Изобр(тение относится к аналитической химии, в частности к методам идентификации и определения нано- и микрограммовых ко:1ичеств (или содержаний) веществ с совместным применением новой комбинации двух ана 1итических методов: тонкослойной хроматографии (ТСХ) и рентгеновской флуоресценции (РФ), и может найти широкое применение при анализе состава природных и промышленных микрообъектов и малых количеств вешеств (например, минералов, микровключений, материалов радиоэлектроники, полупроводниковой техники), а также при исследовании механизма сорбции, комплексообразования, поведения разновалентных и разных по составу форм элементов.
Известны различные способы инструментального определения и идентификации химических соединений в зонах тонкослойных хроматограмм, например, денситометрированием зон окрашенных соединений 1 , флуориметрическим сканированием зон флуоресцирующих соединений 2, радиометрическим сканированием пятен радиоактивных веществ 3.
Однако указанные способы либо недостаточно селективны, либо имеют ограниченную область применения. Так, например, денситометрический способ без дополнительных манипуляций применим только в случае анализа окрашенных соединений, а при анализе неокрашенных веществ необходимо предварительно опрыскивать хроматограмму раствором реагента, дающим с определяемым ионом цветную реакцию. Если эта реакция недостаточно селективна, то не удается определить на хроматограмме неполностью разделенные элементы. Недостаточно чувствительная цветная реакция понижает чувствительность определения веществ при денситометрическом сканировании. Радиометрическое сканирование, которое применяется, например, в случае использования радиоактивных индикаторов, требует предварительного активирования анализируемой пробы или ее «метки, что обычно достаточно сложно.
Наиболее близким к изобретению является способ идентификации и определения содержаний химических соединений в веществе, заключающийся в разделении анализируемого вещества на компоненты методом тонкослойной хроматографии, облучении хроматограммы электромагнитным излучением и регистрации интенсивности флуоресценции определяемых компонентов, по которым судят о их содержаниях. Хроматограмму облучают ультрафиолетовым излучением и измеряют либо интенсивности возникающей в зоне флуоресценции (хроматограмма предварительно обработана флуресцентным реагентом), либо гашение определяемым веществом флуоресценции подложки, когда
последняя содержит ультрафиолетовый индикатор 4.
Однако согласно известному способу при измерении интенсивности флуоресценции можно определять только ограниченный круг соединений, обладающих ультрафиолетовой лю.минесценцией, либо предварительно путем химической модификации необходимо переводить нефлуоресдирующее вещество во флуоресцирующее введением в его состав флуоресцирующих групп. При тущении необходимо предварительно вводить в сорбционный слой флуоресцирующий индикатор, что усложняет технику работы, а также может отрицательно влиять на хроматографическое разделение определяемых веществ, изменяя свойства используемого сорбента.
Кроме того, для избирательного определения элементов необходимо четкое их разделение на хроматограммах. В случае более размытых хроматограмм и перекрывающихся зон метод не обладает избирательностью.
Целью изобретения является повышение избирательности и расщирение области применения способа.
Поставленная цель достигается тем, что согласно способу идентификации и определения содержаний химических соединений в веществе, заключающемуся в разделении анализируемого вещества на компоненты методом тонкослойной хроматографии, облучении хроматограммы электромагнитным излучением и регистрации интенсивности флуоресценции определяемых компонентов, по которым судят о их содержании, на хроматографическую пластинку в точку перед стартовой линией на расстоянии 3-5 мм от нее наносят известное количество вещества, служащего внутренним стандартом, в качестве которого используют химическое соединение, характеризующееся параметром удерживания, не превышающим 0,01, сканируют сорбционный слой с разделенными компонен тами по прямой перемещения компонентов, определяемой подвижной фазой, облучая его коллимированным рентгеновским излучением и регистрируя характеристическое рентгеновское излучение одного из элементов, входящих в определяемый компонент.
Кроме того, ширину пучка рентгеновского излучения выбирают не более 0,2 ширины зоны определяемого компонента, характеризующегося на хроматограмме наиболее узкой зоной, и производят интегрирование измеряемого сигнала с учетом формы зоны хроматограммы.
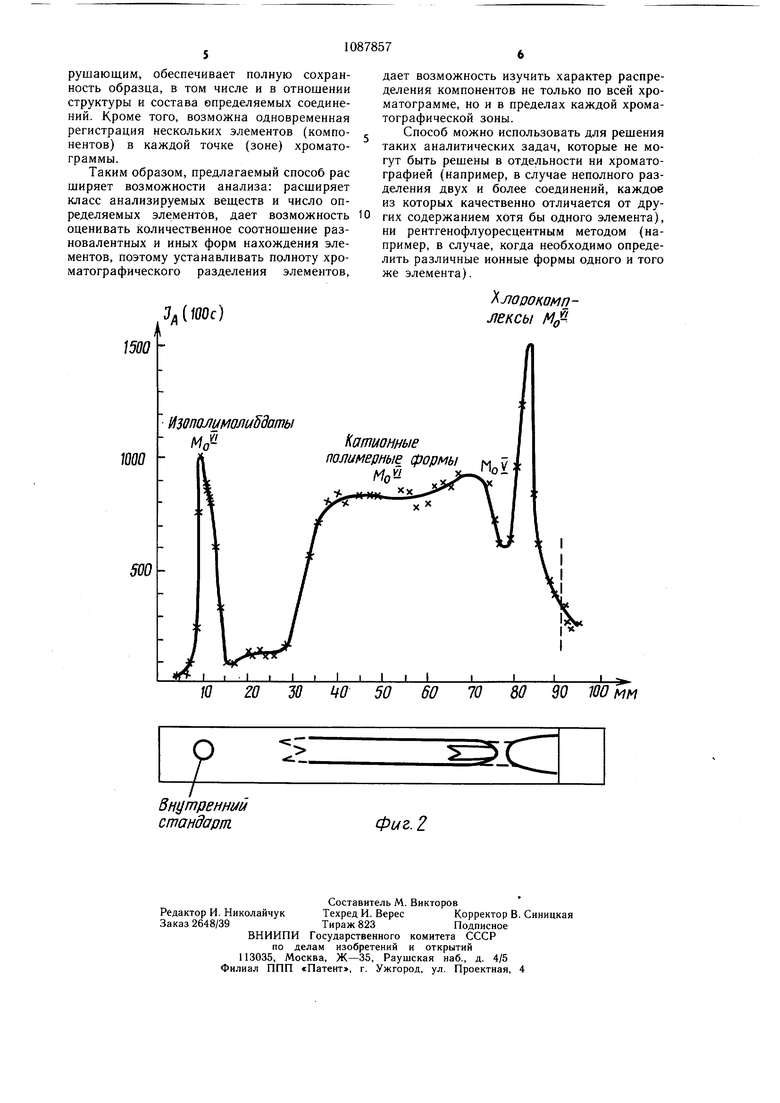
В зависимости от содержания элемента в зоне сканирование хроматограммы может проводиться автоматически путем ее перемещения под рентгеновским зондом с одновременной записью интенсивности аналитической линии на диаграмме самопишущего потенциометра или (при малом содержании) путем установки отдельных «точек хроматограммы под зонд и регистрации интенсивности за определенный интервал времени (обычно 10-100 с) для накопления сигнала, обеспечивающего требуемую точность. Объектный столик с микроприводом обеспечивает установку образца с погрешностью не более 0,01 мм, причем положение и перемещение образца может контролироваться визуально с помощью встроенного в прибор микроскопа. Это позволяет детально изучить распределение элементов по всей поверхности хроматограммы. В соответствии с требуемым пространственным разрешением, обусловленным размерами хроматографических зон, рентгеновский зонд коллимируется диафрагмой диаметром 0,1 - 1 мм. В данном случае размеры зон на хроматограммах (в частности, никеля и кобальта) составляют 10-20 мм (в продольном направлении) и 2-3 мм (в поперечном направлении) поэтому зонд диаметром 1 мм является оптимальным. Для количественной и качественной оценки полученный плоскостной хроматограммы важным является использование метода внутреннего стандарта. Однако традиционный метод внутреннего стандарта, при котором стандартное вещество вводится в анализируемую пробу перед анализом, не может быть использован, так как стандарт может (особенно при анализе неорганических соединений) реагировать с растворителем или компонентами анализируемой смеси, а зона стандарта может налагаться на зоны анализируемых соединений. Для преодоления этих затруднений в общем случае необходимо провести специальное исследование, равное или превышающее по сложности рещаемую задачу. Кроме того, метод внутреннего стандарта должен был в данном случае учитывать требования как тонкослойной хроматографии, так и ренгенофлюоресцентного анализа. Предлагаемый метод внутреннего стандарта заключается в том, что раствор с внутренним стандартом необходимо наносить на расстоянии 3-5 мм до стартовой линии. Кроме того, необходимо, чтобы стандартное вещество (вещества) практически не перемещалось в ходе хроматографического разделения на слое, т.е. чтобы параметр удерживания стандарта (Rf) не был больше 0,01. В противном -случае возможно перекрывание зон определяемых веществ зоной стандарта. Предлагаемым способом могут исследоваться плоскостные хроматограммы на любых подложках (стекло, полимерная пленка, алюминиевая фольга). Подложка не оказывает влияния на результаты анализа, так как при определении концентрации элементов по рентгеновским линиям учитывается фон от сорбционного слоя и подложки. Апробирование способа проведено на флуоресцентном микроанализаторе. Пример 1. Исследование возможности раздельного определения микрограммовых содержаний никеля и кобальта. На фиг. 1 представлены кривые распределения никеля и кобальта после их хроматографического разделения из растворов их хлоридов при содержании каждого из элементов по 1 мкг, а также схематическое изображение хроматограммы в масштабе записи концентрационных кривых и интенсивности сигнала от внутреннего стандарта (1-10 г кобальта и 1-10 г никеля), нанесенного на расстоянии 4 мм от стартового пятна. Кривые сняты по аналитическим линиям кобальта (СоК«) и никеля ( при облучении хроматограммы рентгеновским излучением трубки с вольфрамовым анодом (диаметр коллимирующей диафрагмы 1 им). Эти кривые иллюстрируют избирательность метода, его разрещающую способность и чувствительность. При, суммарном содержании нанесенных на слой элементов 1 мкг определяются их нанограммовые количества в отдельных зонах хроматограмм. Пример 2. Исследование возможности определения различных ионных форм молибдена, разделенных предварительно хроматографически при исследовании солянокислых растворов. Общая концентрация молибдена в растворе . Наносят на старт микролитровые объемы исследуемого раствора. На фиг. 2 показано распределение молибдена, снятое по интенсивности аналитической линии молибдена MoKot, в сорбционном слое (вдоль направления движения потока подвижной фазы) с разрешением 1 мм. Это распределение выявляет различные ионные формы молибдена, которые можно идентифицировать как изополимолибдаты молибдена (IV) в нижней зоне, катионные полимерные формы молибдена (V и VI) в центре хроматограммы и хлорокомплекса молибден (VI) у фронта растворителя. На фиг. 2 приведено также схематическое изображение хроматограммы. Зона изополимолибдатов молибден (Vi) имеющих в выбранных условиях R О, используется в качестве внутреннего стандарта. При использовании изобретения объектами анализа могут быть вещества как неорганической, так и органической природы, в том числе и элементоорганические соединения, при этом в принципе возможно определение широкого круга элементов с атомным номером от 3 до 105. Способ характеризуется полной избирательностью, так как каждый элемент может быть определен независимо от присутствия других элементов. Абсолютный предел обнаружения 10 -10 г. Локальность метода характеризуется возможным диаметром анализируемой зоны 0,1 -1,0 мм. Метод анализа является неразрушающим, обеспечивает полную сохранность образца, в том числе и в отношении структуры и состава определяемых соединений. Кроме того, возможна одновременная регистрация нескольких элементов (компонентов) в каждой точке (зоне) хроматограммы.
Таким образом, предлагаемый способ рас ширяет возможности анализа: расширяет класс анализируемых веществ и число определяемых элементов, дает возможность оценивать количественное соотношение разновалентных и иных форм нахождения элементов, поэтому устанавливать полноту хроматографического разделения элементов.
3fy(WOcl
то
H30noJ}UMonuSdambi М/
то
500
Ю 20 30 W 50 60 10 80 90 100 мм
дает возможность изучить характер распределения компонентов не только по всей хроматограмме, но и в пределах каждой хроматографической зоны.
Способ можно использовать для решения таких аналитических задач, которые не могут быть решены в отдельности ни хроматографией (например, в случае неполного разделения двух и более соединений, каждое из которых качественно отличается от других содержанием хотя бы одного элемента), ни рентгенофлуоресцентным методом (например, в случае, когда необходимо определить различные ионные формы одного и того же элемента).
У лоооммпжксы М

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СОСТАВА ГЛИФОСАТСОДЕРЖАЩИХ СМЕСЕЙ | 2022 |
|
RU2787117C1 |
| СПОСОБ ХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА В ЗАКРЫТОМ ТОНКОМ СЛОЕ СОРБЕНТА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2011 |
|
RU2494391C2 |
| СПОСОБ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ В ЗАКРЫТОМ ТОНКОМ СЛОЕ СОРБЕНТА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2011 |
|
RU2483303C2 |
| СПОСОБ РАЗДЕЛЕНИЯ И ОПРЕДЕЛЕНИЯ СОЕДИНЕНИЙ МЕТОДОМ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2008 |
|
RU2410680C2 |
| Способ определения патулина в пищевых продуктах | 1988 |
|
SU1585754A1 |
| СПОСОБ ХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА МНОГОКОМПОНЕНТНОГО ВЕЩЕСТВА | 2010 |
|
RU2439552C1 |
| СПОСОБ СПЕКТРАЛЬНОГО АНАЛИЗА ХИМИЧЕСКОГО СОСТАВА ВЕЩЕСТВА | 2006 |
|
RU2319137C1 |
| СПОСОБ ДЕТЕКТИРОВАНИЯ В ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ | 1976 |
|
SU671503A1 |
| Способ определения патулина в пищевых продуктах | 1982 |
|
SU1103146A1 |
| Способ определения кодеина в сложных лекарственных смесях | 1983 |
|
SU1133546A1 |
1. СПОСОБ ИДЕНТИФИКАЦИИ И ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЙ ХИМИЧЕСКИХ СОЕДИНЕНИЙ в веществе, заключающийся в разделении анализируемого вещества на компоненты методом тонкослойной хроматографии, облучении хроматограммы электромагнитным излучением и регистрации интенсивности флуоресценции определяемых компонентов, по которым судят о их содержаниях, отличающийся тем, что, с целью повышения избирательности и расширения области применения способа, на хроматографическую пластинку в точку перед стартовой линией на расстоянии 3- 5 мм от нее наносят известное количество вещества, служащего внутренним стандартом, в качестве которого используют химическое соединение, характеризующееся параметром удерживания, не превышающим 0,01, сканируют сорбционный слой с разделенными компонентами по прямой перемещения компонентов, определяемой подвижной фазой, облучая его коллимированным рентгеновским излучением и регистрируя характеристическое рентгеновское излучение одного из элементов, входящих в определяемый -компонент. 2. Способ по п. 1, отличающийся тем, что ширину пучка рентгеновского излучения выбирают не более 0,2 ширины зоны определяемого компонента, характеризующегося на хроматограмме наиболее узкой зоной, и производят интегрирование измеряемого сигнала с учетом формы зоны хроматограммы. S W к П 16 L,«M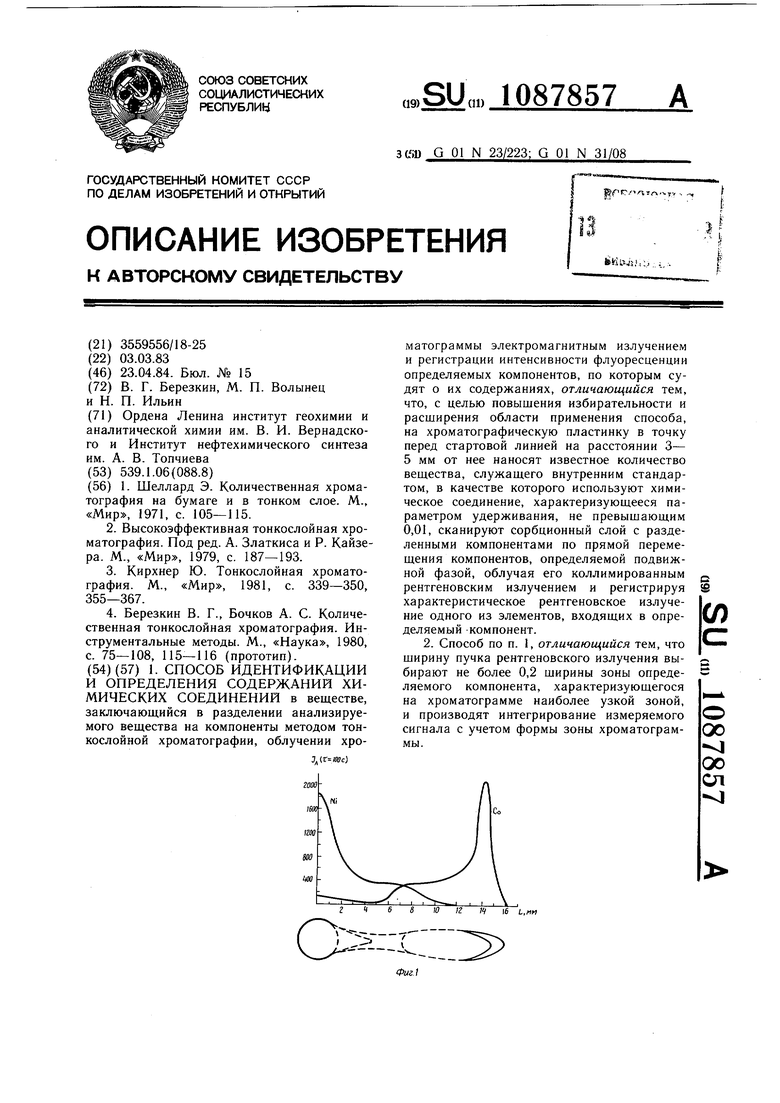
Внутренний стандарт.
Фиг. 2
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Шеллард Э | |||
| Количественная хроматография на бумаге и в тонком слое | |||
| М., «Мир, 1971, с | |||
| Транспортер для перевозки товарных вагонов по трамвайным путям | 1919 |
|
SU105A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Под ред | |||
| А | |||
| Златкиса и Р | |||
| Кайзе ра | |||
| М., «Мир, 1979, с | |||
| Индукционная катушка | 1920 |
|
SU187A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Тонкослойная хроматография | |||
| М., «Мир, 1981, с | |||
| Ручной ткацкий станок | 1922 |
|
SU339A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Г., Бочков А | |||
| С | |||
| Количественная тонкослойная хроматография | |||
| Инструментальные методы | |||
| М., «Наука, 1980, с | |||
| Фальцовая черепица | 0 |
|
SU75A1 |
