э 1 Изобретение относится к способам регенерации катализаторов, предстал ляющих собой пористый нерастворимый носитель с нанесенным на него метал лом или оксидом металла, или гидроо сидом металла, в частности алюмоникелевых катализаторов, применяе№1 /для конверсии углеводородов, и может быть использовано для очистки сбросных газов от оксидов азота в химической промьтленности. Известен способ извлечения цветных металлов, например никеля, путем обработки отработанного катализатора водным раствором азотной кис лоты в присутствии хлорид- и йодатионов СП. Недостатками такого способа явля ются использование товарных реактивов и сложность композиционной смеси. Наиболее близким по технической сущности и достигаемому результату К предлагаемому является способ извлечения цветных металлов из металлосодержащего катализатора путем об работки отработанного катализатора оксидами азота в присутствии воды. При этом обработку ведут газом, Содержащим 0,5% оксидов азота и |8% Кислорода при температуре окружающе среды в течение 18 ч .при скорости барботирования газа 3 л/мин. Одновременно с извлечением метал ла из катализатора имеет место очистка барботируемого газа от окси дов азота С2Д. Недостатком известного способа является недостаточно высокая степе извлечения никеля из-за малой скорости растворения металла, находящегося в порах носителя. Так, в течение 60 ч обработки катализатора - никеля на оксиде алюминия - степень, извлечения никеля составляет только 85%. Цель изобретения - повьшение сте пени -извлечения металлов. Указанная цель достигается способом извлечения цветных металлов и металлосодержащего катализатора путем обработки отработанного катал,изатора вначале карбамидом при 3D-140°Q, а затем оксидами азота в присутствии воды. 01 Предпочтительным является предварительная обработка раствором, содержащим 600-1150 г/л карбамида. Использование изобретения дает возможность получить следую1Щ1й положителы{ый эффе кт. Степень извлечения металлов увеличивается до 99,5% (против 85% по прототипу) для никелевого катализатора. Цля алюмопалладиевогоо катализатора она составляет 98,3% (против 75% для катализатора, не обработанного карбамидом у. Сущность способа заключается в следунж1ем. Газ, содержащий оксиды азота, например топочный газ со стадии прокалки в производстве алюмоникелевого катализатора, подают в слой отработанного пропитанного карбамидом алюмоникелевого катализатора, орошаемого водой, при этбм оксиды азота взаимодействуют с водой 2NQ2 + HjO HNOj + HNOj (О Образующаяся азотная кислота реагирует с никелем, присутствующим в катализаторе Ni + 2HNO Ni(NOj),2 + Hj (2) Образующаяся азотистая -кислота взаимодействует с карбамидом 00()2 + 2ШЮ2 2N2 + COj + + Выделякмциеся газы способствуют быстрому удалению азотнокислого никеля из пор, доступу свежих порций азотной кислоты к никелю, находящемуся в порах, а значит и более полному извлечению металла в раствор. Пример 1. Отработанный алюмоникелевый катализатор конверсии метана, в состав которого входят 92% А1г,0 и 8% , приготовленный в виде колец с наружным диаметром 12 мм, внутренним диаметром 5 мм и высотой 12 мм, в количестве 272 г предварительно обрабатывают при раствором, содержащим 800 г/л карбамида, и после охлаждения до комнатной температуры Загружают в сосуд типа Дрексель объемом 6,5 л. Туда же заливают 200 мл воды. Газ, содержащий 0,3 - 0,4% (N0 + N0,j) и 18% 0, барботйруют с расходом 2 л/мин через слой пропитанного карбамидом отработанного катализатора в поглотите.льном сосуде в течение 60 ч. Концентрация извлеченного азо нокислого никеля составляет 264,2 г а степень извлечения 99,5%. Пример 2. В отличие от при мера 1 в поглотительный сосуд загружают катализатор, обработанный п раствором, содержащим 600 г/л карбамида. Условия ведения процесса аналогичны условиям примера 1. Концентрация извлеченного в раствор азотнокислого никеля составляет 264,2 г/л, а степень извлечения 99,5%. Пример 3. В отличие от при мера I в поглотительный сосуд загру жают отработанный алюмоникелевый ка тализатор, обработанный при 140°С плавом карбамида. Условия ведения процесса аналогичны примеру I. Концентрация извлеченного азотнокислого никеля составляет 264,2 г/л, а степень извлечения 99,5%. Пример 4. В отличие от при мера I в поглотительный сосуд загружают катализатор, обработанный при 100°С раствором, содержащим 1150 г/л карбамида. Условия ведения процесса аналогичны условиям пр мера 1. Концентрация извлеченного азотнокислого никеля 264,2 г/л а степень извлечения 99,5%. В табл. I приведена зависимость степени извлечения никеля в раствор от продолжительности процесса в условиях прототипа и примера 1. Из анализа данных табл. 1 следует, что степень извлечения никеля при условии предварительной обработ ки катализатора карбамидом достигает 99,5%, т.е. на 14,5% вьше, чем в известном способе за время 5060 ч. В условиях прототипа для достижения высокой степени извлечения W потребуется большая продолжительность времени. Примеры 5-9. Отработанный алюмопапладиевый катализатор АПК-2, в состав которого входят 97, 7%, 2, 3% PdO,приготовленный в вице цилиндров с наружным диаметром и высотой 12 мм, в количестве 272 г пропитывают карбамидом в течение 15 мин. Условия пропитки и извлечения палладия аналогичны условиям примеров 1 - 4. В табл. 2 приведены данные по извлечению палладия из отработанног катализатора. 014 Примеры 10-14. OrpaOciтанны пинкосодержатий катапизлтор , в состав которого входят 15% Zn(CII)2 , и 85% активированного угля, обрабатывают карбамидом в описанных условиях (примеры 1 - 8). Данные по извлечению из отработанного катапизатора цинка приведешл в табл. 3. Примеры 15- 19. В отличие от примера 1 в поглотительный сосуд загружают отработанньп алюмоникелевый катгшизатор, содержащий 92% А120а|И 8% N , пропитанный карбамидом. Пропитку катапизатора и извлечение никеля проводят при тех же условиях, что и в примерах 1 - 3, 5 - 8 и 10 - 13. Данные по извлечению никеля представлены в табл. 4. В примерах 20-21 показано использование карбамида в концентрациях и при температурах, выходянщх за выбранные пределы. Пример 20. В отличие от примера I в поглотительный сосуд загружают катализатор, в состав которого входят 92% А120 и 8% Hi О, обработанный при в течение 15 мин раствором, содержащим 500 г/л карбамида. Условия ведения процесса аналогичны условиям примера 1. Концентрация извлеченного азотнокислого никеля 225,6 г/л, степень извлечения 85%, время изгшечения 60 ч. Пример 21. В отличие от примера 13 в поглотительный сосуд загружают 272 г отработанного цинксодержащего катализатора состава 15% Zn(OH)2 и 85% активированного угля, обработанного плавом карбамида в течение 15 мин при 150С. Условия ведения процесса извлечения цинка аналогичны условиям примера 13. Концентрация извлеченного азотнокислого цинка составляет 203 г/л, степень извлечения 99,6% время извлечения 60 ч. Раствор карбамида при содержании его в воде выше верхнего предела {1150 г/л), например 1200 г/л, представляет собой пересыщенный раствор. При таком содержании карбамида и повьш1енной температуре (вьше }30°С) обработка, по существу, осуществляется плавом карбамида. Как подтверждают примеры при обработке плавом
$
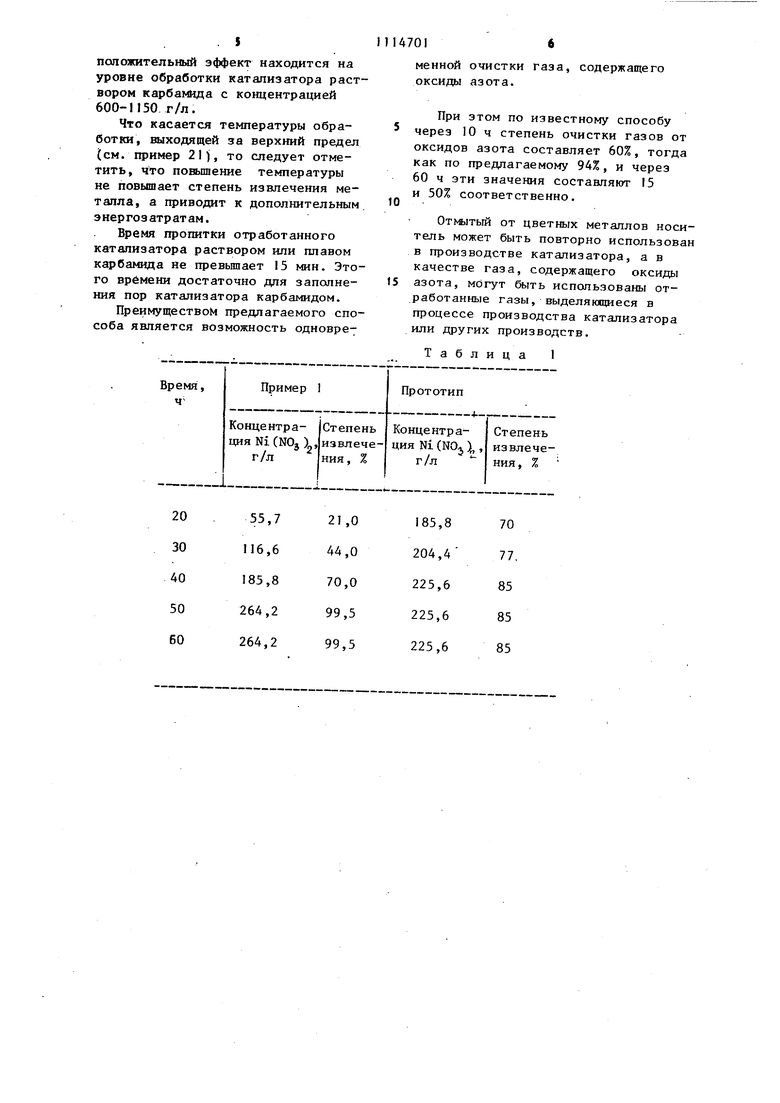
положительный эффект находится на уровне обработки катализатора раствором карбамида с концентрацией 600-1150 г/л.
Что касается TBNmepaTypbi обработки, выходящей за верхний предел (см. пример 21), то следует отметить, что повышение температуры не повышает степень извлечения металла, а приводит к дополнительным энергозатратам.
Время пропитки отработанного катапизатора раствором или плавом карбамида не превьшает 15 мин. Этого времени достаточно для заполнения пор катализатора карбамидом.
Преимуществом предлагаемого способа является возможность одновре)147016
менной очистки газа, содержащего оксиды азота.
При зтом по известному способу через 10 ч степень очистки газов от оксидов азота составляет 60%, тогда как по предлагаемому 94%, и через 60 ч эти значения составляют 15
и 50% соответственно. 10
Откытый от цветных металлов носитель может быть повторно использован в производстве катализатора, а в качестве газа, содержащего оксиды 15 азота, могут быть использованы отработанные газы, выделяющиеся в процессе производства катализатора или других производств.
Таблица 1

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ очистки газов от окислов азота | 1977 |
|
SU778752A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНОГО РАСТВОРА НИТРАТА НИКЕЛЯ | 1993 |
|
RU2100278C1 |
| Способ очистки газов от оксидов азота | 1982 |
|
SU1156720A1 |
| Способ получения концентрата драгоценных металлов из продуктов переработки руды и вторичного сырья | 2017 |
|
RU2673590C1 |
| Способ переработки отработанного никель-хромового катализатора | 1977 |
|
SU730849A1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ МЕТАЛЛИЧЕСКОГО ПАЛЛАДИЯ | 2023 |
|
RU2817811C1 |
| Способ очистки газа от сероводорода | 1980 |
|
SU929182A1 |
| СПОСОБ ЭКСТРАКЦИОННОГО АФФИНАЖА УРАНА | 2005 |
|
RU2295168C1 |
| СПОСОБ ОБРАБОТКИ ОТРАБОТАННОГО АБСОРБЦИОННОГО МАТЕРИАЛА НА ОСНОВЕ ОКСИДА НИКЕЛЯ НА НОСИТЕЛЕ, ИСПОЛЬЗОВАННОГО ДЛЯ ОЧИСТКИ ЖИДКИХ УГЛЕВОДОРОДОВ ОТ ПРИМЕСЕЙ | 1991 |
|
RU2022997C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ЦЕРИЯ | 2012 |
|
RU2495147C9 |
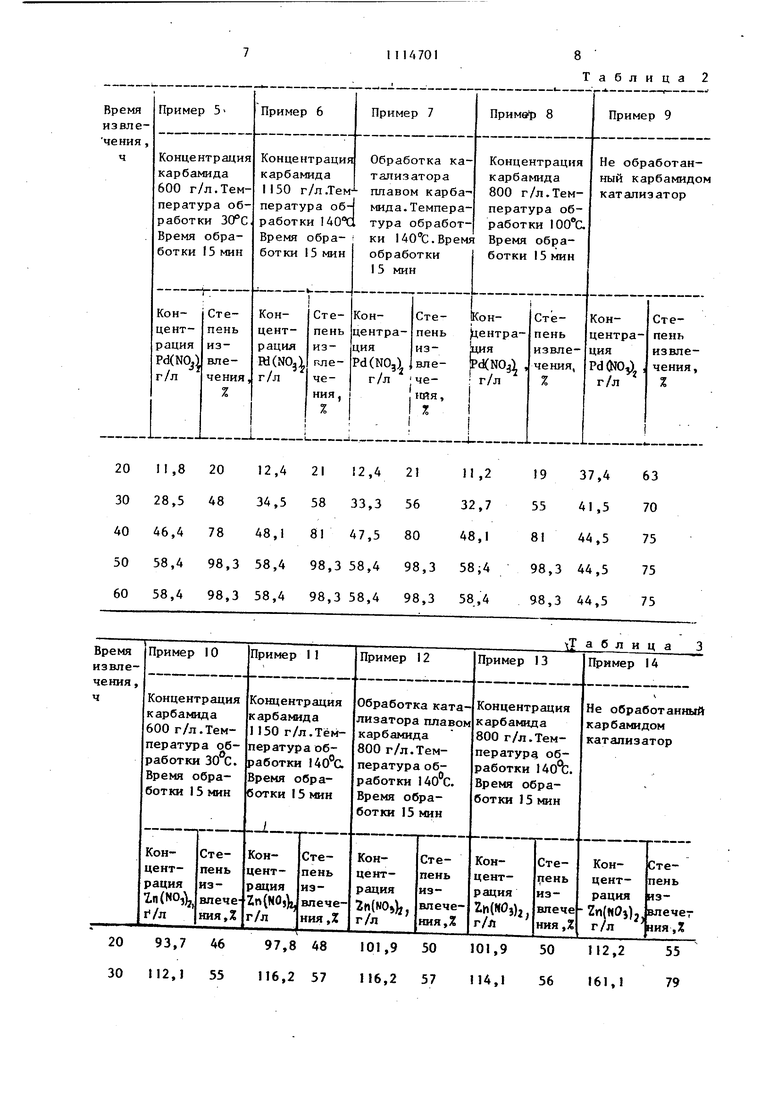
I. СПОСОБ ИЗВЛЕЧЕНИЯ ЦВЕТНЫХ МЕТАЛЛОВ ИЗ МЕТАЛЛОСОДЕРЖАЩЕГО КАТАЛИЗАТОРА путем обработки отработанного катализатора оксидами азота в присутствии воды, отличающий-, с я тем, что, с целью повышения степени извлечеиия металлов, катализатор предварительно обрабатывают, карбамидом при 30-140 С. 2. Способ по п. I, отличающий с я тем, что предварительную обработку ведут раствором, содержащим 600-1150 г/л карбамида.
20 30 40 50 60 20 93,7 46 97,8 48 101,9 50 30 112,1 55 116,2 57 116,2 57
Таблица 2 101,9 50 112,2 55 «14,1 56 161,1 79
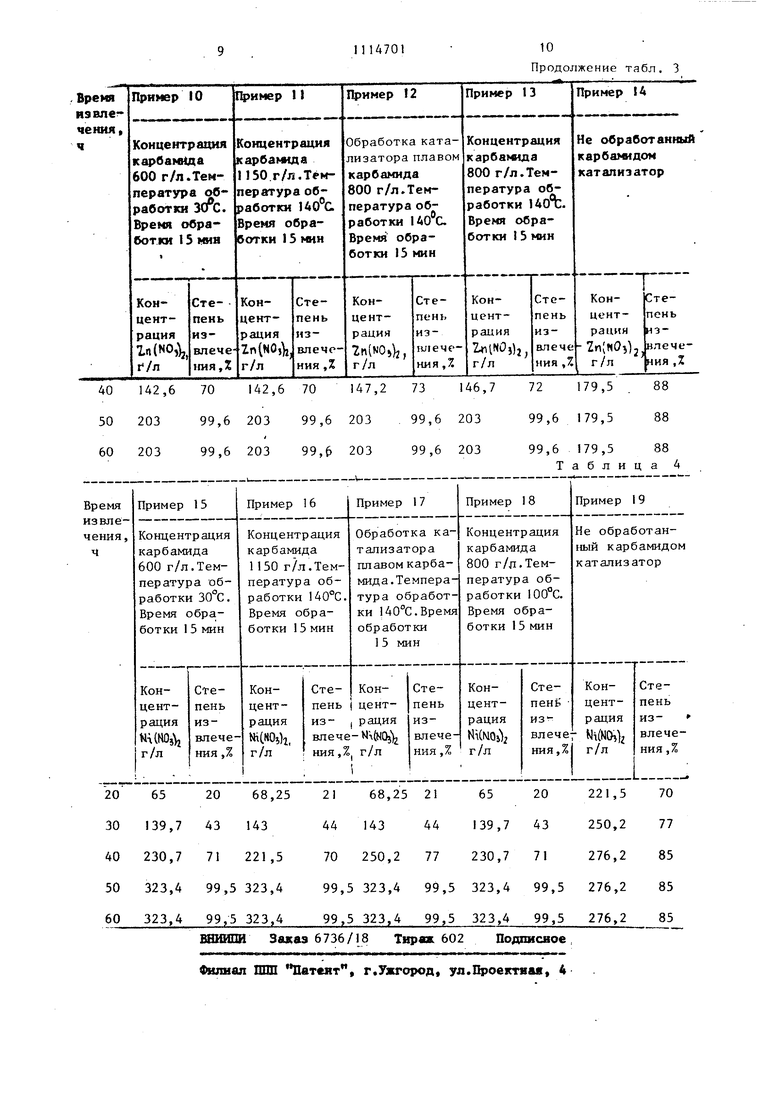
40 142,6 70 142,6 70 147,2 73 146,7 50 20399,6 203 99,6 20399,6 203
60 20399,6 203 99,6 20399,6 203
Время извлечения, ч
72 179,5 . 88 99,6 179,588
99,6 179,588
Таблица 4
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Патент ФРГ 1196495, кл | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Кинематографический аппарат | 1923 |
|
SU1970A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ очистки газов от окислов азота | 1977 |
|
SU778752A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| .АЯ йй.-.Ш-:-: - V T Krw--J - БйВ йОТМА | |||
