Изобретение относится к новому биологически активному соединениюциклическому аналогу энкефалина, обладающему анальгетической активностью, которое может найти примейение в медицине.
Известны природные аналоги энкефалина: метионин-энкефалин (H-Tyr-GlyGly-Phe-Met ОН) и лейцин-энкефалнн (H-Tyr-Gly-Gly-Phe-Leu-OH), проявляющие морфиноподобную активность в опытах на специфических моделях опиатного рецептора и подавляющие стереоспецифическое связьгаание с рецептором опиатного антагониста налоксона в гомогенатах мозга. Однако и метионин и лейцин-энкеф лины проявляют слабую и кратковременную (5-15 мин) анальгезию при внутрижелудочковом введении в мозг мыши и неактивны лри других сп собак введения. Известны циклические аналоги энкефалина например, -Tyr-Gly-Gly-Phe Leu), не проявляющие специфической активности энкефалинов. Наиболее близким по химической структуреявляется циклический аналог энкефапина Tyr-D-A,, y-Phe,Leu.. однако анальгетическая активность этого циклоаналога не исел дована.
tyr
РЬе
Cly 8 Цель изобретения - расширение арсенала средств воздействия на живой организм. Поставленная цель достигается описьшаемым циклическим аналогом энкефалина формулы H-Tyr-D-Orn-Gly-Pheобладающим анальгетической активностью. Описываемое соединение синтезировано по приведенной схеме ступенчатым наращиванием пептидной цепи с использованием активированных эфиром трет-бутилоксикарбониламинокислот.. Для защиты о -амйнофункдии D-оонитина используют .бензилоксикарбонильную группу гидроксила тирозина - трет-бутилоксикарбонилвнзпо .группу.
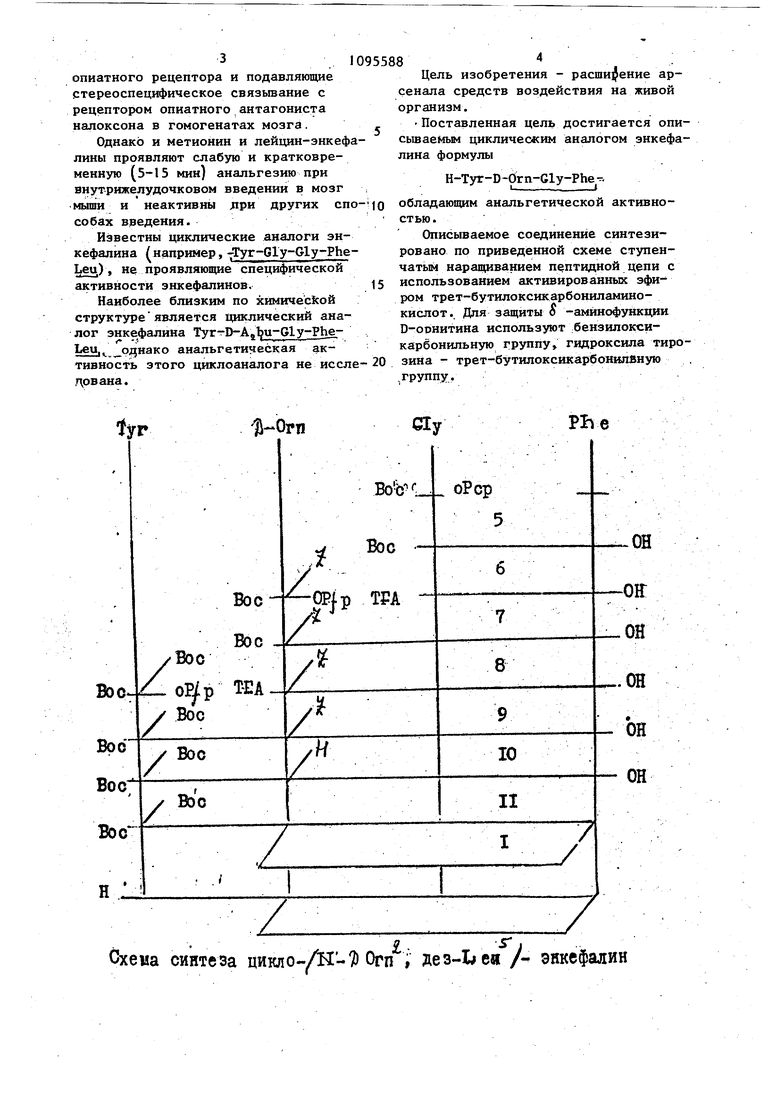




Циклический аналог энкефалина формулы Н-Туг-Р -prn-Gly-Phe обладающий анальгетической активностью..
Схема синтеза цикпо- - v7) Огп i дез-Ьеи / энкефалин П р им е р. Для Синтеза описываемого аналога энкефалина используют аминокислоты и их производные фирмы Reanal (Венгрия). Все амино кислоты, кроме D-орнитина, имеют L-конфигурацию. Температуру плавления веществ определяют в капилляре . (без коррекции). Индивидуальность промежуточных соединений контролируют с помощью тех на пластинках Silufо1 (ЧССР), аналог энкефалина хроматографируют на Стеклянных плас тинках с закрепленным слоем силикагеля Silika Gel 60 F-254 фирмы Merck (ФРГ). Исйользуют следующие системы рас ворителей: хлороформ-этанол - уксусная кислота (АсОН), 85:10:5 (А), н-бутанол-пиридин - АсОН - Н 15: :10:3:6 (В)j н-бутанол-АсОН-НзО, 4:1:1 (С), хлороформ - метанол АсОН - , 30:20:4,6 (fl)j 60:18:2: (Е), н-бутанол - пиридин-АсОН-Н О 15:12:3:10 (Е). Удельное оптическое вращение пептидов измеряют на поляриметре Perkin Elmer 14/М (США). Ки лотный гидролиз проводят при 120 С в течение 24 ч. Аминокислотный состав определяют .на анализаторе Jeol-3 Для всех соединений данные элементного анализа удовлетворительно совпадают с вычисленным содержанием Спектры ЯМР -н описываемого ана лога энкефалина получены на спектро;метре Ш-360 в растворе при рН температуре 23°С с внутренним стандартом DSS. Молекулярный вес описываемого цик лопептида определяют масс-спектромет ;рически наприборе СН-5 VarianMAT .(США)-при температуре приблизительно ; 250 С и ионизирующем напряжении 70 Э N-трет-бутилоксикарбонил-глицил-фенйлаланин (5). 6,6 г (40 ммоль) L-фенилаланина jрастворяют в 40 мл 1 н.раствора гидроокиси натрия, добавляют 3,34 г бикарбоната натрия, 50 мл диметилформамида (ДМФА) и 16,94 г (40ммоль) пентахлорфенилового эфира трет-бутилоксикарбонил-глицина, растворен.ного в 20 мл ДМФА. Смесь перемешивают в течение нескольких часов. После завершения реакции(хроматографический контроль) реакционную массу разбавляют этилацетатом и водой (до разделения слоев), охлаждают до О С и подкисляют 0,5 н. соляной кисло- той до рН 3. Этилацетатный слой отделяют, водную фазу экстрагируют повторно. Объединенный этилацетатный слой промывают водой, насыщенным раствором хлористого натрия и сушат над безводным сульфатом натрия. Кристаллический осадок, полученный после отгонки растворителя, перекристагшизовывают из этилацетата с небольшой добавкой петролейного эфира. Выход дипептвда (5) - 11,4 (86%), т.пл. 144-135С, oi +15,7° (с.1, ДМФА), Rf 0,73 (А); Rf.0,80 (Д) N -трет-бутилоксикарбонил, N -бензилоксикарбонил-О-орнитилглидил- , -фенилаланин (7); 10,9 г (33,8 ммоль) дипептида (5)i растворяют в 50 мл 70%-ного раствора трифторуксусной кислоты (TFA) в дихлорметане и выдерживают 30 мин. Растворитель отгоняют, остаток растирают эфиром, отфильтровьшают, промывают эфиром на фильтре и сушат в вакууме над гидроокисью калия. Получают 10,4 г (91%) трифторацетата дипептида 6) 0,63 (Д). 5,2 (15,5 ммоль) трифторацета- , та (Ь) растворяют в 50 мл ДМФА, охлаждают, до 0С и при охлаждении добавляют 2,65 мл (15,5 ммоль)диизопропилэтиламина в 15 мл ДМФА и 8,2 г (15,5 ммоль) пентафторфенияового эфира К -трет-бутилоксикарбонил, N бензилоксикарбонил-В-орнитина, растворенного в 15 мл ДМФА. Реакци-онную массу перемешивают несколько часов и после завершения р еакции обрабатывают аналогично соедине - . нию (5). Продукт, полученный после отгонки растворителя, очищают на колонке с силикагелем фирмы Merck.Элюирование проводят в системе: хлороформ - этанол - этилацетат - АсОН 285:5:8:2:0,25. Соответствующие фракции собирают, упаривают и сушат в вакууме. Получают 5,9 г (67%) масла защищенного трипептида (7)с Rf 0,65 (А), Rf 0,72 (В). 0-ди-трет-бутилоксикарбонил-тирозил Ы -бензилоксикарбонш1-В-орнитил-глицил-фенилаланин (9). 20,0 г (35 ммоль). трипептида (7) растворяют в 100 мл 50%-ного раствора трифторуксусной кислоты в дихлорметане и выдерживают в течение 20 мин. Растворитель отгоняют, остаток растирают с эфиром. Образовавшийся осадок отфильтровьюают, вновь промьшают эфиром и высушивают в вакууме над гидроокисью калия. По лучают 19,2 (94%) трифторадеЧ-ата трипептида (8) с Rf 0,62 (Д). 18,1 г (31 ммоль) трифторадетата трипептида/ (8) р астворяют в 100 мл ДМФА, охлаждают до , добавляют при перемешивании 5,3 мл (31 ммоль) диизопропилэтиламина в 10 мл ДМФА и 16,6 г (31 ммоль) пентафторфенклового эфира N,0-ди-третбутилоксикарбонил-Ъ-тирозина. Реакционкую массу перемешивают 1,5-2 ч (хроматографический контроль) и после завершения реакции обрабатывают аналогично соединению (5). Поо дукт, полученный после отгонки этил ацетата, размешивают с эфиром и охлаждают. Выпавший осадок отфильтровывают, промывают эфиром и сушат в эксикаторе. Выход: 16,0 г (63%) с Rf 0,75 (А); Rf. 0,55 (В) i i/D +5,6 (с 1, ДМФА). Н,0-ди-трет-бутилоксикарбонил-тирозил.-цикло(К - Д-орнитилглицил-фенил аланид-) (II). К 1,8 г (2,2 ммоль) защищенного тетрапептида (9) в 50 мл метанола, 5 мл АсОН и 2 мл добавляют пал лодиевую чернь и гидрируют в течение нескольких часов. Катализатор о фильтровьюают, фильтрат упаривают. Остаток растирают с эфиром, затем дважды переосаждают из спирта эфиро вновь промьшают эфиром и сушат в ва кууме над гидроокисью калия. Получа 1,2 г (81%) тетрапептида (10)-с Rf 0,83 (Д), Rf 0,77 (I). 500 мг (0,7 ммоль) тетрапептида (10) растворяют в 500 мл очищенного .ДМФА, охлаждают до -20°С, прибав ляют триэтиламин до рН 7,2 и медлен но прикапьшают 0,45 мл (2,1 ммоль) дифенилфосфорилазида в 50 мл ДМФА. Раствор перемешивают в течение 7 дней и поддерживают рН 7 периодическим добавлением триэтиламина к реакционной массе. По завершении реакции растворитель отгоняют при 40 к остатку добавляют 1-1,5 мл этилацетата и небольшое количество эфира. Выпавший осадок отфильтровьгоают прокатают эфиром, сушат в вакууме. лучают 355 мг белого порошка защищенного циклического тетрапептида (II) Ввиду плохой растворимости . (II) очистку не проводят. , Тирозил-цикло(Ы -В-орнитил-глицил-фенйлаланил-) 350 мг (0,5 ммоль) защищенного циклического тетрапептида (II) растворяют в 5 мл 70%-ного TFA в хлористом метилене и выдерживают 30 мин. Растворитель отгоняют, остаток растирают с эфиром и сушат в вакууме. Полученный продзост очищают высокоэффективной жидкостной хроматографией на хроматографе Du Pont-830. Элюирование осуществляют смесью: этанол 0,1 М раствор ацетата аммония, 20:80. Соответствующие фракции собирают и лиофилируют., , I Выход 70 мл (28%) циклотетрапептида энкефалина (1), (с 1; 0,2 Н.АСОН); Rf 0,82 (D), Rf 0,83 (F), аминокислотный состав: Gly l,05i Phe 0,98, Туг 0,92; Orn 1,07 EHis 0,72 (1,0 M раствор/AcOH, 950 в). При изменении f)H от 5 до 2наблюдается отсутс рвие изменения химического сдвига Н Phe, что указывает на отсутствие свободной карбоксильной группы у тетрапептида, а следовательно, подтверждает циклическое строение. Во-вторых, большая неэквивалент- ; ность J -протонов Orn (iS .«0,45м.д.) также подтверждает циклическое строение полученного соединения. Величина расчетного молекулярного веса совпадает с его экспериментальным значением,,определенным масс- . спектрометрически (М 481). Исследована биологическая активность описьшаемого циклического аналога энкефалина в опытах in vivo. Анальгетическую активность определяют по методу H.Ueda.. В работе использованы беспородные мыши-самцы массой , 18-22 г. Исследуемое вещество, растворенное в стерильном физиологическом растворе, вводят при помощи У-образной иглы в usterna magna мозга неанестезированным мьш1ам в количестве 10 мкл. Исследовался диапазон, доз от 0,075 до 1,0 мкг/жив. Каждая экспериментальная группа состояла из 8-10 мьшгей. Анальгетический эффект оценивают по методу tail pineh при помощи зажима, накладываемого на основание хвоста. Определение болевой реакции проводят через 5,15,30,60, 90 мин после введения. Результаты выражены альтернативным методом в проценте мышей, показавших анальгети ческую реакцию. При вычислении Efljj, (эффективная доза исследуемого вещества, которая вызывает анальгетиче.скйй эффект у 50% подопытных животных) используют метод Литчфилда и :Уилкоксона. : Анальгетическая активность- иссле дуемых соединений приведена в табл. и 3. Как видно из табл.2 описьгеаемый цикло-(К -D Огп, дез-Leu) энкефалин обладает выраженной анальгетической активиостьк). Максимальный эффект наблюдается на 5 минуте после введения. Анальгетическая активнрсть описываемого циклического аналога энкефалина (1) значительно превьппает активность природных метионин и лейцин-энкефалйнов ив 7,5 раз превьпнаёт анальгезию йорфина яа молярной основе (см.табл.2)« Более того, описываемьй циклический аналог энкефалина в отличие от природных энкефалинов активен при
Химические сдвиги Н циклического тетрапептида
С1у,
4,1,1; 3,40
4,58 3,п; 2,96 Phe
3,24; 2,79
7,27-7,30 внутривенном введении. Его активность (ЕДдд 32 (20-52) ммоль/кг) -. составляет более половины (56% активности морфина (ЕД g.jj 18 (15-21) ммоль/кг)(см.табл.З). Продолжительность;анальгетической реакции описываемого соединения составляет 60 мин против 15 мин у природных энкефалинов (2,3) при интрацистернальном введении (см.табл.2) и равна продолжительности действия морфина при обоих способах введения (см.табл.2,3). Ведение описываемого соединения в дозе 200 мг/кг не вызывает токси- ческих эффектов. Учитывая, что эффективная доза описываемого аналога э.нкефалина - 32 мкмол/кг, что составляет 20 мг/кг, рассчитан терапевти200 мг/кг .ei. ческий индекс: 200 мг/кг описьюаемое соединение обладает достаточно безопасным интервалом для терапевтического действия. Результаты проведенных биологических испытаний позволяют сделать вывод о возможности использования предлагаемого соединения в медицине. Таблица 1
| Hughes J., Smith Т | |||
| W.,, Koster,litz H.W | |||
| ,Forthergill L | |||
| A., Morgan В | |||
| A., Morris H.R | |||
| Identification related pentafeptides from the Groin with potent Opiate agonist activity Nature, 1975, V..253, 547 | |||
| Biiescher H.H., Hill R.C., Romer D., Cardinaux F.,Closse A., Nauser D., Pless I | |||
| Evideme for -anailgesic activity of enkephalin in phe mouse | |||
| Планшайба для точной расточки лекал и выработок | 1922 |
|
SU1976A1 |
| Одновальный, снабженный дробителем, торфяной пресс | 1919 |
|
SU261A1 |
| Hudson D., Sharf e ..R., Szelke M., Methionins enkephalin and isosteric analogues Int.J | |||
| Peptide Protein Res | |||
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
| Kessler H., Holzemann G., CycloLeu -Enkephaiip Angew Chem | |||
| Int | |||
| Ed | |||
| Engl., 1981, 20, № 1, 124 | |||
| Di MaioJ., Schiller P.W | |||
| A cyclic enkephalin analog with high in vitro opiate activity | |||
| Proc | |||
| Natl.Sei | |||
| USA, 1980,:77, № 12, 7162-7166. | |||
