Изобретение относится к способам получения концентрированных оксидов азота и касается извлечения оксидов азота из нитрозных газов.
. Цель изобретения - интенсификация процесса поглощения оксидов азота путем увеличения емкости сорбента и подавления в нем процесса кислото- образования.
Во всех случаях поглощение оксидов азота проводят под давлением 730 кПа.
Нитрозный газ, получаемый конверсией аммиака кислородом воздуха, после выделения кислого конденсата и окисления N0 до N0. имеет следунщий состав:
кмоль
об.%
N0
N0,
Ог
N.
2982,3 100,0 На выходе из абсорбера состав газа также постоянен:
N0
О. N.
H,j.O
2774,6
100,0
Пример 1 (прототип). На орошение абсорбера,подают 19,2 т/ч
раствора ШО, в ТБФ, содержащего 4% азотной кислоты, при температуре 10 С. Из колонны выходит раствор, содержащий 27% N0 (в пересчете на NOj). Для обеспечения содержания N0 в отходящих газах 0,01% требует.ся 23 тарелки абсорбера. КПД процес
5
0
5
0
5
0
5
0
S
.са абсорбции на единичной тарелке составляет 48%. Выделение N0 из сорбента проводят нагреванием его до 50-60 с при атмосферном давлении с получением N0 концентрацией 90- 95% с последующей продувкой воздухом при скорости 0,1 м/с для удаления остатков растворенных N0, которые затем присоединяется к исходному нитрозному газу. На выходе из десорбера сорбент содержит 14% азотной кислоты. При подаче сорбента без регенерации в абсорбер скорость поглощения оксидов азота уменьшается в 1,5 раза в сравнении с первоначальной. Поэтому сорбент требует регенерации от азотной кислоты и воды, которую проводят путем водной промьшки с последующим выпариванием воды из ТБФ. После охлаждения до сорбент возвращают на орошение абсорбера. Кислый конденсат, образующийся при охлаждении нитрозного газа, используется в производстве HNOj промежуточной, концентрации.
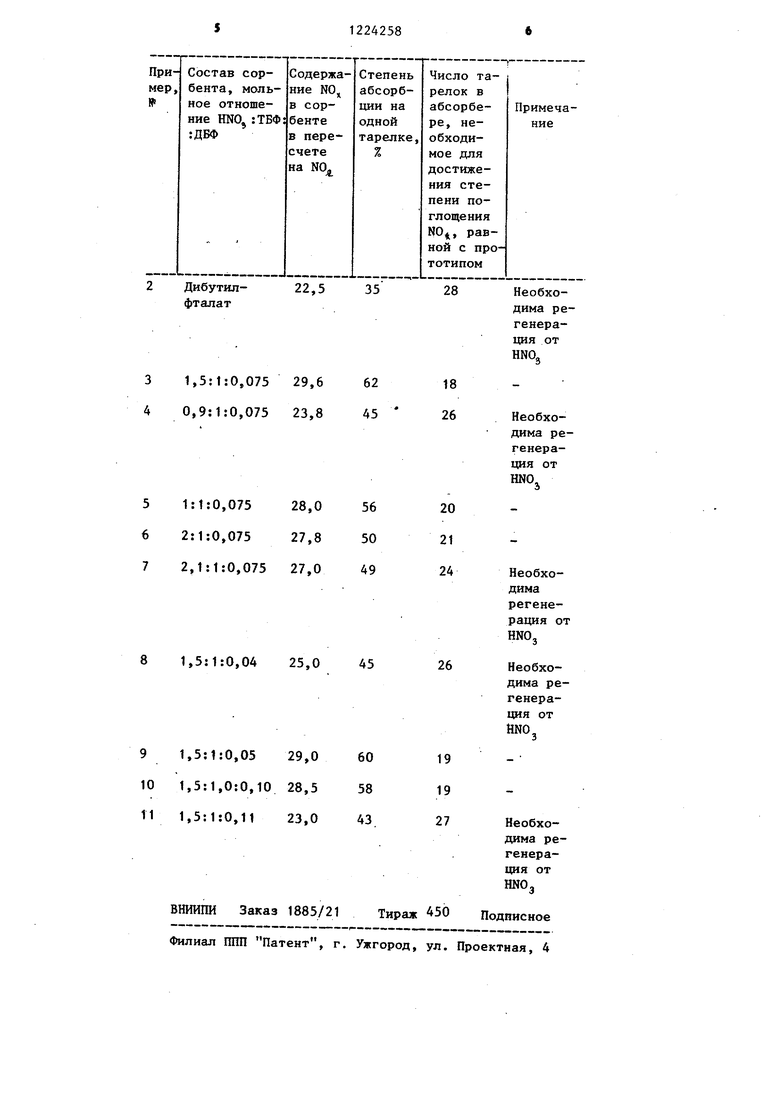
Другие примеры осуществления способа приведены в таблице (организация технологии аналогична привет денной в примере 1).
Как следует из таблицы, при составе сорбента вне указанных пределов снижается емкость поглотителя и уменьшается ЮВД процесса поглощения на единичной тарелке. Кроме того, возникает необходимость в регенерации сорбента, так как он не способен подавлять кислотообразо)ва- ние.
Интенсификация процесса осуществляется за счет того, что сорбент предложенного состава поглощает оксиды азота с КПД процесса на единичной тарелке, в 1,2-1,6 раза превышающей КПД поглощения N0 на единичной тарелке трибутилфосфатом (прототип) или дибутилфталатом. Это ведет к уменьшению числа тарелок в абсорбере при сохранении степени использования N0 на уровне прото- .типа. Кроме того, предлагаемый сорбент подавляет образование азотной кислоты в жидкой фазе, поэтому ее содержание в сорбенте согласно равновесным данным не может быть более двух молей, что позволяет отказаться от многостадийной регене- .рации сорбента, так как незначительное количество азотной кислоты, поп дающее в сорбент при поглощении нитрозного газа, вьщеляется из сорбента при получении концентрированных оксидов нагреванием всего до 50-60 с,что приводит к упрощению технологической схемы получения концентрированных N0 и снижению энергетических затрат, и в частности греющего пара.
Такие свойства сорбента обусловлены синергетическим эффектом растворения N0 в комплексах типа ТБФ-ННОз,.ТБФ-2 HNO ДБФ-ШОз , а также более сложного состава.
При мольном отношении ШОз-ТБФ в сорбенте больше 2:1 происходит разложение HNOj в растворе, вследствие чего снижается движущаяся сила поглощения N0 и соответственно уменьшается скорость процесса (увеличивается число тарелок в абсорбере, необходимое для достижения степени использования N0, равной прототипу, пример № 7). При мольном отношении HNOj: ТБФ в сорбенте менее t:1 резко падает его емкость по отношению к ся скорость мер 4) .
NOj, а также уменьшает- поглощения NOy (приПрисутствие в сорбенте дибутил- фталата вне указанных пределов не обеспечивает сохранения емкости сорбента по NOj, на уровне прототипа. Скорость поглощения оксидов азота в таких случаях уменьшается по сравнению со. скоростями поглощения N0
5
0
5
0
5
трибутилфосфатом (прототип) и дибу- тилфталатом (приме14ы № 1,2, 8 и 11).
Технологическая схема предлагаемого способа получения концентри- рованньк оксидов азота состоит в следующем.
Нитрозный газ, получаемый каталитическим окислением аммиака кислородом воздуха, проходит холодильник-конденсатор, где вьщеляется реакционная вода, и поступает вниз абсорбера, орошаемого жидким сорбентом - раствором HNQj, ТБФ и ДБФ при мольном отношении (1-2):1:(0,05- 0,1) соответственно. Поглощение N0, идет при 10-20 С давление 730 кПа. Раствор оксидов азота в сорбенте направляют в десорбер, где раствор нагревают до 50-60 с и выделяют оксиды в газовую фазу под вакуумом с концентрацией близкой к 100% или подачей горячего воздуха с расходом, обеспечивающим получение оксидов азота концентрацией 90-95%. После вьзделения концентрированных оксидов азота, направляемых потребителю, жидкий сорбент охлаждают до указанных выше температур и возвращают на абсорбцию нит- розных газов.
Предложенное техническое решение позволяет интенсифицировать процесс поглощения оксидов азота путем увеличения КПД процесса поглощения на единичной тарелке, упростить технологическую схему путем подавления кислотообразования в сорбенте на стадии поглощения и снизить энергетические затраты.

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения азотной кислоты | 1981 |
|
SU1133229A1 |
| Способ улавливания окислов азота | 1978 |
|
SU806598A1 |
| Способ получения азотной кислоты | 1984 |
|
SU1239093A1 |
| Способ получения концентрированныхОКиСлОВ АзОТА | 1976 |
|
SU831724A1 |
| Способ получения нитрита натрия | 1983 |
|
SU1155561A1 |
| Массобменная колонна для очистки газов | 1983 |
|
SU1125029A1 |
| Способ очистки выхлопных газов | 1959 |
|
SU129193A1 |
| Способ очистки газов от окислов азота | 1982 |
|
SU1011205A1 |
| УСТАНОВКА ДЛЯ ПРОИЗВОДСТВА АЗОТНОЙ КИСЛОТЫ | 2001 |
|
RU2203851C2 |
| Способ получения сложных удобрений | 1977 |
|
SU842080A1 |

| Способ очистки газов от окисловАзОТА | 1976 |
|
SU814848A1 |
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| Способ получения концентрированныхОКиСлОВ АзОТА | 1976 |
|
SU831724A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
