ы
Изобретение относится к способу получения новых производных пиразола общей формулы
соннг
COR
где , RI и Rj - галоген, кокси, С -С -алкил или водород, R4 окси, С,-С -алкокси, С -€4-аЛкенилок- си или фитологически приемлемый остаУказанный промежуточный продукт растворяют в 13 л хлороформа и обрабатывают путем пробулькивания хлористого нитрозила через раствор при температуре окружающей среды. Хлористый нитрозил получают путем медленного добавления 1,23 кг нитрита натрия в водном растворе к 7,2 г концентрированной хлористоводородной кислоты. Прибавление нитрита
4с контролируют так, чтобы его добавле- ток, образующий соль карбоновои кис-
лоты, которые обладают способностью ингибировать образование пыльцы и могут быть использованы в сельском хозяйстве для получения гибридных злаковых семян.
20
ние шло 2ч, а максимальная температура реакционной смеси составляла 35°С. Смесь перемешивают при температуре окружающей среды в течение 1 ч после завершения вьщеления газообразного хлористого нитрозила, а затем смесь нагревают до температуры кипения и 1 ч перемешивают при этой температуре. Смесь охлаждают,
Цель изобретения - получение-новых производных пиразола, обладающи способностью ингибировать образовани пыльцы, и тем самым способствовать получению гибридных семян злаковых культур, более высококачественных, чем при использовании для ингибиро- вания образования пыльцы известного соединения.
Пример 1. Получение 4-карбо- кси-1-(З-хлорфенил)-5-пиразолкарбо- ксамида.
В 22-литровую колбу, снабженную обогреваемым кожухом и холодильником добавляют 2 л деионизированной воды, 7,55 л уксусной кислоты, 1,37 кг ацетата натрия, 1,923 кг хлоргидрата 3- хлорфенилгидразина и 1,854 кГ этилового эфира (этоксиметилен)-циануксус ной кислоты. Смесь медленно подо- гревают до температуры кипения с обратным холодильником и перемешивают при кипении в течение 4 ч. Смесь охлаждают до , добавляют немного воды, чтобы поддерживать смесь при густоте, достаточной для перемешивания. Смесь перемешивают при 10°С.в течение 1 ч и фильтруют. Твердый .продукт промьшают водой и высушивают Твердый продукт растворяют затем в 13л денатурированного этанола и раствор нагревают до температуры кипения с обратным холодильником. Реакционную смесь фильтруют при повьш1енной температуре, охлаждают при перемешива- НИИ и фильтруют. Получают 2,195 кг продукта. Фильтрат концентрируют и получают 170 г продукта при последовательных кристаллизациях. Общий вы
ход 2,3ь5 кг этилового эфира 5-амино- 1-(З-хлорфенил)-4-пиразолкарбоновой кислоты.
Указанный промежуточный продукт растворяют в 13 л хлороформа и обрабатывают путем пробулькивания хлористого нитрозила через раствор при температуре окружающей среды. Хлористый нитрозил получают путем медленного добавления 1,23 кг нитрита натрия в водном растворе к 7,2 г концентрированной хлористоводородной кислоты. Прибавление нитрита
контролируют так, чтобы его добавле-
ние шло 2ч, а максимальная температура реакционной смеси составляла 35°С. Смесь перемешивают при температуре окружающей среды в течение 1 ч после завершения вьщеления газообразного хлористого нитрозила, а затем смесь нагревают до температуры кипения и 1 ч перемешивают при этой температуре. Смесь охлаждают,
перемешивают в течение ночи, сушат сульфатом натрия и фильтруют. Растворитель удаляют под вакуумом и твердый остаток кристаллизуют из денатурированного этанола, получают 2,002 кг этилового эфира 5-хлор-1- (З-хлорфенил)-4-пиразолкарбоновой кислоты.
5 0
промежуточный продукт
f
безводного диме5
0
Указанный
растворяют в 13л тилформамида и к раствору добавляют 686 г безводного цианида натрия. Смесь перемешивают при 100°С ;В течение 4 ч, оставляют стоять в течение ночи при температуре окружающей среды при перемешивании. Затем смесь порциями при перемешивании добавляют к 40 л ледяной воды. Водную смесь перемешивают 45 мин и фильтруют, твердый остаток промывают несколько раз водой. Твердый остаток перекристаллизовьшают из безводного этанола, получают 1,591 кг этилового эфира 1-(З-хлорфенил) -5-циано-4-пиразолкарбоновой кислоты.
Порцию указанного промежуточного продукта массой 1,445 кг добавляют к 14 л денатурированного этанола и нагревают до 0°С, К смеси добавляют 5 л этанола, 1 л воды и 1,035 кг 85%-ной гидроокиси калия. Смесь перемешивают при температуре кипения с обратным холодильником (76-77 С)
2 ч. Смесь оставляют стоять в течение ночи при комнатной температуре. Затем смесь заново нагревают примерно до , чтобы растворить все твердые продукты, и после этого добавляют 25-30 л смеси льда с водой, а рН понижают до 1-2 доб авлением концентрированной хлористоводородной кислоты. Смесь перемешивают 1 ч при и фильтруит. Твердый остаток промывают водой и сушат при , получают 1,024 кг целевого продукта, т.пл. 223-225 С.
Вычислено, %: С 49,73; Н 3,04; N 15,82.
C HgClNjO,
Найдено, %: С 49,81; Н 2,82; N 15,61.
Приме р 2. Получение 4-карбо- кси-1 - (3-фторфешш) -5-пиразолкарбо- ксамида.
5,5 г этилового эфира 5-циано-1- (З-фторфенил)-4-пиразолкарбоновой кислоты и 4,7 г гидроокиси калия растворяют в 100 мл этанола при температуре кипения с обратным холодильником и смесь перемешивают при этой температуре 2 ч. Смесь разбавляют до 350 мл хлорной водой и подогрева- ют до растворения, раствор подкисляют концентрированной хлористоводородной кислотой. Продукт реакции выпадает в осадок добавлением малых количеств льда, осадок отфильтровывают и кристаллизуют из водно-этанольной смеси, получают 3,6 г целевого продукта, т.пл. 215-216 С.
. Вычислено, %: С 53,01; Н 3,21; N 16,87.
C HsFNjOj .
Найдено, %: С 53,16; Н 3,27; N 16,91.
П р и м е р 3. Получение 4-карбо кси-1-фенил-5-пиразолкарбоксамида..
4 г этилового эфира 5-циано-1-фе нил-4-пиразолкарбоновой кислоты обрабатывают 2 г гидроокиси калия в 60 мл денатурированного этанола, ка это описано в примере 2, получают 2,7 г целевого продукта, т.пл. 234- 235°С.
Вычислено, %: С 57,14; Н 3,92; N 18,17.
C,iH9NjO,
Найдено, %: С 57,38; Н 3,93; N 18,37.
П р и м е р 4. Поя чение 4-карбо- кси-1-(4-хпорфенил)-5-пиразолкарбо- ксамида.
Как описано в примере 2, исходя из 3,7 г этилового эфира 1-(4-хлор- фенил)-5-циано-4-11иразолкарбоновой кислоты и 2 г гидроокиси натрия в 60 мл денатурированного этанола и 60 мл воды, полз ают 2,3 г целевого продукта, т.пл. 249-250 С.
Вычислено, %: С 49,73; Н 3,04; N 15,82.
С,Н„С1Н,0,
Найдено, %: С 49,94; Н 3,32; N 15,78.
П р и м е р 5. Получение 4-карбо- кси-1-(2,4-дихлорфенил)-5-пиразол- карбоксамида.
2,5 г этилового эфира 1-(2,4-д1Г- хлорфенил)-5-циано-4-пиразолкарбоно- вой кислоты обрабатьшают 1 г гЧДро- окиси калия в 60 мл 50%-ного водного этанола, получают 1,2 г очищенного продукта, т.пл. 239-240 С. .
Вычислено, %: С 44,03; Н 2,35; N 14,01.
C,,H-,
Найдено, %: С 44,05; Н 2,64; N 13,83.
П р и м е р 6. Получение 1-(4- бромфенил)-4-карбокси-5-пиразолкар- боксамида.
3,5 г этилового эфира 1-(4-бром- фенил)-5-циано-4-пиразолкарбоновой кислоты обрабатьшают 2,15 г гидроокиси калия в 50 мл этанола при температуре кипения с обратным холодильником в течение 2 ч. Получают 2,45 г целевого продукта, т.пл. 251-252,5 С
Вычислено, %: С 42,61; Н 2,60; N 13,55.
C.HjBrNjOj
Найдено, %: С 42,84; Н 2,72; ,N 13,29.
П р и м е р 7. Получение 4-карбо- кси-1 - ( 3,4-дихлорфенил) -5-Шфазол- карбоксамида.
15 г этилового эфира 1-(3,4-ди- хлорфенш1)-5-циано-4-пиразолкарбоно- вой кислоты растворяют в 200 мл этанола и добавляют 5,6 г гидроокиси калия. Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 1 ч, выливают в лед и подкисляют концентрированной хлористоводородной кислотой Смесь фильтруют, твердьй остаток, как было установлено методом ЯМР,
Состоит из частично гидролкзованного А-карбокси-5-цианопиразола. 2 г этого промежуточного продукта отбирают для дальнейшего исследования, а остальное количество твердого остатка заново растворяют в 200 мл этанола и перемешивают при температуре кипения с обратным холодильником в течение 18 ч с 5,6 г. гидроокиси калия. Смесь выливают на лед, подкисляют и фильтруют. Твердый остаток перекрис- таллизовывают из этанола и маточник хроматографируют на силикагелевой колонке с метанолом в качестве элю- ента. Фракции, содержащие продукт, выпаривают досуха и остатки перекрис таллшзовывают из метанола с получением целевого продукта, т.пл. 249- 250°С.
Вычислено, %: С 44,03; Н 2,35; N 14,00,
С.,,Н 7Cl.,N OsНайдено, %: С 43,93; Н 2,37; N 13,9Ь
Примерз. Получение 4-карбо- кси-1-(4-метилфенил)-5-пиразолкар6о- ксамида.
К 10 г этилового эфира 5-циано-1 (4-метилфенил)-4-пиразолкарбоновой кислоты добавляют 5,6 г гидроокиси калия и 200 мл воды, а затем дополнительное количество воды, достаточное для растворения смеси. Смесь перемешивают при температуре кипения с обратным холодильником в течение 8 ч и оставляют стоять в течение 3 дней. Затем смесь вьшивают на лед, подкисляют и фильтруют. Отфильтрован
на лед, подкисляют и фильтруют, твердый остаток кристаллизуют из этаноль- ной смеси, сушат при 168°С и получа- ют 10 г целевого продукта, т.пл.215- 217 С с разложением. Продукт реакции идентифицируют ЯМР-анализом в CDClg (DMCO-d. (f : 7,6б (с, 1), карбокса- мид; 7,30-7,64 (м, 4), ароматические; 8,08 (с, 1), пиразол; 9, 10 (с, 1), карбоксамид.
Пример 10. Получение 4-карбо- кси-1-(З-метилфенил)-5-пиразолкарбо- ксамида.
9 г этилового эфира 5-циано 1-(3- метилфенил)-4-пиразолкарбоновой кислоты растворяют в 200 мл этанола и добавляют 5,8 г гидроокиси калия. Смесь перемешивают при температуре кипения с обратным холодильником в
течение 4ч, охлаждают, разбавляют
0
водой и подкисляют. Смесь фильтруют и твердый осадок промывают этилаце- татом, как показано в примере 8, по- 5 лучают 3,8 г целевого продукта, т.пл. 209-211°С с разложением.
Вычислено, %: С 58,77; Н 4,52; N -17,13.
.NiOj.
Найдено, %: С 58,58; Н 4,63; N 16,85. .
Пример 11. Получение 4-карбо- кси-1-(4-фторфенил)-5-пиразолкарбо- ксамида.
2,5 г этилового эфира 5-циано-1- (4-фторфенил)-4-пиразолкарбоновой кислоты и 2,6 г гидроокиси калия перемешивают при кипении с обратным холодильником в 100 мл денатурирован5









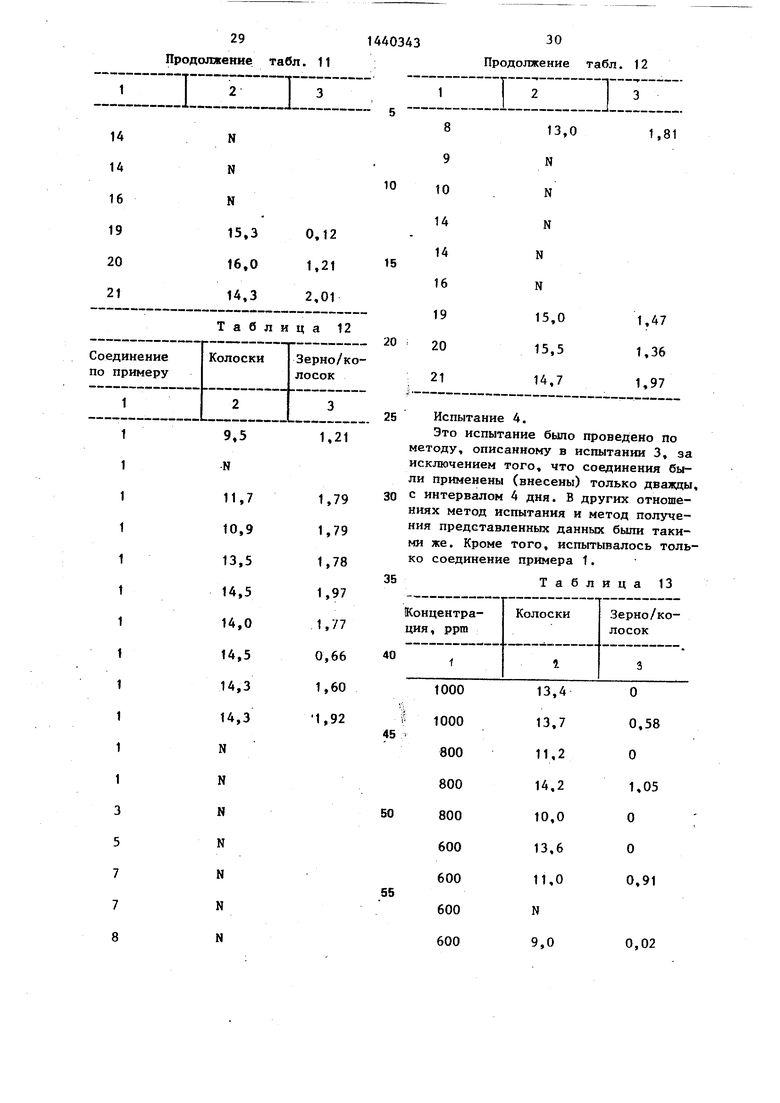
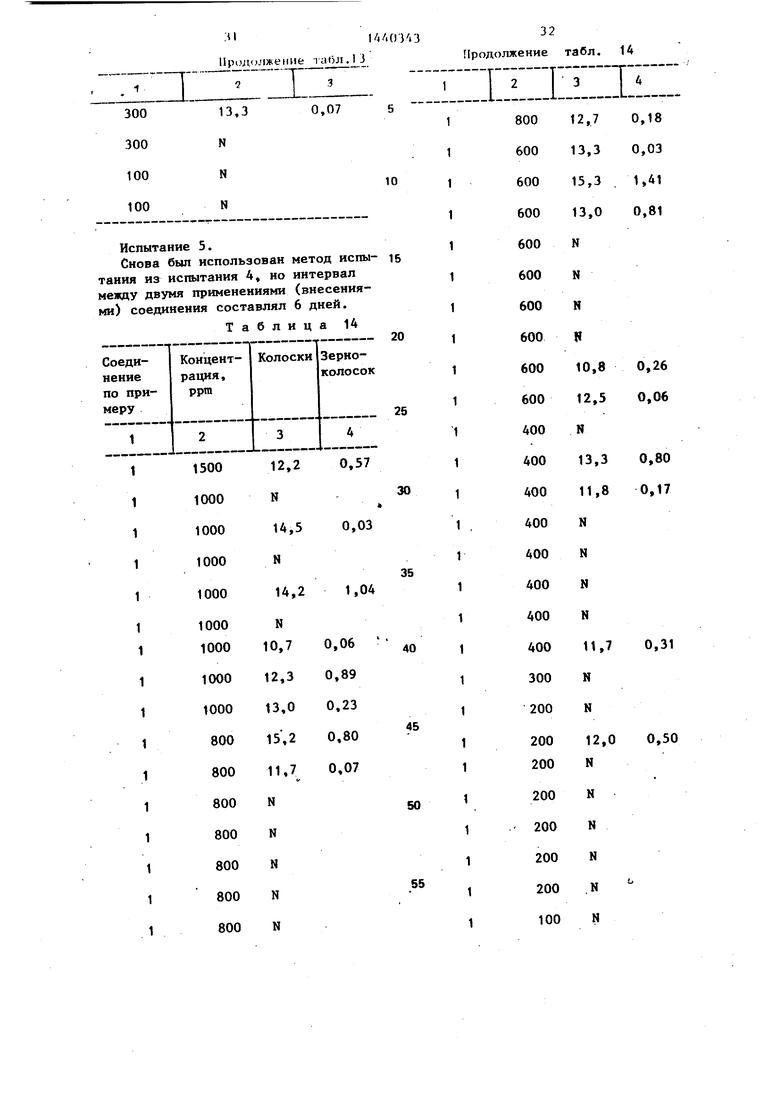

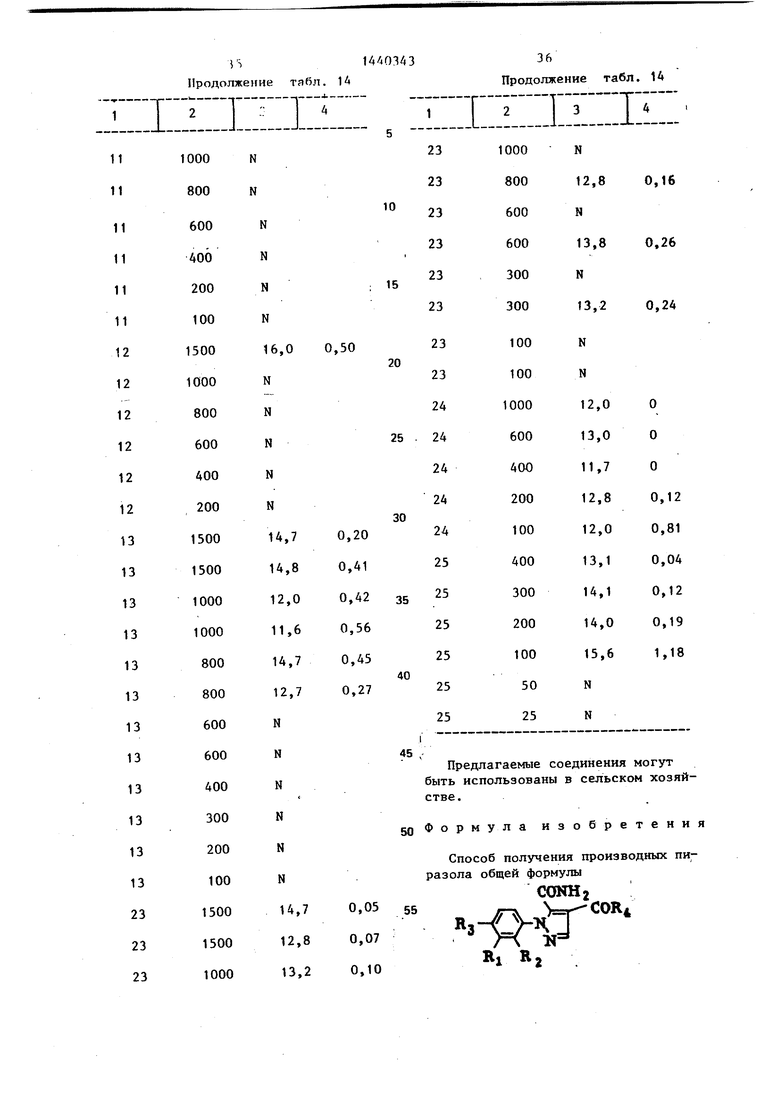
Изобретение касается гетероциклических соединений, в частности производных пиразола общей формулы: ( ,-CR СН-СН С - N-N-CH-C C(0)R C-C(Q)-NH2, где R,, R независимо - галоген, С -алкокси, С . алкил или Н, при условии, что хотя бы один из R.f, RI и RI Н и что RI Н только когда один из R и Rj Са не оба ff, , С ,. -алкокси, С -алкеняпокси или фитологически приемлемьй остаток, образующий соль карбоновой кислоты, обладающих способностью ингибировать образование пыльцы, что может быть использовано в сельском хозяйстве для получения гибридных злаковых семян. Цель изобретения - создание новых более активных веществ указанного класса. Синтез целевых соединений ведут гидрр- лизом нитрила общей формулы; Ra-C Е,-СНз СН-СН й - (0)OR53 C-CN, где R, RlиRз- см,вьппе, R.f С,-алкил, в присутствии основания при кипении и выделяют целевой продукт, где R4 ОН, или в виде эфира, где R 4 С -алкокси, С ,-4-алке- нилокси или в виде фитологически приемлемой соли. Новые соединения позволяют получать гидридные семена с нормальным внешним видом, время как при использовании известных соединений образуются г ибридные семена деформированной формы. 14 табл. (Л о со 4 оо
ный остаток нагревают в 100 мл этил- до кого этанола в течение 4 ч. Смесь охацетата, нерастворившийся твердый остаток отделяют, сушат, полз т1ают 4,25 г целевого продукта, т.пл.260 С с разложением. Продукт идентифицируют с помощью ЯМР-спектра, растворитель - дейтерированный диметилсульфо- ксид DMCO-dg. d ; 2,36 (с, 3), метил; 7,32 (д, 2), ароматические; 7,45 (с, 1), карбоксамид; 8,33 (с, 1), карбоксамид .
П р и м е р 9. Получение 1-(3- бромфенил)-4-карбокси-5-пиразолкар- боксамида.
14 г этилового эфира 1-(3-бромфелаждают, подкисляют, разбавляют водой и концентрируют под вакуумом, получают 2,8 г целевого соединения, т.пл. 232°С с разложением после пе- 45 рекристаллизации из ацетона.
Вычислено, %: С 53,02; Н 3,24; N 16,86.
Ci HeFNjGj
Найдено, %: С 53,27;
50
Н 3,02; N 16,69.
Пример 12. Получение 4-карбо- кси-1-(2,З-дихлорфенил)-5-пиразолкар- боксамида.
15 г этилового эфира 1-(2,3-динил)-5-циано-4-пиразолкарбоновой кис- хлорфенил)-5-циано-4-пиразолкарбоно- лоты гидролизуют при перемешивании вой кислоты перемешивают при темпера,- при кипении с обратным холодильником |туре кипения с обратным холодильни- в 200 мл этанола с 6 г гидроокиси ком в течение ночи в 200 мл этанола калия в течение 2ч.. Смесь вьшивают с 6 г гидроокиси калия. Реакционную
лаждают, подкисляют, разбавляют водой и концентрируют под вакуумом, получают 2,8 г целевого соединения, т.пл. 232°С с разложением после пе- 5 рекристаллизации из ацетона.
Вычислено, %: С 53,02; Н 3,24; N 16,86.
Ci HeFNjGj
Найдено, %: С 53,27;
0
Н 3,02; N 16,69.
Пример 12. Получение 4-карбо- кси-1-(2,З-дихлорфенил)-5-пиразолкар- боксамида.
71
смесь хроматографируют на силикагеле объединяют фракции,.содержащие продукт, и выпаривают досуха. Остаток перекристаллизовывают из водно-этанольной смеси, получают 1,31 г целевого продукта, т.пл. 228-230 С.
Вычислено, %: С 44,03; Н 2,35; N 14,00,
C.H CljNjO
Найдено, %: С 44,04; Н 2,39; N 13,88.
П р и м е р 13. Получение 4-карбо кси-1-(З-хлор-4-метилфенил)-5-пира- золкарбоксамида.
9,5 г этилового эфира 1-(З-хлор- 4-метилфенил) -5-циано-4-пиразолкар- боновой кислоты растворяют в 250 мл этанола и добавляют 2 г гидроокиси калия. Смесь перемещивают при темпе- ратуре кипения с обратным холодильником в течение 30 мин. Добавляют 100 мл воды и перемешивают при температуре кипения с обратным холодиль НИКОМ в течение еще 4 ч. При попытке выделить продукт реакции обнаружено, что он является трудно разделяемой смесью. Поэтому всю реакционную смес возвращают обратно в колбу, заново растворяют в водном этаноле и кипя- тят с обратным холодильником в течение 1 ч на паровой бане с 6 г дополнительного количества гидроокиси калия. Затем смесь разбавляют водной уксусной кислотой и фильтруют, твер- дый остаток сушат, получают 5,2 г целевого продукта, т.пл. 228-232 С с разложением.
Вычислено, %: С 51,53; Н 3,60; N 15,02.
С„Н,оС1НзОз
Найдено, %: С 51,80; Н 3,36; N 14,92.
Приме р14. Получение метилового эфира 5-аминокарбонил-1-(3-хлор фенил)-4-пиразолкарбоновой кислоты.
2,2 г 4-карбокси-1-(3-хлорфенил)- 5-пиразолкарбоксамида суспендируют в 40 мл метанола и через смесь про- булькивают клористьй водород в тече- ние 1 мин. Смесь перемешивают при температуре кипения с обратным холо дильником в течение 2ч, выливают в лед и подщелачивают разбавленным водным растйором гидроокиси натрия. Смесь фильтруют и твердьш остаток высушивают и кристаллизуют из толуо ла, получают 1,7 г целевого продукт т.пл. 191-192°С.
8
Вычислено, %: С 51,53; Н 3,60; N 15,02.
C,jH ,сС1К,Оз
Найдено, %: С 51,23; Н 3,71; N 14,83.
Пример 15. Получение аллило- вого эфира 5-аминокарбонил-1-(3-хлор- фенил)-4-пиразолкарбоновой кислоты.
3,33 г 4 карбокси-1-(3-хлорфенил)- 5-пиразолкарбоксамида суспендируют в 35 мл метанола и добавляют 0,68 г ме- токсида натрия. Метанол удаляют под вакуумом, получают натриевую соль исходного соединения. Эту соль смешивают с 1,26 г триэтиламина и 1,51 г аллилбромида в 35 мл толуола и Лолу- ченную смесь перемешивают при температуре кипения с обратным холодильником в течение ночи. Смесь вьшивают в 150 мл смеси льда с водой, подщелачивают с помощью насыщенного раствора бикарбоната натрия и экстрагируют 150 мл этилацетата. Органический слой промывают рассолом, сушат и выпаривают под вакуумом с получением твердого продукта, который перекрис- таллизовьгоают из толуола, получают 1,48 г очищенного продукта, т.пл. 132-133°С.
Вычислено, %: С 55,00; Н 3,96; N 13,74.
С„Н .,СШзОз
Найдено, %: С 55,15; Н 3,96; N 13,70.
Пример 16. Получение этилового эфира 5-аминокарбонил-1-(3-хлорфе- нил)-4-пиразолкарбоновой кислоты.
2,5 г 4-карбокси-1-(3-хлорфенил)- 5пиразолкарбоксамида суспендируют в 50 мл абсолютного этанола, подкисляют, этерифицируют и вьщеляют, как это описано в примере 14. Получают 1,64 г целевого продукта, т.пл..
Вычислено, %: С 53,16; Н 4,12; N 14,31.
С,зН С1ЫзОз
Найдено, %: С 53,37; Н 4,04; N 14,61.
Пример 17. Получение метилового эфира 5-аминокароонил-1-(3-ме- тилфенил)-4-пиразолкарбоновой кислоты.
Газообразный хлористый водород пробулькивается в течение 1 мин в суспензию 3 г 4-карбокси-1-(3-метил- фен1т)-5-пиразолкарбоксамида в 30 мл метанола. Смесь перемешивают при температуре кипения с обратным холодильНИКОМ 2ч, охлаждают и оставляют на ночь. Затем выливают в 150 мл льда и воды и подщелачивают разбавленной гидроокисью натрия. Выпавший продукт реакции отфильтровьшают, высушивают и перекристаллизовывают из толуола, обрабатьгеая древесным углем, получают 1,16 г целевого продукта, т.пл.
ley-ies c.
Вычислено, %: С 60,23; Н 5,05; N 16,21.
,эНзОз-.
Найдено, %: С 60,18; Н 4,99; N 16,08.
П р и м е р 18. Получение натриевой соли 4-карбокси-1-(3-метилфенил) 5-пиразолкарбоксамида.
3 г 4-карбокси-1-(3-метилфенил)- 5-пиразолкарбоксамида суспендируют Б 30 мл метанола и добавляют 0,66 г метоксида натрия. Смесь перемешивают в течение непродолжительного времени фильтруют и выпаривают досуха. Остаток растворяют в метаноле, обрабатывают древесным углем и перекристаллиэовывают. Продукт реакции очень гигроскопичен, его высушивают в течение 8 ч при .
Вычислено, %: С 53,94; Н 3,77; N 15,72.
Ci4H,,NjNaOjНайдено, %: С 54,11; Н 3,73; N 15,52..
Приме р19. Получение натриевой соли 4-карбокси-1-(3-хлорфенил) 5-пиразолкарбоксамидаk
6,14 г 4-карбокси-1-(3-хлорфенил) 5-пиразолкарбоксамида суспендируют в 60 мл метанола и добавляют 1,25 г метоксида натрия. Смесь перемешивают в течение нескольких fflнyт и фильтруют. Фильтрат выпаривают досуха под вакуумом, остаток растворяют в 50 мл метанола и кристаллизуют добавлением диэтилового эфира. Твердый остаток отфильтровывают, высушивают и получают 4,55 г целевой соли, т.пл. 274С 45,93; Н 2,45;
276°с:
Вычислено, N 14,61.
C i H-jClNj-NaO.
Найдено, %: С 46,10; Н 2,26; N 14,58.
П р и м е р 20. Получение калиевой соли 4-карбокси-1-(3-хлорфенил)- 5-пиразолкарбоксамида.
4,25 г 4-карбокси-1-(3-хлорс1)енил) 5-пиразолкарбоксамида суспендируют
в 40 мл абсолютного этанола вместе с 1,03 г 85%-ного гидроксида калия и смесь нагревают до температуры кипения с обратным холодильником. К кипящей смеси добавляют незначительное количество воды для полного растворения всей смеси, затем охлаждают до температуры окружающей среды. Затем смесь охлаждают в холодильнике, фильтруют и получают 3,32 г целевого продукта, т.пл. выше 300°С с разложением.
Вычислено, %: С 43,50; Н 2,32;
N 13,83.
Ci H- ClKNgOj,
Найдено, %: С 43,26; Н 2,09; N 13,55.
П р и м е р 21. Получение изопропиламиновой соли 4-карбокси-1-(3- хлорфенил ) -5-пиразолкарбоксам11да.
Вычислено, %: С 52,10; Н 4,68; N 17,36.
С„Н,,С1Ы,Оз
Найдено, %: С 5Z,16; Н 4,77;
IN 17,19П р и м е р 22. Получение тетра- бутиламмонийной соли 4-карбокси-1- (3-хлорфенил)-5-пиразолкарбоксамида.
в 50 МП метанола и прибавляют 12 мл в 1 М раствора гидроокиси тетрабутнл- аммония. Смесь перемешивают в течение 30 мин и выпаривают досуха под вакуумом для получения 5,2 г целевого продукта., т.пл. 120-121 С.
Вычислено, %: N 11,05.
С.Н4зС1К40э
Найдено, %: N 11,07.
П р и м е р 23. Получение 4-карбо- кси-1-(3,4-диметилфенил)-5-пиразолкарбоксамида.
2,5 г этилового эфира 5-циано-1- (3,4-диметилфенил)-4-пиразолкарбоно- вой кислоты гидролизуют в 50 мл этанола и 12 МП воды с помощью 1,3 г гидроокиси калия при температуре кипения с обратньм холодильником в течение 2,25 ч. Смесь охлаждают и выли
вают в 300 мл воды. Водную смесь фильтруют, подкисляют концентрировной хлористоводородной кислотой и фильтруют. Твердый остаток сушат и перекристаллизовывают из этанол-воной смеси. Получают 1,62 г целевог продукта, т. пл. 231-232,5°С.
Вычислено, %: С 60,23; Н 5,05; N 16,21.
C,,H,,
Найдено, %: С 60,47; Н 4,94; N 16,05.
П р и м е р 24. Получение 4-карбо- кси-1 - ( З- Этилфенил) -5-пиразолкарбо- ксамида.
Порцию 3-этиланилина весом 60,6 г прибавляют к 132 г концентрированной хлористоводородной кислоты и 67 г льда при , добавляют еще 67 г льда и смесь снова охлаждают до . К смеси по каплям добавляют 36,3 г нитрита натрия, растворенного в 75мл воды в течение 1 ч, поддерживая температуру ниже 6 С. За это же время получают раствор сульфита калия, про булькивая двуокись серы через раствор 163,8 г гидроокиси калия в 750м воды. Двуокись серы продолжают пропускать до достижения рН 4,7. Затем добавляют 67 г льда и раствор охлаждают до 0°С.
Два препарата смешивают вместе, по возможности быстро, в результате чего температура повьшзается до 8°С. Затем смесь нагревают на паровой бане до 70°С и перемешивают при этой температзфе в течение 1 ч. Затем смесь охлаждают до 0°С и выпадаюпщй в осадок продукт реакции отделяют фильтрованием и высушивают. Твердые остатки перекристаллизовывают из большого количества этанола, получают 70,9 г 3-этилфенилгидразинсульфоната калия, разлагается при температуре вьш1е, чем 195°С.
15,0 г указанного промежуточного продукта перемешивают в 150 мл воды и 75 мл хлористоводородной кислоты
вместе с малым количеством древесно- 50 Ра, органический слой высушивают и
го угля при и смесь фильтруют в горячем состоянии. Фильтрат охлаждают до температуры окружающей среды в течение ночи и после завихрения раствора начинает немедленно выпадать осадок. Твердые остатки отделяют фильтрованием, высушивают и получают г хлоргидрата 3-этилфенил гидразина, т.пл. 147-157 С.
выпаривают, получают 7,9 г маслообразного продукта, который очищают жидкостной хроматографией высокой производительности, элюируя 1,2-ди- 55 хпорэтаном. Выпаривание фракций, со держащих продукт реакции, дает 6,1 г этилового эфира 5-циано 1-(3-этилфе нш1)-4-пиразолкарбоновой кислоты в виде масла.
I 2
10,7 г указанного промежуточного продукта смешивают с 10,5 г эт1шово- го эфира (этоксиметилен)-цианоуксусной кислоты и 10,2 г ацетата натрия в 100 мл этанола, и смесь перемешивают при температуре кипения с обратным холодильником в течение 20 ч. Смесь ме.опенно выливают в 400 мл смеси льда с водой при перемешивании и твердый остаток отделяют фильтрацией и высушивают. Твердый остаток перекристаллизовывают из водного этанола, получают 12,7 г этилового эфира 5-амино 1-(3-этилфенил)-4-пираэол- карбоновой кислоты, т.пл. 79-79,. 10,2 г указанного промежуточного продукта растворяют в минимальном количестве хлороформа и через раствор
в течение 1 мин при комнатной температуре пробулькивают газообразный хлористый водород. Затем через эту смесь в течение 20 мин пробулькивают газообразный хлористый нитрозил, под
держивая при этом температуру в пределах 20-35°С с помощью ледяной банн. Затем смесь нагревают на паровой бане, чтобы удалить избыток хлористого нитрозила, сушат с помощью фазоразделяющей бумаги и органическую часть вьшаривают под вакуумом. Остаток очищают с помощью Ж5вдкостной хроматографии высокой производительности, элюи- руя 1,2-дихлорэтаном. фракции, содержащие продукт реакции, собирают и выпаривают под вакуумом, получают 2,9 г этилового эфира 5-хлор-1-(3-этилфе- нил)-4-пиразолкарбоновой кислоты в виде масла.
8,9 г указанного промежуточного продукта смешивают с 35 мл диметил- формамида и 3,4 г цианида натрия и смесь нагревают в течение 6 ч примерно при 100°С. Смесь охлаждают, добавляют дополнительное количество цианида натрия и смесь нагревают при 100 С в течение более 2 ч. Смесь охлаждают, выливают в 300 мл смеси льда с водой, экстрагируют 300 мл диэтилово-го эфивыпаривают, получают 7,9 г маслообразного продукта, который очищают жидкостной хроматографией высокой производительности, элюируя 1,2-ди- хпорэтаном. Выпаривание фракций, содержащих продукт реакции, дает 6,1 г , этилового эфира 5-циано 1-(3-этилфе- нш1)-4-пиразолкарбоновой кислоты в виде масла.
2,5 г указанного промежуточного продукта добавляют к 25 мл этанола, содержащего 1,6 г гидроокиси калия, и смесь нагревают при температуре кипения с обратным холодильником в течение 20 мин. Добавляют 5 мл воды, смесь нагревают при кипячении с обратным холодильником в течение 1,5 ч а затем выливают в 100 мл воды. Смесь подкисляют концентрированной хлористоводородной кислотой, охлаждают при в холодильнике и фильтруют, получают 2,0 г целевого продукта, т1пл 176-177,.
Вычислено, %: С 60,23; Н 5,05; N 16,21.
nHliNgO..
Найдено, %: С 60,03; Н 4,83; N 15,93о
П р и м е р 25. Получение 4-карбо кси-1-(3-метоксифенил)-5-пиразолкар- боксамида.
34,9 г хпоргидрата 3-метоксифенил гидразина прибавляют к 300 мл уксус- ной кислоты, 100 мл воды, 36 г ацета та натрия и 37,2 г этилового эфира (этоксиметилен)-цианоуксусной кислоты. Смесь нагревают в течение ночи на паровой бане, а затем охлаждают и выпивают в 1 л смеси льда с водой при энергичном размешивании. Смесь фильтруют и твердый остаток сушат воздухом и перекристаллизовьгаают из водного этанола с древесным углем, получают 27,4 г э- штового эфира 5- амино-1-(3-метоксифенил)-4-пиразол- карбоновой кислоты, т.пл. 66г67 С.
13,3 г указанного промежуточного продукта растворяют в 80 мл бромо- форма и смесь охлаждают до 5 С. К смеси добавляют по каплям 10,5 г трет-бутилнитрита, оставляют до достижения температуры окружающей среды и нагревают на паровой бане в тече- ние 15 мин. Смесь вьшаривают под вакуумом, получают 21,1 г темного масла, которое растворяют в этилацетате Раствор промывают 1 N раствором хло- ристоводородной кислоты, водой, насыщенным раствором бикарбоната натрия и рассолом, а затем высушивают и выпаривают под вакуумом, получая темное масло, которое очищают жидкост- ной хроматографией высокой производи тельности, элюируя 1:3 смесью этил- ацетата и гексана, получают 10,6 г этилового эфира 5-броя-1-(3-метокси-,
фенил)-4-пиразолкарбоновой кислоты, т.пл. 77-79°С.
3,6 г указанного промежуточного продукта смешивают с 1,2 г цианида натрия в 20 мл диметилформамида и смесь нагревают в течение 10 ч при . Добавляют дополнительное количество (0,3 г) цианида натрия и смесь нагревают в течение ночи при , затем выпивают в 100 мл смеси льда с водой. Выпадающий осадок отделяют фильтрованием и сушат, затем перекристаллиэовывают из этанола с древесным углем, получгиот 1,0 г этилового эфира 5-циано-1-(3-метоксифе- нш1)-4-пиразолкарбоновой кислоты, т.пл. 84-85°С.
0,95 г указанного промежуточного продукта прибавляют к 25 мл этанола и 0,6 г гидроокиси калия и смесь нагревают до кипения с обратным холодильником. Добавляют 10 мл воды и смесь нагревают при кипении с обратным холодильником в течение 1,5 ч. Затем ее охлаждают и фильтруют, фильтрат выливают в 100 мл воды. Водную смесь подкисляют концентрированной хлористоводородной кислотой и энергично перемешивают, охлаждают в холодильнике. Выпавший осадок отделяют фильтрованием, высушивают, перекрис- таллизовывают из водного этанола, получают 0,4 г целевого продукта, т.пл. 213-216°С.
Вычислено, %: С 55,17; Н 4,24; N 16,08.
С,.Н,, N,0,.
Найдено, %: С 55,12; Н 3,99; N 15,83.
Предлагаемые соединения тщательным образом испытаны, чтобы продемонстрировать их активность при ингиби- ронании образования пыльцы. Результаты ЭТ1-1Х испытаний показаны ниже.
Испытание 1.
Зарегистрированный здесь эксперимент был полевым экспериментом, проведенным в центральной Индиане, США. Эксперимент начинали с засева узкими полосками пшеницы Обена и Боу осенью Некоторые узкие полоски были засеяны 4 октября, а другие 14 октября. Во время посева поле удобряли сочетанием удобрений, подходящих для выращивания шпеницы. Соседние узкие полоски пшеницы Обена и Боу были засеяны поставляющими пыльцу делянками смешанных сортов пшеницы Кэлдвила, Обена и Титана в соотношении 1:1:2. Поставляющие пыльцу делянки были засеяны 1 октября при норме 113,6 кг зерна на 1 га (100 фунтов на акр).
Ис1тытуемым соединением в этом эксперименте было соединение из примера 1. Соединение вводят в состав для применения в объемном соотношении 1:1 смеси ацетона и денатурированного спирта. Органический раствор разбавляют водой, содержащей 0,25% поли- сорбата 20, при применении. Все внесения (применения) осуществляли с объемной нормой 4728 л на 1 га (500 галлонов на акр) и применяли путем опрыскивания листвы испытуемых де- лянок.
Первое применение соединения бьшо осуществлено 26 апреля после посева пшеницы. Если соединение применяли многократно, то более поздние применения осуществляли с интервалами в одну неделю.
мой делянки составлял 4 ряда г 396,5 см (4 ряда х13 фут).
Во время появления метелки шпени- g цы некоторые метелки в каждой обработанной делянке укрьтали в мешочки с помощью зерновых опьшйтельных целлофановых изоляционных мепючков. В мешочки укрывали пять нетелок в каж- 10 дом ряду каждой испытуемой делянки.
Когда образовались зерна, предусматривали подсчет содержания коли чества зерен в метелке в растениях,
15 укрытых в мешочки и у которых инги- бировали образование пыльцы, потому что эти зерна могут образовываться только путем самоопыления. Количество зерен в метелке у неукрытых расте20 НИИ в каждой испытуемой делянке под- считьгоали так же, как и количество зерен в метелке у необработанных контрольных делянок.
Две делянки были обработаны согИспытуемые делянки были расплани- 25i ласно каждому режиму обработки и ре- рованы по зонам пшеницы Обена и Boy, зультаты представлены в усредненных которые становятся женскими делянка- значениях в табл. 1, где дана актив- ми для выращивания .гибридной пшеницы, ность соединения по примеру 1 в отношении ингибирования образования пыльопыляемой поставляющими пыльцу делянками шпеницы. Размер каждой,испытуе- 30, ЦЫ пшеницы.
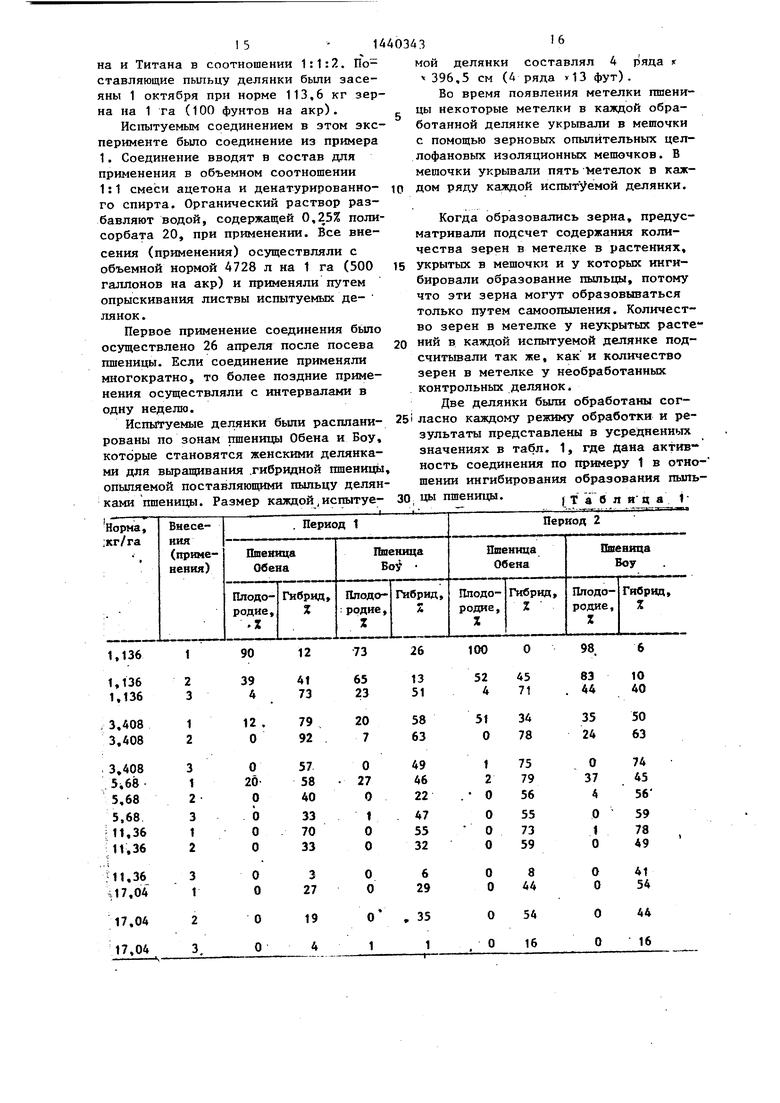
Т а б л н ц а i
1,136
90
12
мой делянки составлял 4 ряда г 396,5 см (4 ряда х13 фут).
Во время появления метелки шпени- цы некоторые метелки в каждой обработанной делянке укрьтали в мешочки с помощью зерновых опьшйтельных целлофановых изоляционных мепючков. В мешочки укрывали пять нетелок в каж- дом ряду каждой испытуемой делянки.
Когда образовались зерна, предусматривали подсчет содержания коли чества зерен в метелке в растениях,
укрытых в мешочки и у которых инги- бировали образование пыльцы, потому что эти зерна могут образовываться только путем самоопыления. Количество зерен в метелке у неукрытых растеНИИ в каждой испытуемой делянке под- считьгоали так же, как и количество зерен в метелке у необработанных контрольных делянок.
Две делянки были обработаны согТ а б л н ц а i
73
26
100
98
Данные представляются отдельно для каждого вида пшеницы и для каждой даты посева. Более рано засеянные делянки описьгеаются как период I, а более поздние как период 2. Столбцы, озаглавленные словом плодородие, представляют количество зерен в метелке обработанных, укрытых в мешочки, растений, в виде процента к необработанным контрольным растениям, и поэтому представляют непосредственную меру образования пыльцы. Столбцы, озаглавленные словом гибрид, представляют разницу мелзду количеством зерен в метелке, обработанных, неукрытых в мешочки, растений и зерен в метелке обработанных, укрытых в мешочки, растений
в виде процента к необработанным кон- 20 нократной обработке.
Испытание 2.
Соединение примера 1 было применено к 61 разновидности пшеницы. Каждая испытуемая делянка состоит из трех по 61 см (2 фута) вручную засеянных рядов,с расстоянием 61 см (2 фута) между группами из трех рядов. Делянки бьши засеяны в центральной Индиане 5 октября. На следуюпц1Й год 5 мая два из трех рядов были опрыскатрольньп растениям. Таким образом, этот столбец представляет количество .выращенных гибридных зерен по сравнению с зернами, выращенными из необработанных контрольных растений. Чистота выращенных гибридных зерен может быть установлена сравнением процента плодородия и процента
гибрида.
Дополнительные делянки в той же группе делянок пшеницы Обена и Боу бьти обработаны однократно, либо 4 мая, лЬбо 10 мая, после посева шпе
ницы в октябре.Результаты этих экспериментов представлены ниже в табл. 2, где дана активность соединения по примеру 1 в отношении инги- ;бирования образования пыльцы при одТаблицу,
ны соединением примера 1 при норме 5,68 кг на 1 га (5 фунтов на акр) и 11,36 кг на 1 га (10 фунтов на акр), а третий ряд оставляли в качестве необработанных контрольных растений.
Растения были укрыты в мешочки и образование зерен у укрытых в мешочки, неукрытых в мешочки и необработанных растений бьшо подсчитано, как
19
это описано в испытании 1. Пьтьца для этих экспериментов бьша обеспечена зонами той же смеси пшенищ 1, поставляющими пыльцу, что было описано в испытании 1.
Данные из этой группы экспериментов представляются в табл. 3 в суммированной форме, в виде ряда разновидностей пшеницы, которые показьгоа- ют плодородие укрытых обработанных растений в различных пределах по сравнению с плодородием необработанных контрольных растений.
Таблица 3
Испытание 3.
Стандартизованное тепличное испытание было использовано, чтобы оценить предлагаемые соединения. Испытание было начато посевом шпеницы Воддрона в делянках по 10 см (4 дюй- мй), по 4 зерна на делянку в стерильной песочно-суглинистой почве. ПшениПредпочтительными соединениями на стоящего изобретения, которые являют ся также предпочтительными соединени ями для практического применения ос пред ставленных способов, ингибирова- Ния образования пыльцы, является 4- карбокси-1-(3-хлорфенил)-5-пиразол- карбоксамид, 4-карбокси-1-(3-метил- фенил)-5-пиразолкарбоксамид, 4-карбо
це было позволено расти в благоприят- до кси-1-(3-этил Ьенил)-5-пиразолкарбоксамид, 4-карбокси-1-(3,4-дихлорфе- нил)-5-пиразолкарбоксамид и 4-карбокси- 1-(3-метоксифенил)-5-пиразолкарбоксамид. Соли щелочных металлов аммонийные соли и моно-, ди- и три (С -С4-алкил)-аминовые соли только что названных соединений также явля ются предпочтительными соединениями настоящего изобретения.
ной тепличной среде, а растения бьши обработаны трижды испытуемым составом. Первое применение (внесение было осуществлено спустя примерно 22 дня после посева зерен, а второе и третье внесение было осуществлено спустя примерно 3 и 10 дней после первого внесения.
Каждое соединение было введено в состав для испытания путем растворения необходимого количества для двух одинаковых рядов в делянке и для трех внесений (применений) в зависимости от концентрации, которая должна быть испытана в 5 мл 1:1 объемной смеси ацетона и денатурированного спирта, содержащего 10 об.% полисорбата 20. Нерастворяющиеся соединения были тонко диспергированы в растворителе.
Затем органическая смесь была разбавлена до 30 мл с помощью деионизиро- ванной воды при температуре окружающей среды, и водная дисперсия была равномерно опрыскана на листву двух делянок пшеницы.
В каждом эксперименте были предусмотрены необработанные контрольные растения.
Результаты экспериментов представляются в виде количества колосков, вьфащенных из обработанных растений, и в виде количества зерен на колосок Среднее количество колосков на нормальное растение составляет примерно 15, а количество зерен на колосок изменяется в пределах примерно 1,5- 2,5 м.
В табл. 4-14 усреднены результаты скопированных экспериментов. Если результаты эксперимента не отличались от результатов, совпадающих во вре- мени необработанных контрольных растений, то неактивность обозначается просто буквой N.
Предпочтительными соединениями настоящего изобретения, которые являются также предпочтительными соединениями для практического применения пред ставленных способов, ингибирова- Ния образования пыльцы, является 4- карбокси-1-(3-хлорфенил)-5-пиразол- карбоксамид, 4-карбокси-1-(3-метил- фенил)-5-пиразолкарбоксамид, 4-карбокси-1-(3-этил Ьенил)-5-пиразолкарбо
ксамид, 4-карбокси-1-(3,4-дихлорфе- нил)-5-пиразолкарбоксамид и 4-карбокси- 1-(3-метоксифенил)-5-пиразолкарбоксамид. Соли щелочных металлов, аммонийные соли и моно-, ди- и три (С -С4-алкил)-аминовые соли только что названных соединений также являются предпочтительными соединениями настоящего изобретения.
При применении по данному назначению известных соединений, а именно 1-арил-1,4-дигидро-4-оксо(ш1И тио) пиридазинов, обр зуются гибридные семена деформированной формы, в то время как гибридные семена, полученные при использовании предлагаемых соединений, имеют нормальный внешний вид.
В табл. 4 приведены данные о личном испытании соединений.
Таблица 4
В табл. 5-12 приведены зависимости от концентрации соединений. Концентрация ррт: в табл. 5 - 1200; в табл. 6 - 1000; в табл. 7 - 800; в табл. 8 - 600; в табл. 9 - 400; в табл. 10 - -300; в табл. 11 - 200; IB табл. 12 - 100.
22
Таблица 5
Колоски
Зерно/колосок
1 1 2 5
10,6 11,7 N 13,5
Т а б л
О 0,22
0,01 и ц а 6
5
0
5
0
5
0
5
9,0 9,4 12,0 11,2 N N N
12,9 13,8 13,5 12,3 13,6 13,7 15,3 12,7 15,5 15,6 14,0 15,3 15,0
О О
0,16 0,27
О
О
0,50
0,41
0,68
0,84
1,28
I
0,89 0,81 0,69 0,15 0,08 0,98
29 Продолжение табл. 11
I
Таблица 12
9,5
N
11,7 10,9 13,5 14,5 14,0 14,5 14,3 14,3 N N N N N N N
1,21
.1.77 0,66 1,60
1,92
14А0343
30 Продолжение табл. 12
Испытание 4.
Это испытание было проведено по методу, описанному в испытании 3, за исключением того, что соединения были применены (внесены) только дважды,
с интервалом 4 дня. В других отношениях метод испытания и метод получения представленных данных были такими же. Кроме того, испытывалось только соединение примера 1.
35
13
40
45
ВО
5S
.и
11р1)Д(.чжемне liitbji.l 3
Испытание 5.
Снова был использован метод испытания из испытания 4, но интервал между двумя применениями (внесениями) соединения составлял 6 дней.
Таблица 14
1/4/,(Г)ДЗ
15
20
33 Продолжение табл. 1A
600 600
600
U,5 13.7
;14,2
0,09 0,18
0,18
1А403АЗ
34
37
где R, RguR. - независимо друг
от друга, галоген.
С -С -алкокси,
при условии
2
С,-С.-апкш1 или водород, что по крайней мере один из Ri,, R Rg - водород и при условиии что R может представлять группу, отличную от водорода, только когда один из R и Rj, а не оба, представляют группу, отличную от водорода; Rч оксиС,.-С4 алкокси, С2-С4-алкенш1окси или фитологически приемлемый остаток, образующий соль карбоновой киcлotы, отличающийся тем, что осуществляют гидролиз нитрила общей формулы
.
,10
где К
;-f
034338
CN. COORs
Я, R,
R i и R 5 имеют указанные значения, Rу - низший С -С -алкил, в присутствии основания при температуре кипения растворителя с вьщелением целевого продукта, где R4 является оксигруппой, или с вьщелением целевого продукта в виде эфира, где R представляет собой С1,-С -алкокси или 5 С;2-С4-алкенилокси, или в виде фитологически приемлемой соли.
| Физер Л., Физер М | |||
| Органическая химия | |||
| - М.: Химия, 1970, т.1, с.429. |
