Область, к которой относится изобретение
Данное изобретение касается противоопухолевых соединений. Более конкретно, изобретение предусматривает новые перорально активные производные паклитаксела, фармацевтические препараты на их основе и их применение в качестве перорально вводимых противоопухолевых агентов.
Предшествующий уровень техники
Паклитаксел представляет собой природный продукт, экстрагируемый из коры тиса тихоокеанского, Taxus коротколиственного, и активный компонент противоракового агента TAXOL®. Было показано, что он имеет превосходную противоопухолевую активность in vivo моделях животных, последние исследования выявили уникальный метод его действия, который включает аномальную полимеризацию тубулина и разрыв митоза. Он используется клинически для борьбы с рядом раковых болезней у людей. Он является важным противораковым агентом с терапевтической и коммерческой точек зрения. Продолжаются многочисленные клинические испытания для расширения областей применения этого агента при лечении пролиферативных заболеваний у людей. Результаты клинических исследований TAXOL® описаны многими авторами. Последний обзор статей ряда различных авторов содержится в полном собрании материалов Семинаров по онкологии, 1999, 26 (1, Suppl. 2). Другими примерами являются: Rowinsky et al., in TAXOL®: A Novel Investigational Antimicrotubule Agent, J. Natl. Cancer Inst, 82: pp 1247-1259, 1990; Rowinsky and Donehower in “The Clinical Pharmacology and Use of Antimicrotubule Agents in Cancer Chemotherapeutics”, Pharmac. Ther., 52: 35-84, 1991; Spencer and Faulds in “Paclitaxel, A Review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Potential in the Treatment of Cancer”, Drugs, 48(5) 794-847, 1994; by К.С.Nicolaou et al. in “Chemistry and Biology of TAXOL®”, Angew. Chem., Int. Ed. Engl., 33: 15-44, 1994; by F.A.Holmes, A.P.Kudelka, J.J.Kavanaugh, M.H.Huber, J.A.Ajani, V.Valero in the book “Taxane Anticancer Agents Basic Science and Current Status” edited by Gunda T.Georg, Thomas T. Chen, Iwao Ojima and Dolotrai M. Vyas, 1995, American Chemical Society, Washington, DC, 31-57; by Susan G. Arbuck and Barbara Blaylock in the book “TAXOL® Science and Applications” edited by Mathew Suffness, 1995 CRC Press Inc., Boca Ration, Florida, 379-416, а также источники, процитированные в данном описании.
Было обнаружено, что полусинтетический аналог паклитаксела, называемый доцетакселом, также имеет хорошую противоопухолевую активность и является активным ингредиентом коммерчески доступного противоракового агента TAXOTERE®. Смотри Biologically Active Taxol Analogues with Deleted A-Ring Side Chain Substitutents and Variable C-2' Configurations, J. Med. Chem., 34, pp 1176-1184 (1991); Relationships between the Structure of Taxol, Analogues and Their Antimitotic Activity, J. Med. Chem., 34, pp 992-998 (1991). Обзор клинических исследований TAXOTERE® Jorge E. Curtes и Richard Pazdur появился в Journal of Clinical Oncology, 1995, 13(10), 2643-2655. Структуры паклитаксела и доцетаксела приведены ниже вместе с обычной для этого класса соединений системой нумерации атомов; такая система применяется также в данной заявке.
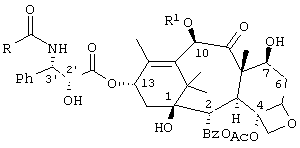
паклитаксел (TAXOL®): R = Ph; R' = ацетил;
доцетаксел (TAXOTERE®) R = трет-бутокси; R' = водород
Свидетельство того, что паклитаксел не обладает активностью при пероральном введении, можно найти в следующем абзаце из заявки WO 98/53811, Samuel Broder, Kenneth L. Duchin and Sami Selim и в цитируемых в этом абзаце ссылках, который звучит следующим образом: “Паклитаксел очень плохо абсорбируется при пероральном введении (менее 1%); см. Eisemann et al., Second NCI, Workshop on Taxol and Taxus (Sept. 1992); Suffness et al., Taxol Science and Applications (CRC Press 1995). Eisemann и другие указывают, что при пероральном введении биодоступность паклитаксела равна 0%, a Suffiiess и др. сообщают, что пероральное введение паклитаксела не представляется возможным, так как при пероральном введении с дозой 160 мг/кг/день не было обнаружено противоопухолевой активности. Более того, не было разработано эффективного метода перорального введения паклитаксела (а именно метода повышения биодоступности паклитаксела при пероральном введении) или других таксанов или аналогов паклитаксела, таких как доцетаксел, которые проявляют противоопухолевую активность. По этой причине до сих пор паклитаксел не вводят людям перорально и в курсе лечения заболеваний, реагирующих на паклитаксел”. Другое сообщение J. Terwogt et al., The Lancet, July 25 th, 1998, vol.352, p.285 также описывает низкую биодоступность паклитаксела при пероральном введении. В нашей работе мы вводили паклитаксел перорально в дозах до 160 мг/кг в модели опухолей у мышей (sc M109) и не наблюдали никаких признаков активности, мы сделали, как Suffness, вывод, что дальнейшее введение не будет эффективным, несмотря на то, что токсичные дозы не были достигнуты. Более того, наши попытки обнаружить активность вводимого перорально паклитаксела в отношении ксенотрансплантатов опухолей человека, имплантированных в организм антимических мышей или антимических крыс, оказались безуспешными.
Цель данного изобретения заключается в создании С-4 метилкарбонатных аналогов таксана, которые обладают неожиданной активностью при пероральном введении и поэтому смогут применяться против пролиферативных заболеваний. Некоторые из известных источников, имеющих отношение к данному изобретению, приведены ниже.
Некоторые производные таксана с модификацией по С-4 гидроксилъной группе известны из уровня техники.
В патенте США 5808102, Poss et al. и международной заявке WO 94/14787 содержится описание аналогов таксана, модифицированных в положении С-4.
Gunda I Georg et al. описывают синтез С-4 эфирного аналога в Tetrahedron Letters, 1994, 35(48), 8931-8934.
S. Chen et al. описывают получение С-4 циклопропилового эфира в Journal of Organic Chemistry, 1994, 59(21), 6156-6158.
Патент США 5840929, Chen, Shu-Hui, описывающий С-4 метоксиэфирные производные, был выдан 24 ноября 1998 года. На эту же тему появилась публикация: Chen, Shu-Hui, First syntheses of C-4 methyl ether paclitaxel analogs and unexpected reactivity of 4-deacetyl-4-methyl ether baccatin III, Tetrahedron Lett. 1996, 37(23), 3935-3938.
В следующем источнике обсуждается ряд C-4 эфирных или карбонатных аналогов: Chen, Shu-Hui, Wei, Jian-Mei; Long, Byron H.; Fairchild, Craig A.; Carboni, Joan; Mamber, Steven W.; Rose, William C.; Johnston, Kathey; Casazza, Anna M.; et al. Novel C-4 paclitaxel (Taxsol) analogs: potent antitumor agents. Bioorg. Med. Chem. Lett. 1995, 5(22), 2741-2746.
Получение C-4 азиридинилкарбаматных аналогов было описано в Chen, Shu-Hui, Fairchild, Craig; Long, Byron H. Synthesis and Biological Evaluation of Novel C-4 Aziridin-Bearing Paclitaxel (Taxol) analogs. J. Med. Chem. 1995, 38(12), 2263-2267.
Следующие источники описывают реакции или превращения, которые можно использовать для получения C-4 аналогов.
Новый метод модификации в C-4 положении 10-деацетилбаккатина-III Uoto, Kouichi; Takenoshita, Haruhiro; Ishiyama, Takashi; Terasawa, Hirofumi; Soga, Tsunehiko. Chem. Pharm. Bull, 1997, 45(12), 2093-2095.
Samaranayke, Gamini; Neidigh, Kurt A.; Kingston, David G.I. Modified taxols, 8 Deacylation and reacylation of baccatin III. J. Nat. Prod. 1993, 56(6), 884-898.
Datta, Apurba; Jayasinghe, Lalith R.; Georg, Gunda I. 4-Deacetyltaxol and 10-Acetyl-4-deacetyltaxotere: Synthesis and Biological Evaluation. J. Med. Chem. 1994, 37(24), 4258-4260. Несмотря на вышеупомянутые примеры C-4 аналогов или методов их получения, нет сведений об орально активных C-4 аналогах. И TAXOL®, и TAXOTERE® не проявляют оральной активности на моделях людей или животных, как указано в нижеследующих источниках о таксанах и оральных модуляторах. Таким образом, в известном уровне техники не содержится предположения о том, что C-4 таксаны отличаются от других таксанов и, следовательно, они не должны быть активными при пероральном введении. Как нам известно, в предшествующем уровне техники не указаны никакие C-4 аналоги, которые можно было бы вводить перорально.
Данное изобретение описывает новые C-4 аналоги, которые благодаря их уникальному замещению обладают активностью при пероральном введении.
В нижеследующих источниках описаны способы или возможные методы использования таксанов, активных при пероральном введении.
Существуют методы введения таксанов в присутствии модуляторов, которые, как сообщалось, позволяют повысить содержание таксанов в плазме при пероральном введении: Terwogt, Jetske M., Meerum; Beijnen, Jos H., Ten Bokkel Huinink, Wim W.; Rosing, Hilde; Schellens, Jan H.M. Coadministration of cyclosporin enables oral therapy with paclitaxel. Lancet (1998), 352(9124), 285.
Terwogt, Jetske M., Meerum; Malingre, Mirte M., Beijnen, Jos H., Huinink, Wim W. ten Bokkel; Rosing, Hilde; Koopman, Franciska J.; Van Tellingen, Olaf, Swart, Manha; Schellens, Jan H.M. Coadministration of oral cyclosporin A enables oral therapy with paclitaxel. Clin. Cancer Res. (1999), 5(11), 3379-3384.
Hansel, Steven В. A method of making taxanes orally bioavailable by coadministration with cinchonine. Заявка WO 9727855, опубликованная 7 августа 1997 года.
Broder, Samuel; Duchin, Kenneth L.; Sclim, Sami. Method and compositions for administering taxanes orally to human patients using a cyclosporin to enhance bioavailability. Заявка WO 9853811, опубликованная 3 декабря 1998 года. В этих сообщениях нет данных о противоопухолевой активности, но наличие таксанов в плазме экстраполируется, что свидетельствует об их потенциальной полезности при лечении раковых заболеваний.
Опубликовано, по меньшей мере, одно сообщение об активности пролекарств при пероральном введении при использовании доклинических моделей животных: Scola, Paul M.; Kadow, John F.; Vyas, Dolatrai M. Preparation of paclitaxel prodrug derivatives. Заявка EP 747385, опубликованная 11 декабря 1996 года.
Биодоступность пролекарства при пероральном введении лекарства не описана и нет сообщений о дальнейшем изучении этих соединений.
Недавно появился реферат, в котором описан аналог таксана (IDN-5109) с оральной активностью по отношению к опухолям у мышей, он стал известен American Association of Cancer Researchers in Philadelphia в 1999 году. Источник этого сообщения:
Pratesi G, Polizzi D, Totoreto M, Riva A, Bombardelli E, Zunino F.: IDN5109 a new taxane active after oral administration. Proc. Am. Assoc. Cancer Res. 1999, 40, Abs 1905, Istituto Nazionale Tumori, 20133 Milan and Indena SpA, 20139, Milan, Italy. Структура этого соединения очень отличается от структуры соединений по изобретению. В отличие от соединений согласно настоящему изобретению IDN-5109 получен на основе 14-бета-гидроксибаккатина III и содержит ацетатную группу у гидроксильной в положении С-4.
Имеются два источника об iv активности этого соединения:
Nicoletti ML, Rossi С, Monardo С, Stura S, Morazzoni P, Bombardelli E, Valoti G., Giavazzi R.: Antirumor efficacy of a paclitaxel analogue, IDN5109, on human ovarian carcinoma xenografts with different sensitivity to paclitaxel. Proc. Am. Assoc. Cancer Res. 1999, 40 Abs 1910 [Evals+citations].
Polizzi Donatella; Pratesi, Graziella; Totoreto Monica; Supino, Rosanna; Riva, Antonella; Bombardelli, Ezio; Zunino Franco. A novel taxane with improved tolerability and therapeutic activity in a panel of human rumor xenografts. Cancer Res. 1999, 59(5), 1036-1040.
Паклитаксел является лекарством, действие которого сильно зависит от схемы приема и которое обычно выигрывает от продолжительного действия на опухоль. Это относится к механизму действия паклитаксела, поскольку таксаны только распознают и связываются с тубулином в полимеризованном состоянии, что происходит только во время короткого периода цикла раковой клетки. Используемые в настоящее время внутривенные вливания (1-3 часа) являются эффективньми и исключают рутинное применение продолжительных (>24 часов) непрерывных схем. Однако таксан, вводимый перорально, для обеспечения такого длительного периода воздействия может привести к увеличению стоимости лечения и усложнению его. В последнее время проводились клинические испытания при повторяющемся введении один раз в неделю умеренных (а именно отличающихся от максимально переносимых) доз TAXOL®, и таксан, вводимый перорально, будет идеальным для такой схемы. Другие клинические показания к применению таксанов (например, ревматоидный артрит, рассеянный склероз) выигрывают от доступности таксана, вводимого перорально. Вводимый перорально эффективно действующий таксан является как привлекательной альтернативой, используемой в настоящее время в парентеральной схеме введения таксана, так и лекарством с потенциальным терапевтическим преимуществом, так как предстоит изучение многих схем введения.
Таким образом, очевидно, что существует необходимость в таксанах с хорошей биодоступностью и высокой эффективностью при пероральном введении, сравнимых с паклитакселем, вводимым парентерально.
Сущность изобретения
Настоящее изобретение относится к новым противоопухолевым соединениям формулы I или их фармацевтически приемлемым солям:
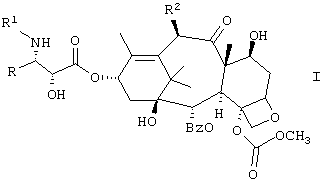
где R обозначает фенил, изопропил или трет.бутил;
R1 обозначает -С(О)RZ, где RZ является (СН3)3СО-, (СН3)3ССН2-, СН3(СН2)3О-, циклобутилом, циклогексилокси или 2-фурилом;
R2 обозначает СН3С(O)O-.
Другой аспект данного изобретения относится к способу ингибирования роста опухолей в организме млекопитающего, который включает введение указанному млекопитающему эффективного в отношении опухоли количества соединения формулы I или его фармацевтически приемлемых солей. Предпочтительно, чтобы применялся пероральный метод введения.
Еще один аспект данного изобретения относится к фармацевтической композиции, которая содержит эффективное по отношению к опухоли количество соединения формулы I или его фармацевтически приемлемых солей в сочетании с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами, разбавителями или адъювантами.
Подробное описание изобретения
В нижеследующем описании, если иное не оговорено специально или в контексте, применяются следующие определения. Обозначения, использованные однажды, сохраняют то же значение по всему описанию, если не указано иное.
Цифры, указанные после символа “С”, обозначают число атомов углерода, которое может содержать конкретная группа. Например, “С1-6алкил” означает линейную или разветвленную углеродную цепь, имеющую от одного до шести атомов; примеры включают: метил, этил, н-пропил, изопропил, н-бутил, втор.бутил, изобутил, трет.бутил, н-пентил, втор.пентил, изопентил и н-гексил. В зависимости от контекста “С1-6алкил” может также относиться к С1-6алкилену, который соединяет две группы; примеры включают: пропан-1,3-диил, бутан-1,4-диил, 2-метилбутан-1,4-диил и т.д. “С2-6алкенил” означает линейную или разветвленную углеродную цепь, имеющую, по меньшей мере, одну углерод-углеродную двойную связь и содержащую от 2 до 6 атомов углерода; примеры включают: этенил, пропенил, изопропенил, бутенил, изобутенил, пентенил и гексенил. В зависимости от контекста “С2-6алкенил” может также относиться к С2-6алкендиилу, который соединяет две группы; примеры включают этилен-1,2-диил (винилен), 2-метил-2-бутен-1,4-диил, 2-гексен-1,6-диил и т.д. “С2-6алкинил” означает линейную или разветвленную углеродную цепь, содержащую, по меньшей мере, одну углерод-углеродную тройную связь и от 2 до 6 атомов углерода; примеры включают: этинил, пропинил, бутинил и гексинил. Используемые здесь термины трет.бутилокси и трет.бутокси являются взаимозаменяемыми.
“Арил” означает ароматический углеводородный радикал, содержащий от 6 до 10 атомов углерода; примеры включают фенил и нафтил. “Замещенный арил” означает арил, замещенный одним-пятью (но предпочтительно, одним-тремя) заместителями, выбранными из С1-6алканоилокси, гидрокси, галогена, С1-6алкила, трифторметила, C1-6алкокси, арила, С2-6алкенила, С1-6алканоила, нитро, амино, циано, азидо, С1-6алкиламино, ди-С1-6алкиламино и амидо. “Галоген” означает фтор, хлор, бром и йод.
“Гетероарил” означает пяти- или шестичленное ароматическое кольцо, содержащее, по меньшей мере, один и до 4 атомов, отличающихся от углерода, выбранных из кислорода, серы и азота. Примеры гетероарила включают: тиенил, фурил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, тетразолил, тиатриазолил, оксатриазолил, пиридил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил и т.п.
“Группы, защищающие гидроксил”, включают, без ограничения, эфирные, такие как метил, трет.бутил, бензил, п-метоксибензил, п-нитробензил, аллил, тритил, метоксиметил, метоксиэтоксиметил, этоксиэтил, 1-метокси-1-метоксиэтил, тетрагидропиранил, тетрагидротиопиранил, диалкилсилилэфиры, такие как диметилсилиловый эфир, и триалкилсилиловые эфиры, например триметилсилиловый эфир, триэтилсилиловый эфир и трет.бутилдиметилсилиловый эфир, диалкилалкоксисилиловые эфиры, такие как диизопропилметоксисилиловые эфиры; сложноэфирные группы, такие как бензоил, ацетил, фенилацетил, формил, моно-, ди- и тригалоидацетил, например хлорацетил, дихлорацетил, трихлорацетил, трифторацетил; и карбонатные, такие как метил, этил, 2,2,2-трихлорэтил, аллил, бензил и п-нитрофенил. Дополнительные примеры групп, защищающих гидроксил, можно найти в известных работах, например, в Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed, 1999, John Wiley and Sons, New York.
“Ph” означает фенил; “ipr” означает изопропил.
Заместители у замещенных алкила, алкенила, алкинила, арила и гетероарила и фрагментов, описанных в данном описании, могут представлять собой алкил, алкенил, алкинил, арил, гетероарил и/или могут содержать азот, кислород, серу, галогены и включают, например, низший алкокси, такой как метокси, этокси, бутокси, галоген, такой как хлор или фтор, нитро, амино или кето.
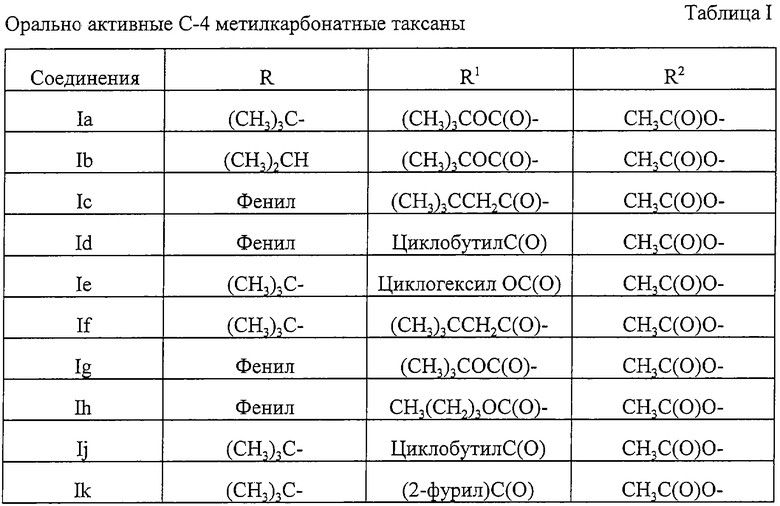
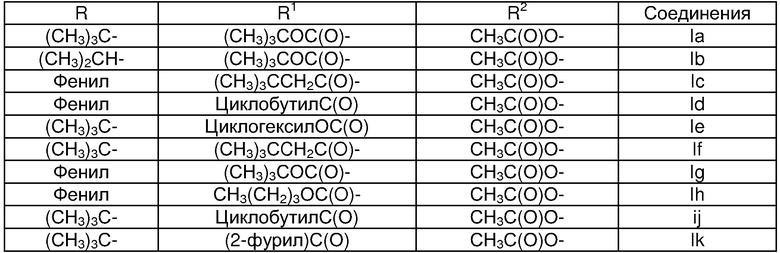
Предпочтительный вариант относится к соединениям формулы I или их фармацевтически приемлемым солям, которые описаны в таблице I ниже.
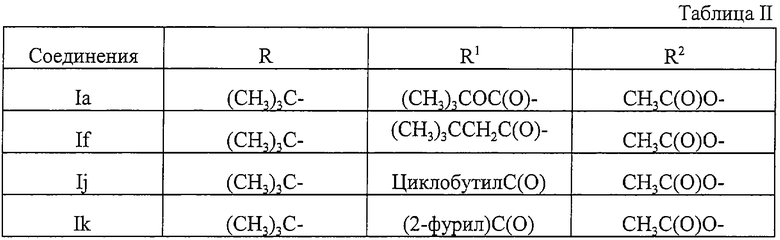
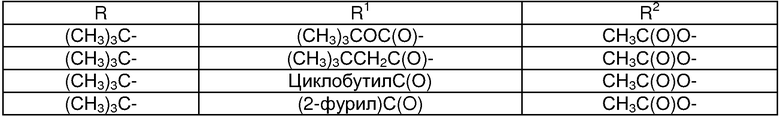
Еще более предпочтительными являются соединения формулы I или их фармацевтически приемлемые соли, представленные в таблице II.
Новые соединения, имеющие общую формулу I, обладают значительным ингибирующим действием в отношении аномальной пролиферации клеток и имеют терапевтические свойства, позволяющие осуществлять лечение пациентов с патологическими состояниями, связанными с аномальной пролиферацией клеток. Кроме того, эти соединения характеризуются значительной оральной биодоступностью и таким образом проявляют положительное терапевтическое действие после перорального введения. Патологические состояния включают аномальную пролиферацию злокачественных и незлокачественных клеток в различных тканях и/или органах, включая, без ограничения, мускулы, кости и/или соединительные ткани, кожу, мозг, легкие и половые органы; лимфатическую систему и/или почечную систему; клетки молочной железы и/или гемоциты; печень, пищеварительную систему и поджелудочную железу; щитовидную железу и/или надпочечники. Эти патологические состояния также включают псориаз, твердые опухоли; рак яичников, груди, мозга, простаты, толстой кишки, желудка, почек и/или тестикулярный рак, саркому Капоши, холангиокарциному, хориокарциному, нейробластому; опухоль Вильмса, болезнь Ходжкина, меланомы, множественную миелому, хронический лимфолейкоз и острые или хронические гранулоцитарные лимфомы.
Новые соединения по изобретению особенно пригодны для лечения лимфомы, не являющейся лимфомой Ходжкина, множественной миеломы, меланомы и рака яичников, мочевого пузыря, oesophageal, легких и рака груди. Соединения можно использовать для профилактики или задержки признаков болезни или повторного проявления этих признаков, или для лечения этих патологических состояний. Соединения можно применять в качестве ингибиторов задержки развития кровеносных сосудов как в случае ракового заболевания, так и в случае аномального заживления ран или других гиперпролиферативных заболеваний, зависимых от образования кровеносных сосудов.
Кроме того, соединения формулы I пригодны для лечения и/или профилактики полицистоза почек (PKD) и ревматоидного артрита. Соединения по изобретению могут также применяться для лечения болезни Альцгеймера, или болезни Паркинсона, или рассеянного склероза. Хотя некоторые продукты общей формулы I представляют интерес благодаря их преимуществам по сравнению с коммерческими таксанами при iv введении, их основное достоинство заключается в их уникальных свойствах после перорального введения.
Соединения по изобретению могут быть получены обычными методами органической химии. Схемы 1-3, описывающие получения соединений формулы I, приведены только для иллюстрации и не ограничивают способы получения соединений только этими методами.
Соединение формулы I может быть получено способами, отображенными на нижеследующих схемах 1-3. Эти методы можно легко изменить для получения соединений формулы I не описанными здесь методами. Дальнейшие вариации этих методов получения этих же соединений по несколько иным схемам очевидны для специалиста. Нумерация атомов в производном баккатина III формулы II такая же, как показано выше для структуры таксана.
Один из способов получения соединений по изобретению показан на Схеме 1. На стадии (а) азетидинон IV реагирует с соединением формулы II (производное баккатина III). Класс азетидинонов (β-лактамов) формулы IV хорошо известен. Способы получения соответствующим образом замещенных β-лактамов можно найти в патенте США 5175315, заявке ЕР 0590267, других патентах США или в литературных источниках, упомянутых выше или в ссылках, приведенных в: Ojima et al. in Tetrahedron, 48, №34, pp 6985-7012 (1992); Journal of Organic Chemistry, 56, pp 1681-1683 (1991) and Tetrahedron Letters, 33, №39, pp 5737-5740 (1992); by Brieva et al. in J. Org. Chem., 58, pp 1068-1075; by Palomo et al., Tetrahedron Letters, 31, №44, pp 6429-6432 (1990) и в Rey, Allan W.; Droghini, Robert; Douglas, James L.; Vemishetti, Purushotham; Boettger Susan D.; Racha, Saibaba; Dillon, John L. Can. J. Chem. 72(10), 2131-2136 (1994).
Все приведенные здесь источники указаны в качестве ссылок. Методы, которые можно адаптировать для получения других азетидинонов формулы IV, не описанные в данной заявке, или в приведенных ссылках, или в других источниках, очевидны для специалиста.
Производные баккатина III (формула II) могут быть присоединены к боковой цепи с использованием методов, уже хорошо известных из уровня техники. Многие указанные здесь источники и Tetrahedron, 48, №34, рр 6985-7012 (1992) описывают способы, когда класс азетидинонов формулы IV реагирует с (С)13-гидроксилом производных баккатина III или его алкоксидом металла с получением аналогов таксана с различными (С) 13-боковыми цепями. На стадии (а) схемы 1 предпочтительно превратить гидроксильную группу в (С)13-положении в алкоксид металла до сочетания. Образование алкоксида металла можно осуществить реакцией соединения формулы II с сильным основанием металла, таким как диизопропиламид лития, С1-6алкиллитий, бис(триметилсилил)-амид лития, натрия или калия, фениллитий, гидрид натрия, гидрид калия, гидрид лития и т.п. Например, когда требуется алкоксид лития, соединение формулы II может реагировать с н-бутиллитием в инертном растворителе, таком как тетрагидрофуран. Например, присоединение замещенных баккатинов к соответствующим образом замещенным лактамам по методу Холтона см. в патентах США 5175315, США 5466834, США 5229526, США 5274124, США 5243045, США 5227400, США 5336785 и патенты США 5254580, США 5294637 или ЕР 0590267 А2. Некоторые примеры применения β-лактамов для получения других замещенных производных таксана приведены в заявке WO 94/14787. В ней также описан альтернативный метод присоединения цепей замещенного изосерина к замещенным баккатинам, который можно использовать для соединений по изобретению. Этот же альтернативный метод описан в публикации Kingston et al., Tetrahedron Letters (1994), 35(26), 4483-4484. Дальнейшая информация об альтернативных методах присоединения боковых цепей к баккатинам содержится в Thottathil et al., заявка ЕР 735036, опубликованная 10/2/96.
Схема 1
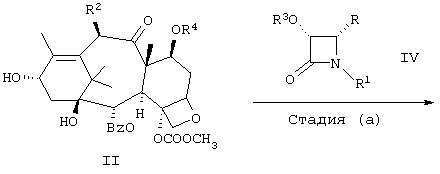
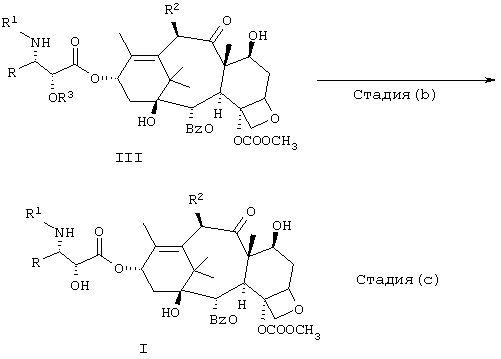
Схема 2
В данном описании R3 и R4 обозначают обычные группы, защищающие гидроксильные группы. Обычные группы, защищающие гидроксилы, представляют собой группы, которые можно использовать для блокировки или защиты гидроксильной функции, и они хорошо известны специалистам в данной области. Предпочтительно, чтобы эти группы были такими, которые можно удалить способами, которые не вызывают деструкции оставшейся части молекулы. Примеры таких легко удаляемых групп, защищающих гидроксилы, включают хлорацетил, метоксиметил, 1-метил-1-метоксиэтил, тетрагидропиранил, тетрагидротиопиранил, диалкилсилиловые эфиры, такие как диметилсилиловый эфир, и триалкилсилиловые эфиры, такие как триметилсилиловый эфир, триэтилсилиловый эфир и трет.бутилдиметилсилиловый эфир, диалкилалкоксисилиловые эфиры, такие как диизопропилметоксисилиловые эфиры; 2,2,2-трихлорэтилоксиметил, 2,2,2-трихлорэтилоксикарбонил (или просто трихлорэтилоксикарбонил), бензилоксикарбонил и т.п. Другие подходящие группы для защиты гидроксильных групп, которые могут быть использованы, можно найти в Главе 2 “Protecting Groups in Organic Synthesis”, 3rd Ed, by Theodora W. Greene and Peter G. M. Wuts (1999, John Wiley and Sons, New York). Защитная группа для соединений формулы IV, которая используется часто в известных публикациях, представляет собой триалкилсилил. Наиболее предпочтительные группы для R3 включают 1-метил-1-метоксиэтил (МОР), триалкилсилиловый эфир или диалкилалкоксисилиловый эфир, такой как диизопропилметоксисилиловый эфир. Наиболее предпочтительной группой для R4 является диалкилалкоксисилиловый эфир, такой как диизопропилметоксисилиловый эфир, но предпочтительными являются также триалкилсилиловый эфир или карбонат, такой как бензилкарбонат. На стадии (b) защитная группа R3 или R4 или возможно обе удаляются. Если R3 или R4 представляют собой защитные группы на основе силила, удаление осуществляется триэтиламинтригидрофториодом в среде тетрагидрофурана. Можно использовать и другие фторидные источники. Например, применение могут найти тетрабутиламмонийфторид, пиридинийгидрофторид, фторид калия или фторид цезия. Фторид калия можно использовать в сочетании с комплексообразующим агентом, таким как 18-краун-6 или т.п. для облегчения десилилирования. При этих условиях обычно используют растворитель, такой как ацетонитрил. Для удаления силильных групп можно применять и другие условия, например соляную кислоту умеренной концентрации или трифторуксусную кислоту и сорастворитель, такой как ацетонитрил или ТГФ. Такие же кислые условия подходят для удаления 1-метил-1-метоксиэтильной (МОР) защитной группы.
Используемые в действительности условия зависят от защитных групп R3 и R4. Например, согласно одному предпочтительному методу можно применять группу МОР в качестве R3 и диизопропилметоксисилилэфирную группу в качестве R4. В этом случае на стадии (b) используют умеренные кислые условия с применением соляной кислоты и органического растворителя. Полученное 2'-депротектированное соединение подвергается воздействию источника фторида, такого как триэтиламинтригидрофторид, в ТГФ на стадии (с) с получением соединения I после хроматографической или кристаллографической очистки.
Схема 3
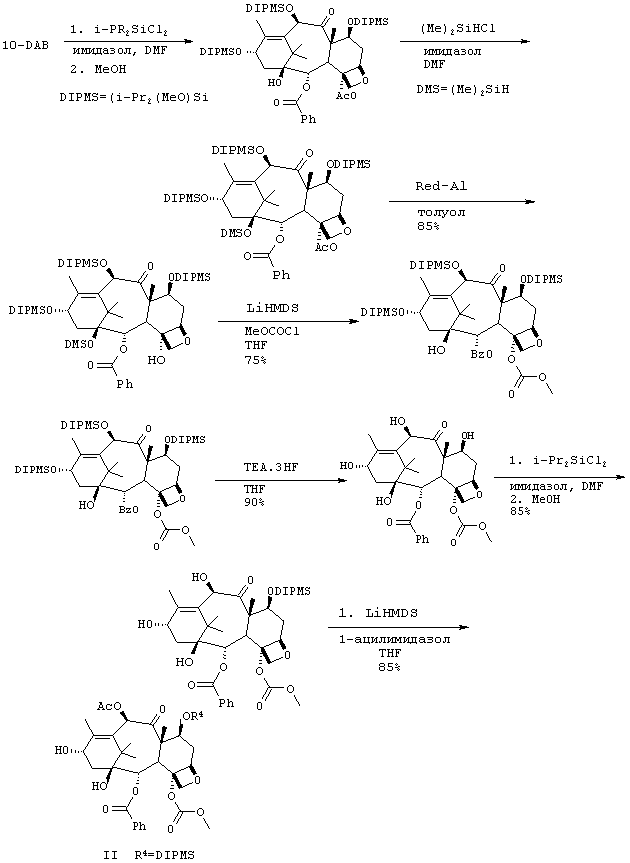
Предпочтительный способ синтеза защищенного С-4 метилкарбонатного баккатина.
Другой аспект данного изобретения включает синтез соединений I с новыми заместителями R2 в С-10 положении. Эти соединения могут быть получены присоединением альтернативной сложноэфирной группы, а не ацетатэфирной группы, которая показана на схеме 3.
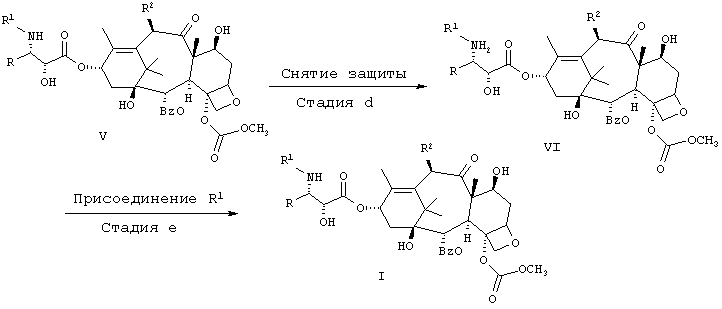
Альтернативный способ получения соединений I представлен на схеме 2. Он включает конверсию одного соединения V, в котором R' образует защищающую азот группу, например трет.ВuО(СО)-(трет.Вос) или РhСН2ОС(O)-(СВZ). Эти группы могут быть удалены путем кислотного гидролиза или в случае CBZ-гидрогенолиза. Получение аминного промежуточного VI описано в примерах и осуществляется хорошо известными методами. Промежуточный амин VI растворяют в инертном растворителе, таком как этилацетат, и добавляют основание, такое как бикарбонат натрия. Для получения соединения I добавляют стехиометрическое или немного большее количество хлорангидрида кислоты (то есть R'-С(O)Сl), хлорформиата или же ангидрида кислоты.
Получение производных баккатина формулы II (как показано на схеме 1, где R2 обозначает АсО-) показано на схеме 3 и проиллюстрировано в препаративном примере 7. Конкретные примеры, следующие ниже, иллюстрируют способы получения соединений по изобретению и не ограничивают объем изобретения. Способ может быть адаптирован для получения соединения, охватываемого данным изобретением, но конкретно не описанного. Специалистам в данной области также очевидно, что можно изменить способы получения одного и того же соединения.
В следующих примерах все температуры указаны в градусах Цельсия, если не оговорено иное. Данные ядерного магнитного резонанса (ЯМР) относятся к химическим сдвигам, выраженным в ч/млн (ррm) при использовании в качестве эталона тетраметилсилана (TMS). Относительные величины, указываемые для различных сдвигов в протонном ЯМР, соответствуют числу атомов водорода конкретного функционального типа в молекуле. Природа сдвигов описывается в виде широкого синглета (bs или br s), широкого дублета (bd или br d), широкого триплета (bt или br t), широкого квадруплета (bq или br q), синглета (s), мультиплета (m), дублета (d), квадруплета (q), триплета (t), дублета дублета (dd), дублета триплета (dt) и дублета квадруплета (dq). В качестве растворителя для ЯМР используют дейтерированный ацетон-d6, ДМСО-d6 (пердейтеродиметилсульфоксид), D-20 (дейтерированная вода), СDСl3 (дейтерированный хлороформ) и другие обычные дейтерированные растворители. Данные ИК включают только длины волн (см-1), характеризующие функциональные группы.
Целит является товарным знаком для диатомовой земли, зарегистрированным фирмой Johns-Manville Products.
В нижеследующих экспериментах использовали силикагель 60 с размером частиц 230-400 меш, поставляемый ЕМ Separations Technology.
Сокращения, используемые в данном описании, являются общепринятыми. Некоторые из них: ДАВ (деацетилбаккатин III); MS (масс-спектроскопия); HRMS (масс-спектроскопия высокой степени разрешения); Ас (ацетил); Ph (фенил); v/v (объем/объем); FAB (бомбардировка быстрыми электронами); NOBA (м-нитробензиловый спирт); мин (минута, -ы); ч или час (час, -ы); DCC (1,3-дициклогексилкарбодиимид); ВОС (трет.бутоксикарбонил); CBZ или Cbz (бензилоксикарбонил); Вn (бензил); Bz (бензоил); Тrос (2,2,2-трихлорэтилоксикарбонил); DMS(диметилсилил); TBAF (тетрабутиламмонийфторид); DMAP (4-диметиламинопиридин); TES (триэтилсилил); DMSO (диметилсульфоксид); THF (тетрагидрофуран); HMDS (гексаметилдисилилазан); MeOTf (метилтрифлат); NMO (морфолин-N-окись); (DHQ)2PHAL (гидрогинин-1,4-фталазиндииловый диэфир); Tf = трифлат = трифторметансульфонат; LRMS (масс-спектроскопия с низкой степенью разрешения); ESI (ионизация электрораспылением); TEMPO (2,2,6,6-тетраметил-1-пиперидинилокси, свободный радикал); DBU (диазобициклоундекан); МОМСl (хлорметилметиловый эфир); Ас (ацетил); Аr (арил); DCI (десорбционная химическая ионизация); DMF (диметилформамид); LiHMDS (гексаметилдисилазан лития или бис(триметилсилил)амид лития); i-РrОН (изопропиловый спирт); rt (комнатная температура); tBu (трет.бутил); TES (триэтилсилил); TLC (тонкослойная хроматография); Y (выход); ТРАР (тетрапропиламмонийперутенат); МСРВА (мета-хлорпероксибензойная кислота); LDA (диизопропиламид лития); TBS (трет.бутилдиметилсилил); 18-краун-6 (1,4,7,10,13,16-гексаоксациклооктадекан); DEAD (диэтилазодикарбоксилат); Red-Al® (Aldrich Catalogue) представляет собой 65 вес.%-ный раствор бис(2-метоксиэтокси)алюминийгидрида в толуоле; DCM означает дихлорметан; “sat” означает насыщенный.
Препаративные примеры
Препаративный пример I
(+)-цис-4-трет.бутил-1-трет.бутилоксикарбонил-3-триэтилсилилокси-азетидин-2-он
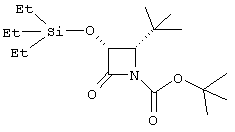
Триметилацетальдегид (20,3 мл, 1,25 экв) добавляли к перемешиваемой суспензии п-анизидина (18,4 г, 0,150 моль) и безводному Na2SO4 (150 г) в безводном DCM (250 мл) при комнатной температуре. Через 2 часа смесь отфильтровывали и промывали твердый остаток дополнительным количеством безводного DCM. Удаляли из фильтрата растворитель и растворяли кристаллический остаток в безводном DCM (750 мл) и помещали в атмосферу азота. Добавляли триэтиламин (48,0 мл, 2,3 экв) и охлаждали реакционную смесь до -78°С. Добавляли по каплям бензилоксиацетилхлорид (27,2 мл, 1,15 экв) и затем давали реакционной смеси нагреться до комнатной температуры. Через 24 часа смесь промывали 0,5 М НСl (дважды), насыщенным водным раствором NaHCO3, рассолом и высушивали (Na2SО4). Удаляли растворитель и остаток подвергали хроматографии на колонке с силикагелем (градиент элюирования с 20% DCM в гексане, содержащем 0-20% EtOAc), получая (+)-цис-4-трет.бутил-3-бензилокси-1-п-метоксибензилазетидинон в виде кристаллического твердого вещества (46,9 г, 92%). 1Н ЯМР (CDCl3) δ 1,09 (s, 9H), 3,81 (s, 3Н), 4,15 (d, 1H, J=5,5 Гц), 4,77 (d, 1H, J=11,9 Гц), 4,81 (d, 1H, J=5,5 Гц), 5,03 (d, 1H, J=11,9 Гц), 6,87-7,43 (m, 9H); LRMS (ESI) 340 ([M+H]+). Раствор аммонийнитрата церия (60,4 г, 3,6 экв) в 900 мл воды добавляли к хорошо перемешиваемому раствору азетидинона (10,38 г, 30,6 ммоль) в ацетонитриле (600 мл) на ледяной бане в течение 1 ч. Реакционную смесь затем экстрагировали EtOAc (дважды) и объединенные органические экстракты промывали насыщенным водным раствором NаНСО3 (дважды), 20% водным раствором NаНSО3, насыщенным раствором NаНСО3 и рассолом. После сушки (Na2SO4) удаляли растворители и осадок подвергали хроматографии на колонке с силикагелем (градиент элюирования порциями гексана, содержащего 10-40% EtOAc) с получением 5,64 г слегка загрязненного (+)-цис-3-бензилокси-4-трет.бутилазетидина. 1Н ЯМР (CDCl3) δ 1,04 (s, 9H), 3,51 (d, 1H, J=5,2 Гц), 4,71 (m, 2H), 4,96 (d, 1H, J=11,9 Гц), 6,10 (brs, 1H), 7,35 (m, 5H). Суспензию этого продукта (5,54 г, 23,8 ммоль) и 2,5 г 10% Pd на угле в абсолютном EtOH (100 мл) гидрировали (34 ф/дюйм2 (234,4 кПа) H2, аппарат Парра) в течение 23 ч. Добавляли еще 2 г Pd-катализатора и продолжали гидрирование еще 17 ч при давлении 50 ф/дюйм2 (344,7 кПа). Катализатор отделяли при фильтровании и удаляли из фильтрата растворитель с получением сырого (+)-цис-3-гидрокси-4-(трет.бутил)азетидин-2-она. 1Н ЯМР (СDСl3 + 1 капля D2O) δ 1,05 (s, 9H), 3,48 (d, 1H, J=5,0 Гц), 4,98 (d, 1H, J=5,0 Гц). Этот продукт растворяли в сухом ДМФ (40 мл) и добавляли имидазол (3,24 г, 2 экв) и триэтилсилилхлорид (4,0 мл, 1 экв). Через 10 минут реакционную смесь распределяли между водой и смесью EtOAc и гексана (1:1). Органическую фазу промывали водой (дважды), рассолом и затем сушили (Na2SO4). Растворители удаляли и остаток подвергали хроматографии на колонке с силикагелем (градиент элюирования с 20-25% EtOAc в гексане) с получением (+)-цис-4-трет.бутил-3-триэтилсилилоксиазетидин-2-она (3,86 г): 1Н ЯМР (CDCl3) δ 0,70 (m, 6H), 0,98 (m, 18H), 3,39 (d, 1H, J=5,0 Гц), 4,88 (dd, 1H, J=2,1, 5,0 Гц), 6,08 (br s, 1H). Раствор этого азетидинона (2,04 г, 7,92 ммоль), диизопропилэтиламин (1,66 мл, 1,2 экв), ди-трет.бутилдикарбонат (1,90 г, 1,1 экв) и п-диметиламинопиридин (194 мг, 0,2 экв) в сухом DCM (24 мл) перемешивали при комнатной температуре 3 часа. Реакционную смесь разбавляли DCM, промывали рассолом и сушили (Na2SO4). Удаление растворителя с последующей хроматографией на силикагеле (элюирование 0-20% EtOAc в гексане) привело к получению 2,71 г (96%) соединения, указанного в заголовке в виде масла: 1Н ЯМР (СDСl3) δ 0,70 (m, 6Н), 1,00 (m, 9H), 1,09 (s, 9H), 1,53 (s, 9H), 3,90 (d, 1H, J=6,5 Гц), 4,93 (d, 1H, J=6,5 Гц).
Препаративный пример 2
(+)-цис-1-трет.бутилоксикарбонил-4-изопропил-3-триэтилсилилоксиазетидин-2-он
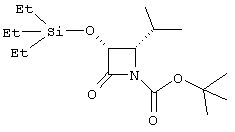
Изобутиральдегид (4,62 мл, 1,25 экв) добавляли к перемешиваемой суспензии п-анизидина (5,00 г, 40,7 ммоль) и безводного Na2SO4 (25 г) в безводном DMC (80 мл) при комнатной температуре. Через 1 час реакционную смесь фильтровали и твердое вещество промывали дополнительным количеством безводного ДХМ. Из фильтрата удаляли растворитель и остаток растворяли в безводном ДХМ (200 мл) и помещали в атмосферу азота. Добавляли триэтиламин (13,1 мл, 2,3 экв) и реакционную смесь охлаждали до -78°С. По каплям добавляли ацетоксиацетилхлорид (5,00 мл, 1,15 экв) и оставляли смесь нагреваться до комнатной температуры. Через 20 ч смесь промывали 0,5 М НСl (дважды), насыщенным водным раствором NaHCO3, рассолом и сушили (Na2SO4), растворитель удаляли и остаток подвергали хроматографии на колонке с силикагелем (градиент элюирования смесью 20-30% EtOAc с гексаном) с получением (+)-цис-3-ацетокси-4-изопропил-1-п-метоксибензилазетидин-2-она в виде твердого вещества (7,15 г, 63%): 1Н ЯМР (СDСl3) δ 0,99 (d, 3Н, J=7,0 Гц), 1,02 (d, 3Н, J=7,0 Гц), 2,20 (s, 3H), 3,82 (s, 3H), 4,24 (t, 1H, J=5,6 Гц), 6,06 (d, 1H, J=5,3 Гц), 6,88-7,38 (m, 4H). Раствор аммонийнитрата церия (51,3 г, 3,6 экв) в 750 мл воды добавляли к хорошо перемешиваемому раствору азетидинона (7,2 г, 26,0 ммоль) в ацетонитриле (500 мл) на ледяной бане в течение 1 часа. Затем реакционную смесь экстрагировали EtOAc (дважды) и соединенные органические экстракты промывали насыщенным водным раствором NaHCO3 (дважды), 20% водным раствором NаНSО3, насыщенным водным раствором NаНСО3 и рассолом. После сушки (Na2SO4) растворители удаляли с получением 4,26 г технического (+)-цис-3-ацетокси-4-изопропилазетидин-2-она: 1H ЯМР (СDСl3) δ 0,86 (d, 3H, J=6,6 Гц), 0,99 (d, 3H, J=6,6 Гц), 1,89 (m, 1H), 2,17 (s, 3H), 3,52 (dd, 1H, J=4,8, 9,0 Гц), 5,96 (dd, 1H, J=2,5, 4,6 Гц), 6,38 (brs, 1H); LRMS (negative ESI) 170[(M-H)-]. Суспензию этого продукта (4,26 г, 24,9 ммоль) и К2СО3 (102 мг, 0,03 экв) в МеОН (40 мл) оставляли при комнатной температуре при перемешивании в течение 1,5 ч. Затем для нейтрализации добавляли Amberlite IR-20. Смесь фильтровали и удаляли растворитель из фильтрата с получением технического (+)-цис-3-гидрокси-4-изопропилазетидин-2-она. Этот продукт растворяли в сухом DMF (40 мл) и добавляли имидазол (3,39 г, 2 экв) и триэтилсилилхлорид (4,19 мл, 1 экв). Через 10 минут реакционную смесь распределяли между водой и смесью EtOAc и гексана (1:1). Органическую фазу промывали водой (дважды), рассолом и затем сушили (Na2SО4). Растворители удаляли и остаток подвергали хроматографии на колонке с силикагелем (градиент элюирования, смесь 25-35% EtOAc с гексаном), получая (+)-цис-4-изопропил-3-триэтилсилилоксиазетидин-2-он (4,63 г, 77%): 1Н ЯМР (СDС13) δ 0,65-1,03 (m, 21H), 1,93 (m, 1H), 3,29 (dd, 1H, J=4,8, 9,1 Гц), 4,87 (dd, 1H, J=2,8, 4,7 Гц), 6,05 (br s, 1H). Раствор этого азетидинона (1,05 г, 4,32 ммоль), диизопропилэтиламина (0,90 мл, 1,2 экв), ди-трет.бутилдикарбоната (1,04 г, 1,1 экв) и п-диметиламинопиридина (106 мг, 0,2 экв) в сухом DCM (10 мл) оставляли при перемешивании при комнатной температуре в течение 30 минут. Смесь разбавляли DCM, промывали рассолом и сушили (Na2SO4). Удаление растворителя с последующей хроматографией на колонке с силикагелем (градиент элюирования 10-20% EtOAc в гексане) позволило получить 1,31 г (88%) соединения, указанного в заголовке, в виде масла: 1Н ЯМР (CDCl3) δ 0,66-1,07 (m, 21H), 1,53 (s, 9H), 2,15 (m, 1H), 3,87 (t, 1H, 1=6,4 Гц), 4,88 (d, 1H, J=6,1 Гц); LRMS (ESI) 344 [(M+H)+].
Препаративный пример 3
(+)-цис-1-бензоил-4-изопропил-3-триэтилсилилоксиазетидин-2-он
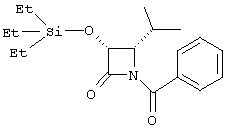
Раствор (+)-цис-4-изопропил-3-триэтилсилилоксиазетидин-2-она (486 мг, 2,00 ммоль), бензоилхлорида (0,255 мл, 1,1 экв), диизопропилэтиламина (0,346 мл, 1,2 экв) и п-диметиламинопиридина (244 мг, 1 экв) в сухом DCM (6 мл) оставляли при перемешивании в течение 6 часов при 0°С. Удаляли баню и реакционную смесь оставляли при перемешивании в течение ночи. Затем ее разбавляли DCM и промывали водой, водным раствором НСl (0,1 N), насыщенным водным раствором NaHCO3, рассолом и высушивали (Na2SO4). Удаление растворителя с последующей хроматографией на колонке с силикагелем (элюирование 0,5% EtOAc в гексане) позволило получить соединение, указанное в заголовке: 1Н ЯМР (CDCl3) δ 0,47-0,94 (m, 21H), 2,09 (m, 1H), 4,07 (m, 1H), 4,75 (m, 1H), 7,24-7,76 (m, 5H).
Препаративный пример 4
(3R,4R)-1-неопентилкарбонил-4-фенил-3-триэтилсилилоксиазетидин-2-он
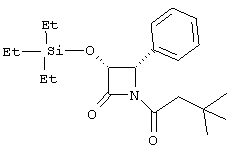
По вышеописанной методике с использованием неопентилкарбонилхлорида (3R,4R)-4-фенил-3-триэтилсилилоксиазетидин-2-он превращали в указанное в заголовке соединение:
1H ЯМР (СDСl3) δ 0,19-0,62 (m, 15Н), 0,88 (s, 3Н), 2,43 (d, 1H, J=13,8 Гц), 2,62 (d, 1H, J=14,1 Гц), 4,90 (d, 1H, J=5,7 Гц), 4,95 (d, 1H, J=6,0 Гц), 7,05-7,17 (m, 5H).
Препаративный пример 5
(3R,4R)-1-циклобутилкарбонил-4-фенил-3-триэтилсилилоксиазетидин-2-он
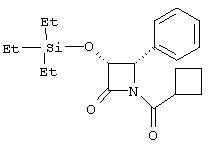
По вышеописанной методике, используя циклобутилкарбонилхлорид, превращали (3R,4R)-4-фенил-3-триэтилсилилоксиазетидин-2-он в соединение, указанное в заголовке: 1Н ЯМР (CDCl3) δ 0,18-0,61 (m, 15Н), 1,66-2,22 (m, 6Н), 3,61 (m, 1H), 4,89 (d, 1H, J=5,7 Гц), 4,94 (d, 1H, J=5,7 Гц), 7,03-7,18 (m, 5H).
Препаративный пример 6
(3R,4R)-1-неопентилоксикарбонил-4-фенил-3-триэтилсилилоксиазетидин-2-он
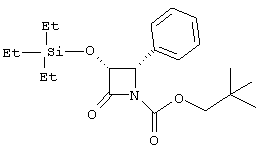
По вышеописанной методике, используя неопентилхлорформиат, (3R, 4R)-4-фенил-3-триэтилсилилоксиазетидин-2-он превращали в соединение, указанное в заголовке: 1Н ЯМР (СDСl3) δ 0,39-0,97 (m, 24Н), 3,73 (d, 1H, J=10,2 Гц), 3,90 (d, 1H, J=10,2 Гц), 5,10 (m, 2Н), 7,31 (m, 5H).
Препаративный пример 7
1) Синтез производного баккатина 1
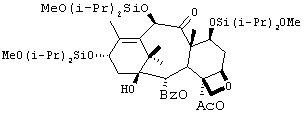
К раствору 10-дезацетилбаккатина (47,4 г, 87 ммоль) в безводном N,N-диметилформамиде (DMF) (500 мл) добавляли имидазол (47 г, 691 ммоль) при комнатной температуре. Раствор перемешивали в течение 10-15 минут до образования прозрачного раствора. К реакционной смеси добавляют 58 мл, 332 ммоль, диизопропилдихлорсилана. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли к раствору дополнительное количество диизопропилдихлорсилана (6 мл) и реакционную смесь перемешивали в течение 60 мин. Жидкостная хроматография высокого разрешения показывает завершение реакции. К этой смеси добавляли метанол (36 мл) и перемешивали в течение 60 мин. Реакцию прекращали и разбавляли смесью трет.бутилметилкетона (ТВМЕ) (500 мл) и воды (200 мл). Слои разделяли, промывали органическую фазу рассолом (250 мл), высушивали (сульфат натрия) и выпаривали, получая трисилилированное производное баккатина I (91 г, >100% выход) в виде белого аморфного продукта, который использовали на следующей стадии без дальнейшей очистки.
ESELRMS M+, вычислено для C50H84O13Si3: 977, найдено 977.
2) Синтез производного баккатина 2
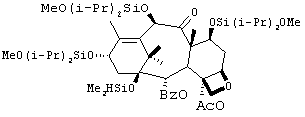
К раствору производного баккатина I (90 г, 92 ммоль) в (DMF) (500 мл) добавляли при 0°С имидазол (22 г, 320 ммоль). При 0°С по каплям добавляли диметилхлорсилан (35 мл, 320 ммоль). Наблюдалось осаждение соединения. Реакционную смесь (суспензию) перемешивали в течение 0,5 часа при температуре 0°С. Твердый продукт фильтровали и промывали холодным DMF (3 × 150 мл). После сушки воздухом твердый продукт вновь растворяли в ТВМЕ (700 мл) и промывали полученный раствор водой (3 × 200 мл), рассолом (250 мл) и сушили (сульфат натрия). Раствор фильтровали через кремнеземный фильтр. Удаление растворителя под вакуумом позволило получить 2 с выходом 77% (70 г).
ESELRMS M+ вычислено для C50H90O13Si4: 1035, найдено 1035.
3) Синтез производного баккатина 3
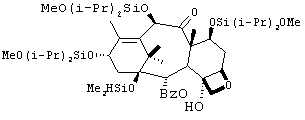
К перемешиваемому раствору 2 (66,3 г, 64 ммоль) в толуоле (680 мл) при температуре -34°С добавляли по каплям Red-Al (50 мл, 160 ммоль, 65 вес.%-ный раствор бис(2-метоксиэтокси)алюминийгидрида в толуоле) в течение 10 мин. Реакционную смесь нагревали до -25°С и перемешивали в течение 1,5 ч. К реакционной смеси по каплям добавляли метанол (62 мл) при внутренней температуре от -20 до -25°С. Раствор разбавляли ТВМЕ (500 мл), добавляли 1 N раствор гидроокиси натрия (60 мл) и рассол (60 мл). Раствор перемешивали в течение 30 минут. К смеси добавляли целит (12 г), перемешивали в течение 10 минут и фильтровали через целит. Разделяли полученные слои. Органический слой промывали водой, рассолом и сушили (сульфат натрия). Затем раствор пропускали через прокладку, выполненную из кремнезема, перед удалением растворителя. Было получено белое твердое вещество с выходом 97% (62 г).
ESILRMS M+ вычислено для C50H88O12Si4: 993, найдено: 993.
4) Синтез производного баккатина 4
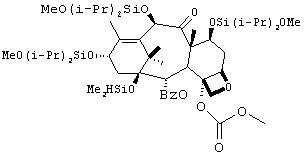
В атмосфере аргона к раствору 3 (62 г, 62 ммоль) в безводном тетрагидрофуране (THF) (600 мл) при -60°С по каплям добавляли LHMDS (бис(триметилсилил)амид лития) (125 мл, 125 ммоль, 1М раствор в THF). Раствор перемешивали в течение 15 минут с последующим добавлением метилхлорформиата (9 мл, 116 ммоль), внутреннюю температуру раствора поддерживали равной -60°С. Реакционную смесь медленно нагревали до 0°С и перемешивали в течение 3 часов. После завершения реакции добавляли насыщенный хлорид аммония (300 мл). Реакционную смесь экстрагировали ТВМЕ (100 мл). Органический слой промывали насыщенным хлоридом аммония (200 мл), водой (200 мл), рассолом (200 мл), сушили (сульфат натрия) и выпаривали с получением 4 в виде масла (67 г, > 100%). Технический продукт использовали на следующей стадии без дополнительной очистки.
ESILRMS M+ вычислено для C52H90O14Si4: 1051, найдено: 1051.
5) Синтез производного баккатина 5
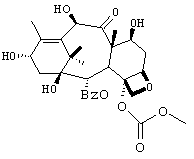
К раствору производного баккатина 4 (62 г, 59 ммол) в сухом THF (260 мл) добавляли комплекс - триэтиламин-фтористоводородная кислота (56 мл, 344 ммоль) при комнатной температуре. Реакционную смесь перемешивали 3 часа. Реакционную смесь разбавляли этилацетатом (350 мл) и промывали водой (200 мл), рассолом (200 мл), сушили (сульфат натрия) и выпаривали с получением 5 (43 г, выход >100% технического продукта). Повторное суспендирование технического соединения в смеси горячего этилацетата (350 мл) и гексанов (50 мл) приводило к получению чистого 5 с выходом 90%.
ESILRMS М+ вычислено для С29Н36О11: 560, найдено: 560.
6) Синтез производного баккатина 6
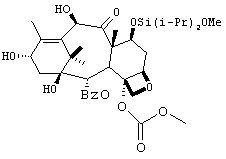
К перемешиваемому раствору производного баккатина 5 (32 г, 57 ммоль) и имидазола (11,7 г, 172 ммоль) в DMF (220 мл) при -65°С добавляли диизопропилдихлорсилан (26,8 мл) в атмосфере аргона. Температуру реакционной смеси поддерживали равной -60°С и перемешивали смесь в течение 2 часов. После завершения реакции (HPLC) добавляли раствор имидазола в метаноле (11,7 г имидазола, растворенного в 35 мл метанола) и перемешивали раствор при 0°С в течение 30 мин. Смесь экстрагировали ТВМЕ (500 мл). Органическую фазу промывали водой (4 × 150 мл), высушивали (сульфат натрия) и выпаривали с получением технического 6 (45 г). Технический продукт затем растворяли в ацетонитриле (150 мл) и промывали раствор гексанами (3 × 100 мл). Удаление ацетонитрила привело к получению чистого 6 в виде белого твердого вещества (34 г, выход 84%).
ESILRMS М+ вычислено для C96H57O12Si: 704, найдено: 704.
7) Синтез производного баккатина 7
4-деацетил-7-[бисизопропил(метокси)]силилокси-4-метоксикарбонил]-баккатин
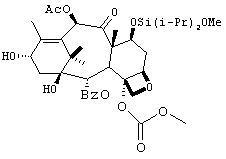
К раствору производного баккатина 6 (33,2 г, 47 ммоль) в DMF (200 мл) добавляли по каплям LHMDS (61,2 мл, 61,2 ммоль) при -43°С. Реакционную смесь перемешивали в течение 15 минут с последующим добавлением уксусного ангидрида (5,8 мл, 63 ммоль). Смесь перемешивали в течение 30 минут при -40°С. Добавляли уксусную кислоту (3,6 мл) и удаляли охлаждающую баню. Реакционную смесь экстрагировали ТВМЕ (300 мл). Органический слой отделяли и промывали водой (3 × 150 мл), рассолом (150 мл), высушивали (сульфат натрия) и выпаривали с получением технического продукта. Очистка этого соединения осуществлялась кристаллизацией из смеси ТНР:гептан (1:6). Из 40 г было получено 21 г кристаллического производного баккатина 7 (выход 60%).
ESILRMS М+ вычислено для С38Н54О13Si: 746, найдено: 746.
Пример 1. Соединение Ia
3'-трет.бутил-3'-N-трет.бутилоксикарбонил-4-деацетил-3'-дефинил-3'-N-дебензоил-4-O-метоксикарбонилпаклитаксел
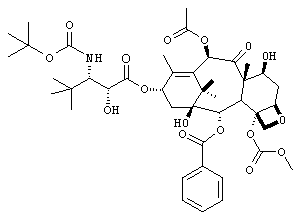
Раствор (+)-цис-4-трет.бутил-1-(трет.бутилоксикарбонил)-3-триэтилсилилоксиазетидин-2-она (2,71 г, 5 экв) и 4-деацетил-7-[бисизопропил(метокси)]силилокси-4-метоксикарбонилбаюкатина (1,13 г, 1,52 ммоль) в сухом THF (100 мл) в атмосфере азота охлаждали до -50°С и добавляли раствор LiHMDSA (1,97 мл, 1,3 экв, 1,0 М в ТГФ). Через 5 минут помещали эту смесь в баню, которую выдерживали при температуре от -35 до -30°С в течение 20 часов и затем при -25°С в течение 24 часов. Затем реакцию обрывали насыщенным водным раствором NН4Cl и экстрагировали смесью EtOAc и гексана (1:1). Органические экстракты промывали рассолом и сушили (Na2SО4). Растворители удаляли и подвергали остаток хроматографии (радиальная хроматография на пластине из силикагеля толщиной 6 мм; градиент элюирования 5-20% EtOAc в гексане) с получением 1,55 г 3'-трет.бутил-3'-N-трет.бутилоксикарбонил-7-[бисизопропил-(метокси)]силилокси-4-деацетил-3'-дефинил-3'-N-дебензоил-4-O-метоксикарбонил-2'-триэтилсилиллксипаклитаксела в виде смеси 2',3'-диастереомеров. Эту смесь растворяли в сухом THF (60 мл) и добавляли триэтиламинтригидрофторид (0,92 мл, 4 экв). Через 22 часа при комнатной температуре реакционную смесь нейтрализовали насыщенным водным раствором NаНСО3 и затем экстрагировали EtOAc. Органические экстракты промывали рассолом, сушили (Na2SO4) и удаляли растворители. Остаток подвергали хроматографии (радиальная хроматография, пластинка из кремнезема, 2 мм; градиент элюирования от 10 до 50% EtOAc в гексане) с получением (по порядку элюирования) 210 мг (18%): 2'S,3'R-3'-трет.бутил-3-N-трет.бутилоксикарбонил-4-деацетил-3'-дефинил-3'-М-дебензоил-4-О-метоксикарбонилпаклитаксела {1Н ЯМР (CDCl3) δ l,04(s,9H), l,13(s,3H), l,20(s,3H), 1,37 (s, 9H), 1,65 (s, 1H), 1,66 (s, 3Н), 1,84-1,93 (m, 2H), 2,17 (s, 3Н), 2,25 (s, 3H), 2,55 (m, 3Н), 3,00 (d, 1H, J=6,5 Гц), 3,74 (d, 1H, J=10,8 Гц), 3,79 (d, 1H, J=6,9 Гц), 3,92 (s, 3Н), 4,16 (d, 1H, J=8,5 Гц), 4,33 (d, 1H, J=8,5 Гц), 4,42 (m, 1H), 4,54 (d, 1H, J=6,5 Гц), 4,87 (d, 1H, J=10,6 Гц), 5,01 (d, 1H, J=7,7 Гц), 5,68 (d, 1H, J=7,0 Гц), 5,76 (m, 1H), 6,32 (s, 1H), 7,44-8,05 (m, 5H); LRMS (ESI) 846 [(M+H)+]} и 668 мг (56%) соединения, указанного в заголовке {1Н ЯМР (CDCl3) δ 1,07 (s, 9Н), 1,14 (s, 3Н), 1,24 (s, 3Н), 1,33 (s, 9H), 1,66 (s, 4H), 2,23 (s, 3Н), 2,38-2,59 (m, 4H), 3,11 (d, 1H, J=5,8 Гц), 3,77 (d, 1H, J=11,1 Гц), 3,82 (d, 1H, J=7,0 Гц), 3,96 (s, 3Н), 4,20 (d, 1H, J=8,6 Гц), 4,33 (d, 1H, J=8,6 Гц), 4,39 (m, 1H), 4,53 (d, 1H, J=5,4 Гц), 4,88 (d, 1H, J=10,6 Гц), 4,98 (d, 1H, J=7,9 Гц), 5,69 (d, 1H, J=7,1 Гц), 6,03 (m, 1H), 6,28 (s, 1H), 7,40-8,11 (m, 5H); LRMS (ESI) 846 [(M+H)+]}.
Пример 2. Соединение Ib
3'-N'-трет.бутилоксикарбонил-4-деацетил-3'-дефинил-3'-N-дебензоил-3'-изопропил-4-O-метоксикарбонил-паклитаксел
По вышеописанной методике (+)-цис-1-трет.бутилоксикарбонил-4-изопропил-3-триэтилсилилокси-азетидин-2-он сочетали с 4-деацетил-7-[бисизопропил-(метокси)]силилокси-4-O-метоксикарбонилбаккатином.
После снятия защиты и хроматографического выделения получали соединение, указанное в заголовке.
1Н ЯМР (CDCl3+D2O) δ 1,03 (d, 3Н, J=6,7 Гц), 1,09 (d, 3H, J=6,7 Гц), 1,14 (s, 3Н), 1,24 (s, 3H), 1,31 (s, 9H), 1,66 (m, 3H), 1,83-2,02 (m, 5H), 2,24 (s, 3H), 2,25-2,59 (m, 3H), 3,68 (dd, 1H, J=2,0, 9,2 Гц), 3,82 (d, 1H, J=6,9 Гц), 3,98 (s, 3Н), 4,19 (d, 1H, J=8,6 Гц), 4,34 (d, 1H, J=8,6 Гц), 4,39 (m, 1H), 4,43 (d, 1H, J=2,0 Гц), 4,82 (br s, 1H), 4,98 (d, 1H, J=7,8 Гц), 5,69 (d, 1H, J=7,0 Гц), 6,11 (m, 1H), 6,28 (s, 1H), 7,45-8,12 (m, 5H); LRMS (ESI) 832 [(М+Н)+].
Пример 3. Соединение Iс
3'-N-неопентилкарбонил-4-деацетил-3'-N-дебензоил-4-O-метоксикарбонил-паклитаксел
Раствор (3R,4R)-1-неопентилоксикарбонил-4-фенил-3-метилсилилоксиазетидин-2-она (525 мг, 1,4 экв) и 4-деацетил-7-[бисизопропил(метокси)]силилокси-4-O-метоксикарбонилбаккатина (523 мг, 0,7 ммол) в сухом THF (15 мл) охлаждали до температуры -50°С и при перемешивании добавляли раствор LiHMDSA (0,84 мл, 1,2 экв, 1,0 М в THF). Через 40 мин реакционной смеси давали нагреться до 0°С. Через 1,5 часа реакцию обрывали насыщенным водным раствором NН4Сl, реакционную смесь экстрагировали EtOAc. Органический экстракт промывали насыщенным водным раствором NH4Cl, водой, рассолом и высушивали (Na2SO4). Удаление растворителей с последующей хроматографией на колонке с силикагелем (элюирование смесями от 0 до 20% EtOAc в гексане) позволило получить 2,78 мг (54%) 3'-N-неопентилоксикарбонил-7-[бисизопропил(метокси)]-силилокси-4-деацетил-3'-N-дебензоли-4-O-метоксикарбонил-2'-триэтилсилилоксипаклитаксел. Это вещество отбирали и обрабатывали триэтиламинтригидрофторидом (0,161 мл, 4 экв) в сухом THF (6 мл) и оставляли стоять при комнатной температуре в течение ночи. После нейтрализации насыщенным водным раствором NaHCO3 реакционную смесь экстрагировали EtOAc. Органические экстракты промывали рассолом и сушили (Nа2SO4). Удаление растворителей и хроматография на колонке с силикагелем (градиент элюирования, смеси 20-50% EtOAc в гексане) привело к получению 151 мг (71%) соединения, указанного в заголовке; 1Н ЯМР (СDСl3) δ 0,96-2,58 [32Н, включая 0,96 (s, 9Н), 1,14 (s, 3H), 1,24 (s, 3Н), 1,66 (s, 3Н), 1,84 (s, 3Н), 2,23 (s, 3Н)], 3,58 (br s, 1H), 3,77 (s, 3H), 3,80 (d, 1H, J=5,5 Гц), 4,19 (d, 1H, J=8,3 Гц), 4,33 (d, 1H, J=8,7 Гц), 4,36 (m, 1H), 4,65 (d, 1H, J=2,0 Гц), 4,95 (d, 1H, J=8,5 Гц), 5,58 (dd, 1H, J=2,3, 8,8 Гц), 5,69 (d, 1H, J=7,0 Гц), 6,11 (d, 1H, J=8,9 Гц), 6,16 (m, 1H), 6,27 (s, 1H), 7,29-8,12 (m, 10H); LRMS (ESI) 864 [(М+Н)+].
Пример 4. Соединение Id
3'-N-циклобутил-4-деацетил-3'-N-дебензоил-4-O-метоксикарбонилпаклитаксел
По вышеописанной методике, применяя (3R,4R)-1-циклобутил-4-фенил-3-триэтилсилилоксиазетидин-2-он, 4-деацетил-7-[бисизопропил(метокси)]силилокси-4-O-метоксикарбонилбаккатин превращали в указанный в заголовке продукт. 1Н ЯМР (CDCl3) δ 1,14-2,53 [m, 27H, включая 1,14 (s, 3Н), 1,25 (s, 3Н), 1,67 (s, 3Н), 1,84 (s, 3Н), 2,24 (s, 3H)], 301 (m, 1H), 3,56 (br s, 1H), 3,81 (s, 3H), 3,82 (m, 1H), 4,20 (d, 1H, J=8,4 Гц), 4,34 (d, 1H, J=8,5 Гц), 4,37 (m, 1H), 4,68 (d, 1H, J=2,3 Гц), 4,96 (d, 1H, J=8,6 Гц), 5,58 (dd, 1H, J=2,4, 9,0 Гц), 5,70 (d, 1H, J=7,0 Гц), 6,16 (m, 2H), 6,27 (s, 1H), 7,29-8,14 (m, 10Н); LRMS (ESI) 848 [(М+Н)+].
Пример 5. Соединение Iе
3'-трет.бутил-3'-N-циклогексилкарбонил-4-деацетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонилпаклитаксел
Используя ту же методику для циклогексилоксихлорформиата, 3'-трет.бутил-3-N-трет.бутилоксикарбонил-4-деацетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонилпаклитаксел превращали в продукт, указанный в заголовке: 1Н ЯМР (CDCl3+D2O) δ 1,10-2,61 [38Н, включая 1,10 (s, 9H), 1,16 (s, 3H), 1,26 (s, 3Н), 1,69 (s, 3H), 1,95 (s, 3Н), 2,26 (s, 3H)], 3,84 (m, 2Н), 3,99 (s, 3Н), 4,23 (d, 1H, J=8,6 Гц), 4,40 (m, 3Н), 4,57 (s, 1H), 5,02 (m, 2H), 5,7 (d, 1H, J=7,08 Гц), 6,06 (m, 1H), 6,30 (s, 1H), 7,46-8,13 (m, 5H); LRMS (ESI) 872 [(М+Н)+].
Пример 6. Соединение If
3'-трет.бутил-4-деацетил-3'-дефенил-3'-N-дебензоил-3'-N-неопентилкарбонил-4-O-метоксикарбонилпаклитаксел.
Используя вышеописанную методику и трет.бутилацетилхлорид, 3'-трет.бутил-3-N-трет.бутилоксикарбонил-4-деацетил-3'-дефенил-3'-N-дебензоил-4-O-метоксикарбонилпаклитаксел превращали в соединение, указанное в заголовке: 1Н ЯМР (CDCl3+D2O) δ 1,00-2,56 [39Н, включая 1,00 (s, 9H), 1,11 (s, 9H), 1,16 (s, 3H), 1,26 (s, 3H), 1,69 (s, 3H), 1,91 (s, 3H), 2,26 (s, 3H)], 3,83 (d, 1H, J=7,1 Гц), 3,98 (s, 3H), 4,17 (d, 1H, J=10,1 Гц), 4,26 (d, 1H, J=8,8 Гц), 4,37 (m, 2H), 4,55 (s, 1H), 5,00 (d, 1H, J=7,5 Гц), 5,73 (m, 2H), 6,02 (m, 1H), 6,29 (s, 1H), 7,45-8,13 (m, 5H); LRMS (ESI) 844 [(М+Н)+].
Пример 7. Соединение Ig
4-деацетил-3'-N-дебензоил-3'-N-трет.бутоксикарбонил-4-O-метоксикарбонил-паклитаксел.
По вышеописанной методике, используя (3R,4R)-1-трет.бутоксикарбонил-4-фенил-3-триэтилсилилоксиазетидин-2-он, получали соединение Ig: 1Н ЯМР (300 МГц, CDCl3) δ 8,13-8,10 (m, 2H), 7,67-7,26 (m, 8H), 6,27 (s, 1H), 6,19 (m, 1H), 5,68 (d, J=6,9 Гц, 1H), 5,35-5,29 (m, 2H), 4,97 (d, J=7,7 Гц, 1H), 4,63 (d, J=3,9 Гц, 1H), 4,27-4,37 (m, 1H), 4,25 (AB q, J=8,8 Гц, J=47,7 Гц, 2Н,), 3,85-3,81 (m, 4H), 3,40 (d, J=5,1 Гц, 1Н), 2,59-1,03 (m, 30Н, включая синглеты при 2,24, 1,87, 1,71, 1,27, 1,14, 3Н каждый, 1,32, 9Н).
Пример 8. Соединение Ih
4-деацетил-3'-N-дебензоил-3'-N-н-бутоксикарбонил-4-O-метоксикарбонилпаклитаксел.
По вышеописанной методике, используя (3R,4R)-1-н-бутоксикарбонил-4-фенил-3-триэтилсилилоксиазетидин-2-он, получали соединение Ih: 1H ЯМР (300 МГц, СDСl3) δ 8,11 (d, J=7,4 Гц, 2Н), 7,62-7,29 (m, 10Н), 6,27 (s, 1H), 5,69 (d, J=7,0 Гц, 1H,), 5,41 (abq, J=47,4 Гц, 9,4 Гц, 2Н), 4,97 (d, J=7,0 Гц, 1H), 4,66 (br s, 1H), 4,38-4,32 (m, 1H), 4,26 (abq, J=45,0 Гц, 8,6 Гц, 2Н), 3,83 (s, 3Н), 3,42 (brd, J=4,1 Гц, 1H), 2,59-2,35 (m, 4H), 2,24 (m, 3Н), 1,86 (s, 3H), 1,67 (s, 3Н), 1,65 (d, J=33,0 Гц, 3Н), 1,67 (s, 3Н), 1,51-1,47 (m, 2H), 1,24 (s, 3Н), 1,14 (s, 3H), 0,83 (m, 3H).
Данные элементного анализа: вычислено для C45H55NO16: С 62,42; Н 6,40; N 1,62; найдено: С 62,28; Н 6,45; N 1,55.
Пример 9. Соединение Ij
3'-N-дебензоил-3'-N-циклобутилкарбонил-3'-дефенил-3'-трет.бутил-4-деацетил-4-метоксикарбонилпаклитаксел.
Раствор 3'-N-дебензоил-3'-N-трет.бутил-3'-дефенил-3'-трет.бутил-4-деацетил-4-метоксикарбонилпаклитаксел (2,30 г, 2,72 ммоль) в DCM (15,0 мл) обрабатывали трифторуксусной кислотой (15,0 мл) и перемешивали при температуре 0°С в течение 1,5 ч. Смесь разбавляли 100 мл DCM и выливали в холодный раствор (0°С) 50 г NаНСО3 в 150 мл воды. Фазы разделяли и концентрировали органический слой в вакууме. Продукт может быть очищен хроматографией на колонке с силикагелем с элюированием смесью 4% метанола/DCM, но обычно используется без очистки. Технический 3'-N-дебензоил-3'-дефенил-3'-трет.бутил-4-деацетил-4-метоксикарбонилпаклитаксел растворяли в этилацетате (15,0 мл) и обрабатывали насыщенным раствором NaHCO3 (15,0 мл). Добавляли циклобутанкарбонилхлорид (460 мкл, 4,08 ммоль, 1,5 экв) и перемешивали энергично двухфазную смесь при комнатной температуре в течение 20 минут. Смесь разбавляли этилацетатом и разделяли фазы. Органическую фазу промывали насыщенным NаНСО3 и рассолом. Органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали в вакууме. Очистка препаративной хроматографией с обращенной фазой (элюирование смесью 20% ацетонитрила/вода в течение 5 мин, затем смесью 60% ацетонитрила/вода в течение 45 мин, затем в изократических условиях в течение 45 мин при скорости истечения 250 мл/мин) привело к получению соединения, указанного в заголовке (1,47 г, выход 65%, степень чистоты 97% (ЖХВР)) в виде белого аморфного вещества со следующими физическими свойствами: 1H ЯМР (СDСl3, 300 МГц) δ 8,08 (d, J=7,1 Гц, 2Н), 7,62-7,55 (m, 1H), 7,48-7,43 (m, 2H), 6,27 (s, 1H), 5,99 (dd, J=7,8 Гц, J=9,0 Гц, 1H,), 5,69 (m, 2H), 4,98 (dd, J=2,0 Гц, J=9,5 Гц, 1H), 4,55 (dd, J=1,1 Гц, J=5,2 Гц, 1H), 4,40-4,32 (bm, 2H), 4,22 (d, J=8,4 Гц, 1H), 4,14 (d, J=10,2 Гц, 1H), 3,98 (s, 3Н), 3,80 (d, J=7,0 Гц, 1H), 3,30 (d, J=5,1 Гц, 1H), 2,97 (p, J=7,9 Гц, 1H), 2,58-2,36 (bm, 4H), 2,23 (s, 3Н), 2,19-2,03 (bm, 4H), 1,92-1,76 (bm, 3Н), 1,88 (s, 3H), 1,66 (s, 3Н), 1,24 (s, 3Н), 1,14 (s, 3Н), 1,06 (s, 9H); 13С ЯМР (CDCl3, 75 МГц) δ 203,70, 174,99, 174,93, 171,46, 166,89, 153,21, 142,54, 133,74, 133,20, 130,22, 129,81, 128,72, 84,13, 83,21, 78,92, 76,09, 75,68, 74,94, 73,39, 72,06, 70,17, 58,36, 57,73, 56,03, 51,07, 45,74, 43,34, 39,91, 35,94, 35,47, 27,37, 26,85, 25,60, 25,44, 22,12 20,95, 18,29, 14,95, 9,72; LRMS (ESI) 828,51 ((М+1)+, 100%); 886,57 ((M+NH4ACN)+, 15%); 826,48 ((М-1)-; 100%).
Пример 10. Соединение Ik
3'-N-дебензоил-3'-N-(2-фуроил)-3'-дефенил-3'-трет.бутил-4-деацетил-4-метоксикарбонилпаклитаксел.
Получают это соединение, как описано выше. Его получают (2,13 г, 73% выход, степень чистоты 98% (ЖХВР)) в виде белого аморфного продукта со следующими физическими свойствами: 1Н ЯМР (СDСl3, 300 МГц) δ 8,15-8,08 (m, 2H), 7,64-7,56 (m, 1H), 7,52-7,25 (m, 3Н), 7,03 (dd, J=0,6 Гц, J=3,4 Гц, 1H), 6,78 (d, J=10,2 Гц, 1H), 6,48 (dd, J=1,8 Гц, J=3,5 Гц, 1Н), 6,25 (s, 1H), 6,06 (dd, J=7,6 Гц, J=8,9 Гц, 1H), 5,70 (d, J=7,0 Гц, 1H), 5,00 (dd, J=1,9 Гц, J=9,4 Гц, 1H), 4,40-4,32 (m, 3Н), 4,23 (d, J=8,6 Гц, 1H), 4,05 (s, 3Н), 3,48 (d, J=4,5 Гц, 1Н), 2,60-2,50 (m, 2H), 2,38 (dd, J=3,3 Гц, J=8,8 Гц, 1H), 2,23 (s, 3Н), 2,05 (s, 1H), 1,95-1,85 (m, 1H), 1,81 (s, 3Н), 1,68 (s, 3H), 1,21 (s, 3H), 1,14(s, 12H); 13С ЯМР (CDCl3, 75 МГц) δ 203,71, 174,23, 171,55, 167,06, 158,22, 153,06, 147,65, 144,30, 142,53, 133,93, 133,36, 130,32, 129,42, 128,88, 114,95, 112,51, 84,20, 83,48, 79,06, 77,65, 76,20, 75,81, 75,05, 72,96, 70,55, 58,50, 57,97, 56,40, 46,07, 43,33, 36,09, 35,55, 27,53, 27,05, 22,00, 21,25, 21,06, 14,97, 14,39, 9,83; LRMS (ESI) 840,43 ((М+1)+ 100%); 838,43 ((М-1)-, 100%).
Биологические свойства
Другой аспект данного изобретения касается способа ингибирования опухолей у человека и/или другого млекопитающего путем перорального введения в организм, содержащий опухоль, противоопухолевого эффективного количества соединения формулы I.
Материалы и методики, использованные при in vivo испытаниях перорально вводимых таксанов, указаны ниже.
Материалы
Животные. Обычные или атимические (“голые”) мыши и голые крысы, им имплантировали подкожно (sc) измельченную опухоль или фрагменты опухоли. Мышиная опухоль была имплантирована обычным мышам, человеческие опухоли - голым мышам или крысам. Опухоли. Использованные опухоли включали, чаще всего, мышиную легочную карциному, M109, мышиную карциному молочной железы, МАМ 16/С, человеческую карциному яичника, А2780, опухоли прямой кишки человека, НСТ-116 и HCT-116/pk. Соединения I проявляли противоопухолевую активность после перорального применения в одной или нескольких вышеупомянутых моделях опухолей.
Методы получения результатов, показанных в таблице III
Опыты проводили с использованием атимических (“голых”) мышей. Обычно курс лечения начинали, когда опухоли имели размер между 100 и 500 мг (обычно 7-12 дней после имплантации опухоли) в случае модели ксенографта опухоли яичника человека. В группе обычно было 8 животных, в контрольных группах также было по 8 мышей. Соединение вводили в дозе, указанной в таблице III, а именно 1 раз в день через день пять раз (то есть q2d×5).
Лечение таксанами осуществляли перорально при помощи зонда, используя носитель, состоящий из 10% этанола + 10% Cremophor EL + 80% воды. Объем вводимой жидкости составлял 0,01 мл на грамм веса тела мыши. Типичный опыт на мышах включает оценку каждого испытуемого соединения, вводимого в трех различных дозах. Противоопухолевую активность оценивали путем определения размера опухолей у всех подвергшихся лечению и контрольных мышей во времени. Каждое животное идентифицировалось, рост опухоли, имплантированной в каждое животное, измеряли один или два раза в неделю штангенциркулем. Определялась разница во времени, требующемся для достижения заданного размера (например, 500 или 1000 мг) опухолями у подвергшихся лечению (Т) и контрольных (С) групп, и оценка абсолютного и относительного противоопухолевого эффектов (например, между соединениями) производилась на основе задержек во времени достижения опухолями заданных размеров. Животные с опухолями весом 35 мг или менее в конце эксперимента назывались “вылеченные”. Опыты прекращали обычно после истечения периода времени, которое было, по меньшей мере, в 10 раз больше времени, в течение которого размер опухоли увеличивался вдвое (TVDT) для опухолей у контрольных животных в каждом эксперименте. Активность в испытуемой группе определялась как активность, вызвавшая задержку роста опухоли (среднее время достижения опухолью заданного размера) относительно роста контрольной опухоли (а именно, Т-С) в 3,32 раза по сравнению с TVDT. Активность выражалась в “log убийства клеток”, который был равен (T-C)/(TVDT × 3,32). Токсичность определялась путем измерения среднего веса тела всех животных во время эксперимента до и вскоре после любого введения лекарства. Кроме того, считалось, что животные погибли вследствие вреда, нанесенного лечением, если они погибли до любой смерти у животных контрольной группы, имеющих опухоли, размер которых меньше заданного. Никакие результаты лечения, никакие декларации об активности не учитывались для определенной группы, если более одного животного в этой группе погибало в результате лечения.
Все соединения, указанные в таблице III, характеризовались при пероральном введении противоопухолевой активностью в модели опухоли ScM109, имплантированной мышам, которая считалась эквивалентной противоопухолевой активностью паклитаксела, вводимого внутривенно в соответствии с оптимальной схемой введения и дозировкой.
In vivo активность орально активных С-4 метилкарбонатных таксанов в отношении Sc А2780.
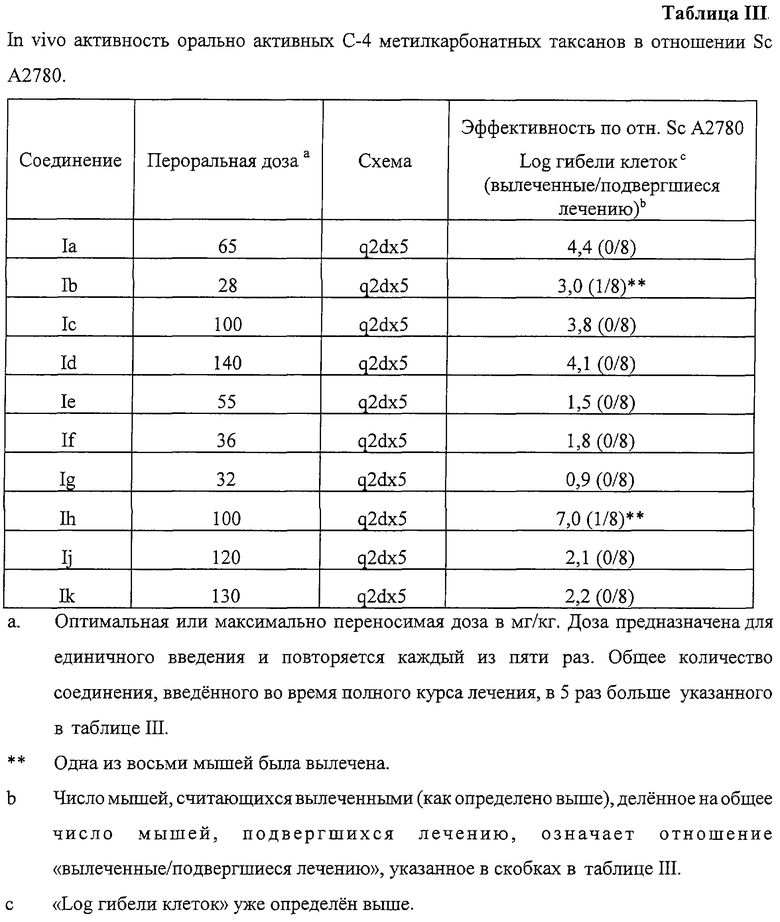
Как видно из таблицы III, все соединения Ia-Ik обладали значительной оральной противоопухолевой активностью. Значительная противоопухолевая активность определяется как приблизительно один log гибели клеток. Это противоположно результатам, которые наблюдались бы для соединений, не имеющих оральной активности, таких как паклитаксел, который является активным ингредиентом коммерчески доступного противоопухолевого лекарства TAXOL®, вводится внутривенно и не вводится перорально, поскольку не является активным при таком введении.
При лечении различных опухолей соединение формулы I по изобретению может быть использовано методом, сходным с тем, который применяется для паклитаксела, например, см. Physician's Desk Reference, 49th Edition, Medical Economics, p. 682, 1995. Дозировка, метод и схема введения соединения по изобретению не ограничены; онколог, специалист по лечению опухолей сможет без дополнительных экспериментов подобрать схему введения соединения по изобретению. Так, соединение формулы I может быть введено любым методом, парентерально или перорально. Парентеральное введение включает внутривенное, внутрибрюшинное, внутримускульное и подкожное введение.
Дозы, используемые для осуществления методов по изобретению, являются дозами, которые позволяют проводить профилактическое лечение или дать максимальный терапевтический результат. Эти дозы меняются в зависимости от вида введения, конкретного соединения и личных характеристик пациента. В общем используются дозы, которые являются терапевтически эффективными для лечения расстройств, вызванных аномальной пролиферацией клеток. Продукты согласно изобретению можно вводить так часто, как это необходимо, для получения желаемого терапевтического эффекта. Некоторые пациенты могут быстро реагировать на сравнительно высокие или низкие дозы и затем им вводятся небольшие дозы или же вообще прекращается введение соединения. При внутривенном введении дозы могут, например, составлять от примерно 20 до примерно 500 мг/м2 в течение 1-100 ч. При пероральном введении дозы могут равняться 5-1000 мг/кг/день. Используемая доза будет меняться в зависимости от конкретного состава композиции, метода введения, конкретного места, организма и типа опухоли. При определении дозы следует принимать во внимание многие факторы, модифицирующие действие лекарства, включая возраст, вес, пол, способ питания и физическое состояние пациента.
Данное изобретение предусматривает также фармацевтические препараты (композиции), содержащие противоопухолевое эффективное количество соединения формулы I в сочетании с одним или более фармацевтически приемлемыми носителями, эксципиентами, разбавителями или адъювантами. Композиции могут быть приготовлены обычными методами. Примеры композиций на основе паклитаксела или его производных можно найти, например, в патентах США 4960790 и 4814470, по приведенным в них примерам можно получить композиции с соединениями по изобретению. Дополнительные примеры композиций паклитаксела можно найти в ссылках, указанных выше. Например, соединение формулы I может быть в составе композиций в виде таблеток, пилюль, порошковых смесей, капсул, препаратов для инъекций, растворов, суппозиториев, эмульсий, дисперсий, пищевых премиксов и других подходящих форм. Можно также готовить стерильные твердые композиции, например, высушенные при замерзании, и, если желательно, в сочетании с другими фармацевтически приемлемыми эксципиентами. Такие твердые композиции могут быть воссозданы добавлением стерильной воды, физиологического раствора или смеси воды и органического растворителя, такого как пропиленгликоль, этанол и т.п., или другой стерильной среды, вводимой сразу же перед использованием путем парентерального введения.
Типично фармацевтически приемлемыми носителями являются, например, маннит, мочевина, декстраны, лактоза, картофельный и маисовый крахмал, стеарат магния, тальк, растительные масла, полиалкиленгликоли, этилцеллюлоза, поливинилпирролидон, карбонат кальция, этилолеат, изопропилмиристат, бензилбензоат, карбонат натрия, желатин, карбонат калия, кремниевая кислота. Фармацевтический препарат может также содержать нетоксичные вспомогательные вещества, такие как эмульгаторы, стабилизаторы, смачиватели и т.п., например, сорбитанмонолаурат, триэтаноламинолеат, полиоксиэтиленмоностеарат, глицерилтрипальмитат, диоктилсульфосукцинат натрия и т.п.
Изобретение относится к новым С-4 карбонатсодержащим таксанам формулы 1 и их фармацевтическим солям:
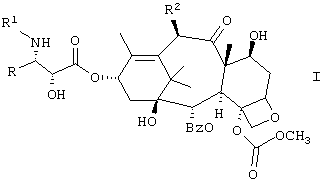
где R обозначает фенил, изопропил или трет.бутил; R1 обозначает -C(O)RZ, в котором RZ обозначает (СН3)3СО-, (СН3)3ССН2-, СН3(СН2)3О-, циклобутил, циклогексилокси или 2-фурил и R2 обозначает СН3С(O)O-, а также и к фармацевтическим композициям на их основе и их использованию в качестве протиопухолевых агентов для лечения болезней людей и животных. Технический результат - получение новых производных таксана, обладающих ценным противоопухолевым действием. 3 н. и 10 з.п., ф-лы, 3 табл.
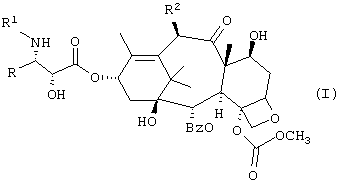
где R обозначает фенил, изопропил или трет-бутил;
R1 обозначает -С(O)RZ, в котором RZ обозначает (СН3)3СО-, (СН3)3ССН2-, СН3(СН2)3О-, циклобутил, циклогексилокси или 2-фурил;
R2 обозначает СН3С(O)O-.
| US 5399726 А, 21.03.1995 | |||
| WO 9414787 A, 07.07.1994 | |||
| ТАКСОИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2144920C1 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |
| RU 2059631 С1, 10.05.1996 | |||
| ТАКСОИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2139864C1 |

