4 00
-AW,
Ш2
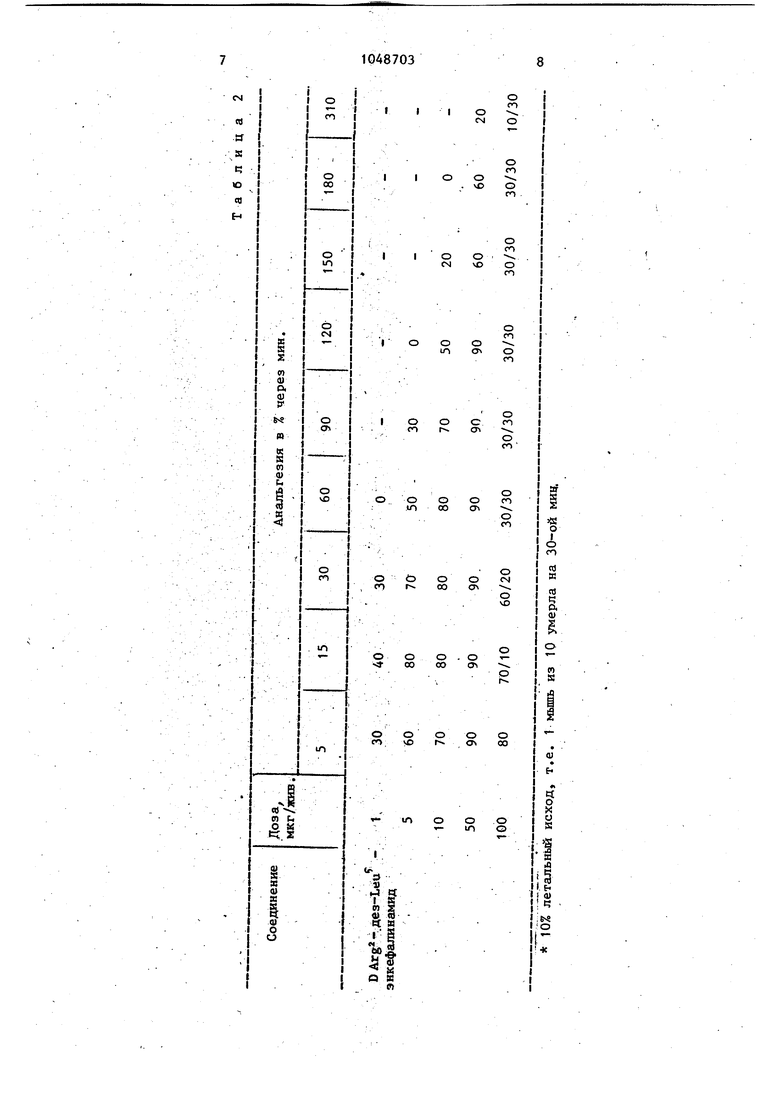
О 00 Изобретение относится к новому биологически активному соединению аналогу энкефалина, обладающему анальгетическим действием,которое может найти применение в медицине. .Природные энкефалины - метионин и лейцин Н Tyr-Gly-Gly-Phe ОН и Н Tyr-Gly-Gly-Phe-Leu ОН, соответственно проявляют морфиноподобную активность в опытах на специфических моделях оциатного рецептора и подавляют стереоспецифическое связывание с рецептором опиатного антагониста налоксона в гомогенатах мозга, Однако эти энкефалины проявляют слабую и кратковременную (5-15 мин) анальгезию при внутрижелудочковом введении в мозг мыши и не активны при внутривенном перрорйльном и дру гих способах введения. Наиболее близким структурным аналогом описываемого соединения .яв ляется (D-Arg) - лейцин.энкефалин, обладающий анальгетической активностью при внутримозговом введении. При внутривенном введении этот аналог анальгезией не обладает. Цель изобретения - расширение арсенала средств воздействия на живой организм, обладающих анальгетическим Действием, Цель достигается предлагаемым аналогом энкефалина формулы Н Tyr-D Arg-Gly-Phe Ш обладающим анальгетическим действие Отесываемое соединение. - (D-Arg дез-Ьеи) - энкефалинамид синтезиро ван с испо.пьзованием известных мето дов пептидной химии по схеме с использованием в качестве конденсирую щего агента дициклогексилкарбодиими да, а также пентафторфениловых эфиров трет-бутилоксйкарбониламинокислот, Гуанйдиновая функция D-аргинина быпа защищена нитрогруппой, гидр ксия тирозина - бензильным радикало Карбоксильную группу глицина защища ли этерификацией, Вос-группы в синтезе пептида удаляли 50%-ным раство ром трифторуксусной кислоты (TFА) в хлористом метилене. Нитро- и бензил ную защиты снижали каталитическим гидрогенолизом. Общая схема синтеза приведена на чертеже. Для синтеза (D-Arg, дез-Lei энкефалинамида используют аминокислоты и их производные фирмы Reanal. Все аминокислоты, кроме D-аргинина имеют L-конфигурацию. Температуру плавления веществ определяют в капилляре (без коррекции). Индивидуальность промежуточных соедгшений контролируют с помощью тех на пластинках Silufol (ЧССР) в следующих системах растворителей: хлороформ - этанол уксусная КИСЛОТА АсОН), 85:10:5 (А), Н. бутанол - пиридин - АсОН - , 15:10:3:6 (В), н. бутанол - АсОН - HjO, 4:1:1 (С). Аналог энкефалина хроматографируют на пластинках фирмы Meric в системах: хлороформ - метанол - АсОН - HjO, 30:20:4:6 (D), н. бутанол - пиридин - АсОН - , 15:12:3:10 (Е). Удельное., оптическое вращение пептидов измеряют на поляриметре Pefkin-Elmer- 141 М C1IIA) . Кислотный гидролиз проводят в запаянных ампулах в 6 ц.соляной кислоте при в течение 24 ч. Аминокислотный состав определяют на анализаторе Jeol-3. Для всех соединений данные элементного анализа удовлетворительно совпадают с вычисленным содержанием С. Н, N. Этиловьй эфир ,ет-бутилоксикарбонил-Н -нитро-1)аргинил-глици- на (1). К раствору 6,38 г (20 мяоль) N -трет-бутилоксикарбонил-К -нитроD-аргинина и 3,24 г (24 ммоль) 1оксибензотриазрла в 20 мл д1Ю1етйпформамида (ДМФА) при охлаждении до -15°С прибавляют 4,12 г (20 ммоль) дициклогексш1карбоди1 ида (ДСС) .в 8 мл ддаА, перемешивают 3 ч при ОС, затем добавляют 3,07 г (22 ютоль) этилового глицина гидрохлорида, 3,08 мл (22 ммоль) триэтиламияа и перемешивают 24 ч при 2р°С. Реакционную массу разбавляют этилацетатом и водой (до разделения слоев). Этилацетатшлй слсий отделяют и промывают последовательно 5%-ным раствором KHSO, 5%-ным раствором NaHCO,, водой, насыщенным раствором NaCl, сушат над безводным . Растворитель отгоняют, остаток перекристал лизовывают из этилацетата. Выход 6,24 г (О (77%), т.пл. 119 С (размягчается при 96 С), М1 - 13,5 (с 1. ДМФА): ,68 (В). Этиловый эфир N-трет-бутилоксикapбoнил-o-бeнзил-L-тиpdзил-N -нитpo-D-аргинил-глицина. А,04 г(10ммол дипептида (1) растворяют в 15 мл 50%-ного раствора TFA в дихпорметане и выдерживают 30 мин. Раствор1итель отгоняют, остаток растирают с эфиром и сушат в вакуум-эксикаторе над КОН. Получают 4,01 г (96%) трифторадетата (11), ,48 (В). . 2 г (5 ммоль) трифторадетата (11) растворяют в 10 мл ДМФА, охлаждайт до ,и при перемешивании добавляют 0,6 мл (5 ммоль) триэтиламина в 2 мл ДМФА и 2,46 г (5 ммоль) н-нитро фенилового эфира N-трет-бутилоксикарбонйл-о-бензшт-Ъ-тирозина, растворенного в 10 мл ЛМФА. Смесь выдер живают 1 ч при -5 С, 24 ч при 0°С и еще 24 ч при комнатной температуре. Затем реакционную массу выливают в 500 мл воды со льдом, выделивше еся- в осадок вещество отфильтровываю Осадок растворяют в этилацетате и . промывают/ 5%-ным раствором KHSQ, водой,5%-ным раствором KHCO, водой, насыщенным раствором NaCl и высзшивают над безводным Na 804. Растворитель отгоняют, продукт этиловый эфир Н-бутилоксикарбонил-о-бензил-Ьтирозил-к -нитро-В-аргинил-глицина(III) пёрекристаллизовывают из Этилацетата. Вызсод t,77 г (54%) трипептида (III), Т.Ш1. 162-164®С,,1° . (с 1, даФА); R.|.0,65 (А). К-трет-бутилоксикарбонил-о-бензщ1 Ь-тирозил-Ы нитро-О-аргинил-гли.Щ1н (IV), 9,66 г (1 ммоль) трип птида (III) растворяют в 15 мл метанола прибавляют 1 и. раствор NaOH до рН 10-11 и выдерживают несколько минут до окончания гидролиза (хроматографический контроль). Отгоняют растворитель, добавляют воды и подкисляют 1 н. HG1 до рН 3. Выпашший осадок отфильтровывают и хроматографируют на колонке с силикагелем в системе хлороформ-этанол-этилацетатAeOH-Hj 0,85:5:8:2:0,25. Получают 0,34 г (54%) т рипептида (IV), т.пл. ,,3° (с. 1, ДМФА) 5 ,60 (С). . . . / Ы-трёт-бутилок:сикарбонш1-о-бензил Ь-тирозил-Ы -.нитро-В-аргинил-глицилL-фенилаланинамид. 3,15 г (5 ммоль) трипептида (IV) растворяют в 10 мл этилацетата, охлаждают до О С и при 03 .4 перемешивании добавляют 1,01 г (5,5 ммоль) пентафторфенола и 1,13 г (5,5 ммоль) лес, растворенных в этилацетате. Через 2 ч смесь этилацетате, охлаждают до -10 С, вьтавшую дициклогексилмочевину отфильтровывают, растворитель отгоняют, маслообразный остаток растирают с гексаном до затвердения. Получают 3,54 г пентафторфенилового эфира (V); ,90 (С). 0,89 г (4,45 ммоль) гидрохлррида L-фенилаланинамида (VI) растворяют в 6 мл ДМФА и при перемешивании добавляют 0,62 мл (4,45,ммоль) триэтиламина в 2 мл ДМФА и 3,54 г (4,45 ммоль) пентафторфенштового эфира (V), растворенного в 4 мп ДМФА. Реакционную массу перемешивают несколько часов, ДМФА отгоняют при 40 С, остаток растворяют в этилаце- тате и обрабатывают аналогично соединению (III). Продукт К-трет-бутилoкcикapбoнил-o-бeнзил-L-тиpoзил-N нитро-В-аргинил-глицил-Ъ-фенилаланинамид (VII), полученный после отгонки растворителя, растирают с теплым этилацетатом, отфильтровывают и сушат в вакуум-эксикаторе. Выход 1,44 г (42%) защищенного амида тетрапептида (VII), т.пл. 173175°С, -6,3°(с.1, ДМФА), ,43 (А), ,74 (В). Ъ-тирозил-В-аргинил-глицил-Ь-фенилаланинамид. 0,5 г (0,64 ммоль) тетрапептида (VII) растворяют в 7 мл 50%-ного раствора TFA в дихлорметане и выдерживают в течение 30 мин. Раствор упаривают, остаток растирают с эфиР° сушат в вакуум-эксикаторе над КОН. Получают 0,46 г трифторацетата (VIII); ,85 (Д). К раствору 0,46 г (0,58 ммоль) соединения (VIII) в 5 мл метанола добавляют палладиевую чернь, 0,2 мл уксусной кислоты и гидрируют 6-ч при 20 С. Катализатор отфильтровывают, фильтрат упаривают досуха, остаток растирают с эфиром, отфильтровывают, промывают эфиром на фильтре, сушат над КОН Ь-тирозил-В-аргинилглицил-2-фенилаланин (IX) в вакууме. Полученный продукт (0,42 г) очищают на КМ-целлюлозе. Соответствующую фракцию собирают и лиофилизуют. Выход 0,20 г (57%) амида тетрапептида энкефалина (IX),uilD +23,7° бс 1, метанол), +43,3 (с,1. 0,2 н АсОН); ,75 (Д), ,62 (Е аминокислотньй состав: Туг 0,87; Phe 1,04; Gly 1,00; Arg 0,97. Биологическая активность (D-Arg, двз-Leu)-энкефаЛИНамида исследована в опытах in vivo. В работе использованы беспорбдньш мьпни-самцы массой 20 г. Описываемое вещество, растворенное в стерильном физиологическом растворе, вводят при помощи 1-образной иглы в cisterna magna мозга неанестезнррванньм мьшам в количестве 10 мкл. Исследован диапазон доз: t,0 - too,О мкг/жив. Контрольным животным вводят to мкл стерильного физиологического раствора, ,Каждая акспериментальная группа состоит из to мышей. Анальгетичёский эффект соединения оценивают по тесту tail pinch при помощи артериального зажима, который накладывают на основание хвоста. Определение болевой реакции проводят через 5, 15, 30, 60,90 мин после введения и затем каждые 30 мин до исчезновения анальгетической реакции. Результаты выражают альтернативным методом в проценте мьнвей, пока 3 авших ан аль ге тическую реакцию. При вычислении ЕД (эффективная доза исследуемого вещества, которая вьГзывает анальгетический эффект у 50% подопытных живот ных) используют метод Литчфилда и Уилкоксойа. Анальгетическая активность исследованных соединений приве дены в табл. 1. (D-Arg, дез-Leu)-энкефалинамид проявляет значительный и пррдолжительньй анальгетическнй эф|1ект. Максимальный эффект наблюдают на 530 мин после введения. Анальгетическая активность предл гаемого аналога энкефалина в 2 раза превышает анальгетическую активност известного аналога энкефалина(D-Arg -лейцин-энкефалина на молярной основе (табл. 1). Кроме того, описываемый аналог энкефалина в отличие от своего структурного прототипа активен при внутривенном введении(ЕД 28,0, 17,9-АЗ,7 мг/кг). Характерной особенностью описанного аналога является значительная, пролонгация анальгетического эффекта. Продолжительность его анальгезии составляет 4 ч против нескольких минут (5-15) природных энкефалинов. Анальгетическая активность природных эн :ёфалинов и сопоставила по величине к продолжительности с анальгетической активностью морфина (табл. 1). Таблица 1 223(160-310) Лейцин-энкефалин 146( 99-215) Метионин-энкефалин (D-Arg)-лейцин-эн5,3(2,9-9,3) кефалин (D-Arg, дез-Leu )2,5(0,9-7,2) энкефалинамид 1,8(0,5-5,6) Морфин В табл.. 2 представлен анальгетический эффект описываемого (D-Arg, дез-Leu)-энкефалинамида, полученный в опытах на мьшах при интерацистернальном введении в дозах 1-100 мкг/животных. 91 Эффективная доза соединения () вычисленная на основании данных таблиц, составляет 2,5 нмоль/жив, или 1,7 мкг/жив. Из данных табл. 2 видно чт;о токсические признаки соединения начинают проявляться в дозах, -йревышающих эффективную в 59 раз ( . ). следовательно. 1,7 мкг/жив, терапевтический индекс соединения /JlfljQ /ЕДуо/ больше 59 и является достаточно безЬпасным интервалом для лекарственного средства. Интервал между дозой, вызывающей минимальный анальгетический эффект. 3 и дозой, вызывающей первые признаки токсичности (терапевтическая широта), также достаточно велик. Он составляет более сотни , 100 мкг/жив. ) и является безопас1,О мкг/жив. ным для лекарственного средства. Результаты биологических испытаНИИ описываемого {B-Arg, дез-Leu )энкефалинамида показали, что предлагаемое соединение малотс ксично и обладает ярко выраженным анальгетическим действием при интрацистернальном введении.



Аналог энкафалина Tyr-D Arg-Gly-Phe NH , обладающий анальгетическим действием. i he-NH-i Gly
