточное подкисление водного диметилформамида (т.е. рН выше ..8) приводит к неполному.. из влечению алифатического втоеич ного амина. Так, при рН 6, в водный дйметилформамид переходит 0,0 высшего алифатического третичного амина и 70% высшего алифатического вторичного амина. В углеводородной фазе соответственно остается 99,9б высшего алифатического третичного и 30 высшего алифатяческого вторичного амина. Подкисление до рН, меньших чем 5,, приводит к значительному переходу высшего алифатического третичного амина в водный дйметилформамид. В водный дйметилформамид переходит 99,951 высшего алифатического вторичного и 30% высшего алифатического третичного амина. В углеводородной фазе остается 0,05% высшего алифатическо- го вторичного и 70 высшего алифатического третичного амина. Таким образом, для эффективного разделения смесей этих аминов необходим выбор оптимальной величины рН, т.е. 5,5-5,8. Применение повторной экстракции углеводорода и водного дйметилформамид а , содержащих высший алифатический третичный и высший алифатический вторичный амины (по 93,6% от исходного количества в каждой фазе смесью водным диметилформамидом и углеводородом при соотношении объемов i: и 1: (с целью экономии растворителей) при рН 5. и 6,16,2 соответственно, позволяет локализовать в углеводородной фазе не менее 99«9% от исходного количества высшего алифатического третичного амина, а в водном диметилформамиде не менее 99.8% от исходного количества высшего алифатического вторичного амина. Выделение высшего алифатичес г кого вторичного амина из водного диметилформамида достигается путем добавления избытка концентрированного водного раствора аммиака до .рН 10-11 с целью разрушения нитрэтной соли высшего алифатического вторичного амина с последующей его экстракцией петролейным эфиром. П р и м е р 1 . Очистка диоктиламина. В 100 мл водного диметилформамидз соде{ кащего 20 об.% воды, вносят 5 г диоктиламина содержащего 5% триоктиламина. При интенсивном перемешивании добавляют 50 мл петролёйного эфира и добавлением по каплям концентрированноговодного раствора азотной кислоты дoвoдяt рН водного диметилформамида до 6,0. После встряГхивания углеводородную фазу отделяют) а оставшиеся примеси экстрагируют еще ТО мл петролёйного эфира. Углеводородную фазу отделяют, а в водный дйметилформамид вводят избыток кон- , центрированного водного раствора аммиака до рН 10-11. Выделившийся амин экстрагируют 20 мл петролёйного эфира, прО1М1Ь1вают экстракт водой до полного удаления диметилформамида и отгоняют петролейный эфир. После отгонки петрояейного эфира получают k,S г диоктиламина, содержащего 99,8 основного вещества и менее 0,1% триоктиламина. При очистке описанн 1М способом 5. г диоктиламина, содержаП«. . . . щего 10 триоктиламина, при рН 6,5 получают 3,9 г продукта, содержащего оснвного вещества и менее 0,1 триоктиламина. Светопоглощение исходного и очищенного продук ов в области 500- 10 н представлены в табл. 5. Из табл. 5 видно, что степень осветления достигает 20 раз. Таблица П р и м е р 2 . Очистка дидециламина. В 100 мл водного диметилформамид содержащего 10 об. воды, вносят Ц г дидециламина, содержащего 10% трид циламина. При интенсивном пере мешивании доводят 50 мл гептана или октана и добавлением азотной кислот доводят рН водного диметилфбрмамйда до 6,5. После встряхивания фазы раз деляют, ставшиеся примеси экстрагируют еще 10 мл гептана или октана и выделяют дидециламин из водного диметилформамида аналогично примеру 1. Получают 3,2 г дидецламина, содержащего 99,8 основного вещества и менее 0,1 тридециламина. При очи ке k г дидециламина, содержащего 5t тридециламина, при рН 6,0 получают г продукта, содержащего основного вещества и менее 0,1% тридециламина. Светопоглощение исходного и очищенного продуктов 8 области 500410 нм представлено в табл. 6. Из табл. 6 видно, что степень осветления составляет 10-15 раз. Таблиц 3; 6 П р и м е рЗ . Очистка дидодециламина. В 50 мл октана и нрнана растворяют 1,5 г дидодециламина, сбдержащего 10% тридодециламина. К полученному раствору приливают 100 мл водного диметилформамида, содержащего 5 об.% воды, и при интенсивном пере- мешивании доводят рН водного диметилформамида азотной кислотой до После встряхивания фазы разделяют, оставшиеся примеси экстрагируют 10 мл октана или нонана и выделяют из водного диметилформамида дидодециламин аналогично примеру 1. Выделяют 1,2 г дидодециламина, содержащего основного вещества и менее 0,1% тридодециламина. Светопоглощение исходного и очищенного продуктов, в области 500410 нм представлено в табл. 7.. Из табл. 7 видно, что осветление достигает 11 раз. Т а б л и ц а 7 . П р и м е р 4 .Очистка триоКтиламина. В 100 мл водного диметилформамида, содержащего 20 об.% воды, вносят 3,0 г триоктиламина, содержащего 5% диоктиламина. При интенсивном перемешивании, добавляют 50 мл петролейного эфира и по каплям добавляют концентрированный водный раствор азотной кислоты до рН 2,5. После расслаивания углеводородную фазу, содержащую окрашенные примеси, отделяют, а оставшиеся примеси экстрагируют еще 10 мл петролейного эфира. Фазы раэделяют, а к водно-диметилформамидному раствору солей аминов по каплям добавляют концентрированный водный раствор аммиака в присутствии 50 мл петролейного эфира до рН 5.5. При этом происходит процесс разрушения нитратной соли триоктиламина и его экстракция в углеводородную фазу.. Фазу петролейного эфира, содержащую триоктиламин, промывают водой до полного удаления диметилформамида и отгоняют петролейный эфир. Получают 2,7 г триоктиламина, содержащего основного вещества и менее 0,1 дйоктиламина. При очистке 3.0 г триоктиламина, содержащего 10 дйоктиламина, при рН 2,5 1(1 5,0/COOTвет ственно, получают 2,35 г продукта, содержащего 99.8 основного вещества и менее 0,1% дйоктиламина. Светопоглощение исходного и очищенного продуктов представлено в табл. 8. Из табл. 8 видно, что осветление достигает 25 раз. Таблица8 П р и м е р 5 . Очистка тридецил амина. В 100 мл водного диметилформамида, содержащего 5 об.% воды, вносят 4,0 г тридециламина, содержащего 10 дидециламина. При интенсивном перемешивании добавляют 50 мл гептана или октана и доводят рН водного диметилформамида до 2,0. После рассла вания углеводородную фазу отделяют, а оставшиеся окрашенные примеси экстрагируют 10 мл гептана или окта на. Фазы разделяют, добавляют 50 мл гептана или октана и доводят рН водного диметилформамида до 5,0. Углеводородную фазу, содержащую тридециламин,.о тде ля ют, п ромы за ют водой до полного удаления диметилфо амида и отгоняют углеводород. Полу51IчаЮт 3,05 г тридециламина, содержащего 99,9 основного вещества и менее 0,1% дидециламина. При очистке 3,0 г тридециламина, содержащего 5% дидециламина при рН 2,0 и 5,5, соответственно, получают 2,6 продукта, содержащего 99,9% основного вещества и менее 0,1% дидециламина. Светопоглощение исходного и очищенного продуктов приведено в табл. 9. Таблйца9 Из табл. 9 видно, что осветление достигает 16 раз. П р и м е р 6 . Очис,тка тридодециламина. В 100 мл диметилформамида вносят 5 г тридодециламина, содержащего 10% дидодециламина. При интенсивном перемешивании добавляют 50 мл рктана или нонана и доводят рН диметилформамидной фазы до 2,5. После расслаивания углеводородную фазу отделяют.и экстрагируют оставшиеся примеси 10 мл октана или нонана. Фазы разделяют, добавляют 50 мл октана или нонана и доводят рН диметилформамидной фазы до 6,0. Углеводородную фазу, содержащую тридодециламин, промывают водой до полного удаления диметилформамида и отгоняют углеводород. Получают ,0 г тридодециламина, содержащего 99,8% основного вещества и 0,1% дидодециламина. При очистке 5 г тридодециламина, содержащего 5% дидодециламина при рН 2,5 и 6,0, соответственно, получают k,k г тридодециламина, содержащего 99,8% основного и менее X), 1 % дидодециламина. Светопоглощение исходного и очищенного продуктов приведено в табл. 10. Из табл. 10 видно, что осветление достигает 15 раз. Т а б ли ц а 10 Приме р 7 . Разделение смесей tpиoктил (С и диоктиламина (С В 100 мл петролейного эфира растворяют 8 г смеси, содержащей г триоктиламина и 2,5 г диоктиламина. К полученному раствору приливают 100 мл водного диметилформамида, соде жащего 20 об.% воды, и при интенсивном перемешивании доабдят рН в систе ме азотной кислотой до . После расслаивания фазы разделяют, а к углеводородной фазе добавляют 25 мл йодного диметилформамйда, содер)(ащего 20 об,% воды fсоотношение объемов фаз А:1), и при интенсивном пере мешивании доводят рН до 5- При этом происходит удаление оставшегося диоктиламина . Водно-дйметилформамидный слой отделяют и объединяют его с раствором, полученным при Первой экстракции, добавляют 30 мл петролей ного эфира (соотношение объемов фаз 125:30 :) и при интенсивном перемешивании ДОВОДЯТ рН водным раствором аммиака до 6,2. При этом происхо дит процесс, разрушения нитратной соли третичного амина и его переход в фазу петролейного эфира. Фазу петролейного эфира, содержащую примеси третичного амина, отделяют, а в водный диметилформамид вводят избыток . концентрированного раствора аммиака. Выделившийся диоктиламин экстрагируют петролейным, эфиром. После отмывки фаз петролейного эфира, содержащих триоктил и диоктиламин, соответственно, и отгонки петролейного эфиравыделяют 5,3 г триоктилам на с содержанием основного вещества 99.9 и 2,32 г диоктиламина с содержанием основного вещества 39.85% При разделении указанным способом смеси, содержащей по 2,5 г ами нов, выделяют 2,4 г триоктиламина с содержанием основного вещества 99,9 и 2, г диоктиламина с содержанием основного вещества . Примео8 .- Разделение смесей тридецил () и дидециламина ( 100 мл гептана или октана растворяют 3,0 г смеси, содержащей 2,0 г тридециламина и 1,0 г дйдециламина. К полученному раствору приливают 100 мл водного диметилформамйда.содержащего 10 об.% воды, и при интенсивном перемешивании доводят рН водного диметилформамйда азотной кислотой до . Фазы разделяют, а к гептановой фазе приливают 25 мл водного диметилформамйда, содержащего 10 об.% воды. При интенсивном перемешивании доводят рН азотной кислотой до 5.0. При происходит процесс извлечения оставшегося дидециламинд в водг ный диметит«})ормамид. фазы разделяют водно-дйметилформамидный слой сливают и объединяют с ратвором, полученным при предыдущей экстракции. К полученному раствору добавляют 30 мл гептана и при перемешивании доводят рН до 6,1-аммиаком. Лри этом происходит процесс разрушения нитратной соли тридециламина и его переход в гептановую фазу. Дидециламин выделяют , из водного диметилформамйда аналогично примеру 1. После отмывки углеводородных растворов аминов водой и отгонки углеводорода выделяют 1,90 г тридециламина, содержащего 99.9 основного вацества, и 0,93 г дидециламина, содержащего 99. основ-, ного вещества. При разделении указанным способом смеси, содержащей 2,Ц г тридецил- и 1, 6 г дидециламина, получают 2,30 г дидециламина- с содержанием основного вещества 99.9 и 1,5 г дидециламина с содержанием основного вещества 99.7. П р и м е р 9 . Разделение смесей тридодецил (с5) и дидециламина (с 2.4). В 1.00 мл октана или нонана растворяют k г смеси, содержащей 2,5 г тридодециламина и 1,5 г дидодециламина. К полученному раствору приливают 100 мл водного диметилформамйда, содержащего 5 об. воды. При интенсивном перемешивании доводят рН водного диметилформамйда азотной кисло17той до Фазы затем разделяют а к октановой фазе добавляют 25 мл водного диметилформамида, содержащего 5 об при интенсивном перемешивании доводят азотной кислотой .до 5iO. При этом происходи процесс извлечения из октана оставш гося дидодециламина. Затем отделяют водно-диметилформамидный слой и объединяют его с раство0ом, получен ным после предыдущей экстракции. К полученному раствору добавляют 30 мл октана и доводят рН водным ра вором аммиака до 6,1. При этом протекает процесс разрушения нитратной соли тридодеииламина и его переход в октановую фазу. Дидодециламин выд ляют из водного диметилформамида аналогично примеру 1.,После выде ления аминов из углеводородных piacT воров получают 2,kB г тридодецилами на, содержащего основного вацестёа, и I,АО г дидодециламина« содержащего основного вещества. При разделении указанным способом 3 f смеси, содержащей по 1,5 г аминов, получают 1,А5 г тридодецил51амина с содержанием основного вещества 99,9 и г дидодециламина с содержанием основного вещества 99,8%. Таким образом, предлагаемый спог соб позволяет быстро ( время экстракции 2-3 мин, полное время процесса разделения с учетом всех стадий процесса - не более мин) и эффективно (степень чистоты целевых продуктов достигает 99,9 разделить высшие алифатические вторичные и третичные амины. Возможность регене рации экстрагентов путем отгонки делает этот способ экономичным. Использование дешевых и доступных 3KCtpareHToe (петролейный эфир, алифатические углеводороды, диметилформамцд) позволяет осуществлять его в промышленном масштабе. Данный способ целесообразно использовать для очистки смесей, состав которых или близок к эквимолярному, или с относительно небольшим содержанием (не более 10-20%) примесного амина либо высшего алифатического третичHoro, либо вторичного амина).
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Патент США (f 3337630, кл | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
