Предлагается новое производное имидазола, а именно гидрохлорид 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5-[бис-(2-хлорэтил)аминоэтил] тиоимидазола формулы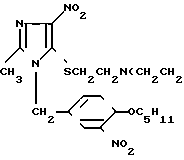 Cl)2•HCl обладающее противоопухолевой активностью.
Cl)2•HCl обладающее противоопухолевой активностью.
Цель изобретения поиск новых соединений в ряду имидазола, обладающих улучшенной противоопухолевой активностью.
П р и м е р 1. 1-(4-Амилокси-3-нитробензил)-2-метил-4-нитроимидазол.
К алкоголяту калия, полученному из 2,0 г (0,05 г·ат) металлического калия и 50 мл абсолютного этанола, прибавляют 6,4 г (0,05 моль) 2-метил-4(5)-нитроимидазола, 12,9 г (0,05 моль) 4-амилокси-3-нитробензилхлорида, и смесь кипятят в течение 7-8 ч. Осадок хлористого калия отфильтровывают, растворитель отгоняют в вакууме, добавляют воду, выпавший осадок отфильтровывают и перекристаллизовывают из смеси этанол вода (2:1). Выход 10 г (57,5% ), т.пл. 115-117оС, Rf 0,53 (ацетон гексан, 1:1).
Найдено, C 55,00; H 5,84; N 15,85.
C16H20N4O5
Вычислено, C 55,16; H 5,79; N 16,08.
ИК-спектр, ν, см-1: 1620, 1575 (ароматика); 1540, 1340 (NO2).
ПМР-спектр (CD3OD + DMCO-d6), δ, м.д. протоны бензольного кольца 7,65 (1Н, д, I 2 Гц); 7,36 (1Н, д-д, I1 8 Гц, I2 2 Гц); 7,10 (1Н, д, I 8 Гц); протон ядра имидазола 8,0 (1Н, с); 5,15 (2Н, с, СН2); 4,03 (2Н, т, ОСН2); 2,30 (3Н, с, СН3); 0,66-2,0 (9Н, м. С4Н9).
М 348 (масс-спектрометрически).
П р и м е р 2. 1-(4-Амилокси-3-нитробензил)-2-метил-4-нитро-5-бромимидазол.
К 7,0 г (0,02 моль) 1-(4-амилокси-3-нитробензил)-2-метил-4-нитроимидазола в 50 мл диметилформамида при 60оС прикапывают 1,1 мл (0,022 моль) брома. Смесь перемешивают при этой температуре в течение 2 ч и выливают в воду. Выпавший осадок отфильтровывают и перекристаллизовывают из этанола. Выход 6,5 г (76,1%), т.пл. 120-122оС; Rf 0,58 (ацетон гексан, 1:1).
Найдено, C 44,77; H 4,33; N 12,96; Br 18,54.
C16H19BrN4O5.
Вычислено, C 44,98; H 4,48; N 13,11; Br 18,70.
ИК-спектр, ν, см-1: 1610, 1570 (ароматика); 1540, 1330 (NO2).
ПМР-спектр (DMCO-d6), δ, м.д. протоны бензольного кольца 7,78 (1Н, с); 7,40 (2Н, с); 5,40 (2Н, с, СН2); 4,15 (2Н, т, ОСН2); 2,43 (3Н, с, СН3); 2,0-0,66 (9Н, м, С4Н9).
М 426/428 (масс-спектрометрически).
П р и м е р 3. Аммонийная соль 1-(4-амилокси-3-нитробензил-2-метил-4-нитро-5-ти- оимидазола.
Через смесь 6,4 г (0,015 моль) 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5- бромимидазола, 80 мл раствора аммиака в этиловом спирте при перемешивании пропускают ток сероводорода в течение 15-20 мин и продолжают перемешивание еще 10 мин. Выпавший осадок отфильтровывают, промывают на фильтре ацетоном. Выход 4,5 г (75,5% ), т.пл. 174-176оС (с разложением); Rf 0,49 (ацетон-гексан, 1:1).
Найдено, C 48,39; H 6,10; N 17,35; S 7,83.
C16H23N5O5S
Вычислено, C 48,35; H 5,83; N 17,62; S 8,07.
ИК-спектр, ν, см-1: 1610, 1570 (ароматика); 1535, 1330 (NO2).
ПМР-спектр (DMCO-d6 + D2O), δ, м.д. протоны бензольного кольца 7,70 (1Н, д, I 2 Гц); 7,46 (1Н, д-д, I1 8 Гц; I2 2 Гц); 7,22 (1Н, д, I 8 Гц), 5,28 (2Н, с, СН2); 2,18 (3Н, с, СН3); 2,00-0,66 (9Н, м, С4Н9).
П р и м е р 4. Гидрохлорид 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5-[бис-(2-хлорэтил)аминоэтил] тиоимидазола.
Смесь 2,0 г (0,005 моль) аммонийной соли 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5-тиоимидазола, 1,2 г (0,006 моль) свежеперегнанного трис-(2-хлорэтил)амина и 40 мл абсолютного этилового спирта кипятят в течение 6-7 ч. Этиловый спирт отгоняют и прибавляют хлороформ. Выпавший осадок хлористого аммония отфильтровывают, фильтрат промывают водой, сушат сернокислым натрием и насыщают безводным хлористым водородом до кислой реакции. Растворитель отгоняют, добавляют абсолютный эфир, выпавший осадок отфильтровывают. Выход 1,5 г (51,4% ), т.пл. 86-88оС; Rf 0,45 (н-бутанол уксусная кислота вода, 4:1:5).
Найдено, C 45,41; H 5,77; N 11,67; Cl 18,45; S 5,74.
C22H32Cl3N5O5S
Вычислено, C 45,17; H 5,51; N 11,97; Cl 18,18; S 5,48.
ИК-спектр, ν, см-1: 1605, 1560 (ароматика); 1545, 1340 (NO2).
Биологические испытания токсичности и противоопухолевой активности гидрохлорида 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5-[бис-(2-хлорэтил)аминоэтил] тиоимидазола (соединение I), сарколизина и структурного аналога (дигидрохлорида 1-(4-метокси-3-нитробензил)-2-[бис-(2-хлорэтил) аминометил] имидазола) проведены на белых беспородных мышах и крысах обоего пола массой 18-20 и 90-110 г соответственно, а также на мышах линии C57BI.
Соединение I нерастворимо в воде, в связи с чем при изучении противоопухолевой активности его вводили перорально в подсолнечном масле, а структурный аналог и сарколизин внутрибрюшинно. Такие пути применения являются оптимальными для исследуемых соединений (пероральное применение структурного аналога и сарколизина на некоторых штаммах: саркоме 45, Лейкозе Швеца, лимфорсаркоме Плисса, Са-755, приводит к значительному снижению их противоопухолевой активности).
Острую токсичность соединения I, структурного аналога и сарколизина изучали на мышах при пероральном введении. Установлены абсолютно смертельная (ЛД100) и максимально переносимая (МПД) дозы.
Противоопухолевую активность соединения определяли на 7 моделях перевиваемых опухолей мышей и крыс. Лечение животных с лимфосаркомой Плисса, лейкозом Швеца и асцитной карциномой Эрлиха проводили через 24 ч после перевивки, аденокарциномой молочной железы (Са-755) через 48 ч, а с саркомами 45, 180 и карциносаркомой Уокера на 4-5 сутки роста опухоли. Соединения вводили ежедневно в течение 5-6 дней (мышам) и 8 дней (крысам) в дозе, равной 1/15-1/20 от ЛД100. Подопытные и контрольные группы составляли из 8-10 животных.
В химиотерапевтических опытах через день после последнего введения соединения животных забивали эфиром и определяют массу животного и опухоли. О терапевтическом эффекте судили по проценту торможения роста опухоли (Т), а об общетоксическом действии по коэффициенту роста опухоли животного (Кр).
В опытах с Са-755 противоопухолевый эффект оценивали торможением роста опухоли, определяемым по ее объему как непосредственно, так и через 7 дней после окончания курса лечения. Полученный цифровой материал подвергали статистической обработке по методу Стъюдента-Фишера.
В результате проведенных исследований установлено, что соединение I по токсичности значительно отличается от сарколизина и структурного аналога оно соответственно в 12,5 и 1,5 раза менее токсично). Так, абсолютная смертельная доза соединения I, структурного аналога и сарколизина при пероральном применении составляет 750, 500 и 60 мг/кг соответственно. При внутрибрюшинном введении ЛД100 структурного аналога и сарколизина составляет 186 и 30 мг/кг. Результаты приведены в табл. 1.
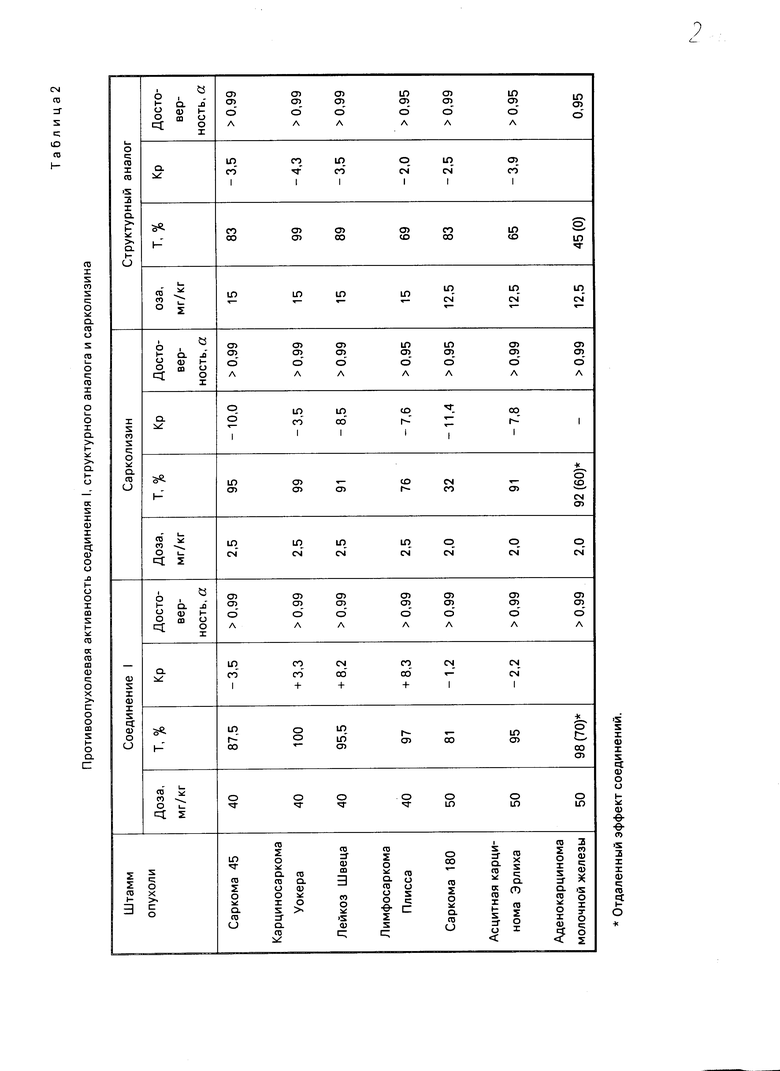
В химиотерапевтических опытах выявлено, что соединение I, подобно сарколизину и структурному аналогу, проявляет высокую противоопухолевую активность в отношении саркомы 45, карциносаркомы Уокера и лейкоза Швеца, угнетая рост опухолей на 87-100% (см. табл. 2). На лимфосаркоме Плисса соединение I по эффективности несколько превосходит сарколизин и структурный аналог, по ингибирующему действию на рост саркомы 180 он не отличается от структурного аналога, но существенно превосходит сарколизин.
Так, в адекватных дозах соединение I и структурный аналог угнетают рост данной опухоли на 81 и 83% соответственно, а сарколизин на 32%
Выраженное преимущество соединения I по сравнению со структурным аналогом установлено при лечении мышей с асцитной карциномой Эрлиха и Са-755. Так, соединение I и сарколизин угнетают рост асцитной карциномы Эрлиха и Са-755 на 91-98% а структурный аналог на 45-65%
В химиотерапевтических экспериментах выявлено также, что соединение I оказывает более слабое общетоксическое действие на организм животных, чем сарколизин и структурный аналог (см. табл. 2, Кр).
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИГИДРОХЛОРИД 1-(3-НИТРО-4-МЕТОКСИБЕНЗИЛ)-2-[БИС-(2-ХЛОРЭТИЛ)АМИНОМЕТИЛ]ИМИДАЗОЛА, ПРОЯВЛЯЮЩИЙ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 1981 |
|
SU976654A1 |
| 4(5)-(3-НИТРО-4-(3,3-ДИМЕТИЛТРИАЗЕНО-1)ФЕНИЛ)ИМИДАЗОЛ, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1982 |
|
SU1074091A1 |
| ГИДРАЗОН-4-АМИЛОКСИ-3-НИТРОАЦЕТОФЕНОНА, ПРОЯВЛЯЮЩИЙ АНТИМУТАГЕННУЮ АКТИВНОСТЬ | 1989 |
|
SU1626614A1 |
| СТЕРОИДНЫЕ ЭФИРЫ N-ДИ(2-ХЛОР-ЭТИЛ)-АМИНОФЕНИЛ-N-АЦЕТИЛАЛАНИНА, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ, АНДРОГЕННОЙ И АНАБОЛИЧЕСКОЙ АКТИВНОСТЬЮ | 1984 |
|
SU1266111A1 |
| С-4 КАРБОНАТСОДЕРЖАЩИЕ ТАКСАНЫ | 2000 |
|
RU2243223C2 |
| Способ получения активного тиоэфира производных (Z)-2-(2-амино-4-тиазолил)-2-алкоксикарбонилалкоксииминоуксусной кислоты | 1985 |
|
SU1380612A3 |
| N-(4-[БИС-(2-ХЛОРЭТИЛ)-АМИНО]-ФЕНИЛАЦЕТИЛ)-2-(4-ФТОРФЕНИЛ)-DL-АЛАНИН, ПРОЯВЛЯЮЩИЙ ПРОТИВООПУХОЛЕВОЕ ДЕЙСТВИЕ | 1988 |
|
SU1568480A1 |
| Способ получения 4,5-диарил-2-нитроимидазолов | 1979 |
|
SU940647A3 |
| N-НИТРОЗО-N-[(2-ХЛОРЭТИЛ)КАРБАМОИЛ]-L-ОРНИТИН | 2012 |
|
RU2503657C1 |
| ПРОИЗВОДНЫЕ БЕНДАМУСТИНА, РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2695383C2 |

Изобретение относится к производному имидазола, в частности гидрохлориду 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5-[бис-(2-хлорэтил)аминоэтил] тиоимидазола(1), обладающему противоопухолевой активностью, и может найти применение в медицине. Для выявления улучшенной активности в ряду имидазола получено новое соединение 1. Синтез ведут бензилированием 2-метил-4(5)-нитроимидазола 4-амилокси-3-нитробензилхлоридом в присутствии эквимолярного количества этилата калия с последующим бромированием и переводом в аммонийную соль 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5-тиоимидазола. Последний обрабатывают свежеперегнанным трис-(2-хлорэтил)амином в присутствии этилового спирта при кипячении с последующим отделением осадка NH4CI, промыванием фильтрата и насыщением его HCI. Выход соединения I 51,4%; т. пл. 86-88oС. Вещество I вызывает торможение роста опухоли равноценно с аналогом - дигидрохлоридом 1-(4-метокси-3-нитробензил)-2-бис-(2-хлорэтила) на 81 и 83%, но соединение I значительно лучше сарколизина (на 32%). Токсичность соединения I (LD5 0) 750 мг/кг против 60 мг/кг ( для сарколизина). 2 табл.
Гидрохлорид 1-(4-амилокси-3-нитробензил)-2-метил-4-нитро-5-[бис-(2-хлорэтил)аминоэт- ил]тиоимидазола формулы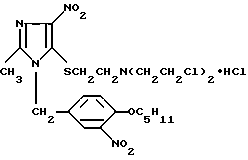
обладающий противоопухолевой активностью.
| ДИГИДРОХЛОРИД 1-(3-НИТРО-4-МЕТОКСИБЕНЗИЛ)-2-[БИС-(2-ХЛОРЭТИЛ)АМИНОМЕТИЛ]ИМИДАЗОЛА, ПРОЯВЛЯЮЩИЙ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 1981 |
|
SU976654A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
