где Y - водород или диметиламиногруппа;
R - 3- или 4-пиперидиламино или 3- ИЛИ 4-пиперидилтйо, в некоторых случаях замещенные по атому азота цикла алкиль- ным радикалом, алкиламино, алкилркси или алкилтиогруппа замещенные одной гидрокси- сульфонильной, алкиламино, одной или двумя диалкил- аминогруппами, в некоторых случаях замещенной диалкил- аминогруппой, триалкиламмо- нием или 4- или 5-имидазоли- лом или одним циклом, выбранным среди пиперазино, в некоторых случаях замещенного алкилом, пиперидино, 1-пирро лидинила, 2-, 3- или 4-пипе- ридила и 2-пирролидинш1а, в некоторых случаях замещенног по атому азота алкильным ра- дикалом, при условии, что алкильньй радикал содержит 1-4 атома углерода в прямой или разветвленной цепи,
или их фармацевтически приемлемых
солей.
Соединения формулы проявляют фармакологическую активность.
Целью изобретения явля ется получе ниё новых производных синергистинов, обладающих тем преимуществом, что они растворимы в воде в виде солей в используемых терапевтических дозах, сохраняя основной спектр активности.
Следующие примеры, приведенные не для ограничения, показьшают, как изобретение может быть осуществлено а практике. Спектры ЯМР продуктов.
ЗА
31f
W 4
2NH HN
Все спектры сняты при 250 МГц в дейтерохлороформе, хтдаческие сдвиги выражены в миллионных долях (плм) по отношению к сигналу тетраметипси- лана. Использованные сокращения: с сингле Т , д дублет5 т триплет; мт мультиплет; м массив, дд двойной дублет; длд удвоен- ньй двойной дублетi.дддц удвоение удвоенного двойного дублета.
В примерах 2-7 в скобках даны со- ответственно: химический сдвиг, форма сигнала, интеграция (число про- ;тонов, в некоторых случаях с процен-- ;том изомеров) и отнесение протонов,
В примерах под импульсной хроматографией понимают методику очистки, характеризу1о:(цуюся тем, что ИС польззпот короткую хроматографическую колонку и работают при среднем дав- лении (50 кПа), применяя окись крем- ния с гранулометрией мкм.
Обычно реакцию проводят в органическом растворит эле, таком как стой эфир, напри1чер тетрагидрофуран, спирт, например этанол, или хлорированный растворитель (например, мети- ленхлорид или хлороформ) при темпе ратуре около 20°С. Реакцию проводят в щелочной среде,,. например в присутствии щелочного гидрида или щелочного алкоксида (например, этоксида натрия или третично бутоксида калия).
Когда R не является замещенным алкскси или гетероциклокси, можно также работать в нейтральной среде при 0-50 С,в одном из указанных растворителей или в кислой среде (например, в уксусной кислоте или в Смеси уксусной кислоты и каталитических количеств трифторуксусной кислоты ) в присутствии или в отсутствие растворителя при 0-50 С, предпочтительно около 20°С.
Когда R содержит алкиламиногруп 5 1-(2-меркаптопропил)4-метилпиперази- на в 50 см этанола, содержащего 0,34 г этилата натрия, прибавляют раствор 5,2 г (4-метилфенил)суль фонилоксиметилен пристинамицина 1д в 50 см метиленхлорвда. Реакционную смесь 16 ч перемешивают при тйм- пературе около 20 С, потом разбавляю 500 см метиленхлорида и 100 см дистиллированной воды. После пере20
пу, способную вступать в реакцию, последнюю предпочтительно защищать любым известным способом, которьй не затрагивает остальную часть молекулы
Кроме того, когда исходный продукт содержит вторичную аминогруппу, в некоторых случаях может быть необходимо защищать ее перед реакцией. Защищающий радикал удаляют после реакции. В этом случае используют обьм- 25 мешивания водную фазу 2 раза экстра- ное средство защиты, применяемое гируют 50 см всего метиленхлорида.
базлении водного раствора соответствующей кислоты к соединению.
Соединения, в которых R является радикалом, замещенным одним или двумя оксисул1-.4)онк11ьными радикалами, могут быть превращены в соли метал-, лов или в соли присоединения азотис-, тых оснований по методике, аналогичQ ной приведенной для солей присоединения кислот, но при замене кислоты гидроксидом металла или азотисным основанием.
Пример 1. К раствору 0,87 г
5 1-(2-меркаптопропил)4-метилпиперази- на в 50 см этанола, содержащего 0,34 г этилата натрия, прибавляют раствор 5,2 г (4-метилфенил)суль- фонилоксиметилен пристинамицина 1д в 50 см метиленхлорвда. Реакционную смесь 16 ч перемешивают при тйм- пературе около 20 С, потом разбавляют 500 см метиленхлорида и 100 см дистиллированной воды. После пере0
5 мешивания водную фазу 2 раза экстра- гируют 50 см всего метиленхлорида.







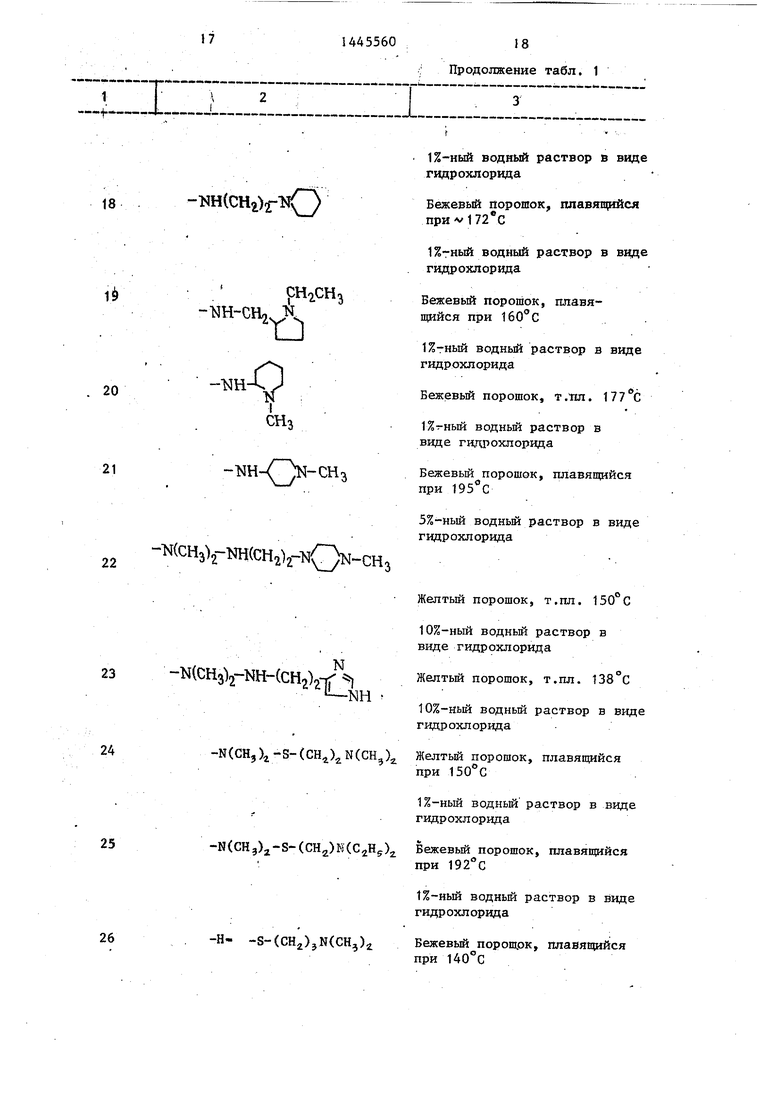


Изобретение касается замещенных гетероциклических веществ, в частности производных синергестинов общей ф-лы 0 -N сн2СНз НГ сн о 3 о н где Y-H, Ы(СНз)г , R-пиперидиламино- или пиперидилтиогруппа, находящиеся в положении.3 или 4 и замещенные в некоторых случаях по азоту в цикле алкилом, или алкиламино-, алкилокси- или алкилтиогруппа, монозамещенные S(0), алкиламино-, одной или двумя диалкиламиногруппами, в некоторых случаях замещенными диалкиламино- грзшпой, триалкиламмонием или 4- или 5-имидазолилом, или одним циклом, выбранным из пиперазино- (в некоторых случаях замещенного алкилом), пи- перидино-, 2-, 3- или 4-пштеридш1а или 1- или 2-пирролидинила (в некоторых случаях замещенного по азоту цикла алкилом) при использовании,,5-С4 -апкила, прямого или разветвленного, или их фармацевтически приемлемых солей, проявляющих фармакологическую активность. Цель - создание .новых, активных и растворимых в воде веществ указанного класса. Синтез новых соединений ведут реакцией соединения общей ф-лы R -Н с соединением общей ф-лы Oy-N О Т СНз о . О, ., Д т 0 .-.;,- Н О где R имеет значения, указанные для R; Y - имеет указанные значения; Z - галоид, диалкилфосфорилоксигруп- па или-0-5(0)2 R O-C(O)R . , R -фенил, замещенный низшим алкилом; R -низший алкил. Вьщеление целевого продукта ведут в свободном виде лли в виде необходимой соли. Новые вещества проявляют активность в отношении золотистого стафилоккока при минимальной ингибирующей концентрации 1-30 мкг/см , лечебной дозе СД-д 4-180 мг/кг и токсичность LDjo 340-1000 мг/кг. 2 табл. I СО « 4:а сл ел оа Ы
для защиты вторичной аминной функци которое не затрагивает остальную часть молекулы и может быть легко удалено. Особенно выгодно использовать в качестве защищающего радикала трифторацетил, затем он.может быть удален с помощью водного раствора щелочного бикарбоната, такого как бикарбонат натрия или калия. .
Соединения могут быть очищены известными методиками - кристаллизация, хроматография или последовательные экстракции в кислой и основной среде. Из-за чувствительности синергистинов к щелочной среде очевидно, что под щелочной средой пнимают среду, способную высвободить вещество-основу из его соли, полученной присоединением кислоты, т.е. среду рН которой не превьппает 7,5-8 Соединения, в которых R является радикалом, содержащим аминнуто функцию, могут быть переведены в СОЛЬ присоединения кислоты при действии кислоты в среде такого органического растворителя, как спирт, кетон, сложный эфир или хлорирован- ньй растворитель. Соль осаждается, в некоторых случаях после концентрирования раствора, ее отфильтровывают или декантируют. Соли присоединения кислоты также могут быть получены в виде водных растворов при до
Объединяют органические фазы, сушат над сульфатом магния, фильтруют, потом концентрируют досуха при пониженном давлении (2,7 кПа) при 30°С. Остаток очищают импульсной хроматографией (элюент: хлороформметанол 97,5-2,5 по объему). Объединяют фракции. 33-80 и концентрируют их досуха при пониженном давлении (2,7кПа при . Таким образом получают 1,25 г 55 1-мeтил-3-(4-мeтилпипepa- зинил) -пропил -тиометиленпристинами- цина 1д в виде бежевого порошка, плавящегося при 195 С.
Готовят 10%-ный водньй раствор 55 З- (4-метил-1 -пиперазишш) -2-про- пил -тиометиленпристинамицина 1д (продукт AAN) в виде гидрохлорида: продукт ААЛ 0,03 г, 0,1 н. соляная кислота 0,3 см .
1 - 2-Меркаптопропил -4-метилпипе разин может быть получен при нагревании при 100° в течение 16 ч смеси
19см пропиленсульфида и 29 см N-ме тилпиперазина. Таким образом получают 32 г бесцветного масла, перегоняющегося при при 1,3 кПа,,
5о- 4-Метилфеншт сульфонш1окси- метиленпристинамицин Получен по следукяцей методике.
К раствору 2,7 г 5 -оксиметш1ен- пристинамицина 1д в 30 см метилен- хлорида прибавляют при температуре около 0,42 см триэт1шамина, потом 0,57 г п -толуолсульфонилхло- розда. Затем реакционную смесь перемешивают 2 ч при температзфе около
20С, концентрируют досуха при пониженном давлении (2,7 кПа) при 30°С. Полученньй остаток очищакГг импульс ной хроматографией (элюент:метилен.хлорид - метанол 96.- 4 по объему). После концентрирования досуха фракций 4-6 при пониженном давлении (2,7 кПа) при 30° получают 2,2 г 58-(4-метилфенил)-сульфонилоксиметиленпристинамицина 1д в ввде белого порошка, плавящегося при 265 С, ЯМР-спёктр: 0,50 (дд, Ш, 5рг),2,35
(с, Ж, -502-(3)-СЩз,30 (дд, Ш,
; 5Si)i 5,25 (д, 1Н, 5oi), 5,30 (дд, Ш, 56,7,35-7,90 (система АВ + М, Н Н
8Н, 4 +46. . 7,85 (дд, 1Н,
Нб).
5 | -Оксиметиленпристинамицин 1д может быть получен следующим обра- . .
При перемешивании прибавляют 420 см водного раствора 0,1 н. соляной кислоты 10,6 г 5о-диметиламино- метиленпристинамицина 1д. Полученный раствор перемешивают 3 ч при температуре около 20 С. Затем прибавляют по каплям 30 см водного насыщенного раствора бикарбоната натрия таким образом чтобы получить рН около 4,
5 О.тфильтровьшают выпавший в осадок продукт, 3 раза промывают 30 см всего дистиллированной воды. После сушки при пониженном давлении , (2,7 кПа) при температуре около
0 20°С 9,5 г 5 -оксиметилен- пристинамицина 1д в виде бежевого порошка. Этот продукт имеет достаточное качество, чтобы его можно бьшо использовать в последующих стаднях. Однако он может быть очищен следующим образом.
9,5 г 55 сырого оксиметиленпри- стинамицина 1д растворяют в 50 см
0 этилацетата, полученньй раствор выливают на 100 г силикагеля, находящегося в колонке диаметром 2,8 см. Элюируют сначала 400 см этилацетата и удаляют соответствующий элюат,
5 затем элюируют 1600 см этилацетата и концентрируют соответствующий элюат досуха при пониженном давлении (2,7 кПа) при . Таким образом получают 6,35 i -оксиметиленg пристинамицина 1д в виде белых кристаллов, плавящихся при 220 С.
ЯМР-спектр: 0,69 (дд, Ш, ,).; 2,43 (д, Ш, 5,), 3,40 (д, 1H,,), 4,0-4,2 (м, ЗН, 4вС+5,+5об); 8, 55 (с,
5 1Н, .СН-ОН); 11,63 (с, широкий, - 1Н, -СН-ОН).
пример 2. Работают по методике примера 1, но исходя из 5,2 г 5 S -(4-метилфенил)-сульфонилоксиме71445560
тиленпристинамицина 1д, 0,6 г 1-ди- метнламинопропантиола-2 и 0,34 эти- лата натрия, после очистки импульсной хроматографией (элюент: хлороформ - метанол 95-5 по объему) и концентрирования досуха фракций 16-38
8
N,Н-Диэтил-4-ацетклтиопентана- мин-1 может быть получен по методике примера 32 для получения N,N-димe тил- -3-ацетш1тио-2-метилпропиламина, но исходя из 32 г Ы,Ы-диэтил-4-хлор-пен- танамина-1 и 15,2 г тиоуксусной кислоты. Таким образом получают 4,31 г продукта в виде желтого масла,
П р и м е р 4. Раствор 7,6 г
при пониженном давлении (2,7 кПа) при получают 1 г 5§-(3-диметШ1амин-2-пропил)тиомети- ю ленпристинамицина 1д в виде желтого порошка, плавящегося при 172°С. . Спектр ЯМР: 0,65 (дд, 1Н, 5),
1,10 (д, зн, -дн-сн); 2,30 (с, 6H-N
(CH5)j), 7,60 (с широкий, 1H,-CH-S-), 15 , прибавляют при этой температуре, раст- 7,85 (дд, 1Н, 1 Hg).
Готовят 5%-ньй водный раствор 5 S -(3-диметиламино-2-пропил)-тиоме5 S (4-метилфенш1) -сульфонштоксимети- лeнj-пристинамицина 1д в 60 см тет- рагидрофзфана охлаждают до тег-тера- туры около -10 С. Туда медленно
вор 0,65 г 2-диметиламиноэтанола в 60 см тетрагидрофурана, содержащего 0,35 г 50%-ной дисперсии гидрида натрия в минеральном масле.По окончании прибавления дают смеси медленно нагреться до температуры около 20 С. Реакционную смесь перемешивают 24 ч при этой температуре, потом разбавляют 500 см метиленхлорида и 2 раза промывают 50 см насьш1ен- ного раствора хлористого аммония. Ор-. ганическую фазу сушат над сульфатом магния, фильтруют, потом концентрируют досуха при пониженном давлении (2,7 кПа) при 40°С. Полученный остаток очищают импульсной хроматографией (элюент: хлороформ-метанол 95-5 по объему).Объединяют фракции 12-17 и концентрируют досуха при по- 35 ниженном давлении (2,7 кПа) при
20
тиленпристинамицина 1д (продукт ААО) в виде гидрохлорида: продукт ААО 0,03 г 0,1 н. соляная кислота 0,3 см дистиллированная вода до 0,6 см .
Пример 3. Работают по методике примера 1, но исходя из 6,3 г 58 -(4-метилфенил)сульфонш1оксимети- 5 ленпристинамицина 1д, 1,05 г 5-ди- этиламинопентатиола-2 и ,0,408 г этилата натрия, после очистки импульсной хроматографией (элюент:хлороформ - метанол 97,5 - 2,5 по объему) и кон- 30 центрирования досуха, фракций 47 - 65 при пониженном давлении (2,7 кПа) при 30°С получают 1,32 г 5о-(5-ди- э тиламино-2-пентш1)-тиометиленпристи- намицина 1 плавящегося при
Спектр ЯМР: 0,65 (дд, 1Н, ) 1,20 (т, 6Н, -N(CH2CHj)2). (д, 3H,.-qH-CH5); 1,70 (с широкий, 4Н, СН/СН)2. -CHjHpi 2,65 (к, 4Н, -N(CH2-CHj)i)-, 3,50 (дд, 1Н, 5ёГ) 7,65 (с широкий, 1Н, -CH-S-), 7,85 (дд, 1Н, I Hg).
Готовят 10%-ный водньй раствор
1д в виде бежевого порошка, 185 С.
вор 0,65 г 2-диметиламиноэтанола 60 см тетрагидрофурана, содержа го 0,35 г 50%-ной дисперсии гидр натрия в минеральном масле.По ок чании прибавления дают смеси мед но нагреться до температуры окол 20 С. Реакционную смесь перемеши ют 24 ч при этой температуре, по разбавляют 500 см метиленхлорид и 2 раза промывают 50 см насьш1е ного раствора хлористого аммония ганическую фазу сушат над сульфа магния, фильтруют, потом концентр руют досуха при пониженном давлен (2,7 кПа) при 40°С. Полученный ос таток очищают импульсной хроматографией (элюент: хлороформ-метано 95-5 по объему).Объединяют фракци 12-17 и концентрируют досуха при 35 ниженном давлении (2,7 кПа) при
25 С. Таким образом, получают 1,5 5о -(2-диметиламиноэтоксиметш1ен) -пристинамицина 1д в виде бежевог порошка, плавящегося при .
Спектр ЯМР: 0,65 (дд, 1Н, 5/Jj) 2,3 (с, 6Н, -N(CH,)2); 2,65 (м,2Н );3.42 (дд, Ш, 56); 4,15 (т, 2Н, -ОСН,-), 5,15 (д, Ш, 5е 7,45 (под ароматикой, 1Н, С-СНО40
5§-(5-диэтш1амино-2-пентш1)-тиомети- 45 7,80 (дд, 1Н, ГН). ленпристинамицина 1д (продукт АРР) Готовят 1%-ный водный раствор в виде гидрохлорида; .прод;укт ААР
5S -(2-диметиламиноэтоксиметш1ен) цристинамицина 1д (продукт ААО) в виде гидрохлорида: продукта ААО 0,03 г , 0,1 н. соляная кислота 0,3 дистиллированная вода до 3 см .
0,05 rj 0,1 н. соляная кислота 0,5 см
5-Диэтиламинопентантиол-2 может быть получен по методике примера 32 для получения 3-димет1шамино-2-метил пропантиола, но исходя из 4,0 г N,N-диэ тил-4-ацетилтиопентанамина-1 и 0,046 г натрия. После очистки импульсной хроматографией (элюент: этилацетат-метанол 70-30 по объему) и концентрирования досуха фракций 16-24 получают 2,0 г 5-диэтиламино- пентантиола-2 в виде желтого,масла.
8
N,Н-Диэтил-4-ацетклтиопентана- мин-1 может быть получен по методике примера 32 для получения N,N-димe тил- -3-ацетш1тио-2-метилпропиламина, но исходя из 32 г Ы,Ы-диэтил-4-хлор-пен- танамина-1 и 15,2 г тиоуксусной кислоты. Таким образом получают 4,31 г продукта в виде желтого масла,
П р и м е р 4. Раствор 7,6 г
5 S (4-метилфенш1) -сульфонштоксимети- лeнj-пристинамицина 1д в 60 см тет- рагидрофзфана охлаждают до тег-тера- туры около -10 С. Туда медленно
прибавляют при этой температуре, раст-
вор 0,65 г 2-диметиламиноэтанола в 60 см тетрагидрофурана, содержащего 0,35 г 50%-ной дисперсии гидрида натрия в минеральном масле.По окончании прибавления дают смеси медленно нагреться до температуры около 20 С. Реакционную смесь перемешивают 24 ч при этой температуре, потом разбавляют 500 см метиленхлорида и 2 раза промывают 50 см насьш1ен- ного раствора хлористого аммония. Ор-. ганическую фазу сушат над сульфатом магния, фильтруют, потом концентрируют досуха при пониженном давлении (2,7 кПа) при 40°С. Полученный остаток очищают импульсной хроматографией (элюент: хлороформ-метанол 95-5 по объему).Объединяют фракции 12-17 и концентрируют досуха при по- ниженном давлении (2,7 кПа) при
25 С. Таким образом, получают 1,5 г 5о -(2-диметиламиноэтоксиметш1ен)- -пристинамицина 1д в виде бежевого порошка, плавящегося при .
Спектр ЯМР: 0,65 (дд, 1Н, 5/Jj)i 2,3 (с, 6Н, -N(CH,)2); 2,65 (м,2Н. );3.42 (дд, Ш, 56); 4,15 (т, 2Н, -ОСН,-), 5,15 (д, Ш, 5е); 7,45 (под ароматикой, 1Н, С-СНО),
7,80 (дд, 1Н, ГН). Готовят 1%-ный водный раствор
7,80 (дд, 1Н, ГН). Готовят 1%-ный водный раствор
5S -(2-диметиламиноэтоксиметш1ен)- цристинамицина 1д (продукт ААО) в виде гидрохлорида: продукта ААО 0,03 г , 0,1 н. соляная кислота 0,3 см, дистиллированная вода до 3 см .
При мер 5. К раствору 0,5 г 5 -(4-метилфенил)сульфонилоксимети-1
лен-пристинамицина
t
1 д В 25 см этанола прибавляют 0,12 г 4-амино-1-ме- тилпиперидина при температуре около . После 16 ч перемешивания при этой температуре реакционную смесь разбавляют 100 см метиленхлорида.
два раза промывают 100 см всего дистиллированной воды. Органическую фазу сушат над сульфатом натрия, потом концентрируют при пониженном давлении (2,7 кПа) при З0 с. Остаток перемешивают с 15 см После фильтрования получают
этилового эфира. 0,42 г
Исходньй продукт ((CH,)2) Z -OCOCH,
(OC.,Hy)
Условия реакции
CHjCOOH 20 С, 6 ч
CHjCOOH, , 20 ч
Использованные продукты могутСпектр ЯМР: 0,55 (дд, 1Н : 5(3),
быть получены следующим образом; . . 1,30 (тд, 6Н : -РО ()-); -55-адетоксиметиленпристинамицин 1.2,40 (д, 1И: 5fi,); 3,40 (дд, 1Н : 56)
К раствору 1,8 г 5 -оксиметилен- 204,25(ддд,4Н: -РО/О-СН СН) | 5, 25(д, 1Н, пристинамицина 1д в 20 см метилен- хлорида, содержащему 0,2 г триэтил- амина, прибавляют при температуре около -20°С 0,14 см ацетилхлорщ1;а, затем оставляют при температуре около 20 С.
Реакционную смесь перемешивают в течение 20 ч при этой температуре, потом концентрируют досуха при .пониженном давлении , (2,7 кПа) при 30°С, полученный остаток очищают флаш- хроматографией (элюент:этилацетат). После концентрирования досуха фракций 4-7 при пониженном давлении (2,7 кПа) при 30 С получают 0,7 г 5 -ацетоксиметиленпристинамицина 1д в виде желтого порошка, плавящегося при НО°С,
Спектр ЯМР: 0,60 (дд, 1Н,,)-, 2,25 (с, ЗН: -СО-СНу, 2,45 (д, 1Н : 5 И,) 3,45 (дд, 1Н : 5Е.,); 5,25 (дд, 1Н, : 5ft:,); 5,45 (д, 1Ht 56,), 7,10 - 7,45 (м, 8Н : 6f + б5 + б + I Hi, + I Hj + -СН-0)5 7,85 (дд, Н,: 1 Нб).
5 -Диэтоксифосфорилоксиметилен- пристинамицин 1д.
Работают по аналогичной методике, но исходя из 1,8 г 55-оксиметилен- пристинам11цина 1д и 0,34 г диэтил- хлорфосфата, после очистки импульсной хроматографией (элюент: зтилацо- тат - метанол) (90-10 по объему) и концентрирования досуха фракций 6-14 при пониженном давлении (2,7 кПа) при получают 0,8 г б8 -диэтокси- фосфорилоксиметиленпристинами1Д1на 1р в виде желтого порошка, плавящегося при 150°С.:,
5Ы.) 5,40(д, 1Н:5,),- 7,10- 7,55 (м, 8H: 6J + 6i + 6Е + СН-0 + 1 Н5+.Г Н,), 7,85 (дд, 1Н X 0,85 ЛГ/Н 1-й изоч., мер); 8 (дд, 1Н X 0,15 : ГН 2-й изомер) .
5 О - оксиметшхенпристинамицин 1 может быть получен, как в примере 1.
Пример 7. Работают по мето- 30 дике аналогичной примеру 5, но перемешивают в течение 20.ч, исходя из 55 -хлорметиленпристинамицина 1д, получают 5 о -(3-диметиламинопропил)- тиометиленпристинамицин 1д в виде бе- 25 жевого порошка, плавящегося при 170 С..
Спектр ЯМР: 0,70 (дд, 1Н : 5р2.), 1,90 (м, 2Н: -S-CHiCHjCHiNO; 2,20 (с, 6Н : Ы(СНз)), 2,40 (д, 1Н: 5 В,), (м, 2Н: )V 3,45 (дд, 1Н: 40 )5 7,65 (широкий с, 1Н: CH-S-).
Готовят 1%-ньм водньш раствор 5 о (3-диметиламинопропил)тиометилен- пристинамицина 1 (продукт AZ) в виде гидрохлорида: продукт AZ 0,03 г 45 Oj 1 н. соляная кислота 0,3 см. дистиллированная вода до 3 см .
Исходньй продукт может быть получен следующим образом.
Через раствор 1,3 см трифенилфос- фита в 25 см метиленхлорида пропускают поток газообразного хлора до по- лз чения стойкой хселто-зеленой окраски при температуре от -20 до -15 С. Затем добавляют 6 капель трифенилфос- фита для обесцвечива1шя раствора, затем 4,1 г оксиметш1ен 5-пристина- шцйнa 1д при поддерживании темпераЛО
туры между -20 С .и -15 С. Перемеши- 7зают полученный раствор в течение 1 ч
50
55
(1-метил-4-пиперидил)-аминомети- ленпристинамицина 1д в виде белого порошка, плавящегося при 208 С,
Пример 6. Работают по указанной методике, следующие производные синергистина:
Продукт определен ранее в примере 5
4,25(ддд,4Н: -РО/О-СН СН) | 5, 25(д, 1Н,
5Ы.) 5,40(д, 1Н:5,),- 7,10- 7,55 (м, 8H: 6J + 6i + 6Е + СН-0 + 1 Н5+.Г Н,), 7,85 (дд, 1Н X 0,85 ЛГ/Н 1-й изоч., мер); 8 (дд, 1Н X 0,15 : ГН 2-й изомер) .
5 О - оксиметшхенпристинамицин 1 может быть получен, как в примере 1.
Пример 7. Работают по мето- дике аналогичной примеру 5, но перемешивают в течение 20.ч, исходя из 55 -хлорметиленпристинамицина 1д, получают 5 о -(3-диметиламинопропил)- тиометиленпристинамицин 1д в виде бе- жевого порошка, плавящегося при 170 С..
Спектр ЯМР: 0,70 (дд, 1Н : 5р2.), 1,90 (м, 2Н: -S-CHiCHjCHiNO; 2,20 (с, 6Н : Ы(СНз)), 2,40 (д, 1Н: 5 В,), (м, 2Н: )V 3,45 (дд, 1Н: )5 7,65 (широкий с, 1Н: CH-S-).
Готовят 1%-ньм водньш раствор 5 о (3-диметиламинопропил)тиометилен- пристинамицина 1 (продукт AZ) в виде гидрохлорида: продукт AZ 0,03 г Oj 1 н. соляная кислота 0,3 см. дистиллированная вода до 3 см .
Исходньй продукт может быть получен следующим образом.
Через раствор 1,3 см трифенилфос- фита в 25 см метиленхлорида пропускают поток газообразного хлора до по- лз чения стойкой хселто-зеленой окраски при температуре от -20 до -15 С. Затем добавляют 6 капель трифенилфос- фита для обесцвечива1шя раствора, затем 4,1 г оксиметш1ен 5-пристина- шцйнa 1д при поддерживании темпераЛО
туры между -20 С .и -15 С. Перемеши- 7зают полученный раствор в течение 1 ч
111445560
при -15°С, затем добавляют по каплям раствор 0,4 см пиридина в 25 см метиленхлорида. Перемешивают реакционную смесь 30 мин при температуре
около 20 с, затем добавляют 0,46 см концентрированной соляной кислоты (ot. 1,19) и 50 см метиленхлорида.
Смесь промывают 4 раза в целом 100см дистиллированной воды, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха при пониженном давлении (2,7 кПа) при 30 С. Остаток очищают импульсной хроматографией (элюант : этилацетат), после концентрирования досуха 7 и 9 фракций при пониженном давлении (2,7 кПа) при , получают 1,2 хлор метилен-5-пристинамицина 1д в виде бежевого порошка с т.пл. 190°С.
Спектр ЯМР: 0,55 (дд; 1Н : 5/32), 2,45 (д-, 1Н, 5,), 3,45 (дц, 1Н,) 5,30 (д, 1Н, ЗоС); 5,45,(д, 1Н, 55,) 7,15 а 7,60 (м; 8Н: 6 + 6 + 65 + + 1 Н5+ СН-С1); 7,85 (дд5 1Н; r-Hg).
Работая по той же методике, могут быть получены следзтощие продукты (см. табл.1). В следующих таблицах.
если нет специального указания, ради- 30 приемлемых солей можно также привес- кал У является диметиламиногруппой. . ти четвертичные аммонийные соли,
когда R является триалкиламмонийИзвестно, что синергистины, полу- ным радикалом, эти соли соответству- ченные ферментацией, представляют со- ют анионам приведенных солей. В ка- бой очень ценные для медицины продук-35 честве фармацевтически приемлемых ты для лечения многих заболеваний, вызьюаег.1ЫХ грамположительными (стафилококки, стрептококки, пневмококсолей можно также упомянуть соли щелочных металлов, такие как соли лития, натрия, калия, соли щелочноземельных металлов, такие соли кальция или магния, соли аммония и соли присоединения органических азотистых оснований: этаноламина,ди- этаноламина,триметиламина,триэтилами- на, метиламина , пропиламина, диизопропил АС амина,N,N-диметилэтаноламина,бензил- амина,диб ензиламина,дициклогексилбен- зиламина, N-бензил-/5-фенетиламина, N,N -дибeнзшIэтилeндиaминa, бензгид- риламйна, аргинина, лейцина, лизина или Ы-метш1Глюкамина.
ки, энтерококки) и грамотрицатель- ными- (в виде гемофилус, гонококки, менингококки) бактериями, Однако эти продукты имеют тот недостаток, что они нерастворимы в водной среде и могут быть введены только оральным путем, обычно в виде таблеток,драже или пилюль. Учитывая эту нерастворимость, невозможно использовать известные синергистины, когда больно не в состоянии глотать, а именно в педиатрии и реанимации, тогда как спектр активйости этих продуктов дает четкое указание на это в большом числе случаев, например при коматозных сепсисах.
Новые продукты согласно изобретению обладают значительным преимуществом из-за их растворимости в воде в виде солей в используемых терапевтических дозах, сох15аняя основной
2
0
5
спектр автивности синергистинов, а именно они являются активными in vitro против золотистого стафилококка Смита при концентрациях (7,1- 125 мкг/мл,
Обычно они малотоксичны. но превьшает 300 мг/кг на мьппах при П.ОДкожном введении.
Для тепапевтического применения можно использовать новые продукты как таковые, т.е. в виде основания, но для применения в виде водного раствора, что является их основным преимуществом, особенно выгодно использовать их фармацевтически приемлемые соли, т.е. нетоксичные при используемых дозах.
В качестве фармацевтически при- 0 емлемых солей можно привести соли, присоединения минеральных кислот, такие как гидрохлориды, гидробромиды, сульфаты, нитраты, фосфаты, или органических кислот, такие как ацетаты, пропионаты, сукцинаты, ма- леаты, фумараты, метансульфонаты, п-толуолсульфонаты, изетинаты или замещенные производные этих соединений. В качестве фармацевтически
5
ным радикалом, эти соли соответству- ют анионам приведенных солей. В ка- 5 честве фармацевтически приемлемых
0
солей можно также упомянуть соли щелочных металлов, такие как соли лития, натрия, калия, соли щелочноземельных металлов, такие соли кальция или магния, соли аммония и соли присоединения органических азотистых оснований: этаноламина,ди- этаноламина,триметиламина,триэтилами- на, метиламина , пропиламина, диизопропил- С амина,N,N-диметилэтаноламина,бензил- амина,диб ензиламина,дициклогексилбен- зиламина, N-бензил-/5-фенетиламина, N,N -дибeнзшIэтилeндиaминa, бензгид- риламйна, аргинина, лейцина, лизина или Ы-метш1Глюкамина.
Фармакологические исследования.
Бактериостатическая активность in vitro.
К серий пластин, содержащих известный объем (20 см ) подходящей культуральной среды (агар Миллера- Хинтона),, прибавляют 1/10 этого объема геометрически прогрессирующих (отношение-2) разбавлений испытуемо-.
0
5
13
144
го продукта. Пластины инокулируют многоточечным инокулятором, который высвобождает пятно 10 образующих колонию единиц микроорганизма в три синовом осевом бульоне, инкубированном в течение 18 ч при 37-С и разбавленном 1/100 этой же среды.После инокуляции пластины инкубируют 24 ч при .
Минимальной ингибирующей концепт рацией является самая нцзкая концентрация, при которой ингибируется развитие микроорганизма.
Активность против интрадеритоне- альной инфекции на мышак.
Мышам делают интраперитоиеальную инъекцию 0,5 см подходя1це л встрлхн ваемой культуры в возрасте 18 ч испытуемого.кмкроорганизма в среде Brain HeartrInfusion (Дифко), соответственно разбавленной hog mucin. Такая инокуляция пызьи ает смерть контрольных животных в течение 24- 48 ч. Испытуемое соединение вводяг подкожно дважды с интервапом 5 ч в день инокуляции, первая доза даемся через час после инокуляции микроорганизма. Использзпот единые дозы, содержащиеся в объеме 50 см /кг,,
50%-ная лечебная доза (СД,д) представляет собой дозу соедннегглтя , взятого в испытание, которая при кахсдом введении позволяег пол овин е обработанрльгк лмвотныз-: зжлть во вре мя периода испытаний (8 /тней) , Результаты прщзедеаь в табл,2 илгюке.
о р
у л
о б
е 1 е
Способ лолучення п оизводны, синергисти1 ав оОндей фг.рмулы
СИ
3
.-Л Ny.-- -Y
- - (I 1
сн™ ()
где Y
- во,п;ород :-ти д нметиламигдэ- группа;
5601
R - 3- или 4-пипериди.ла№1но или 3- или.,4-пиперидилтио, в некоторых случаях замещенные по атому азота цикла ал- . кильным радикалом, алкилами- но,. алкилокси или алкилтио- группа, замещенные одной гкд- роксисульфонмльной, алкил- Oаминор одной или дзумя ди
алкиламиногруппами, в некоторых случаях замещенной ди алкиламиногруппЪйд , триалкил- . аммонием или 4- или 5-ими- 5дазолилом или одним циклом,
выбранньм среди пиперазнно, в некоторых слу-таях замещенного алкигсом, пнпернднно, 1-пирролщцшила5 2- 3 0или 4-пиперид11ла и 2-пирролидини-па, в некоторык слууа- ях замещенного по атому азо- . та алкильньм радикалом, при условии, что aлкIiльный ради- 5- кал содержит 1-4 атома тлерода в прямой или разветвленной цепи,
или их фармацевтически приемлемых солей, о т л и чающийся тем, 0 .то подвергают взаимодействиш сое- Д1п;енН8. общей формулы,
R,-H
где К/ имеет значения, указанные ,-для R,
с с.оедииеннхзм обишй формул1л
,- ш
-, i
.
JcH,cH4 I
«i3 О
iNl t
о
о
. ОН
где I,. - водород или диметила1мино- группа:
,Z -- галоид, диалкил/З осфорилок- CKrpyinia 1Ш11 группа формулы OSOiRgH/nj ОСОКф, где R -- фет-т., замещенный низ- StJ.M алкшюм, . -- низкнш
алки.г1,.
; последуюшим вьиеленнем целевого -игодухта в свободном виде или в вгще 1)армацевтически приемиемой соли.
-NH(CH2)2N(CH,) -NH(CH2,),2,N(CjHy)2
10
-NH(CH2)NHCH,
11
-MCCH), N(CHj)2
12
-NH-CH-CH2N(CH j)2 CH.
13
-NHCH CH-NlCHj CHa
14
- IJH-CH-(СН2)зК (C 2H5)2 СНя
15
16
-NH-(CH2)2--NQ -1SJH(CH2)3-NQ
17-1 H(CH2)2-N(
Таблица 1
Бежевый порошок, плавящийся при 180 1%-ньй водньй раствор Желтый порошок, плавящийся при 150 С
5%-ньш водный раствор в вид гидрохлорида
Желтый порошок, плавящийся при 174°С
1%-ный водньй раствор в вид гидрохлорида
Желтьй порошок, т.пл. 155 С
6, водный раствор в виде гидрохлорида
Желтьй порошок, плавящийся при 160°С
1%-ньй водньй раствор в виде гидрохлорида
Оранжевьй порошок, плавящийся при С
10%-ньй водньй раствор в виде гидрохлорида
Бежевьй порошок, плавящийся при --ТбО С
1%-ньй водньй раствор в виде гидр охл орида
Желтьй порошок, т.пл. 183 С
1%-ньй водньй раствор в виде гидрохлорида .
Желтьй порошок, т.пл.- 170 С 1%-ньй водньй раствор
} {елтьй порошок, плавящийся при Л 16 2° С
1
18-iJHCcHiViiQ
1
рН2СНз -NH-Cli, N
-NH
Шз -КН-(-Шз
-К(СНз)2-Щ(СН2)
23
24
25
26
N(CH3)(CH2)2li Желтьй порошок, т.пл. 138 с
.
-NH
-N(CH,)-S-(CH,,)N(CHp
z
10%-ньй водньй раствор в виде гидрохлорида
Желтьй порошок, плавящийся при 150 С
1%-ньй водньй раствор в виде гидрохлорида
-Ы(СНз)(СН2)К(С2Нд)2 Бежевьй порошок, плавящийся
при 192 С
1%-иьй водньй раствор в виде гидрохлорида
-Н- -S-(CH)jN(CH)2Бежевьй порошрк. Плавящийся
при 140°С
Продолжение табл. 1
..1
3
1%-ный водньй раствор в виде гидрохлорида
Бежевый Порошок, плавящийся прЯА 172 с
1%-ный водный раствор в виде гидрохлорида
Бежевый порошок, плавящийся при
1%-ный водньй раствор в виде гидрохл орида
Бежевьй порошок, т.пл.
1%-гньй водньй раствор в виде гидрохлорида
Бежевьй порошок, плавящийся
при
5%-ньй водньй раствор в виде гидрохлорида
Желтьй порошок, т.пл. 150 С
10%-ный водньй раствор в виде гидрохлорида
KiH .
-NH
27
-KlCHsVS-CHe-CH-CH NtCH l СНз
28
-Н(СНз)2-8-СН2-С-К(СНз)2 СНз СНз
29
м Iи Ltifi 11
-TS(CH3VS-(CH2)2- N(Jпри 180° с
Бежевый п
-S-(CH)
СН, I
N
Т
31
сн.
32
-S
CH2CHj
33
S-(CH2)2N-(CH2)2-N(CH3)3
CHi
34
-S-CH/CH NCCHj), /2
35- S(CH9) N-ГНБежевый порошок, плавящийся .
2 -/ .„„„ 17П°г
при 170 С
и Ltifi 11
при 180° с
10%-ный водный раствор в виде гидрохлорида
О
Бежевый порошок, т.пл. 234 С
1%-ный водный раствор в виде гидрохлорида
Бежевый порошок, плавяищйся при
1%-ньш водный раствор в виде гидрохлорида
Бежевый порошок, плавящийся
1%-ный водньш раствор в вид гидрохлорида
Бежевьй порошок, ллавящийс при 215° С
/
0,6%-ньм водный раствор в виде гидрохлорида
Желтый порошок, плавящийся при 170°С
1%-ный водный раствор в вид гидрохлорида
Бежевый порошок, плавящийся при
1%-ный водный раствор в виде гидрохлорида
Желтый порошок, плавящийся при 160°С
1%-ный водный раствор
Бежевый порошок, плавящийся при 190° С
Т%-ньй водный раствор в виде гидрохлорида
Бежевый порошок, плавящий
„„„ 17П°г
при 170 С
1%-ньй водный раствор в виде гидрохлорида
36
8(СН2) /N-СНтБежевый порошок, плавящийся
3 ,
. при 190 С
37
8-СН2-СН-СН2-К(СНз)з . Шз ®
38
-S(CH2)2S05H
Соединение
для сравнения
Пристинамицин 1.
Бежевый порошок, плавящийся
. при 190 С
10%-ный водный раствор в виде гидрохлорида
Охровый порошок, плавящийся при
1%-ный водный раствор в виде гидрохлорида
Желтьй порошок, т,пл. 280 С 5%-ньй водный раствор
Таблица 2
30
| Bull Sec | |||
| Chem, Fr | |||
| г | |||
| Аппарат для предохранения паровых котлов, экономайзеров, кипятильников и т.п. приборов от разъедания воздухом, растворенным в питательной воде | 1918 |
|
SU585A1 |
