(21)3787351/23-04
(22)07.09.84
(31)3145/83
(32)09.09.83
(33)HU
(46) 15.05.89. Бюл. 18
(71)Эдьт Дьерьсерведьесети, Дьяр (HU)
(72)Йожеф Райтер и Петер Крайчи (HU)
(53)547.491.07(088.8)
(56)Европейский патент № 28483, кл. С 07 D 249/14, опублик. 1981.
(54)СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ДИ- ГИДРОАРИЛОКСИАЖКПАМИНО-1,2-4-ТРКА- ЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИГОДНЫХ СОЛЕЙ ПРИСОЕДИНЕНИЯ КИСЛОТ
(57)Изобретение касается производных диарилоксиалкиламино-1,2,4-триа- зола, в частности получения соединений общей ф-лы
,R,N-CHr d :Hi)rNH4J«N-o(NH1)«N-CH,
где R,R2N - цикл пиперидина или пир- ролидина, или их фармакологически
1
Изобретение относится к усовершенствованным способам получения производных дигидроарилоксиалкиламино- -1,2,4-триазола общей формулы
Rl
iC
H3C-N-N ЖШ-, Ъоедщ-Ч JJ-Ш,
где R( и R вместе с азотом образуют пиперидиновый или пирро- лидиновый цикл,
приемлемых солей, подавляющих секрецию кислоты желудочного сока, стимулированного рецепторами гистамина Н2. Цель - повышение выхода и качества целевых веществ. Синтез ведут восстановлением эквивалентным количеством натрия соответствующего арилоксиами- на при температуре от -83 до -33 С с последующим отделением активного изомера от неактивного дигидроарил- оксиалкиламина с помощью дистилляции, Затем активный изомер обрабатывают N-цианоиминодитиокарбаминовой кислотой при 20-12С°С в среде растворите- . ля (иэопропанола,ди-н-пропилового эфира, хлороформа, бензола, ацетонит рила, диметилформамида) с последующим отделением остатка неактивного изомера и обработкой 1-1,5-кратным молярным избытком метилгидразина при 20-117°С в среде н-бутанола. При этом, полученный продукт переводят в соль обработкой нужной кислотой. Эти условия обеспечивают увеличение выхода до 93,3% с чистотой до 90%.
или их фармацевтически пригодных солей присоединения кислот, подавляющих секрецию кислоты желудочного сока, стимулированного рецепторами гис- тамина-К2.
Цель изобретения,- увеличение выхода и повышение качества целевых продуктов за счет осуществления восстановления арильного ядра арилокси- амина общей формулы
(Л
Јь
00
о vj
О5 J
см
ч
А
NCH2 0(
значения
где R и R имеют указанные
металлическим натрием в
ке при температуре
отделения полугенного дигидроарилоксиалкиламина общей формулы
жидком аммиа- от -83 до -33 С,
-ч.. т„,
(III)
-NCH2 (
и
где R,
П
и К„
имеют указанные значения
неактивного изомера и обработки диметиповым эфиром N-цианиминодитиокар™ баминовой кислоты при 20-120 С в сре14807674
фильтровывают и шесть раз промывают диэтиловым эфиром по 100 мл. Эфирные растворы объединяют и упаривают при с пониженном давлении. Остаток подвергают фракционированной вакуумной дистилляции. Получают 18,6 г продукта с т.кип, 162-168°С (1 мм рт.ст), который состоит из смеси 4:1 целевого соединения и 3-ЦЗ-(1-пирролидинил- метил)-,4-циклогексадиенил -окси - -1-пропанамина. Изомерную смесь при пониженном давлении (1 мм рт.ст) подвергают фракционированной дистилляции. Полученный продукт с т.пл. 166-168 С содержит более 90% целевого соединения. Выход 73,9%.
В. Процесс ведут аналогично приме10
15
ру IA, за исключением того, что применяют металлический натрий в количестве 4,4 г (0,19 моль).
де растворителя, такого как изопропи- ловый спирт, ди-п-пропиловый эфир, 20 хлороформ,, бензол, ацетонитрил, диме- тилформамид, и последующей обработки соединения общей формулы
Hi
NCHn
R,
где R,
н N-CSN (IV) -e(CH2)jN-c-scH3
и R,
. 2 имеют указанные значения после освобождения от остатка неактивного изомера путем кристаллизации 1-1,5-кратным молярным избытком ме- тилгидразина при 20-117°С.
Пример 1. Получение (1 пирролидинилметил)-194-циклогек- садиенил -окси 1-пропиламина.
Аа К 19,9 г (0,085 моль) 3- (1- пирролидинилметил)-фенокси -1-пропил амина добавляют 19,8 г (0,425 моль) этанола, после чего в раствор кон- лансируют 500 мл газообразного аммиака Реакционную смесь охлаждают в бане ацетон-сухой лед до -85 С и добавляют 10,00 г (0,425 моль) металлического натрия порциями так, чтобы вторую и дальнейшие порции добавляли только после полного обесцвечивания раствора. После добавления 1 г натрия реакцию продолжают при -33 С
без охлаждающей бани. После добавления последней порции натрия голубая окраска реакционной смеси остается на 2,5-3 ч. После окончательного исчезновения окраски реакционную смесь нагревают в течение 30 мин до кипения. Добавлением 29,7 г (0,555 моль) хлористого аммония отгоняют аммиак и остаток растворяют в 400 мл диэти- лового эфира. Неорганическую соль от0
5
0
35
Q
45
50
55
ру IA, за исключением того, что применяют металлический натрий в количестве 4,4 г (0,19 моль).
После повторной дистилляции получают 15,1 г целевого продукта с т.пл. 165-167°С. Выход 67,6%.
П р и м е р 2. По лучение метилового эфира Ы-циано-N -Јз- 5-(1-пирролидинилметил) -1,4-циклогексадиенил - окси1-пропилкарбамимид-отиокислоты.
А.. 9 г (0,038 моль) полученного по примеру 1 3- Г 5-1-пирролидинилметил)- ,4-циклогексадкенил -оксич- -1-пропиламина растворяю при комнатной температуре в 10 мл изопропанола„ К полученному раствору добавляют 5.5 г (0,038 моль) диметилового эфира N- цианоиминодитиокарбаминовой кислоты. Реакция начинается при комнатной температуре с образованием меркаптана. Реакционную смесь 0,5 г выдерживают и 0,5 ч нагревают до кипения и затем упаривают при пониженном давлении. Полученный сотового типа остаток при подогревании растворяют в 40 мл ацето- нитрила. Раствор охлаждают, высадив- пптеся кристаллы отфильтровывают и промывают с цетонитрилом. Получают 1,1 г целевого соединения с т. пл. П7-119°С. После сгущения маточника получают 11,5 г целевого соединения. Rf 0,45 (на силикагеле 60 пластин; растворитель:смесь этилацетат - пири дин-вода-уксусная кислота 60:20:11:6).
В. Процесс ведут аналогично примеру 2А, за исключением того, что в качестве растворителя применяют 40 мл ди-н-пропилового эфира и реакционную смесь нагревают до кипения в течение 5ч, Получают 11,4 г продукта сотового типа, RI - 0,44 (растворитель имеет указанный в примере 2А состав). Продукт идентичен полученному по примеру 2А соединению.
C.Процесс ведут аналогично примеру 2А, за исключением того, что в качестве растворителя применяют хлороформ и реакционную смесь нагревают
до кипения в течение 4,5 ч, R 0,45 (при применении описанного в примере 2А растворителя). Продукт идентичен полученному по примеру 2А соединению.
D.Процесс ведут аналогично примефильтровывпют и промывают ацетонитри- лом. Получают 0,8 г метилового эфира М-циано-ы -3- (3-( 1-пирролидинилме- тил) -1 ,4-циклогексадиенил1.оксиЦ -пропилкарбамиминотиокислоты, т.пл. 116- 118°С. После упаривания маточника получают 9,2 г продукта сотового типа, R, 0,46 (растворитель имеет укаэан- Ю ный в примере 2А состав). Продукт идентичен соединению, полученному по прикеру 2А.
ПримерЗ. Получение 1-метил- N (1-пирролидинилметил)-1,4ру 2А, за исключением того, что в ка- 15 циклогексадиенил -окси}-пропил-1Н,1,2- честве растворителя применяют 100 млтриазол-3,5-диамина.
20
25
бензола и реакционную смесь нагревают до кипения в течение 10 мин. Получают 1 1,3 г идентичного полученному по примеру 2А соединению продукта, Rr 0,45 (растворитель имеет указанный в примере 2А состав).
E.Процесс ведут аналогично примеру 2А, за исключением того, что в качестве растворителя применяют
40 мл ацетонитрила и реакционную смесь нагревают до кипения в течение 4,5 ч. Получают 11,5 г продукта, идентичного получаемому по примеру 2А соединению, R, 0,44 (растворитель имеет указанный в примере 2А состав),
F.Процесс ведут аналогично примеру 2А, за исключением того, что в качестве растворителя применяют 40 мл 35 ацетонитрила и реакционную смесь нагревают до кипения в течение 4,5 ч. Получают 11,5 г продукта, идентичного полученному по примеру 2А соединению, RI 0,44 (растворитель имеет 40 указанный в примере 2А состав), Выход практически количественный.
G.9 г (0,038 моль) (1-пирро- лидинилметил)- f4-циклогексадиенил - окси|-1-пропиламина растворяют при комнатной температуре в 10 мл диме- тилформамида. К полученному раствору добавляют 5,5 г (0,038 моль) димети- лового эфира М цианои1$инодитиокарба- миновой кислоты. Реакционную смесь отстаивают при 120°С в течение 5 ч, охлаждают, выливают в 100 мл воды и четырежды экстрагируют 30 мл хлороформа Хлороформенные фазы объединяA.9 г (0,027 моль) полученного по примерам 2A-2G метилового эфира Р1-циано-и -3- 5-(1-пирродилинметил)- -1,4-циклогексадиенилЗ окси}-пропил- карбамиминотиокислоты растворяют при комнатной температуре в 200 мл н-бу- танола. К раствору добавляют 1,24 г (0,02 моль) метилгидразина. Реак ция начинается сразу же с образованием меркаптана. Реакционную смесь нагревают до кипения в течение 20 мин и выдерживают при этой температуре в течение 20 ч. Растворитель отгоняют
30 при пониженном давлении. Сотового типа остаток выливают в смесь 20 мл циклогексана и 5 мл ацетонитрила. Смесь охлаждают, выпавшие в осадок кристаллы фильтруют и промывают аце- тонитрилом, Получают 7,5 г целевого соединения, выход 83,8%, т.пл. 81- 84°Се Сырой продукт перекристалли- зовывают из 60 мл смеси этилацетат циклогексан (1;4), Получают 6,8 г целевого соединения с выходом 76% и т.пл. 86-87°С.
B.Процесс ведут аналогично примеру ЗА, за исключением того, что в качестве растворителя применяют ди45 н-пропиловый эфир. После перекристаллизации получают 5,4 г (60,3%) целевого соединения. Продукт идентичен полученному по примеру ЗА соединению, т.пл. 86-87°С.
C.Процесс ведут аналогично примеру ЗА, эа исключением того, что в качестве растворителя применяют бензол. После перекристаллизации получают 5,1 г целевого соединения, вы50
ют, сушат над безводным сульфатом маг-55 ход 57%, т.пл.85-88 С. Продукт идеи- ния и отфильтровывают. Остаток раст-тичен полученному по примеру ЗА соеворяют в 35 мл ацетонитрила при по-динению.
догреве. Полученный раствор охлажда-D. Процесс ведут аналогично приме07676
фильтровывпют и промывают ацетонитри- лом. Получают 0,8 г метилового эфира М-циано-ы -3- (3-( 1-пирролидинилме- тил) -1 ,4-циклогексадиенил1.оксиЦ -пропилкарбамиминотиокислоты, т.пл. 116- 118°С. После упаривания маточника получают 9,2 г продукта сотового типа, R, 0,46 (растворитель имеет укаэан- Ю ный в примере 2А состав). Продукт идентичен соединению, полученному по прикеру 2А.
ПримерЗ. Получение 1-метил- N (1-пирролидинилметил)-1,415 циклогексадиенил -окси}-пропил-1Н,1,2- триазол-3,5-диамина.
5 циклогексадиенил -окси}-пропил-1Н,1,2- триазол-3,5-диамина.
0
5
5 0
A.9 г (0,027 моль) полученного по примерам 2A-2G метилового эфира Р1-циано-и -3- 5-(1-пирродилинметил)- -1,4-циклогексадиенилЗ окси}-пропил- карбамиминотиокислоты растворяют при комнатной температуре в 200 мл н-бу- танола. К раствору добавляют 1,24 г (0,02 моль) метилгидразина. Реак ция начинается сразу же с образованием меркаптана. Реакционную смесь нагревают до кипения в течение 20 мин и выдерживают при этой температуре в течение 20 ч. Растворитель отгоняют
0 при пониженном давлении. Сотового типа остаток выливают в смесь 20 мл циклогексана и 5 мл ацетонитрила. Смесь охлаждают, выпавшие в осадок кристаллы фильтруют и промывают аце- тонитрилом, Получают 7,5 г целевого соединения, выход 83,8%, т.пл. 81- 84°Се Сырой продукт перекристалли- зовывают из 60 мл смеси этилацетат циклогексан (1;4), Получают 6,8 г целевого соединения с выходом 76% и т.пл. 86-87°С.
B.Процесс ведут аналогично примеру ЗА, за исключением того, что в качестве растворителя применяют ди5 н-пропиловый эфир. После перекристаллизации получают 5,4 г (60,3%) целевого соединения. Продукт идентичен полученному по примеру ЗА соединению, т.пл. 86-87°С.
C.Процесс ведут аналогично примеру ЗА, эа исключением того, что в качестве растворителя применяют бензол. После перекристаллизации получают 5,1 г целевого соединения, вы0



| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 1-алкил-3-окси5-хлор-1,2,4-триазолов или их солей | 1975 |
|
SU682128A3 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛИЛГИДРАЗИДА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИГОДНЫЕ СОЛИ | 1990 |
|
RU2039051C1 |
| Способ получения производных хинолина,их солей или их изомеров | 1974 |
|
SU535034A3 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2026296C1 |
| Способ получения производных пиридо (1,2-а) пиримидина или их фармацевтически приемлемых солей или их оптически активных изомеров | 1978 |
|
SU999973A3 |
| Способ получения производных пиридо /1,2-а/ пиримидина или их оптических изомеров, или их гидратов, или их солей | 1980 |
|
SU980622A3 |
| Способ получения карбапенемов | 1984 |
|
SU1395142A3 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЛАМОТРИДЖИНА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ | 1995 |
|
RU2145602C1 |
| Способ получения дибензо- [а,D-ЦИКЛООКТЕН-6,12-ИМИНОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1978 |
|
SU908248A3 |
| Способ получения производных пиридо[2,1-в] хиназолинона | 1979 |
|
SU1001857A3 |
Изобретение касается производных диарилоксиалкиламино-1,2,4-триазола ,в частности, получения соединений общей формулы R1 R2N-CH2- @ -O-(CH2)3-NH-C=N-C(NH2)=-N-CH3, где R1R2N - цикл пиперидина или пирролидина, или их фармакологически приемлемых солей, подавляющих секрецию кислоты желудочного сока, стимулированного рецепторами гистамина Н2. Цель - повышение выхода и качества целевых веществ. Синтез ведут востановлением эквивалентным количеством натрия соответствующего арилоксиамина при (-83)-(-33)°С с последующим отделением активного изомера от неактивного дигидроарилоксиалкиламина с помощью дистилляции. Затем активный изомер обрабатывают N-цианоиминодитиокарбаминовой кислотой при 20-120°С в среде растворителя (изопропанола, дин-пропилового эфира, хлороформа, бензола, ацетонитрила, диметилформамида) с последующим отделением остатка неактивного изомера и обработкой 1-1,5-кратным молярным избытком метилгидразина при 20-117°С в среде н-бутанола. При этом полученный продукт переводят в соль обработкой нужной кислотой. Эти условия обеспечивают увеличение выхода до 93,3% с чистотой до 90%.
ют, выпавшие в осадок кристаллы отру ЗА,.за исключением того, что применяют 1,86 г (0,041 моль) метилгид- разина„ После перекристаллизации получают 6,6 г целевого соединения, выход 73,9%р т пл. 85-87°С. Продукт идентичен полученному по примеру ЗА соединениюа
П р и м е р 4, Получение -(1-липеридинилметил)-,4-циклогек- садиенда-fj - оксиЧ -1 -пропиламина.
4р62 г (0,1 моль) этанола добавляют к 5S97 г (0,02 моль) (1- пиперидинилметил) -1 -фенокси -1 -пропиламина ч в полученном растворе конденсируют 200 мл аммиака-. Смесь с помощью бани ацетон - сухой лед охлаждают до- ч75 С и добавляют пор-: циями 2„1 т (0,105 моль) измельчен- ного металлического натрия так, что вторую и последующие порции добавляют лишь тогдае когда окраска раствора полностью исчезает
После добавления около 055 г натрия голубая окраска реакционной смеси остается на 2,5-3 ч. После окончательного обесцвечивания раствора реакционную смесь нагревают до кипения в течение 30 мин и добавляют 6,89 г (0,13 моль) хлористого аммония. Аммиак отгоняют9 остаток растворяют в 200 мл диэтилового эфира, неорганическую соль отфильтровывают и шесть раз промьшают по эфира Эфирные растворы объединяют и растворитель удаляют при пониженном давлении. Маслянистый остаток подвергают фракционированному вакуумному процессу дестилляции Получают 4S85 г продук- тя (т,кип, 130 135°С при 0,3ммрт.ст),
10
15
20
25
30
35
с образованием метилмеркаптана, ционную смесь нагревают до - кип в течение 2 ч и затем упаривают лый твердый остаток суспендирую небольшом, количестве эфира и отф ровывают. Получают 3,9 г целево единения, выход 93,3%, т.пл.90После перекристаллизации из изоп нола продукт плавится при 95-97
Примерб. Получение 1-м N -3- 1 5-(1-пиперидинилметил)-1 шклогексадианил -окси|-пропил I- -1Н-1,2,4-триазол-З,5-диамина,
1,74 г ( 0,005 моль) метилово эфира N 4HaHo-N -3)Ј5-(1-пиперид метил)- ,4-циклогексадианшт -окс пропилкарбамидотиокислоты раств в 5 мл бутанола, после чего к р ру добавляют 0,62 г (0,054 мол метилгидразина, Реакция начинае сразу же с образованием меркапт Через 20 мин реакционную смесь вают до кипения и выдерживают п этой температуре в течение 20 м Реакционную смесь охлаждают, ра ритель удаляют при пониженном д нии и остаток растворяют в небо количестве смеси (4{1) циклогек этилацетат Раствор охлаждают, шие в осадок кристаллыефильтрую лучают 1,5 г целевого продукта, ход 86,2%, т.пл, 100-103°С. Пос перекристаллизации из смеси 4: циклогексан - этилацетат продук вится при 105-106 С.
Фумарат 1-метил-К -/Зт (1 перидинилметил)-,4-циклодиенил сиЯ-пропил/-1Н-1,2,4-триаэол-3,
чоторый состоит из смеси (4:1) целе- 40 диамина плавится при 116-120 С.
вого соединения и изомерного -(1 пиперидинилметил)-1,4-циклогекса- диенил -окси|-1-пропиламина Изомерную смесь при мм рт„ств подвергают повторной фракционированной дис- 45 тилляции„ Кипящая при 133-135 С фракция содержит более 90% целевого соединения. Выход 78,0%,
Пример5. Получение метилового эфира N-циано-З- ГГ5-(1-пипериди- сп . .i-Iзи
нилме тил)-1э 4 циклогекнсадиенил|-окси1-пропилкарбамидотиокислоты.
3,0 г полученного по примеру 4 чистого (l-пиперидинилметил)- -1р4-циклогексадиенил -оксиТ-1-про- 55 пиламина растворяют в 30 мл диэтило вого эфира и добавляют 1,75 г (09012 моль) диметилового эфира N- цианоиминоцитиокарбаминовой кислоты
Таким образом, предлагаемый соб позволяет получать соедине формулы (j) с общим выходом 56
ормула изобрете
1 Способ получения произво дигидроарилоксиалкиламино-1,2,4 зола общей формулы
Rl
H3C-N N
Ч /ЧхЧ° I i
иснг о(сн2)
Л2 R, и R,
N 2 вместе с атомом азо
где «. i « г
разуют пиперидиновый или пиррол
новый цикл,
или их фармацевтически пригодны
лей присоединения кислот, включ
восстановление производных арил
0
5
0
5
0
5
с образованием метилмеркаптана, Реакционную смесь нагревают до - кипения в течение 2 ч и затем упаривают. Белый твердый остаток суспендируют в небольшом, количестве эфира и отфильтровывают. Получают 3,9 г целевого соединения, выход 93,3%, т.пл.9095 С. После перекристаллизации из изопропа- нола продукт плавится при 95-97 С,
Примерб. Получение 1-метил- N -3- 1 5-(1-пиперидинилметил)-1,4- шклогексадианил -окси|-пропил I- -1Н-1,2,4-триазол-З,5-диамина,
1,74 г ( 0,005 моль) метилового эфира N 4HaHo-N -3)Ј5-(1-пиперидинил- метил)- ,4-циклогексадианшт -окси|- пропилкарбамидотиокислоты растворяют в 5 мл бутанола, после чего к раствору добавляют 0,62 г (0,054 моль) метилгидразина, Реакция начинается сразу же с образованием меркаптана. Через 20 мин реакционную смесь нагревают до кипения и выдерживают при этой температуре в течение 20 мин. Реакционную смесь охлаждают, растворитель удаляют при пониженном давлении и остаток растворяют в небольшом количестве смеси (4{1) циклогексан- этилацетат Раствор охлаждают, выпавшие в осадок кристаллыефильтруют. Получают 1,5 г целевого продукта, выход 86,2%, т.пл, 100-103°С. После перекристаллизации из смеси 4:1 циклогексан - этилацетат продукт вится при 105-106 С.
Фумарат 1-метил-К -/Зт (1-пи- перидинилметил)-,4-циклодиенил -ок- сиЯ-пропил/-1Н-1,2,4-триаэол-3,50 диамина плавится при 116-120 С.
Таким образом, предлагаемый способ позволяет получать соединения формулы (j) с общим выходом 56%.
ормула изобретения
1 Способ получения производных дигидроарилоксиалкиламино-1,2,4-триа- зола общей формулы
Rl
H3C-N N
Ч /ЧхЧ° I i
иснг о(сн2)
Л2 R, и R,
N 2 вместе с атомом азота обгде «. i « г
разуют пиперидиновый или пирролидиновый цикл,
или их фармацевтически пригодных со«-.
лей присоединения кислот, включающий
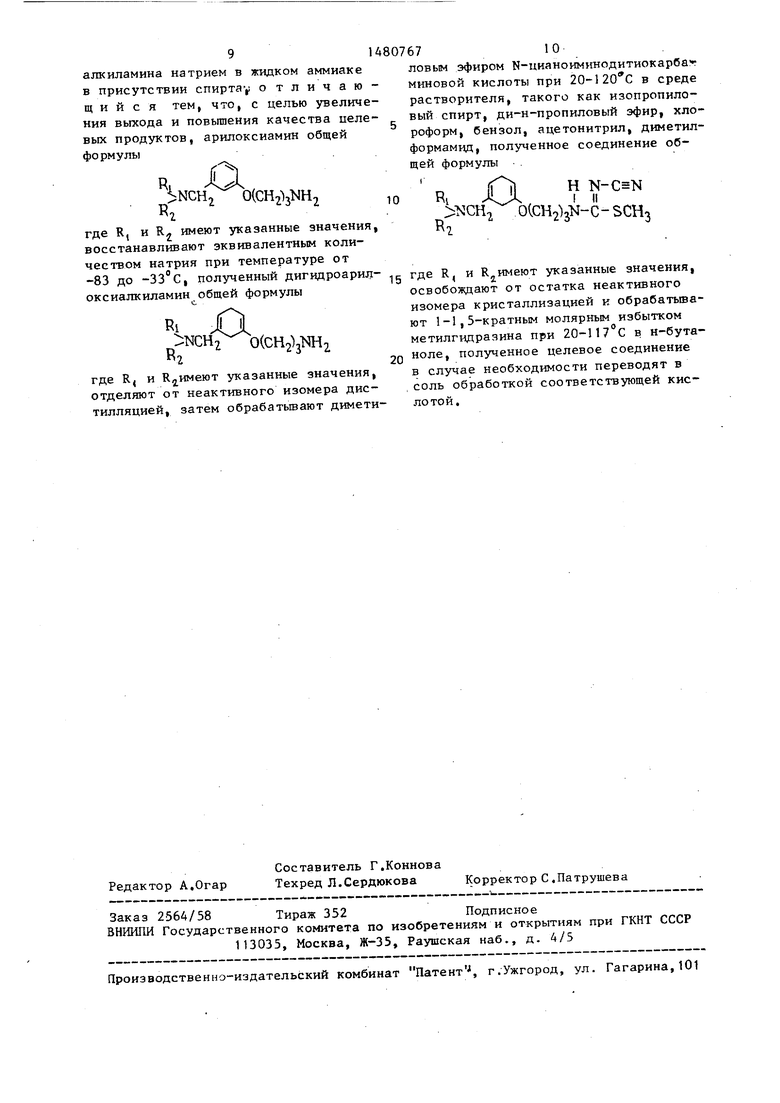
восстановление производных арилоксиалкиламина натрием в жидком аммиаке в присутствии спирта-,; о тличаю- щ и и с я тем, что, с целью увеличения выхода и повышения качества целевых продуктов, арилоксиамин общей фо рмулы
R XX NCH2 0(О%Ш2
нг
где R, и R2 имеют указанные значения, восстанавливают эквивалентным количеством натрия при температуре от -83 до -33°С, полученный дигидроарилоксиалкиламин общей формулы
где R, и Каимеют указанные значения, отделяют от неактивного изомера дистилляцией, затем обрабатывают диметиловым эфиром N-цианоимннодитиокарба миновой кислоты при 20-120 С в среде растворителя, такого как изопропило- вый спирт, ди-н-пропиловый эфир, хлороформ, бензол, ацетонитрил, диметил- формамид, полученное соединение общей формулы
RI
К,
ъ:сн
н
b(CH2)jN-C-SCH3
где R, и R имеют указанные значения, освобождают от остатка неактивного изомера кристаллизацией к обрабатывают 1-1,5-кратным молярным избытком метилгидраяина при 20-117 С в н-бутаноле, полученное целевое соединение в случае необходимости переводят в соль обработкой соответствующей кислотой.