Изобретение относится к биохимии а именно к энзимологии, касается усовершенствования потенциометричес ких методов определения активности ферментов и может быть применено в экспериментальной и клинической биохимии, биохимии сельскохозяйстве ных животных и растений, технической и пищевой биохимии, молекулярно фармакологии и ряде других областей науки и техники, связанных с применением ферментативного анализа. Определение активности ряда ферментов, ускоряющих химические реакции, в результате которых образует,ся один или несколько продуктов, отличающихся от исходных субстратов по кислотно-основным свойствам, может быть осуществлено потенциометри ческим методом по величине сдвига р который является функцией изменения концентрации одного из компонентов исследуемой реакции. Так,для определения активности аденозинтрифосфатаз АТФ-аз) в настоящее время широко применяется потенциометрический метод с использованием.высокрчувствительных рН-метров. Определение аде- нозинтрифосфатаз данным методом возм но благодаря тому, что в реакции АТФ +Н20 АДФ+Н2Р04 высвобождающийся фосфат-ион способен к ограниченной диссоциации(,21 при 25 с) с выделением Н -иона: Кинетика гидролиза АТФ регистрируется рН-метром. Одной из стадий, предществующих определению активности, является приготовление инкубационной среды, включающее взвешивание компонентов, растворение их в буферном,растворе, доведение рН среды до оптимального значения П . Недостатком такого способа являет ся использование буферных систем для растворения компонентов, так как в присутствии даже минимальных количеств буфера изменение рН в ходе реакции зависит не только от скорости реакции, но и от буферной емкоети раствора. Буфер, находя1п;ийся в инкубационной среде, связывает часть освободивщихся Н -ионов, что маскирует ход ферментативной реакции, особенно в начальный период времени, KOI- да емкость буфера максимальна. Это оказывает отрицательное влияние на чувствительность метода (способность улавливать сдвиг рН при исследовании ферментов с низкой активностью), а также на точность измерения важнейшего кинетического параметра начальной скорости реакции (V , характеризующей истинную активность фермента. Кроме того, емкость буфера в ходе реакций непрогнозируемо меняется, что естественно, отражается на конечных результатах эксперимента. Известен способ приготовления инкубационной среды для потенциометрического определения активности Н -ДТФ-азы, включающий взвешивание компонентов, растворение их в дистиллированной воде, доведение рН среды до 8,3±0,1 титрованием сильными кислотами и щелочками 2 . Однако известный способ не обеспечивает достаточной точности в определении активности фермента, так как в присутствии даже минимальных количеств буфера изменение рН в ходе реакции зависит не только от скорости реакции, но и от буферной емкости- раствора. В результате маскирующего действия буфера наблюдаемый сдвиг рН не отражает истинной активности фермента. Поэтому определение активности недостаточно чувствительно и не позгволяет зарегистрировать низкие значения активности из-за связывания буфером части освободившихся в реакции Н -ионов. Целью изобретения является повышение точности определения активности фермента. Указанная цель достигается тем, что согласно способу приготовления инкубационной среды для потенциометрического определения активности Н-АТФ-азы, включающему взвещивание компонентов, растворение их в дистиллированной воде, доведение рН среды до 8,3iO,I, для растворения компонентов используют дистиллированную воду, предварительно обработанную постоянным электрическим током в электролизере с полупроницаемой мембраной при напряжении на электродах 300-600 В и силе тока 10-30 мА в течение 5-30 мин, при этом используют воду из катодной половины электролизера, а доведение рН среды осуществляют добавлением воды из обеих половин электролизера, после чего
инкубационную среду защищают слоем, жидкого индифферентного гидрофобного газонепроницаемого вещества.
При этом для защиты инкубационной среды используют вазелин.
Известно, что в электролизере, разделенном полупроницаемой мембраной, в катодной половине дистиллированная вода приобретает щелочные свойства по реакции
катод ZH O+Ze-ZOH+H fi а в анодной половине - кислотные свойства: анод - е- . .
Смешивая под контролем рН-метра .воду .из,обеих половин электролизе:ра, можно получить дистиллированную воду с любым значением рН. в диапазоне от 3,0 до 11,0. Растворяя в такой воде компоненты, -необходимые для осуществления ферментативной реакции, и доводя рН полученной среды до оптимального значения водой из разных половин электролизера, по лучают готовую инкубационную среду с необходимым значением рН без внесения каких бы то ни было ионов, Iкроме Н и он. Проводить обработку постоянным электрическим током самой инкубационной среды с целью получения оптимальных значений рН.не- .целесообразно, так как во время прохождения электрического тока наблюдаются существенные изменения концентраций компонентов среды, являютщихся электролитами (неорганические соли, диссоциируюр5ие субстраты, кофакторы и т.д. .
Для оптимального течения процесса необходиьаш является напряжение в пределах 300-600 В и ток силой 10-30 мА. Время пропускания тока через дистилированную воду, равное 5-30 мин, достаточно для получения рН в пределах от 3,0 до 11,0 в анодной и катодной половинах соответст- веяна.
Использование напряжения до 200-250 В и тока до 10 мА удлиняет время получения нужных значений рН до 40-60 мин. Напряжение выше 60 В и сила тока выше 30 мА способству|от при прохождении тока высокому разогреву воды дЬ 40-60 С, что существе но влияет на точность измерения рН. Таким образом, получают инкубационкую среду со строго заданным значением рН без добавления каких-либо химических реактивов (буфера, сильных кислот или оснований, оказывающих влияние на сдвиг рН в ходе ферментативной реакции и тем самым вносящих ошибку в измерение, т,е, впервые создана возможность измерения интенсивного сдвига рН в ходе потенциометрического определения активности ферментов.
Готовую инкубационную среду необходимо защитить слоем жидкого инди4гферентного гидрофобного газонепро- ницаемого вещества, например вазелинового масла. В противном случае полученное оптимальное значение рН неуклонно изменяется.
Вещество должно &1ть жидким (для обеспечения защиты всей поверхности жидкой инкубационной сред, индиф-: ферентным (для предотвращения неже- лательного влияния на инкубационную среду -и исследуемый фермент) , гидрофобным (для предотвращения растворения его в инкубационной среде) и газонепроницаемым (для предовращения изменений рН под влиянием рН-активных газов атмосферы СОп, NH« и др.). Перечисленным требованиям удовлетворяет вазелиновое масло. При защите им инкубационная среда сохраняет необходимое рН в течение 8-10 ч,
Изобретение позволяет повысить точность и чувствительности определения активности ферментов при одновременном упрощении способа и эко- 1НОМИИ реактивов и иссследуемого фермента.
Предлагаемый способ осуществляют следующим образом.
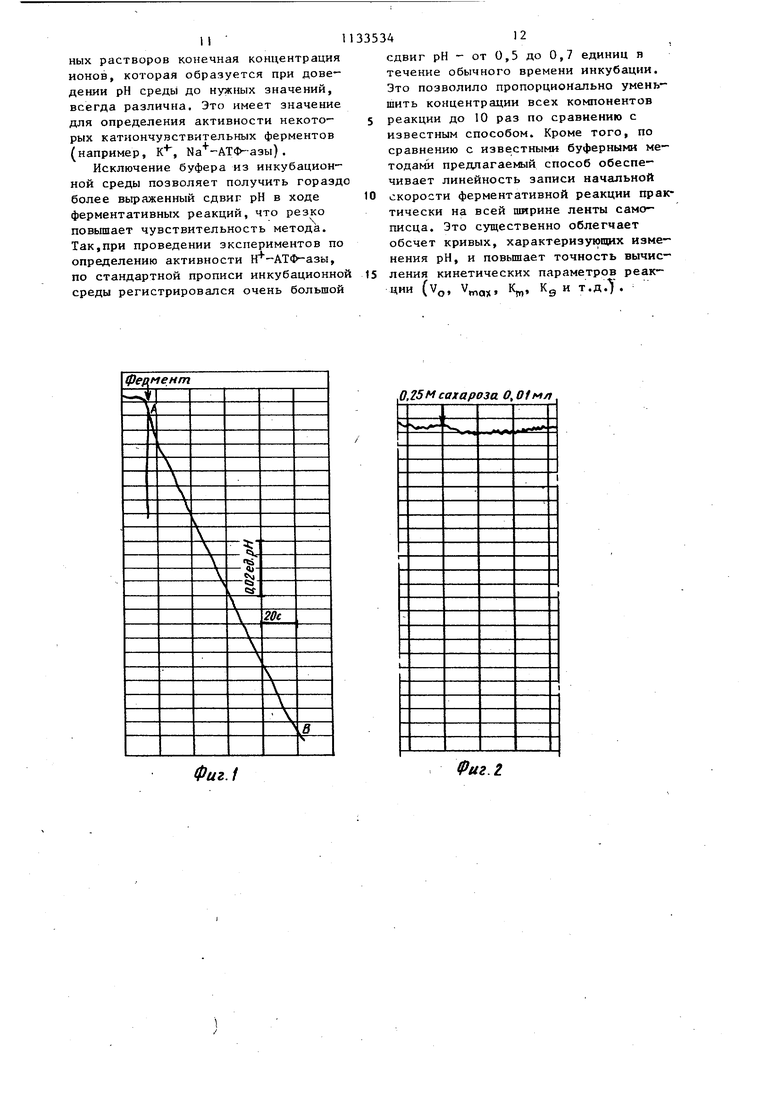
Взвешивают компоненты инкубационной среды в соответствии с ее прописью, исключая буфер. В электролизер, состояшрсй из двух половин etf костью по 100 мл каждая, разделенных полупроницаемой перегорсдкой (целлофан, брезент и т.п.) или соединенных проводником второго рода (.бинт, смоченный дистиллированной водой, заливают дистиллированную воду. В обе половины помещают платиновые электроды и подключают их к источнику.регулируемого постоянного напряжения. Подают на электрод рабочее напр;яженне, равное 300-600 Возникающий при этих условиях ток равен 10-30 мА и рН дистиллированно воды в анодной и катодной, половинах электролизера изменяется в пределах 3,0-4,0 и 10,0-11,0 соответственно в течение 5-30 мин. 5 Для предотвращения изменений рН под влиянием рН-активных газов атмосферы (CO.NH- и др. защищают поверхность анодной и катодной воды споем жидкого, индифферентного гидрофобного газонепроницаемого вещества, например вазелинового масла толщиной 1-2 мм. Такой прием позволяет стабилизировать полученные зна чения рН с точностью до 0,01 в тече ние 8-10 ч, что является достаточны для проведения большого количества экспериментов. Смещивают контролем рН-метра час ти объемов анодной и катодной воды до получения оптимального для ферментативной реакции значения рН и защищают слоем вазелинового масла Эта порция необходима для будущего доведения до конечного объема полученной инкубационной среды с таким же оптимальным значением рН. В мерном стакане на 100 мл, поме щенном на магнитную мешалку, раство ряют приготовленную навеску компонентом инкубационной среды в дистил лированной воде (катодной или анодной ), доводя рН смеси под кон тролем рН-метра до требуемого оптимального значения. Доводят объем данной смеси до метки с помощью во- ды, обработанной электрическим током по предлагаемому способу, и защищают слоем вазелинового масла. Пр готовленная смесь является инкубационной средой с оптимальным значением рН для исследуемой ферментативной реакции (на 10-20 анализов). В электрохимическую ячейку емкостью 5-10 мл, помещенную на магни ную мешалку, отмеривают необходимый объем инкубационной среды, погружают в нее электроды рН-метра, за- щищают слоем вазелинового масла и записывают на самописце, подклю- , ченном.к выходу рН-метра, исходное значение рН (базовую линию). Под слой вазелинового масла в инкубационную среду вводят микрообъем (о,01-0,05 мл) исследуемого ферментного препарата и автоматичес ки записывают динамику сдвига рН, .характеризующего кинетику ферментативной реакции. На период записи магнитную мешалку отключают. По диапазону сдвига рН определяют активность фермента, используя известные методы расчета. 346 Пример. Определение активности Н АТФ-азы митохондрий мозга крыс. Взвешивают 11 мг АТФ (фирмы Reanal, мол.в. 551,15) и 4,9 магния сульфата (MgSO -7H20 марки х.ч., мол.в. 246,3) в мерном стаканчике на 100 МП (навеска на десять проб) , конечная концентрация АТФ и MgS04 в инкубационной среде должна быть 0,2 мМ. В два стеклянных стаканчика емкостью по 200 МП наливают по 90 мл дистиллированной воды, опускают в них концы проводника второго рода - (бинт длиною 20 см, сложенный вчетверо, смоченный той же дистиллированной водой), погружают платиновые электроды (платина марки 99,99%-ной чистоты), представляющие собой проволоку длиной 10 см толщиной 1 мм с общей площадью 314 мм каждый, подключают их к источнику постоянного регулируемого напряжения (УИП-l) и в течение 10 мин подают на электроды рабочее напряжение 600 В. Контролируют значение полученного рН воды в стаканчике, подключенном к катоду, рН-метром марки ЭВ-74. В течение 10-минутного пропускания тока рН должен стать при этих уело- ВИЯХ равным 10,8-11,0. В случае получения меньйих значений рН продолжают электролиз в течение 2-3 мин. Защищают поверхность полученной воды с рН, равным 10,8-11,0, слоем вазелинового масла. В стаканчике емкостью 100 мл смешивают под контролем рН-метра части объемов анодной и катодной воды (общее количество около 60 мл) с тем, чтобы получить значение рН, соответствующее оптимуму (3,3)активности Н -АТФ-зы. Защищают слоем вазелинового масла. Эта порция использована для доведения до необходимого по методике объема приготовленной инкубационной среды. В мерном стаканчике на 100 мл, помещенном на магнитную мешалку, растворяют приготовленную навеску АТФ и магния сульфата в воде с рН 10,8-11,0. Под контролем рН-метра доводят значение рН до 8,3. Оставшийся до метки 100 мл объем доводят водой с рН 8,3. Защищают вазелиновым маслом. В электрохимическую ячейку емкостью 10 МП, помещенную на магнитную мешалку, отмеривают 7 мл инкубационной среды (объем, достаточный для погружения электродов рН-метра), , погружают электроды, защищают поверхность вазелиновым маслом. Записывают на самопишущем ампервольтметре Н-339, подключенном к выходу рН-метра, базовую линию, соответствующую исходному значению рН 0,3ч Под слой вазелинового масла с помощью микрошприца МШ-1 вводят 0,01 суспензии митохондрий мозга крысы, приготовленной на 0,25 М растворе сахарозы стандартным методом дифференциального центрифугирования. Конечная концентрация белка митохон дрий в пробе равна 0,04 мг/мл. Авто матически записывают динамику изменения рН среды,в течение 1-2 мин с выключенной мешалкой (фиг.1). По величине сдвига рН определяют активность Н АТФ-азы в единицах ЛрН/ut, она равна 0,036 за 1 мин (фиг. 1) . После проведения каждого анализа ячейку и электроды обрабатывают спй том и отмывают дистиллированной водой. Как показали многочисленные эксперименты, такая обработка не влияет на точность показаний рНметра. Таким образом, с помощью предлагаемого способа определена активнос Н -АТФ-азы митохондрий мозга крыс. При этом измерен истинный, не замаскированный буфером сдвиг рН в ход ферментативной реакции, который сос тавил 0,086 за 1 мин (при использовании буферной инкубационной среды в контрольном эксперименте сдвиг рН составил 0,017 за 1 мин). Расход исследуемого фермента и реактивов для приготовления инкубационной сре ды снижен в 10 раз. Предлагаемым способом проведено 60 экспериментов по определению активности Н -АТФ-зы митохондрий 113 , 48 мозга крыс. Во всех случаях инкуба- ционная среда с требуемым значением рН (от 7,5 до 8,5) была получена без добавления каких-либо химических реакторов, в частности буфера, и нейтрализующих кислот и щелочей. Инкуба1р1онную среду со значением рН 7,5-8,5 с точностью .до 0,01 ед рН (Получали на основе дистиллированной зоды, обработанной постоянным электрическим током. Полученное значение рН среды отличалось стабильностью и сохранялось 8-10 ч. В приведенном примере использов лись напряжение на электродах 600 В, сила тока 30 мА, время обработки воды 5 мин. Колебания этих параметров в пределах указанного интервала, не влияют на точность и чувствительность метода, а определяют лишьпродолжительность обработки током. Зависимость времени, необходимого для получения рН дистиллированной вода со значением 10,8-11,0 от при- лагаемого напряжения приведена в табл.1. Таблица 1 Время, мин Проведено сравнительное испыта- ние двух способов определения активности Н -АТФ-азы митохондрий мозга крыс: безбуферного (предлагаемого) способа., основанного на использовании электрохимически обработанной дистиллированной воды, и известного (буферного)способа. Результаты не-пытаний приведены в табл.2. Таблица2




| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ ТОКСИЧНОСТИ ВОДНОЙ СРЕДЫ (ВАРИАНТЫ), РЕАГЕНТ ДЛЯ ОПРЕДЕЛЕНИЯ ТОКСИЧНОСТИ ВОДНОЙ СРЕДЫ, ПРИМЕНЕНИЕ Mg-АТФазы ПЛАЗМАТИЧЕСКИХ МЕМБРАН МОЗГА ЖИВОТНЫХ | 2004 |
|
RU2266539C1 |
| СПОСОБ ОЦЕНКИ ТИПОВ МЫШЕЧНЫХ ВОЛОКОН | 2015 |
|
RU2628810C2 |
| Способ определения активности моноаминооксидазы | 1987 |
|
SU1521761A1 |
| АНТИОКСИДАНТНОЕ И МЕМБРАНОСТАБИЛИЗИРУЮЩЕЕ СРЕДСТВО | 2012 |
|
RU2540466C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ДЫХАТЕЛЬНОГО КОНТРОЛЯ У ПОПУЛЯЦИИ БАКТЕРИЙ | 2003 |
|
RU2247780C2 |
| Способ определения активности никотинамидных дегидрогеназ | 1984 |
|
SU1255934A1 |
| Способ получения аденозинтрифосфатазы | 1983 |
|
SU1232679A1 |
| Способ определения активности глутатионтрансферазы | 1990 |
|
SU1759874A1 |
| СПОСОБ ОЦЕНКИ ВЛИЯНИЯ ЦИТОМЕГАЛОВИРУСНОЙ ИНФЕКЦИИ НА АКТИВНОСТЬ NAD-ЗАВИСИМОЙ МАЛАТДЕГИДРОГЕНАЗЫ В СИНЦИТИОТРОФОБЛАСТЕ ВОРСИНОК ПЛАЦЕНТЫ НА ТРЕТЬЕМ ТРИМЕСТРЕ БЕРЕМЕННОСТИ | 2015 |
|
RU2574937C1 |
| Субстанция протеолитического фермента на основе Протосубтилина ГЗХ, иммобилизованного на хитозане, и композиция для лечения гнойно-некротических ран | 2016 |
|
RU2630668C1 |
1. СПОСОБ ПРИГОТОВЛЕНИЯ ИНКУБАЦИОННОЙ СРЕДЫ Д)1Я ПОТЕНЦИОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ АКТИВНОСТИ Н -АТФ-АЗЫ, включающий взвешивание компонентов, растворение их в дистиллированной воде,, доведение рН раствора до 8,3iO,1, отличающийся тем, что, с целью повышения точности определения активности фермента, для растворения компонентов используют дистиллированную воду, предварительно обработанную постоянным электрическим током в электролизере с полупроницаемой мембраной при напряжении на электродах 300-600 В и силе тока 10-30 мА в течение 5-30 мин, при этом использую.т воду из катодной полови- . ны электролизера, а доведение рН среды осуществляют добавлением воды i из обеих половин электролизера, после чего инкубационную среду защищают слоем жидкого индифферентного гидрофобного газонепроницаемого вещества. 2. Способ non.t, отличающийся тем, что для запреты инкубационной Среды используют вазелин. Слд СО СП СО 4
Количество определений
Величина сдвига рН за Гмин
на линейном участке0,,002
Время сдвига рН на 0,01 ед,, с13±1
20
20
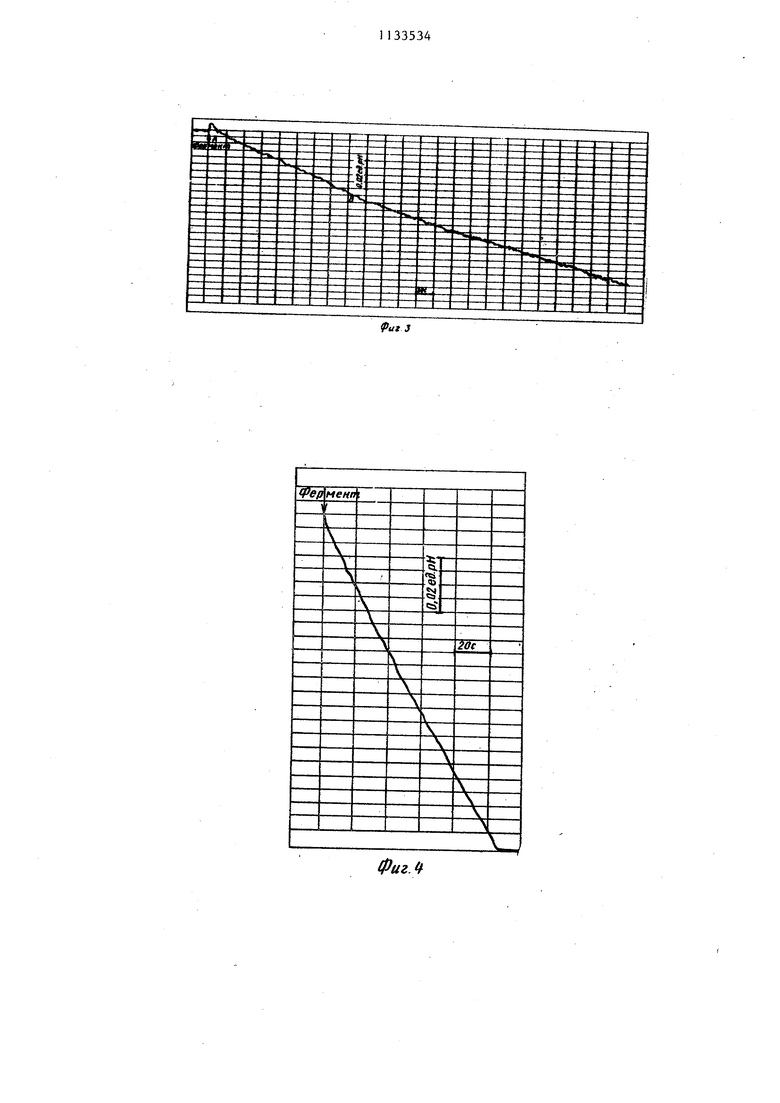
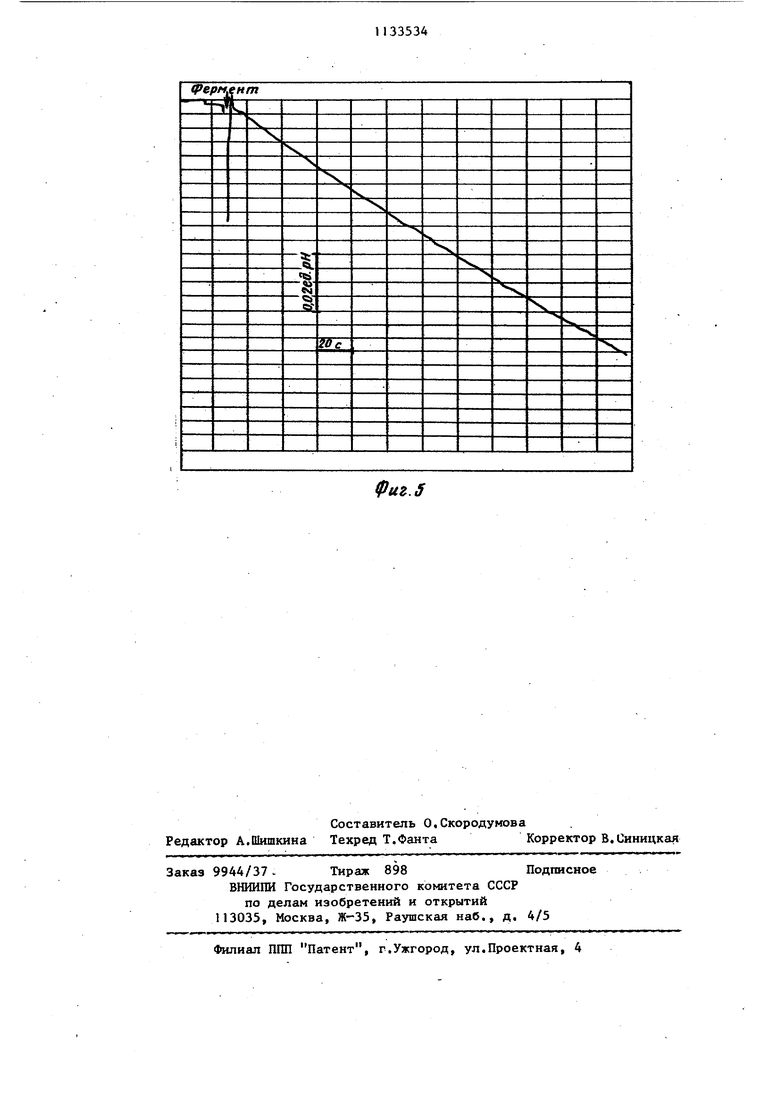
0,018+0,001 7112 Как видно из табл.2, величина сд га рН за один и тот же. произвольно выбранный промежуток времени в 5 ра больше при определении активности ; фермента.безбуферным методом, а вре мя сдвига рН 0,01 ед, рН приблизи тельно во столько же раз меньше, чем при определении.известным потен циометрическим.способом с использо- ваниемтрис НС1 буфера. На фиг. 1--5 приведены наиболее ти пичные примеры определения активности Н АТФ-азы митохондрий мозга предлагаемым и известным способами. Как видно из диаграммы на фиг.1, автоматически записанные изменения рН в ходе энзиматического гидролиза АТФ представляют собой прямую линию практически на всем протяжении диаграммной ленты ;(участок АВ записи) , что является чрезвычайно важным для точности расчета основного кинетического параметра реакции - ее начальной скорости Vg, Метод является весьма чувствительтшш, так как добавление незначительных количеств фермента вызывает большой сдвиг рН, что увеличивает крутизну наклона кинетической кривой. Добавление вместо митохондрии 0,01 мл раствора сахарозьГ.(контроль) не вызывает изменений рН за время регистрации (фиг.2). На фиг.З представлена кинетическая запись активности Н-АТФ-азы митохондрий мозга крыс в инкубацион ной среде, приготовленной на обычной дистиллированной воде, значение рН которой получено при помоп 0,3 мМ|трис-НС1-буфера. Как следует из диаграммы, линейность записи (участок АВ) сохраняется лишь прибли зительно на 1/3 части ширины Диагра мнок ленты, что снижает точность расчета начальной скорости реакции. Кроме того, чувствительность данного способа значительно ниже, о чем свидетельствует существенно более пологий характер кинетической кри- БОЙ. Предлагаемый способ определения активности ферментов может быть использован , для,проведения ингибидного анализа (фиг.4 и З), Введение, в инкубационную среду в процессе ее.приготовления 5 мкг олигоьшцина, я апяющегося известным ингибитором митохондриальной Н -АТО -азы, отчетливс тормозит энзимати- ческий гидролиз АТФ, что видно по резкому снижению крутизны падения рН в присутствии олигомицина (фиг.4 и 5). На фиг.1 показана кинетическая запись активности Н -АТФ-азы митохондрий мозга крыс в безбуферной инкубационной среде, приготовленной на дистилированной воде, подвергнутой электрохимической обработке (состав инкубационной смеси: 0,2 мМ АТФ и 0,2 мМ MgSO 7Е,р, исходное значение рН 8,3, температура 25°С, стрелкой обозначен момент добавления взвеси митохондрий, приготовлен- ный на 0,25 М сахарозе, АВ - линейный участок записи) . На фиг.2 показана контрольная.диаграмма, отражающая изменение рН при введении вместо исследуемого фермента такого же объема сахарозы (стрелкой указан момент добавления 0,25 М раствора сахарозы). На фиг.З показана кинетическая запись активности Н -АТФ-азы митохондрий мозга крыс в буферной инкубационной среде, приготовленной на обычной дистиллированной воде, исходное значение рН которой 8,3 получено при помощи 0,3 мМТрис-НС1- буфера (состав инкубационной среды: 0,2 мМ АТФ,- 0,2 мМ MgS04-7H O, 0,3 мМ трис -НС, стрелкой обозначен момент добавления взвеси митохондрий, приготовленной на 0,25 М сахарозе, АВ - линейный участок записи) . На фиг.4 показана кинетическая запись активности Н-АТФ-азы мито- хондрий мозга крыс в безбуфериой инкубационной среде баз ингибитора (состав среды и условия инкубации те же, что и для фиг.1) . На фиг.5 показана кинетическая запись активности H-АТФ-азы митохондрий мозга в безбуферной инкубационной среде в присутствии 5 мкг олигомицина (состав среды .и условия : инкубации те же, что и в зксперимен-, те, представленном на фиг.1).. Применение предлагаемого способа ; сделало возмозкным приготовление ин- кубационной среды.с заданным значением рН без внесения дополнительных реактивов (буфер, кислоты, щелочи), способных влиять на ионную силу среды и изменять характер ионизации активного центра фермента. Данный способ позволяет получить инкубационную среду с постоянным ионным составом, тогда как пр,и использоваиии буфер-
11
11
ных растворов конечная концентрация ионов, которая образуется при доведении рН среды до нужных значений, всегда различна. Это имеет значение для определения активности некоторых катиончувствительных ферментов (например, К, Ма -АТФ-азы) .
Исключение буфера из инкубационной среды позволяет получить гораздо более вьфаженный сдвиг рН в ходе ферментативных реакций, что резко повышает чувствительность метода. Так,при проведении зкспериментов по определению активности H-АТФ-азы, по стандартной прописи инкубационной среды регистрировался очень большой
353412
сдвиг рН - от 0,5 до 0,7 единиц в течение обычного времени инкубации. Это позволило пропорционально уменьшить концентрации всех компонентов
5 реакции до 10 раз по сравнению с известным способом. Кроме того, по сравнению с известными буферными методами предлагаемый способ обеспечивает линейность записи начальной
10 скорости ферментативной реакции практически на всей ширине ленты самописца. Это существенно облегчает обсчет кривых, характеризующих изменения рН, и повьш1ает точность вычисления кинетических параметров реакKg и т.д. .
(Vo, V
Кп,ции
так
0,25м сахароза 0,01м/1
:s:
:§:
езФиг.1
Фиг. 2.
Фиг.1
го с
фиг. 5
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Диксон М., Уэбб Э | |||
| Ферменты | |||
| М., 1961, 37 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Болдырев А.А., Лебедев ., Ритов Б.В., О методе одновременной регистрации аденозинтрифосфатазных и Са- - насасывающих свойств ферментов саркоплазматического ретикулума.- Вопросы медицинской химии, 1969, т | |||
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
