4
ю
о
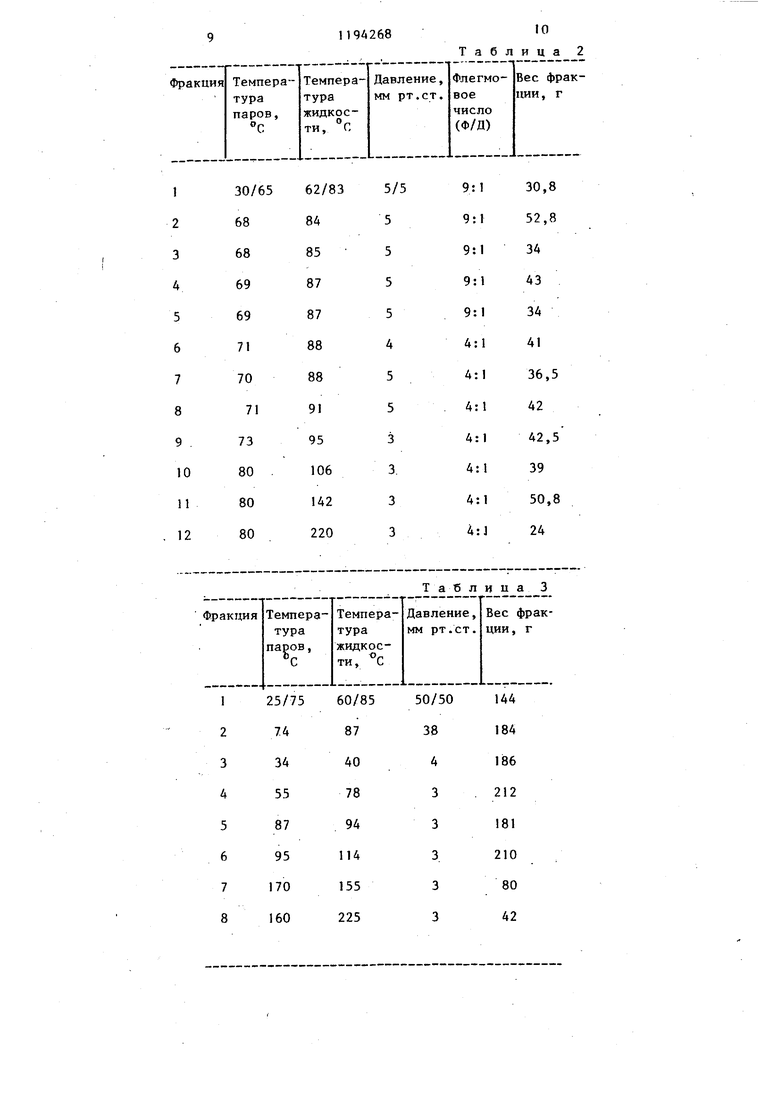
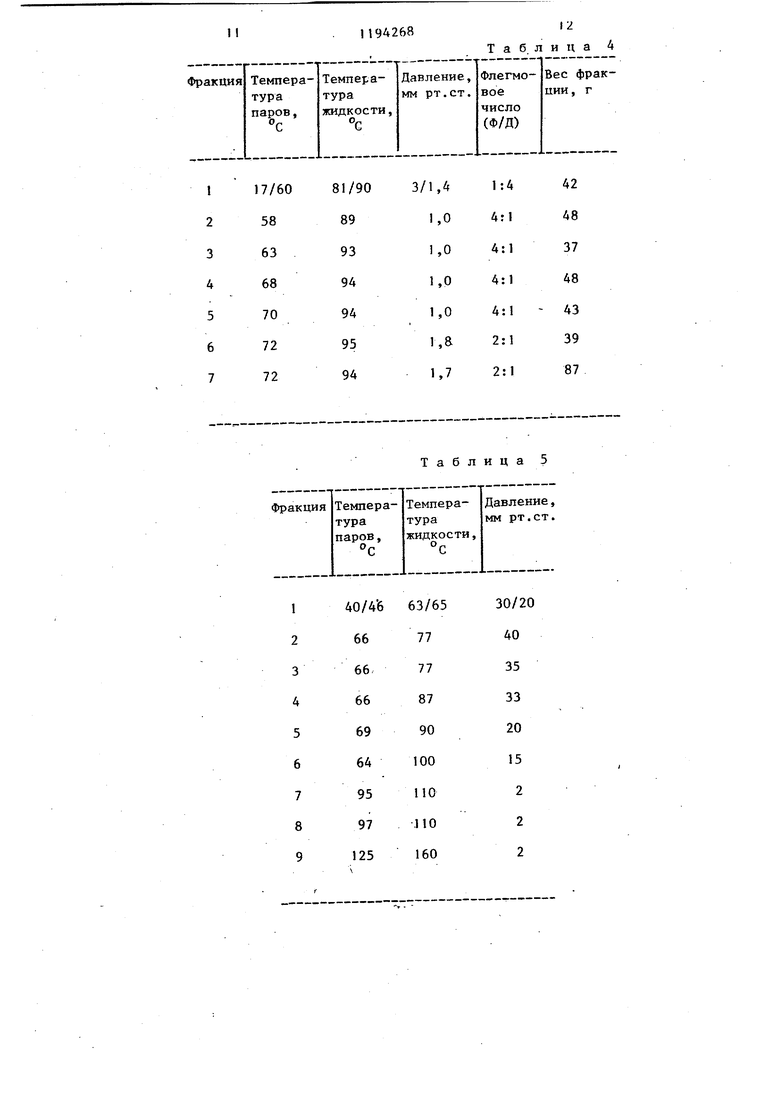
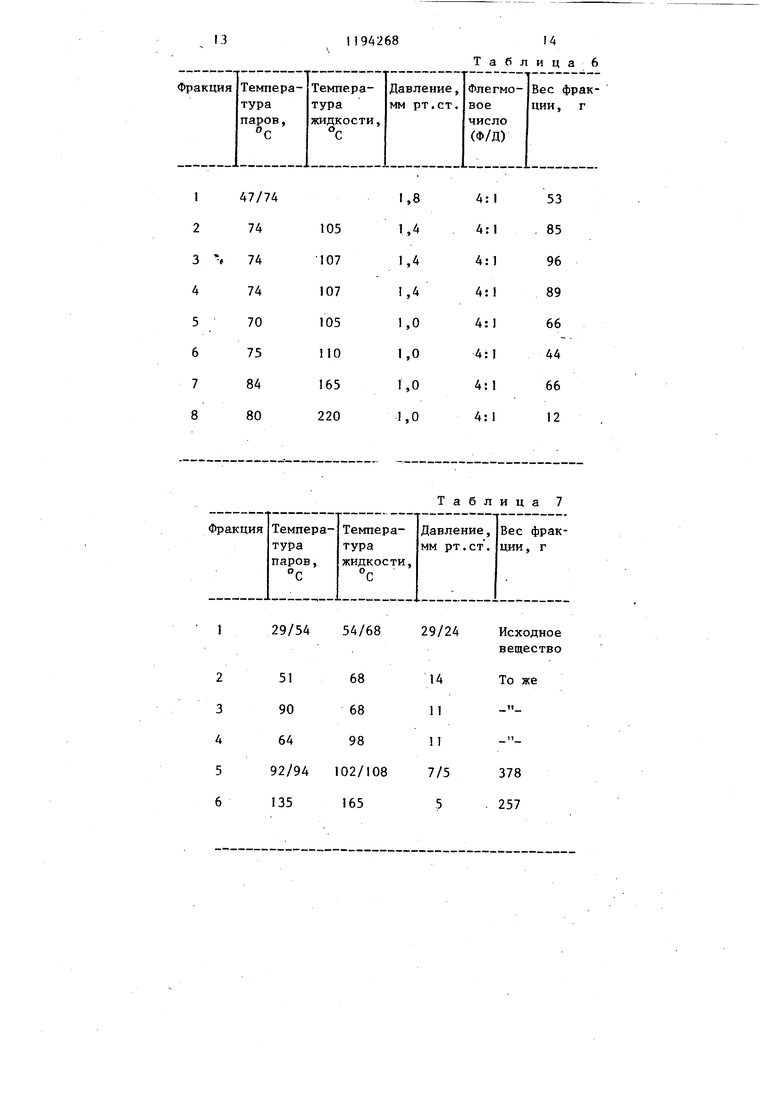
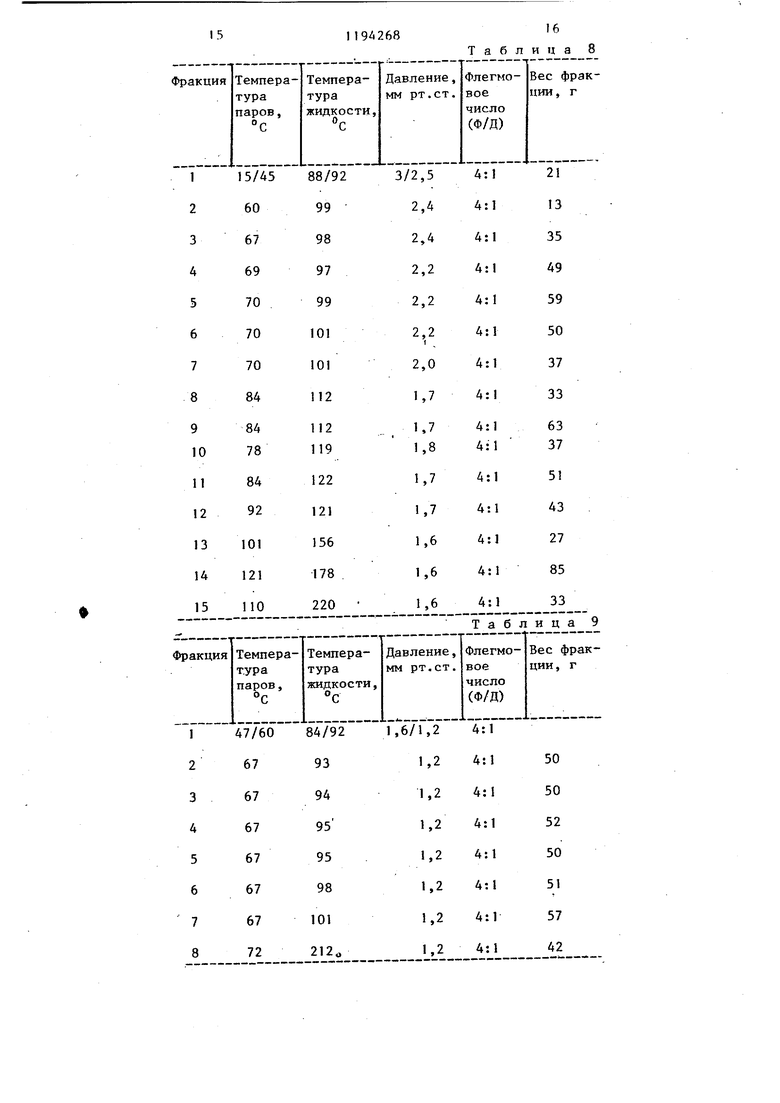
00 1 Изобретени относится к органической химии, в частности к способу получения новых производных диизоамилена общей формулы О где одна из пунктирных линий означает двойную связь углерод-углерод, каждая из остальных пунктирных линий означает простую связь углеродуглерод, а R - С -С -низший алкил, которые могут найти применение в парфюмерных композициях в качестве душистых и ароматических веществ. Целью изобретения является иЬыскание способа получения новых душис тых и ароматических веществ. Пример 1. Получение диизоамилена. 2-Метйл-2-бутен прокачивают в те чение 10 ч через трубку 5x0,625 дюй ма (127 X 16 мм), заполненную 15 г. катализатора - полистиролсульфокислоты, при 100°С и давлении 28,1 атм. . Образовавшийся продукт перегоняют на фракционирующей колонке для того, чтобы отделить диизоамилен от более высокомолекулярньгх полимеротэ, кotopыe образуются в процессе реакции в качестве побочных продук тов. Пример 2, Получение ацетильного производного диизоамилена В двухлитровую реакционную колбу оборудованную мешалкой, термометром, дефлегматором и обогревакяцей рубашкой, помещают 100 г уксусного ангидрида и 80 г эфира трехфтористо го бора. В полученную смесь добавляют 690 г диизоамилена, приготовленного аналогично примеру 1. Реакционную массу вьщерживают при 8285 С в течение 5,5 ч, после чего ее охлаткдают до комнатной температуры. Затем реакционную массу до|бавляют к 1 л воды и образовавгауюся смесь перемешивают, в результате чего получают органическую и водную фазы. Органическую фазу отделяют от водной фазы и нейтрализуют двумя литрами 12,5%-ного раствора гидроокиси натрия, затем промывают 1 л насыщенного раствора хлористого натрия. Полученную органическую фазу затем высушивают над безводным 8 сульфатом натрия и перегоняют на колонке с 1.1ДНОЙ тарелкой, получая фракции, представленные в табл. 1. Полученный материал затем перегог няют на многотарельчатой ректифика-. ционной колонке, получая фракции при флегмовых числах (флегма/дистиллат);, представленные в табл. 2. Методами ГОХ, ЯМР, ИК- и массспектрометрии полученный продукт охарактеризован как смесь цис- и транс-изомеров соединений общей формулы, где . П р и м .е р 3. Получение пропионильного производного диизомилена. В пятилитровую реакционную колбу:, оборудованную дефлегматором, капельной воронкой, термометром, терморегулятором, обогревающей рубашкой и приспособлением для продувки азотом, помещают 1000 г (7,45 моль) пропионового ангидрида и 91,4 мл (0,745 моль) эфирата трехфтористого бора. Полученную смесь вагревают до 65 С. В течение 25 мин добавляют 1501 мл (7,45 моль) диизоамилена, полученного аналогично примеру 1, поддерживая температуру реакIоционной массы при 65-70 С. Затем реакционную Массу перемешивают в . течение 30 мин при 65 С, после чего ее охлаждаюти вьшивают в трехлитровую делительную воронку. Затем добавляют 75 мл воды с по-следующими 75 мл 50%-ного раствора гидроокиси натрия и 25 мл воды, реакционную массу вьшивают в стакан и охлаждают до комнатной температуры, используя баню - сухой лед в изопройиловом спирте. После этого реакционную массу добавляют в пятилитровую делительную воронку, и -нижний водный слой удаляют. Верхнюю органическую фазу промывают 500 мл насыщенного раствора хлористого натрия. Затем органическую фазу промывают 500 мл 5%-ного раствора гидроокиси натрия, 500 мл насыщенного раствора хлористого натрия и 500 мл 5%-ного раствора гидроокиси натрия до достижения значения рН масла в интервале 6-7. Затем масло снова промывают 500 мл насьш енного раствора хлористого натрия. Водную фазу экстрагируют 400 мл диэтилового эфира. Полученный материал затем перегоняют на разбрызгивающей колонке размером 50,8 мм и получают фракции, представленные в табл. 3. 3 Фракции 5-7 объединяют для повторной перегонки на колонке размером 25,А мм и получают фракции, представленные в табл. 4. Методами ГЖХ, ЯМР, ИК- и массспектрометрии полученный продукт охарактеризован как смесь соединени общей формулы, где . Пример 4. Получение н-бути рильного производного диизоамилена. В пятилитровую реакционную колбу, снабженную электрической мешал. кой, нагревающей рубашкой, термомет ром, капельной воронкой и дефлегматором, добавляют 960 г н-маслянного ангидрида и 105 мл трехфтористого бора. Реакционную смесь нагревают до 65°С и по каплям в течение 3,5 ч пр этой температуре добавляют 1725 мл диизоамилена, полученного аналогично примеру 1, причем температуру реакционной смеси поддерживают при . В конце добавления реакционную смесь охлаждают до 38 С и затем пер носят в делительную воронку емкостью 5 л. Затем к реакционной массе добавляют 75 мл 50%-ного водного раствора гидроокиси натрия и 100 мл воды. Органическую фазу промывают 1 л насьщенного раствора хлористого натрия до установления значения рН 4-5. Затем реакционную смесь про мывают 1 л 12,5%-нбго раствора гид.роокиси натрия, перемешивают в течение 15 мин и разделяют. После это го полученную органическую фазу высушивают над безводным сульфатом магния, перегоняют на колонке Стоун размером 25,4 мм и получают фракции представленные в табл. 5. Полученные фракции 7-9 объединяют и перегоняют повторно на кол:онке из нержавеющей стали высотой 616 мм, получают фракции, представленные в табл. 6., Методами ГЖХ и ИК-спектрометрии полученный продукт охарактеризован как смесь соединений общей формулы, где . Пример 5. Получение изобутирйльного производного диизоамилена. В реакционную колбу ёмкостью 5л оборудованную дефлегматором, капель ной воронкой, термометром, терморегулятором, обогревающей рубашкой и приспособлением для продувки 8 азотом, помещают 1361 г (8,6 моль) изобутирильного ангидрида. Затем к изобутирильному ангидриду добавляют 105 мл (0,86 моль) эфирата трехфтористого бора. Образовавшуюся смесь после этого нагревают до . К реакционной смеси добавляют в течение 4 ч 1725 г (8,6 моль) диизоамилена, полученного аналогично примеру 1, при этом температуру реакционной смеси поддерживают равной 83-85 С. Затем реакционную смесЬ охлаждают до комнатной температуры и выливают ее в 5-литровую делительную воронку. Затем к .реакционной смеси добавляют 75 .мл 50%-ного водного раствора гидроокиси натрия и 100 мл воды. Нижнюю водную фазу удаляют, а орга ническую фазу промывают 1 Л насыщенного раствора хлористого натрия, I л 5%-ного раствора гидроокиси натрия, 1 л насьпценного раствора хлористого натрия, 1 л 12,5%-ного раствора гидроокиси натрия, 1 л, 12,5%-ного раствора гид роокиси натрия. Затем реакционную смесь перегоняют на разбрызгивающей колонке (50,8 мм), заполненной каменной мелочью, и получают фракции, представленные в табл. 7. Фракции 5 и 6 полученного дистил лата после перегонки объединяют и повторно перегоняют и получают фрак ции, представленные в табл.. 8.. Затем фракции 3-9 этого дистилjiaTa объединяют, перегоняют повторно и получают фракции, представленные в табл. 9. Методами ГЖХ, ЯМР, ИК-и массспектрометрии полученный продукт охарактеризован как смесь соединений обобщей формулы, где К изо-СяР у. П р м е р 6. Получение ацетильного производного диизоамилена. в реакционную колбу емкостью 5 л, оборудованную электрической 1 1ешалкой, термометром, капельной воронкой, У-трубкой 24/42, холодильником, обогревающей рубашкой и приспособлением для продувки азотом, добавляют 41 мл 70%-ной метансульфокислоты и 30 г пятиокиси фосфора. Полученная смесь экзотермически разогревается до 60°С. На протяжении 7 мин к реакционной смеси добавляют 235 мл уксусного ангидрида, поддерживая температуру при 65°С. Поддерживая температуру реак5НИИ при 80°С, в течение 30 мин по каплям к реакционной массе добавляю 516 кп диизоамилена, полученного аналогично примеру 1. В конце добав ления диизоамилена по данным ГЖХ реакционная смесь содержит 42% продукта. Реакционную смесь выливают в делительную воронку емкостью 15,1 л с открытой крышкой, в которой содер жится 1 л воды. Образовавшуюся смес промывают 1 л 12%-ного раствора гвд роокиси натрия и затем 1 л насыщенного раствора хлористого натрия. Дл облегчения разделения добавляют 100 мл толуола. По данным ГЖХ, ЯМР, ИК- и масс-спектрометрии полученный продукт охарактеризован как смесь соединений обшей формулы 1, где . Затем образовавшийся продукт реакции высушивают над безводным сульфатом магния и перегоняют на ко лонке с каменной мелочью размером 76 мм и получают фракции, представленные в табл. 10. Пример 7. Получение ацетил ного производного дииаоамилена. В реакционную колбу емкостью 500 мм, оборудованную дефлегматором капельной воронкой, термометром, терморегулятором, обогревающей рубашкой, охлаждающей баней и приспособлением для продувки азотом, добавляют 406 мл уксусного ангидрида и 30 мл эфирата трехфтористого бора Реакционную смесь нагревают до 60°С ,и, поддерживая температуру реакцион ной массы при 60 С, в течение 30 мин добавляют диизоамилен, приготовленный .аналогично примеру 1. Затем образовавшуюся реакционную смесь на гревают при 60 С и перемешивании в течение 12 ч. В конце этого периода реакционную смесь перегоняют и получают фракции, представленные в табл.11. По данным ГЭХ, ЙК- и ЯМР-спектро скопии и масс-спектроскопии полученный продукт охарактеризован как смесь соединений общей формулы, где R-CHj. Пример 8. Получение пропионильного производного дииэоамилеВ реакционную колбу емкостью 5 л, оборудованную дефлегматором,, капельной воронкой, термометром, терморегулятором, обогревающей ру86бсШ1кой, охлаждающей баней и приспособлением для продувки азотом, добавляют 415 мл пропионового ангидрида, 11 г метансульфокислоты и 35 мл эфирата трехфтористого бора. Реакционную массу нагревают до 60 С и добавляют в течение 30 мин 1850 мл диизоамилена, полученного аналогично примеру 1. Затем реакционную смесь перемешивают в течение 12ч при 60 С, после чего ее перегоняют на фракционирующей колонке и получают смесь соединений общей формулы, где . Пример 9. Получение изобутирильного диизоамилена. В реакционную колбу емкостью 5л, снабженную дефлегматором, капельной воронкой, термометром, терморегулятором, обогревающей рубашкой, охлаждающей баней и приспособлением для продувки азотом, добавляют 953 мл (6,0 моль) изобутирильного ангидрида, 183 г. полифосфорной кислоты и 135 мл 70%-ной метансульфокислоты. Эта реакционная смесь экзотермически разогревается до . В течение 20 мин, поддерживая температуру реакционной смеси при , добавляют к ней 1725 г (8,6 моль) дииаоамилена, приготовленного аналогично примеру 1. Затем реакционную смесь нагревают до 85 С и вьщерживают при ней в течение 10 ч, после чего ее охлаждают и добавляют к ней100 г ацетата натрия и 1 л воды. Образовавшуюся смесь добавляют в делительную воронку емкостью 5 л и органический слой после этого промывают 1 л 12,5%-ного раствора гидроокиси натрия, 2 л 12,5%-ного раствора гидроокиси натрия, 1 л насьпценного раствора хлористого натрия. . Затем реакционную смесь перегоняют на колонке Гудлоу размером 308 мм, получая фракции, представленные в табл. 12. Методами ГЖХ, ЯМР, ИК- и массспектрометрии полученный продукт охарактеризован как смесь соединений общей формулы, где it3o-C U-f. Пример 10. Получение изобутирильного производного диизоамилена. В реакционную колбу емкостью 5 л, оборудованную деф.пегматором, ка7
пельной воронкой, термометром, тер. морегулятором, обогревающей рубашкой, охлаждающей баней и приспособлением для продувки азотом, добавляют 953 г (6,0 моль) иэобутирильного ангидрида и 105 мл (0,86 моль) эфирата трехфтористого бора. Реакционную смесь нагревают до 65 С в течение 30 мин, добавляют 1725 мл (8,6 моль) диизоамилена, приготовленного аналогично примеру 1. Затем реакционную смесь нагревают до 6365 С и выдерживают при этой температуре в течение 12ч.
Реакционную смесь охлаждают до комнатной температуры и добавляют к ней 82 г ацетата натрия, после чего реакционную смесь выливают в делительную воронку емкостью 5 л и промывают 1 л воды, 1 л 12,5%-нот водного раствора гидроокиси натрия, 1 л 12,5%-ного раствора гидроокиси натрия, 1 л 12,5%-ного раствора гидроокиси натрия, 1 л насьщ1енного раствора хлористого натрия.
Затем органический слой высушивают над безводным сульфатом; натрия и перегоняют на колонке Гудлоу (305 мм), получая фракции, представленные в табл. 13.
Методами ГЖХ, ЯМР, ИК- и массспектрометрии полученный продукт охарактеризован как смесь соединений общей формулы, где R K3o--C }lj,
Пример 11. Получение н-бутирильного производного диизоамилена.
В реакционную колбу емкостью 5л снабженную электрической мешалкой,
942688
термометром, дополнительной воронкообразной трубкой, холодильником, обогревающей рубашкой и приспособлением для продувки азота, загружают 5 55 мл 70%-ной метансульфокислоты и 30 г пятиокиси фосфора. Реакционная масса экзотермически разогревается да , за 10 мин прибавляют АОО мл н-маслянного ангидрида, вьщерживая реакционную смесь при 65 G.Через 40 мин, вьщерживая реакционную смесь при 84°С, прибавляют к ней 400 мл диизоамилена, полученного аналогично примеру 1. Реакционную смесь перемешивают 4 ч при.84с.
Затем реакционную массу переносят в делительную воронку с открытым концом емкостью 5 галлонов (19 л), содержащую 2 л воды. Реакционную смесь промывают 1 л 12%-ной водой гидроокиси натрия, 1 л насыщенного водного раствора хлористого натрияw
Затем реакционную массу перегоняют на колонке Гудлое Сильвер Мир25 pop размером 30,5 см, получают пр дукт, охарактеризованный данными гах, ЯМР, ИК- и масс-спектрометрией как смесь соединений общей формулы, где .
В табл. 14 указаны органолецтические свойства соединений общей формулы.
Предлагаемый способ позволяет новые душистые и ароматические вещества, которые можно использовать в качестве отдушек, ароматических и вкусовых добавок.
.Таблица






| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения парфюмерной добавки-смеси монооксипроизводных терпенов | 1977 |
|
SU671718A3 |
| Сложные эфиры 3-фурантиола в качестве вкусовых добавок к пищевым продуктам | 1975 |
|
SU583133A1 |
| Способ получения изохроманов | 1975 |
|
SU584777A3 |
| 2,6,6-Триметил-1-циклогексен-1винилалканоаты, проявляющие органолептический эффект | 1976 |
|
SU685660A1 |
| Способ получения парфюмерной композиции на основе 3-или-4-(4-метил-4-оксиамил)- -циклогексенкарбоксальдегида | 1976 |
|
SU731882A3 |
| Способ полуяения бицикло/2,2,2/ октана | 1975 |
|
SU587856A3 |
| 3-Тиоалкан-1,4-дикарбонильные соединения в качестве органолептиков | 1975 |
|
SU664959A1 |
| Способ получения замещенных или незамещенных производных 3-тиофурана | 1974 |
|
SU648100A3 |
| Способ получения 2-ацетил-1,2,3, 4,5,6,7,8-октагидро-2,3,8,8-тетраметилнафталина | 1974 |
|
SU576037A3 |
| Способ получения алкилкетонов | 1968 |
|
SU443508A1 |
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ДИИЗОАМШТЕНА общей формулы Лгде одна из пунктирных линий означает двойную связь углерод-углерод, калодая из остальных пунктирных линий означает простую связь углерод-углерод, а R - С -СЗ-ЕИЗШИЙ алкил, отличающийся тем, что 2-метил-2-бутен подвергают димеризации в присутствии кислотного катализатора с последующим ацилированием образовавшихся при этом диизоамилесл нов ацилангидридом в присутствии в качестве .катализатора кислот Льюиса или протонной кислоты при 60 80 С и дистилляцией целевого продукта.
1
2 3 4
161 100 191 189
62/83
30/65
8А 68 68 69 69 71 70 71
85
87
87
88
88
91
95 73
106
142 80 80
220
/5
30,8
9:1
52,8 9:1
34 9:1
43 9:1
34 9:1 А:1
41 А:1
36,5 4:1
42
39 4:1
50,8 4:1
24
Т а б л и ц а 3
Таблица 5
Таблица 7
Таблица 9
17 Таблица 10
18
П94268
Таблица II
Таблица 12
Фруктовый, древесный и амбровый аромат
Сладкий,
фруктовый,
цветочный,
древесный,
амбровый
аромат
Фруктовый,
древесный
аромат
Т а б ti и п а 13
Таблица 14
Древесный,
цитрусовый,
цветочный,
аромат с
древесным,
цитрусовым,
цветочным
вкусом
Фруктовый пряный аромат и вкус как до, так и при курении в основной и в боковой струе
Долгодействующий, свенжий, глубокий, теплый, древесный, подобный ветиверии фруктовый аромат
Теплый, древесный, амбровый аромат с высоким тоном пачулей и мятным обертоном
Подобный
игорому/коньяпо8 ку амбровый древесный (подобный сандаловому дереву) аромат
,
Сладкий, древесный, вфруктовый, приподобный ветиверии, аромат с приятным, подобным жженому сахару ньюансом
Долгодействующий, сильный древесный аромат завершенной формы
Продолжение .табл. 14
Свежий древес ный, восточный, камфаррый аромат до курения и древесноперечный аромат и вкус при курении в основной, а также в боковой струе
Цветочный, ионовый, подобный ладану, и восточный аромат с ионовым и подобным восточному профилем привкуса
Подобный иону и подобный восточному аромат и вкусовой профиль с оттенком горечи.
Подобный жженому сахару ананасовый аромат и вкусовой профиль
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИИНДОЛИЛГЛИОКСИЛАМИДОВ ВЫСОКОЙ ЧИСТОТЫ | 2003 |
|
RU2315046C2 |
| Нефтяная топка для комнатных печей | 1923 |
|
SU568A1 |
| Патент США № 3453317, кл | |||
| Нефтяная топка для комнатных печей | 1923 |
|
SU568A1 |
| Патент США № 2870210, кл | |||
| Нефтяная топка для комнатных печей | 1923 |
|
SU568A1 |
| Whitmore, Mosher, I | |||
| Am | |||
| Chem | |||
| Soc., 1946, 68, 281. | |||
