H3o6peteHHe относится к новому спо- сову получения карбапенемовых антибиотиков, имеющих 2-заместитель формулы и
он. .г
са-..- -сНг-к СНГ- О-Хсоо-;.
(.
где R, - присоединенный через кольцевой атом углерода катион 2- (N-метилпиридиний), или 2- (J,6-диметилпиридиний), или А-(I,3-диметилпиридиний), или Н,Н-диметил-1,2,3-триазо лий, или 2-метил-1,2,3-тио- диазолий,/или калиевая соль 4-или 5-(1-карбоксиметил-З- метил-1,2,3-.триазолия или 5- (I,А-диметилтетразолия); 3- (I,З-диметиппиридин-А-ил), . 3-(1,6-диметилпиридин-2-ил)- бета-метил, 3-(1,6-диметил- пиридин-2-ил), (l,3-ди- метилимидазолил )J (1,2-диметилпиридил) ,(I -метилпиридил )1 , 3- U- (1 - метилпиридил)3 , 3- 2-0 ,А
диметилпиридил) , 3- ГЗ- (1 - метилпиридил)3, (1-ме- тилп иридил)3, 3- 2-(1,4-ди- метилпиридил)3 , 3- 2-{К-ме- тилпиридил) , 3, 5( 1,4-диме- тш1-1,2,4-триазолил), 3-(2,4 Димeтил-l ,2,4-триазолил), 3-(2,4-диметил-,2,4-триазо- лил), 2-;метил-1,2,3-тиоди- азолил, 5-( 1,4-диметид-.1 ,2, 4-тиазолил), 3-(1.З-диметил- З-тетразолил);
R - водород или метильная группа Целью изобретения ЯЕ(ля ется усовершенствование способа синтеза новых производных карбапе1 емов, проявляющих улучшенную фармакологическую активность.
. Пример. Получение (N, К-диметил-1,2,3-триазолий)метилтио - 6et- 1-(н)-оксизтил -7-оксо-1-азабицик ло 3,2,0 гепт-2-ен-2-карбоксилата.
А, Получение изомера А. Метилтри- фторметансульфонат (0,58 мл; 5,16 ммоль по каплян прибавляют к охлажденному льдом, перемешиваемому раствору 4- {метантиоацетат}-1-метил-1,2,3-триазо ла (590 мг; 3,52 ммоль) в сухом мети- ленхлорнде (2 мл), в атмосфере азота.
o
5 0 5
0
j О
с
, ,
Через 0,5 ч Саню убирзют и через г ч- растворитель удаляют аспиратором. Оставшееся масло растворяют в нескольких мл воды и раствор охлаждают на ледяной бане. Затем прибавляют холодный раствор гидроокиси натрия (305 мг; 7,59 ммоль) в нескольких мл воды и реакцию проводят при перемешивании в течение 0,75 ч. Раствор разбавляют до 25 мл водой и рН доводят до 7,5 добавлением твердого моногидрата натрийгидрофосфата. Затем J4 мл этого раствора (около 1,9 ммоль триазолийтиола) прибавляют в охлажденный льдом перемешиваемый раствор енолфосфата (1,0 г; 1,72 ммоль) в
10 мл тетрагидрофурана (ТГФ). Смесь оставляют перемешиваться в течение 0,75 ч (в ходе реакции вькчдает некоторое количество кристаллического соединения, преимущественно ). Суспензию переносят в автоклав с добавкой некоторого количества ТГФ (20 мл) и воды (20 мл). Прибавляют дизтиловый эфир (ЗО мл) и 10%-ный палладий на угле (1,0 г) и смесь гидрируют при давлении 2,8 атм в течение 1 ч. Органическую фазу отделяют и промьшают водой ( мл). Объединенные водные фазы отфильтровьгаа- ют и фильтрат концентрируют под высоким вакуумом (около 0,5 мл, 1,5ч). После зтого желтый раствор хромато- графируют (колонка среднего давления с обращенной фазой 35x90 мм, вода в качестве элюента) и после лиофилиза- ции получают 395 мг карбапенема, слегка загрязненного неорганическими соединениями. Его очищают ЖХВД (колонка Waters Micrcbondapack 0-18, 10x300 мм многократное введение, вода в качестве злюента) и получают ЗЮ.мг (57%) изомера А в виде рыжевато-коричнево- . го порошка: Н-ЯМР (В,о): S 1,23 (ЗН, д., ,4 Гц), 3,10 (2Н, д, 1-9,1 Гц)., 3,24 (1Н, к, 1,7,6,1 Гц), 4,03- 4,71 (ЮН, м), 8,46 (IH, с); ИК (ну- иол): 1760 CM V УФ (фосфатный буфер, рН 7,4, ,05) 296 (-7500)..
В. Получение изомера В и изомера С. МетилтрифторметансульфЬнат (1,60 мл{ 14,0 ммоль) по каплям прибавляют в охлажденный льдом раствор 4-(метантиоацетат)-2-метил-1,2,3-три- азола (1,20 г} 7,02 ммоль) в сухом метиленхлориде (6 мл) в атмосфере азота. Оставляют нагреваться до комнатной температуры и перемешивают
16 ч. Прибавляют дополнительно метил- фторметансульфонат ( мл; 3,56 ммоль) и через 3 ч при комнатной температуре растворитель удаляют аспиратором. Остаточное масло растирают с серным эфиром и результирующее смолистое соединение растворяют в воде (5 мл). Раствор охлаждают на ледяной бане и прибавляют раствор гидроокиси натрия 10 (84А мг; 21,1 ммоль) в воде (5 мл). После перемешивания в течение 0,75 ч этот раствор разводят до 60 мл водой и рН доводят до 8 добавлеиием твердого дигидрофосфата калия. После это- 15 го 40 г этого раствора (около А,7 ммоль смеси изомерных триазолий- тиолов) прибавляют в охлаждаемый льдом, перемешиваемый раствор енол- v фосфата (2,00 г 3,45 ммоль) в ТГФ 20 (60 мл). Эту смесь оставляют перемешиваться на ледяной бане в течение 0,5 ч, после чего ее переносят в автоклав, содержащий суспензию lOZ-aO .
Прим еор 2. 5R,6SJ-6-( )-3-(2-мeтил-l ,2,3-тиадиаз -4-ил-метш1тио)-7-оксо- -азаби f3,2,0 гепт-2-ен-2-карбоксилат
А. Этi лoвый эфир 1,2,3-тиад -4-карбоновой кислоты. Раствор вого эфира ei-N-карбоксигидразо пионовой кислоты (31,2 г, 0,1 5 в тионилхлориде (80 мл) переме при в течение 3 ч и нагре при 70 С в течение 20 мин. Тио рид выпаривают и остаток расти в гексане ( мл). Красное т соединение растворяют в дихлор (150 мл) и раствор промьшают щенным водным раствором бикарб натрия. После высушивания над том натрия раствор концентриру кристаллизации соединения. Пос стаивания при 23 С в течение н рого промежутка фильтррвьгоают iu,u i , 69%). Фильтрат концентрируют и
времени криста (16,8 г, т.пл. 8
30
35
го палладия на угле (2,00 г) и диэти- 25 ют на колонке с силикагелем с левого эфира (60 мл). Смесь гидрируют (2,8 атм) в течение 1 ч. Органическую фаэу отделяют и промывают водой (2x10-мл). Объединенные водные фазы отфильтровьшают и фильтрат концентрируют под высоким вакуумом (око- ла 0,5 мл, 1,5 ч). Затем оставшийся раствор хроматографируют (обращенно- фазовая колонка среднего давления, мм, вода в качестве элюента)(И после лиофилизации получают 595 мг смеси изомерных карбапенемов, загрязненной небольшим количеством неорга нических соединений. Ее затем разделяют и очищают с помощью ВХВД . (колонка Waters Microbondapack G-18, 10 300 мм, многократное введение, вода в качестве элюента) и получают в порядке элюирования изомер В: 153 мг (13%); Я-ЯМР (D,0)S : 1,23 (ЗН, д, 1-6,4 Гц), 3,12 (2Н, к, 1-1,4, 8,9 Гц), 3,39 (Ш, к, 1-2,7, 6,0 Гц), 4,07-4,68 (ЮН, м), 8,19 (1Н, с); Ж (нуйол); 1755 , УФ (фосфатный буфер, рН 7,4, ,05), Л«акс-. 296 нм (г-6700)
40
зованием дихлорметана в качест ента и получают 3,17 г, т.пл. 8 13%; ИК (КВг)мо, 720 ( эфир); Н-ЯМР (CDCl)S : 1,52 триплет, ,1 Гц, CH,CHiO), 4 (2Н, к, ,1 Гц, ), 9, с, Н тиадиазола).
В. 1,2,З-Тиадиазол-4-ил-мет К суспензии этилового эфира 1, таадиазол-4-ип-карбоновой кисл (18,35 г, 0,116 моль) в диэтил эфире (400 мл) по частям в те I ч прибавляют литийалюмогидри (2,47 г, 0,065 моль). Реакцион смесь перемешивают при в 7 ч и обрабатьгоают литийалюмог (2,47 г, 0,065 мл). Перемешива продолжают 24 ч, после чего по вательно прибавляют воду (7 мл 45 15%-ный раствор гидроокиси нат
;(7 мл) и воду (21 мл). После п шивания в течение 15 мин эфирн jCTBOp декантируют и смолистое нение экстрагируют диэтиловым (5100 мл). Эфирные экстракты няют, сушат над сульфатом магн концентрируют (5,4 г). Сырое .с ние очищают на колонке с силик (120 г, 4X16 см), эфир использ качестве элювнта и пол -чают 1, (7%) этилового эфира 1,2,3-тиaд 4-ил-кapбoнoвoн кислоты и 2,45 (18%) 1,2,3-тиадиазол-4-ил-мет ИК (пленка),, 3880 с;м (ОН)
50
: и изомер С: 284 мг (24Z); н-ЯМР (DjO) S: 1,23 (ЗН, д, ,4 Гц), 3,15 (2Н, к, 1-3,7, 9,0 Гц), 3,37 (IH, к, 1-2,6, 6,0 Гц), 3,95-4,65 (ЮН, м), . 8,62 (1Н, с); ИК (нуйол)J 1750 см ; УФ (фосфатный буфер, рН 7,4, ,05) дм1|,с.298 нм (7600).
Прим еор 2. 5R,6SJ-6-(lR-oкcи- )-3-(2-мeтил-l ,2,3-тиадиазолий- -4-ил-метш1тио)-7-оксо- -азабииикло f3,2,0 гепт-2-ен-2-карбоксилат.
А. Этi лoвый эфир 1,2,3-тиадиазол- -4-карбоновой кислоты. Раствор этилового эфира ei-N-карбоксигидразонопро- пионовой кислоты (31,2 г, 0,1 54 моль) в тионилхлориде (80 мл) перемешивают при в течение 3 ч и нагревают при 70 С в течение 20 мин. Тионилхло- рид выпаривают и остаток растирают в гексане ( мл). Красное твердое соединение растворяют в дихлорметане (150 мл) и раствор промьшают насыщенным водным раствором бикарбоната натрия. После высушивания над сульфатом натрия раствор концентрируют до кристаллизации соединения. После выстаивания при 23 С в течение некоторого промежутка фильтррвьгоают iu,u i , 69%). Фильтрат концентрируют и очищавремени кристаллы от- (16,8 г, т.пл. 8б с,
ют на колонке с силикагелем с
0
5
5 ют на колонке с силикагелем с
исполь- элю0
зованием дихлорметана в качестве ента и получают 3,17 г, т.пл. , 13%; ИК (КВг)мо, 720 (сложный эфир); Н-ЯМР (CDCl)S : 1,52 (ЗН, триплет, ,1 Гц, CH,CHiO), 4,57 (2Н, к, ,1 Гц, ), 9,47 (IH, с, Н тиадиазола).
В. 1,2,З-Тиадиазол-4-ил-метанол. К суспензии этилового эфира 1,2,3- таадиазол-4-ип-карбоновой кислоты (18,35 г, 0,116 моль) в диэтиловом эфире (400 мл) по частям в течение I ч прибавляют литийалюмогидрид (2,47 г, 0,065 моль). Реакционную смесь перемешивают при в течение 7 ч и обрабатьгоают литийалюмогидрядом (2,47 г, 0,065 мл). Перемешивание продолжают 24 ч, после чего последовательно прибавляют воду (7 мл), 5 15%-ный раствор гидроокиси натрия
;(7 мл) и воду (21 мл). После переме- шивания в течение 15 мин эфирный ра- jCTBOp декантируют и смолистое соединение экстрагируют диэтиловым эфиром (5100 мл). Эфирные экстракты объединяют, сушат над сульфатом магния и концентрируют (5,4 г). Сырое .соедине-- ние очищают на колонке с силикагелем (120 г, 4X16 см), эфир используют в , качестве элювнта и пол -чают 1,3 г , (7%) этилового эфира 1,2,3-тиaд tagoл- 4-ил-кapбoнoвoн кислоты и 2,45 г (18%) 1,2,3-тиадиазол-4-ил-метаиола; ИК (пленка),, 3880 с;м (ОН);
0
5
Н-ЯМР (CpCl,): 2,31 (Н; с, ОН), 5,22 (2Н, с, ), 8,5и (1Н, с, Н тиадиаэола).
С. 1,2,3-Tиaдиaэoл-A ил-мeтaнoл- мeтaнcyльфoнaт.Pacтвop 1,2,3-тиaдиa- зoл-5-ил-мeтaкoлa (0,75 г, 6,5 моль) в дихлорметане (20 мл) охлаяодают до в атмосфере азота и обрабатывают триэтиламином (1,018 мл} 7,3 ммоль) и метансульфонилхлорядом (0,565 кл; 7,3 ммоль). Через 15 мин ледяную.баню Удаляют и реакционную смесь перемешивают в течение 2 ч. Раствор про10
А,58 (2Н, с, CH4),8,4A(iH, с, Н тиа- диазола).
Вычислено, Z: С 34,47; Н 3,47; N 16,08; S 36,80
Найдено, Z: С 34,48; Н 3,83; N 16,28; S 36,80,
Е. 4-Ацетилтиометил-2-метш1-1,2, 3-тиадиазолийтрифторметансульфонат и 4-ацетилтиометил-3-метил-1,2,3-тиа- диазолийтрифторметансульфонат. К раствору 4-ацетилтиометил-1,2,3-тиа- диазола (0,60 г-, 3,44 ммоль) в смеси диэтипового эфира (4 мл) и дихлорм е «)Шают 1н. раствором хлористоводород- 15 тана (0,4 мл) прибавляют несколько ной кислоты (2x2 мл) и водой, сушат кристаллов целевых соединений и три- над смесью сульфата и окиси магния. . фторметилсульфонат (0,407 мл, и концентрируют. Остаток очищают хро- матографически (колонка с силикаге3,6 ммоль) в течение 5 мин. Реакцион- йую смесь перемешивают при 23°С в
лем, 1, см) с использованием ди- Зтилового эфира в качестве элюента и получают 0,90 г (71%) 1,2,3-тиадиа- зол-4-ил-метанолметансульфрната{ ИК (пленка )|
20
«акс.
1350 см- (SO),
1172 см (SOj )i Н-ЯМР (CDC1, )S 3,09 (ЗН, с, СН,), 5,75 (2Н, с, СН), 8,72 ()Н, с, Н тиадиазола); УФ (хлористый (четилен) .(-1990).
Вычислено, %: С 24,78; Н 3,11; N 14,42; S 33,02
C,,S
Найдено, %: С 24,78; Н 3,09; N 14,66; S 31,94
и 0,13 г (19%) ди-(|,2,3-тиадиаэол- -4-ил-метилового)простого эфира: ИК
25
30
(пленка)9
.
1272, 1242, 1200,
35
986, 805, 728 Н-ЯМР (CDC1,)S 5,16 (с, 4Н, CHj), 8,42 (с, 2Н, Н тиадиазола)..
Д. 4-Ацетилтиометил-1,2,3-тиади- азол. К раствору 1,2,З-тиадиазол-4- ил-метансульфоната (0,90 г; 4,6 ммоль) в тетрагидрофуране (9 мл) прибавляют водный раствор (2 мл) тиолацетата натрия, полученного из :тиолуксусной кислоты (о,38 мл; 5,3 ммоль) и бикарбоната натрия (0,445 г; 5,3 ммоль). Результируюиопо смесь перемешивают при в течение 1 ч и разбавляют диэтиловьм эфиром (75 мл). Органический раствор промьтают.водой (З З мл), сушат над сульфатом магния и концентрируют. Сырую смесь очищают хромато- графически (колонка с силикагелем, 1, см) с использовакием 50%-ного диэтипового эфира а гексане в качестве злюента и получают 0,60 г (75%); ЙК (пленка )мд КС 1675 см (), Н-ДМР (CDC1,): 2,37 (ЗН, с, GHj),
атмосфере азота в течение 6 ч. Белое твердое соединение, являющееся смесью двух целевых соединений, отфильтровывают, промьтают дизтиловым эфиром и получают 1,05 г (90%) ИК(КВг) 1675 см Чс-О);. Н-ЯМР (DMCO d-6)S : 2,43 (ЗН, с, CHjCOS), 3,33 (с, СИ, на N-3), 4,57 (с, СН, на N-2 4,66 (2Н, с, CHj), 9,55 (Н на N-2) тиадиазолия), 9,66 (Н на N-3 тиадиаз лия).
Вычислено, Z: С 20,27; Н 2,38; N 9,45; S 32,46
C,H,N,0,S,F, . . .
Найдено, %: С 24,61; Н 2,57; N 8,47; S 28,21
F.4-Меркаптометил-2-метил-1,2, 3-тиадиазолийтрифторметансульфонат
и 4-меркаптометил-З-метил-1,2, диазолийтрифторметансульфонат.
Раствор смеси 4-ацетилтиометил-2- метил-1,2,3-тиадиазолийтрифторметан- сульфоната и 4-ацетилтиометил-З-метил- 1,2,3-тиадназолийтрифторметансульфон та {1,05 г, 3,1 ммоль) в 6н. хлористодс водородной кислоте (10 мл) нагревают при в атмосфере азота в течение ч. Растворитель выпаривают при пониженном давлении и получают 0,91 желтого сиропообразного соединения. Это соединение используют на следую50 щей стадии без очистки.
G.,(1Н-оксиэтил)-3-(2- метил-I,2,З-тиадиазолий-4-ил-метил- тио)-7-оксо-1-азабицикло 1з,2,0)гепт- 2-ен-2-карбоксилат.
Холодный () раствор 5R, ра-нитробензяп-6-(1К-оксиэтил)-3-(ди фенилфойфоно)-7-оксо-1-азабицикло З, 2,о1- гепт-2-ен-2-карбоксилата (1-7 г
40
55
А,58 (2Н, с, CH4),8,4A(iH, с, Н тиа- диазола).
Вычислено, Z: С 34,47; Н 3,47; N 16,08; S 36,80
Найдено, Z: С 34,48; Н 3,83; N 16,28; S 36,80,
Е. 4-Ацетилтиометил-2-метш1-1,2, 3-тиадиазолийтрифторметансульфонат и 4-ацетилтиометил-3-метил-1,2,3-тиа- диазолийтрифторметансульфонат. К раствору 4-ацетилтиометил-1,2,3-тиа- диазола (0,60 г-, 3,44 ммоль) в смеси диэтипового эфира (4 мл) и дихлорм етана (0,4 мл) прибавляют несколько кристаллов целевых соединений и три- фторметилсульфонат (0,407 мл,
3,6 ммоль) в течение 5 мин. Реакцион- йую смесь перемешивают при 23°С в
атмосфере азота в течение 6 ч. Белое твердое соединение, являющееся смесью двух целевых соединений, отфильтровывают, промьтают дизтиловым эфиром и получают 1,05 г (90%) ИК(КВг) 1675 см Чс-О);. Н-ЯМР (DMCO d-6)S : 2,43 (ЗН, с, CHjCOS), 3,33 (с, СИ, на N-3), 4,57 (с, СН, на N-2), 4,66 (2Н, с, CHj), 9,55 (Н на N-2) тиадиазолия), 9,66 (Н на N-3 тиадиазо- лия).
Вычислено, Z: С 20,27; Н 2,38; N 9,45; S 32,46
C,H,N,0,S,F, . . .
Найдено, %: С 24,61; Н 2,57; N 8,47; S 28,21
F.4-Меркаптометил-2-метил-1,2, 3-тиадиазолийтрифторметансульфонат
и 4-меркаптометил-З-метил-1,2, диазолийтрифторметансульфонат.
Раствор смеси 4-ацетилтиометил-2- метил-1,2,3-тиадиазолийтрифторметан- сульфоната и 4-ацетилтиометил-З-метил- 1,2,3-тиадназолийтрифторметансульфона та {1,05 г, 3,1 ммоль) в 6н. хлористоводородной кислоте (10 мл) нагревают при в атмосфере азота в течение ч. Растворитель выпаривают при пониженном давлении и получают 0,91 г желтого сиропообразного соединения. Это соединение используют на следующей стадии без очистки.
G.,(1Н-оксиэтил)-3-(2- метил-I,2,З-тиадиазолий-4-ил-метил- тио)-7-оксо-1-азабицикло 1з,2,0)гепт- 2-ен-2-карбоксилат.
Холодный () раствор 5R, ра-нитробензяп-6-(1К-оксиэтил)-3-(ди- фенилфойфоно)-7-оксо-1-азабицикло З, 2,о1- гепт-2-ен-2-карбоксилата (1-7 г
2,92 ммоль) в тетрагидрофуране (Ю мл обрабатывают раствором серной смеси А-меркаптометил-2-метш1-1,2,3-тиади- аэолийтрифторметансульфоната и 4-мер- каптометил-З-метил-1,2,3-триадиаэо- лийтрифторметансульфоната (0,9 г) в смеси фосфатного буфера (рН 7,2, 0,ЗМ, 15мл) и тетрагкдрофурана 5 мл).Реакционную смесь перемешива- ют в течение I ч и рН поддерживают при 7,2 с помощью 2н. раствора гидроокиси натрия. Перемешивание поддерживают еще в течение I ч, затем прибавляют диэтиловый эфир (50 мл) и-10%-ный палладий на угле (I г). Результирующую смесь гидрируют при и 3 15 атм в течение 2 ч и фильтруют через цеолит. Органическую фазу отделяют, разбавляют диэтиловым эфиром (50 мл) и фосфатным буфером (рН 7,2, 0,3 М, 20 мл) и -гидрируют (2 г IOZ-ного палладия на угле) в течение 2 ч при 3,5 атм. Водные фазы соединяют (от первого и второго гидрогенолиза), :промьюают диэтиловым эфиром и очищают хроматографически иа Prep Puk 500-C/I8 с использованием воды в качестве элюирующего растворителя и получают 0,22 г сырого соединения. :Его повторно очищают с помощью ЖХВД |с использованием воды в качестве элю- ieHTa и получают 0,040 г (4%) целевого соединения после лиофилизации; ИК(КВг)мокс, 3400 (широкий пик, ОН), 1745 ()р-лактама), 1580 (кар- боксилат) Н-ЯМР (): 1,23 (ЗН, д, 1-6,3 Гц, СН,СНОН), 3,04. . 3,05, 3,16 (2Н, м, Н-4), 3,38.(1Н. двойной дуплет, ,8 Гц, ,0 Гц,- Н-6), 3,9-4,6 (2Н, м, Н-5, iCHjCHOft), 4,51, 4,53 (2Н, с, ЗСНг), 4,61 (с, N®CH,); УФ (вода). 224 (е-4345). 262 (г«4980), .296 (б:-6885),М 18 (,18, вода) Т(,8 п (измерено при концентрации Ю .М в фосфатном буфере рН 7,4 при 36,8 с). ;
П р и м е р 3. Калиевая соль (1-карбоксилатометил-З-метил-1,2,3- тpиaзoлий)-мeтaнтиoЗ-6«i-fl-(R)-oкcи- . этил -7-оксо- J -азабицик ло 3,2, 0 гепт- 2-ен-2-карбоновой кислоты.- .
Литийалюмогидрид ( 2,83 г, 70,9 ммол
небольшими порциями прибавляют в nepig.- мешиваемую суспензию I-метил-1,2,3- триазол-4-карбоновой кислоты (9,00 г, 70,9 ммоль) в сухом ТГФ (200 мл). Смесь оставляют перемешиваться при комнатной температуре в течение 15. ч,
после чего осторожно прибавляют около 20 мл 20%-ного водного раствора гидроокиси натрия порциями по 1 мл. Результирующую грануляционную суспензию отфильтровьгоают и твердый остаток промьгаают дополнительным количеством ТГФ (5x75 мл). Объединенные растворы ТГФ сушат над сульфатом магния и растворитель удаляют. Результирующее желтое масло очищают хроматографией на колонке с снпикагелем 90x35 мм - (порции по 100 мл гексана, смеси этил ацетатгексан 1:1 и 1;3 ив заключение этштацетат - метанол 9:1 используют в качестве элюента). Таким образом получают 4-оксимвтил-1-метил- 1,2,3-триазол (3,18 г, 40%) в виде бесцветного масла: Н-ЯМР (CDCl,) : 4,07 (ЗН, с), 4,73 (2Н, д), 7,52 (1Н, c)j ИК (чистый) 3320 см- .
Метансульфонилхлорвд (3,82 мл , 49,6 ммоль) по каплям1трибавляют в охлажденный льдом, перемешиваемый раствор спирта (5,67 мл| 41,3 ммсль) и тризтиламина (7,47,мл 53,7 ммоль) в метиленхлориде (20 мл). Через 0,5 ч растворитель удаляют и оставшееся твердое соединение растворйют в ацетонитриле (30 мл).-Добавляют тиол- ацетат калия (7,06 г, 62,0 ммоль) и суспензию перемешивают при комнат мой температуре в течение 3 ч. Прибавляют дополнительное количество тиолацетата калия (3,0 г; 26,3 ммоль) и суспензию перемешивают еще в течение 16 ч, затем суспензию, имеющую темный цвет, концентрируют и прибавляют воду (10 мл). Эту смесь экстрагируют метиленхлоРИДОМ (5x40 мл). Объединенные экстракты сушат над сульфатом магния и удаляют растворитель. Оставшееся масло очищают хроматографически на колонке с силикаге- яем мм (в качестве элюента используют гексан, а затем смесь гек- сан - этилацетат 1:1). При этом получают 4-(ме тан тирлаце та т)-1-ме тил- 1,,8-триазол (5,95 г; 84%) в виде бледно-розового твердого соединения: %-ЯМР (CDCl,) ; 2,40 (ЗН, синглет), 4,10 (ЗН, с), 4,20 (2Н, с), 7.53 (1Н, с), ИК (нуйол): 1675 см .
. Раствор триазола (1,00 г; 5,85 ммоль) и этилбромацетата (1,Л8мл 13,3 ммоль) в сухом ацетонитриле (Ю мл) нагревают при п течение 90 ч в атмосфере азота. Рагтворитёль
удаляют и оставгаееся Macrfo растирают с диэтиловым эфиром л мл) и получают 1-метил-З-(этилкарбоксиме- тил)-А-метантиоацетат-1,2,3-триаэолий- бромид в виде коричневатого смолистого соединения, которое испольэзпот непосредственно.
fO
15
Холодный раствор КОН (0,66 г; 12 ммоль) в воде (5 мл) прибавляют в охлаждаемый льдом, перемешиваемый раствор триаэолийбромида в воде (20 мл). Через 20 мин его разводят до 35 мл и прибавляют такое количество твердого дигидрофосфата калия, чтобы рН раствора был равен 8,0. Этот раствор прибавляют затем в перемешиваемый, охлаждаемый льдом раствор фенолфосфата в ТГФ (35 мл). Через JQ 0,5 ч эту смесь перен осят в автоклав, содержащий диэтиловый эфир (35 нл) и 10%-ный палладий на угле (1,5 г). , Проводят гидрирование при 2,8 атм в
течение 55 мин. Органическую фазу от-25 деляют и промьшают водой (2X5 мл). Объединенные водные фазы отфильтровывают и ф,ипьтрат концентрируют под высоким вакуумом. Остаточное соединение хроматографируют на обращенно- JQ фазовой колонке -(35x120 мм) с использованием воды в качестве элюента. Лиофилизацией фракций, содержащих карбапенем, получают 1,20 г зеленого твердого соединения. Его снова хроматографируют с помощью ЖХВД Waters Prep, (колонка Prep. РАК-500/С«9 ), используя в качестве элюента 2% аце- тонитрила в воде. Фракции, содержаI щие карбапенем, объединяют и лиофи- лизуют. Это соединение снова хромато- графируют с помощью ЖХВД (колонка ЮЛГЗОО мм. Waters Microbondapack С-18) с использованием воды в каче- с;тве элюента и получают после лиофи- лизации чистое целевое соединение (190 мг , 17%) в виде бледно-желтого твердого соединения: Н-ЯМР ()S : 1,24 (ЗН, д, ,А Гц), 3,07 (2Н, д, 1-9 Гц), 3,38 (1Н, к, ,7, 6,0 Гц), 4,02-4,30 (ЗН, м), 4,29 (ЗН, с), 5,28 (2Н, с),8,52 (1Н, с); ИК (ну- Йол): 1750 см ; УФ (фосфатный буфер, рН 7,4)„, 296 нм (7520).
П р и м е р 4. Калиевая соль (1-карбоксилатометул-З-метил-1,2,3- 55
35
40
45
50
триазолий)-метантио -6« - 1-(К)-окси- це этилацетатом и получают 1-(
зтилЗ -7-оксо-1-азабицикло 3,2, Oj -гепт- карбсксиметил)-4-окснметнл-1,2,3-триСмесь зтилазидоацетата (30,0 г-, 0,23 моль) и пропилоной кислоты (14,3 мл, 0,23 моль) в толуоле (75 мл) перемегаивают при комнатной температуре. Реакция слабо экзотермична, а через 1,5 ч становится сильно экзотер мической и при этом требуется охлажде ние на ледяной бане. После окончания экзотермической стадии реакцию прово- дятькипячением с обратным холодильником в течение 0,5 ч. После охлаждения на ледяной бане кристаллическое соединение собирают фильтрованием и промывают небольшим количеством толуола. Полученное таким образом сырое соединение (33.3 г{ 72%) состоит из одного изомера: Н-ЯМР (DMCO-dg)5 : 1,20 (ЗН, триплет, Гц), 4,15 (2Н, к, 1-7 Гц), 5,42 (2Н, с), 8,67 flH, З), предположительно 1- (зтилкарбокскме- тнл)-1,2,3-триазол-4-карбоновой кисло ты.
Раствор карбоновой кислоты (5,00rj 25,1 ) и триэтиламина (3,68 мл/ 26,4 ммоль) в сухом хлористом метилене (50 мл) прибавляют в охлаждаемый льдом, перемешиваемый раствор этил- хлорформиата (2,52 мл-, 26,4 ммоль) в сухом хлористом метилене (50 мл). Красный раствор перемешивают в течение 0,5 ч, после чего его промьгаают водой (Ю мл), сушат над сульфатом магния и удаляют растворитель. Сухой смешанный ангидрид растворяют в ТГФ (50 мл).и медленно добавляют в охлаждаемую льдом суспензию боргидрида нат рия (0,72 г; 18,9 ммоль) в ТГФ (50 мл После перемешивания в течение 0,5ч прибавляют дополнительное количество боргидрвда натрия (0,30 г 7,9 ммоль) и реакцию проводят в ледяной бане в течение 1 ч. Затем добавляют воду (5 мл) и через 10 мин прибавляют 10%-ный водный раствор НС1 (3 мл). После дегазирования при перемешивании прибавляют твердый карбонат калия-. (2 г). Затем органическую фазу удаляют и оставшуюся белую пасту дополнительно экстрагируют ТГф. Объединенные органические фазы сушат над сульфатом магния н растворитель удаляют. Проводят очистку хроматографией на силикагеле, элюируют гексаном смесью этилацетат - гексан и в конзтил2-ек-2-карбоновой кислоты.
азол (2,04 г, 44%) в виде белого
O
5
Q
5 Q
5
5
0
5
0
це этилацетатом и получают 1-(
карбсксиметил)-4-окснметнл-1,2,3-триСмесь зтилазидоацетата (30,0 г-, 0,23 моль) и пропилоной кислоты (14,3 мл, 0,23 моль) в толуоле (75 мл) перемегаивают при комнатной температуре. Реакция слабо экзотермична, а через 1,5 ч становится сильно экзотермической и при этом требуется охлаждение на ледяной бане. После окончания экзотермической стадии реакцию прово- дятькипячением с обратным холодильником в течение 0,5 ч. После охлаждения на ледяной бане кристаллическое соединение собирают фильтрованием и промывают небольшим количеством толуола. Полученное таким образом сырое соединение (33.3 г{ 72%) состоит из одного изомера: Н-ЯМР (DMCO-dg)5 : 1,20 (ЗН, триплет, Гц), 4,15 (2Н, к, 1-7 Гц), 5,42 (2Н, с), 8,67 flH, З), предположительно 1- (зтилкарбокскме- тнл)-1,2,3-триазол-4-карбоновой кислоты.
Раствор карбоновой кислоты (5,00rj 25,1 ) и триэтиламина (3,68 мл/ 26,4 ммоль) в сухом хлористом метилене (50 мл) прибавляют в охлаждаемый льдом, перемешиваемый раствор этил- хлорформиата (2,52 мл-, 26,4 ммоль) в сухом хлористом метилене (50 мл). Красный раствор перемешивают в течение 0,5 ч, после чего его промьгаают водой (Ю мл), сушат над сульфатом магния и удаляют растворитель. Сухой смешанный ангидрид растворяют в ТГФ (50 мл).и медленно добавляют в охлаждаемую льдом суспензию боргидрида нат рия (0,72 г; 18,9 ммоль) в ТГФ (50 мл). После перемешивания в течение 0,5ч прибавляют дополнительное количество боргидрвда натрия (0,30 г 7,9 ммоль) и реакцию проводят в ледяной бане в течение 1 ч. Затем добавляют воду (5 мл) и через 10 мин прибавляют 10%-ный водный раствор НС1 (3 мл). После дегазирования при перемешивании прибавляют твердый карбонат калия-. (2 г). Затем органическую фазу удаляют и оставшуюся белую пасту дополнительно экстрагируют ТГф. Объединенные органические фазы сушат над сульфатом магния н растворитель удаляют. Проводят очистку хроматографией на силикагеле, элюируют гексаном смесью этилацетат - гексан и в конзтилкарбсксиметил)-4-окснметнл-1,2,3-т
азол (2,04 г, 44%) в виде белого
кристаллического соединения: H-JMF (CDCl,)5 : 1,28 (ЗН, т, Гц), 4.23 (2Н, к, Гц), 4.75 (2Н, с)/ 4,85 (2Н, с), 7,73 (Ш, с). 5
Дииэопропилазодикарбоксилат (А,II мл; 20,8 ммоль) по каплям прибавляют в охлаждаемый льдом раствор трифенилфосфина (5,47 г; 20,8 ммоль) в сухом ТГФ (100 мл) в атмосфере аэо-10 та. Через 0,5 ч в эту смесь прибавляют охлаждаемый льдом раствор спирта (1,93 г; JO,A ммоль) и тиолуксус- ной кислоты (1,49 мл; 20,8 ммоль) в сухом ТГФ (50 мл) в атмосфере азота. J5 Эту смесь выдерживают в течеиие 2 ч иа ледяиой бане и затем дополнительно 12 ч при комнатной температуре, пойле чего растворитель удаляют. Реак- ционкую смесь хроматографируют на си- 20 ликагеле (40 г, элюируют порциями по 100 мл гексана, 5, 10, 15,.,.,50%-ных смесей зтилацетата и гексаиа). фракции, содержащие тиолацетат объединяют и снова хроматографируют на силика- 25 геле (60 г, элюируют порциями по 200мл гексана, 5, 10, 15 и 20 ;-ных смесей, этилацетата и гексана и 22,5, 25,27, 5,. .,35%-Hb X смесей этилацетата и гексаиа). Таким образом получают зо г (49Z) .1-(этилкарбоксиметил)-4- е1 антиолацетата-,2,3-триазола в вие кристаллического твердого соединения: Н-ЯМР : 1,28 (ЗН, т, 1-7 Гц), 2,37 (ЗН, с), 3,87 (2Н, с), 3,90 (2Н, „ , 1-7 Гц), 5,12 (2Н, с), 7,63 (1Н; ), ИК (яуйол): 1735, 1780см , и доолнительно 1,40 г соединения, загряз- еннрго трифенилфосфиноксидом,
Метилтрифторметансульфонат (0,51 мп,. 4,53 ммоль) по каплям прибавляют в охлаждаемый льдом, перемешиваемый ра- створ триазола (1,00 г 4,12 ммоль) в сухом метиленхлориде (5 мл). Через 0,5 ч удаляют баню и еще через 0,5 ч растворитель удалят под вакуумом. Цо- лучают белое твердое соединение, которое суопендируют в воде (15 мл) , и смесь перемешивают при охлаждении
45
50
иа ледяной бане. Прибавляют раствор
КОН (0,69 Г-, 12,4 ммоль) в воде (5 мл); и проводят реакцию при перемешивании в течение 1 ч. Затем смесь разбавляют до 30 мл водой и прибавляют твердый дигидрофосфат калия, доводя рН до 3,0. Часть этого раствора (около 22 мл; 3,0 ммоль тиолкарбоксилата) прибавляют в охлаждаемый льдом
5
0 5 0 5 о
5
0
5
перемешиваемый раствор енолфосфата (1,60 г; 2,76 ммоль) в ТГФ (ЗО мл). Через 0,5 ч реакцию заканчивают и ТГФ удаляют под высоким вакуумом. Желтый раствор затем хроматографируют на обращенно-фазовой колонке (35 120 мм) с использованием воды в качестве элюента (ЗОО мл), а затем порций по 100 мл 5,10,15,...,30г-ных смесей ацетонитрил - вода. №1офилиза- цией соответствующих фракций получают пара-нитробензиловый сложный эфир в виде желтого твердого соединения (930 мг). Его переносят в автоклав, содержащий диэтиловый эфир (25 мл), ТГФ (25 мл), фосфатный буфер (25 мл), полученный растворением дигидрофос- фата калия (1,36 rj 0,01 моль) в.воде (loo мл) и доведенный до рН 7,4 добавлением 45%-ного водного раствора . КОН, и ЮЛ-ный палладий на угле (900 мг). Проводят гидрирование при 2,8 атм в-течение 1 ч, после чего органическую фазу отделяют и промывают водой (2X5 мл).,0б ьедииенные
водные фазы отфильтровьшают и. концен трируют под высоким вакуумом. Оставшийся раствор хроматографируют на обращенно-фазовой колонке (35x120 мм) с использованием воды в качестве элюента. фракции, содержащие карбапепем, объединяют, лиофилизуют и получают 1,21 г бледно-зеленого твердого соединения. Его оюгщают с помощью ЖХВД (колонка 10x300 мм. Water Microbonda- pack C-18, вода в качестве злюеита) и получают чистый целевой продукт 480 мг (41Я:): Н-ЯМР () : 1,23 (ЗН, д, 1-6,4 Гц), 3,11 (2Н, д, I- 9 Гц), 3,37 (1Н, к, 1-3,0, 6,1 Гц). 4,02 (7Н, м), 5,18 (2Н, с), 8,53 (1Н с); ЮС (нуйол) 1750 УФ (фосфатный буфер, рН 7,4) Я;ио1кс 205 им : :().
П Р и м е р 5. (l,4-ДимeтйЛ- I , 2 , 4-триазолий )метан гио С (R ) аксиэтил -7-оксо-1-азабицикло р,2,03- гепт-2-еи-2-карбоксилат.
А. 1-Метил-5-метантиолацетат-1«2, 4-триазрл.
/Метансульфонилхлорид (0,46 мл; ммоль) по каплям прибавляют к охлаждаемому льдом, перемешиваемому раствору 1-метил-5-оксиме гил-1,2,4 триазола (565 мг{ 5,0 ммоль) и трк- этиламина (0,91 мл; 6,5 ммоль) IB ме- тнпенхлориде (5 мл). Через 20.мин- прибавляют дополнительно трнйтилаМин
(l,05 мл, 7,5 ммоль), a затем тиол- уксусную кислоту (о,53 мл, 7,5 ммоль) и переметивают в течение 45 мин. Затем реакционную смесь разбавляют ме- 5 тнленхлоридом и промьшают водой. Водную фазу экстрагируют метиленхлори- дом ( мл), объединенные органические фазы сушат над сульфатом магйия и растворитель удаляют. Колоночной 10 хроматографией на силикагеле получают чис тый J -метил-5-метактиолаце1 ат-г , 4-триаэол (570 мг) в виде желтого масла. Кроме того, загрязненную фракцию (200 мг) снова хроматографируют (пре-f5 паративная ТСХ, силикагель) и получают еще JOO мг чистого соединения (общий выход 85%): Н-ЯМР (CDCl) : 2,38 (ЗН, с), 3,90 (ЗН, с), 4,25 (ЗН, с), 7,80 (1Н, с). 20
В. 3- 5-(1,4-диметил-1,2,4-триазЬ- лий).-метантио -6е - l-(R)тoкcнэтилJ-7- сксо-1-азабнцикло f3,2.,0 {гепт-2-ен-кар боксилат,
Метилтрнфторметансульфонат( 1,20
10.7ммоль) по каплям прибавляют к охлаждаемому льдом раствору 1-метил-5-. метантидлацетат- ,2,4-триазола (730мг)( 4,27 ммоль) в метиленхлориде (7 мл). Реакционную смесь медленно нагревают 30 до комнатной температуры в течение
3ч, после чего ее концентрируют. Остаточное масло растирают с диэтнповым эфмром и получают сырой 1,4-диметил- 5-метантиолацетат-1,2,4-триазолий- э трифторметансульфонат (1,46 г), ко- торьй используют непосредственно.
Раствор гидроокиси натрия (512 мг; i
12.8ммоль) в воде (5 мл) прибавляют АЛ в охлаждаемый льдом раствор триазоли евой соли (1,45 г;.4,35 ммоль) и воде (5 мл). Через 45 мин смесь разводят до 25 мл водой и рН доводят до
7,6 твердым дигидрофосфатом калия. За тем этот раствор прибавляют к охлаждаемому льдом, перемешиваемому раствору енолфосфата (2,00 г} 3,45 ммоль) в ТГ.Ф {25 мл). Через 30 мин реакционную смесь переносят в автоклав, содержащий дизтиловый эфир (40 мл) и 10%-ный палладий на угле (2,0 г). Смесь гидрируют при 3,15 атм в течение 1,25 ч. Затем реакционную смесь, разбавляют диэтиловым зфиром (25 мл) и Отфильтровывают, Органическую фазу 5 отделяют и промывают водой (2ч5 мл). Объединен1П 1е водные фазы промьшают диэтиловым зфиром (325 мл) и концентрируют под вакуумом. Колоночной хроматографией (обращенная фаза, 45 «|30 мм, вода Б качестве элюента) и последующей лиофилизацией фракций, содержащих карбапенем, получают 650мг сырого соединения. Его снова хроматографируют и получают чистый целевой продукт, 450 мг (39): Н-ШР (D,0). 1,24 (ЗН, д, 1 - 6,4 Гц), 3,19 (2Н, к, 1-2,6, 9,2 Гц), 3,45 (Ш, к, I- -2,8, 6,0 Гц), 3,91 (ЗН, с), 4,06 (ЗН, с), 4,08-4,36 (2Н, м), 4,54 (2Н, д, 1-2,8 Гц), 8,71 (1Н, с), Ж (ну-, иол): 1755 УФ (фосфатный буфер, рН 7,4) -294 нм (г-8202), Т,, (фосфатный буфер, рН 7,4, ,067, Т-37 с) 9,1 ч,
П р и м е р 6. 1 R,5R,(l,3- димeтил-5-тeтJ5aзoлий )-метЯ1:тио -6 ( 1 - оксиэтил) -7-оксо-1 -аз абицикло 3,2 гепт-2-ен-2-карбоксилат,
А, 5-Карбэтокси-2-метилтетразол и 5-карбзтокси-1-метилтетразол,
1а. Метилирование диазометанои. Раствор 5-карбзтокситетразола (9,17 rj 0,064 ммоль) в 80 мл дизтилового зфи- ра (использование смеси зтанола и дизтилового эфира дает то же самое соотношение изомеров) охлаждают до и по каплям прибавляют раствор ди- азометана (З г{ 0,07 ммоль) в дизти- ловом эфире (200 мл) в течение 15 мии. Светло-желтый раствор переметивают в течение 30 мин и избыток диазомета- на разрушают прибавлением уксусной кислоты (1 мл). Выпариванием растворителя и перегонкой остатка получают прозрачное масло; т,кип. 95- 100 С/0,5 мм рт.ст., 9,64 г (96), н-ЯМР зтсазьшает на смесь 1-метилового и 2-метилового изомеров в соотношении 6:: 4. Разделение двух изомеров не достигается ни перегонкой, ни ЖХВД: ИК (пленка)Смс1кс : 1740 (С Ю сложного зфира)-, Н-ЯМР (CDCl) : 1,53 . (ЗН, два перекрывающихся триплета, 1-7,0, CHjjCH,), 4,46 и 4,53 (ЗН, 2S, СИ, I-метил и 2-метилтетразола, соотношение 6:4, 2-метиловый изомер л ежй г ,в более низкой области и является не основным продуктом), 4,5 ппм (2Н, два перекрывающихся квартета, ),
Ib, 5-Карбзтокси-2-метилтетразол, Смесь 5-карбэтокси-2-метилтетразола и 5-карбзтокси-1-метилтетразола (о,252 Г , 1,61 ммоль, соотношение изомеров 1:1) в йодистом метиле (0,5 мл)
герметизируют в стеклянной пробирке и нагревают при течение 15 ч и при в течение 6 ч. Перегонкой реакционной смеси получают целевое « соединение в виде бледно-желтого масла: 0,139 г (55%), т.кип. 95-100 с/ / 0,5 мм рт.ст.(температура воздушной бани); ИК (пленка)„акс 1740 см (С-О сложного эфира), н-ЯМР (CDCl,) Ю S: 1,46 (ЗН, т, Т-7,0, CHjCH), 4,53 (ЗН, с, СН,-2), 4,5 (2Н, к, 1-7,0 CHjCH,).
2. Метилироватсие диметилсульфатом, Раствор 5-карбэтокситетразола (l 42rjt5 0,01 моль) в сухом ацетоне (20 мл) обрабатьшают безводным карбонатом калия (1,36 г, 0,01 моль) и днметил- сульфатом (1,26 г; 0,01 моль). Смесь кипятят с обратным холодильником в 20 течение 12 ч. Карбонат отфильтровы- вают и растворитель вьларивают при пониженном давлении. Остаток разбавляют дихлорметаном (30 мл), промывают насыщенным раствором бикарбоната, кат- 25 рия (10 мл), рассолом (10 мл) и сушат над безводным сульфатом натрия. Выпариванием растворителя и перегонкой под вакуумом получают прозрачное масло: 1,45 г (93%), т.кип. 85-110 с/ ЗО /0,5 мм рт.ст. Н-ЯМР показывает присутствие двух изомеров в соотношении 1:1.35
В, 5-Оксиметил-2-метилтетразол. I. Восстановление смеси сложных эфйров. Смесь 5-карбэтокси-1-метилтет . разола и 5-карбзтокси-2гметилтетра9о- ла в соотношении 6:4 (7,60 г О, 049 моль7 в сухом ТГФ (50 мл) 40 лаждают до О с и обрабатьшают литий- алюмогидридом.1,06 г, 0,049 ммоль), который прибавляют небольшими порциями в течение 15 мин. Смесь ввдержи- вают при еще в течение 30 мни и 45
затем перемешивают при в течение 4 ч. Смесь охлаждают до О с и иэ- быток гидрида осторожно разрушают
прибавлением 6н. НС1 (рН равен 7 после окончания вьзделеНИИ газа). Раст- .
воритель концентрируют под вакуум Зм и оставшееся масло разбавляют дихлорметаном (200 мл), промывают раствором соли (10 мл) и сушат над сульфатом натрия. Концентрированием растворителя и перегонкой остатка под вакуумом 5 получают 1,83 г (33%) прозрачного . масла. н-ЯМР анализ этого соединения показьшает, что продукт представ«
5 0 5 О
5
0 5
.
ляет собой 5-оксиметил-2-метилтетра- зол.
2. Восстановление 5-карбэтОкси-2- метилтетразола. К раствору 5-карбэток- си-2-метилтетразола (0,139 rj 0,89 ммоль{ получен изомеризацией смеси сложных эфиров йодистым метилом) в сухом тетрагидрофуране (I мл) при 1О С прибавляют твердый литийбор- гидрид (0,019 Г), 0,87 ммоль). Смесь медленно нагревают до комнатной тем- пературы и перемешивают в течение 4 ч. Избыток боргидрида разрушают осторожным прибавлением 6н. НС1 при (рН 7). Растворитель выпаривают, остаток растворяют в дихлорметане (25 мл) и сушат над безводным сульфатом натрия. Выпариванием растворителя получают целевое соединение в виде прозрачного .маса: 0,092 г (91%) т.кип. 90-120 С/О,5 мм рт.ст., с разложением, ИК (пленка)дкс : 3350см (широкий пик, ОН), н-ЯМР: 4,4 (2Н, с, СН,-2), 4,93 (2Н, с, СН,-5).
C.5-Ацетилмеркаптометил-2-метил- тетразол.
К раствору 5- Оксиметил-2-метилтвт- разола ( г{ 11,7 ммоль) в сухом дихлорметане (25 мл) при О с прибавляют метансульфонштхлорид (1,47 г 12,9 ммоль), а затем тризтиламия (1,30 г, 12,9 ммоль) по каплям в течение 5 мин. Смесь перемешивают при в течение 1 ч, а затем обрабаты- : вают раствором тиоацетата калий (1,60 г, i4,b ммоль) в сухом N,N-димeтилфopм амиде {Ю мл). Результирующий гель перемешивают при О С в теч ение 3ч, Реакционную смесь разбавляют дихлорметаном (200 мл) ,про№1вают jpacTBOpoM соли (20 мл) и сушат над безводным сульфатом натрия. Выпариванием растворителя под вакуумом и хрОматографи- рованием результирующего масла на си- ликагеле (2Л5 см, элюирование дихлорметаном и смесью дихлорметан - ацетон 5%) получают целевое соединение в виде прозрачного масла: 1,31 г (65%)} ИК (пленка)- мах с : 1696 см ( тиоэфира), Н-ЯМР (CDCl,) : 2,43 (ЗН, с, SaC), 4,36 (ЗН, с, 2- - СН), 4,38 ппм (2Н, с, S-CH,}.
D.5-Меркаптометил-1,3-диметил-, тетразолийтрифторметаисульфат.
Раствор 5-ацетилмеркаптометил-2- метилтетразола (О.АОО г; 2,32 ммолъ)
в сухом дихлорметане (3 йл) обрабатывают метилтрифлатом (0,/6г, 4,64ммоль) и переметивают при 22°С в течение 16 ч. Выпариванием растворителя под - вакуумом получают красное масло. Эту соль растворяют в холодной воде (5 мл из которой удален кислород, и обрабатывают 4 М раствором гидроокиси натрия (о,8 мл1 3,2 ммоль). Смесь пере- 10 мешивают при О С в течение 40 мин, разбавляют водой (7 мл) и рН доводят до 7,8 насыщенным раствором КН,Р04 Прозрачный результирующий раствор хранят в атмосфере азота и сразу ис- (5 пользуют для следующей стадии.
Е. l R,5R,(l,3-димeтил-5- тeтpaзoлий)-мeтилтиoЗ-6-( 1-оксизтил)-. 7-оксо-1-азабицикло 3,2,ОГ|гепт-2-ен- 2-карбоксилат.20
Раствор енолфосфата (0,915 г; 1,S8 ммоль) в ТГФ (8 мл) охлаждают -i до О с и по каплям обрабатьшают раст-.
А. Получение енолфосфата II. В охлаждаемый льдом раствор соответствующего 3-кетопроизводного (3 г, 8,62 ммоль) в ацетонитриле (30 мл) прибавляют этилдиизопропипамин (9 ммоль, 1,04 экв., 1,57 мл) в течение «-2 мин и хлордифенилфосфат (9 ммоль, 1,04 зкв., 1,87 мл) в течение -2 мин. Реакционную смесь перемешивают 45 мин и ТХС-анализ показывает (этилацетат, силикагель) исчезновение кетона-1. Раствор разбавляют этилацетатом (60 мл), промьгоакт холодной водой (2X50 мл) и раствором соли, сушат над сульфатом натрия, концентрируют (температура бани ниже.
вором 5-меркаптометил-1,3-диметиптет- 25 20 С) и получают пенистое соединение.
разолийфторметансульфоната 2,32 ммоль) . в течение 20 мин, рН реакционной смеси равен 6,5 в ходе прибавления. Еще через 20 мин рН раствора доводят до 7,0 насыщенным раствором бикарбоната натрия. Смесь переносят в сосуд для .гидрирования, разбавляют ТГФ (10 мл), длэтиловым эфиром (20 мл) и льдом (20 г). Карбапеном гидрируют над 10%-ным палладием на активированном угле при 3,15 атм и .небольшом повышении температуры до в течение 90 М1Ш. Катализатор отфильтровывают, |проводят промывку холодной водой (5 мл) и диэтиловым эфиром (20 мл). Водную фазу п зомывают эфиром (20 мл) и вьщерживают под вакуумом 20 мин для удаления следов органического растворителя. Хроматографией на Рге РАК 500-C/I8 с использованием воды |в качестве эяюента получают .целевое соединение в виде белого порошка (по- ;сле лиофилизации); 0,0266 г (49%), (с-1,04, вода); УФ (вода, 294 нм (7500); ИК(КВг)
30
35
40
45
макс
И --«-1з
рН 7,4) . ...,-,„ (C 0 i-лактама),1600 см 0 (широкий пик, со карбоксила та); Н-ЯМР ()J : 1,24 ($Н, д, ,4 Гц, СН,СНОН), 3,0-3,3 (2Н, м, Н-4), 3,42 (JH, двойной дуплет, 1«5,8, ,9, Н6), 4-4,2 (2Н, м, Н5 и СН,СНОН), 55 4,34 и 4,57 (, 2$, СН,-1 и 3 Тетразола), 4,49 и 4,51 (2Н, 2с, CHjS). Продукт имеет время полужизкоторое используют непосредственно,
B.Получение Н-метилЛиридин-2-Ш1- метантиола. Через охлажденный льдом раствор тиоацетата соответствующего тиола (3,31 rt 10 ммоль) в воде пропускают азот в течение 5 мин и по каплям прибавляют (примерно в течение 5 мин) холодный раствор гидроокиси натрия (1,75 экв., 17,5 ммоль, 0,7 г) в воде(8 мл). Смесь желтеет. Через
75 мин в атмосфере азота рН доводят до 7,4 яасыщенным водным раствором KHjPO. Реакционную смесь разбавляют водой (15 мл). Этот водйый раствор тиола 4 (50 мл, 0,2 ммоль/мл) используют как таковой.
C.Присоединение. В охлаждаемый льдом раствор соединения II (сырой, получен в А, 8,62 ммоль) в ТГФ (50мл) по каплям прибавляют водный раствор тиола, полученный в В (5 мл раствора каждые 5 мин). При проведении реакции рН реакционной смеси поддерживают около 6,5-7,5 (предпочтительно около 7) прибавлением охлажценного 2н. раствора гидроокиси натрия. После окончания реакции проводят ТСХ;
а - силикагель, этилацетат; b - обращенная фаза Analtech RPSF ацетонит- рйл - буфер с рН 7 (4:6).
В конце используют 1,15 экв. тиола (5 мл раствора). Реакция завершается через 1 ч при н смесь исни 10,5 Ч-(с ГО М н (Зх сфатном буфере с рН 7,4).
Пример. Получение 3-(Н-ме- тилпиридин-2-ил-метантио )-6oi- 1-(Н)- оксиэтш1 -7-оксо-1-азабицикло 3, 2,0 гепт-2-ен-2-карбскс ;лата. способом в одной колбе.
А. Получение енолфосфата II. В охлаждаемый льдом раствор соответствующего 3-кетопроизводного (3 г, 8,62 ммоль) в ацетонитриле (30 мл) прибавляют этилдиизопропипамин (9 ммоль, 1,04 экв., 1,57 мл) в течение «-2 мин и хлордифенилфосфат (9 ммоль, 1,04 зкв., 1,87 мл) в течение -2 мин. Реакционную смесь перемешивают 45 мин и ТХС-анализ показывает (этилацетат, силикагель) исчезновение кетона-1. Раствор разбавляют этилацетатом (60 мл), промьгоакт холодной водой (2X50 мл) и раствором соли, сушат над сульфатом натрия, концентрируют (температура бани ниже
которое используют непосредственно,
B.Получение Н-метилЛиридин-2-Ш1- метантиола. Через охлажденный льдом раствор тиоацетата соответствующего тиола (3,31 rt 10 ммоль) в воде пропускают азот в течение 5 мин и по каплям прибавляют (примерно в течение 5 мин) холодный раствор гидроокиси натрия (1,75 экв., 17,5 ммоль, 0,7 г) в воде(8 мл). Смесь желтеет. Через
75 мин в атмосфере азота рН доводят до 7,4 яасыщенным водным раствором KHjPO. Реакционную смесь разбавляют водой (15 мл). Этот водйый раствор тиола 4 (50 мл, 0,2 ммоль/мл) используют как таковой.
C.Присоединение. В охлаждаемый льдом раствор соединения II (сырой, получен в А, 8,62 ммоль) в ТГФ (50мл). по каплям прибавляют водный раствор тиола, полученный в В (5 мл раствора каждые 5 мин). При проведении реакции рН реакционной смеси поддерживают около 6,5-7,5 (предпочтительно около 7) прибавлением охлажценного 2н. раствора гидроокиси натрия. После окончания реакции проводят ТСХ;
а - силикагель, этилацетат; b - обращенная фаза Analtech RPSF ацетонит- рйл - буфер с рН 7 (4:6).
В конце используют 1,15 экв. тиола (5 мл раствора). Реакция завершается через 1 ч при н смесь используют саму по сеОе для падрирова- ния после того, как рН доводят до 7,
D, Гидрирование. Реакционную смес содержащую целевой продукт I, запти- щенный по карбоксильной группе пара- нитробензолом (получено в с) переносят в колбу Парра с ТГФ (10 мл), фосфатным буфером (рН 7,0, 1 М) (10 мл) эфиром (75 мл) и Pd-C ЮХ-ным (5 г), гидрируют при 3,15, атм и 3-10 С в течение 2 ч. Затем катализатор отфиль- тровьтают, промьшают водой (З.10 мл) и рН доводят до 6,2, осторожно прибавляя холодный 2н. раствор NaOH. Прибавляют эфир, водную фазу отделяют и снова промьшают эфиром.Водную фазу очищают от органического растворителя под вакуумом и затем очищают на колонке Bondapak С-18 (Юб г, А,5x13 см) с помощью холодной дистиллированной воды. Светло-желтые фрак- .ции, содержащие продукт (контроль с помощью УФ и тех), лиофилизуют 1,46 г (50%, выход по бициклкческому кето- ну-6) в виде желтого порошка.
П р и м е р 10. Получение 5R,6S- -3- fC(1 3-диметилпиридиний-4-ил)ме- (R)-оксиэтилЗ -7-оксо- I-азабиТдикло 3,2,о1гепт-2-ен-2-карб- оксилата (10 А) и 4R,5R,6sl-3-(1,3- димeт шпиpидиний-4-ил)мeтил -тиoj-6- tI-(R)-oкcoзтилJ-4-мeтил-7-oкco- I-аза бицикло- 3,2,о1-гепт-2-ен-2-карбокси- лата (10 в)-.
А. Получение 4-оксиметил-З-метил- пиридина. Процесс проводят по схеме получения оксиметилпиридинов по Bocke Iheide. С этой целью раствор свеже- перегнанного 3,4-лутидина (4б,0 0,43 моль) в 120 мл ледяной уксусной кислоты охлаждают до и добавляют к нему по каплям 64 мл Зр%-ной . Полученный раствор нагревают в течение 3 ч при 75 с (температура масляной бани), после чего добавляют к нему еще 20 мл 30%-ной и продолжают нагрев в течение 18 ч. Затем к реакционной смеси снова добавляют 20 мл 30%-ной HjO и выдерживают ее при в течение еще 3 ч. После этого раствор упаривают в вакууме, создаваемом водоструйным насосом, до объема около 100 мл, добавляют к нему 50 мл Н, О и смесь упаривают до
половины объема, после чего охлажда-
ют ее до , подщелачивают с помощью охлажденного 40%-ного водного .раствора NaOH до рН 10 и проводят
o е . 5
«
5
0
экстракцию (5 раз) .l. Экстракт высушивают () и концентрируют с помощью вращаюп1егося испарителя, получая в результате раствор желтого цвета. При добавлении к этому раствору гексана выпадает твердый осадок, который отфильтровывают высушивают в вакууме. В результате получают 48,0 г (выход 83%) 3,4-лутвдин-К- оксида в виде беловатого твердого ведества.
Полученный Nr-оксид порциями добавляют к 60 МП уксусного ангидрида и образующийся в результате оранжевый раствор нагревают на водяной бане в течение 1 ч при . Избыток уксусного ангидрида отгоняют при пониженном давлении и собирают образующийся продукт (39,0 г), кипящий при 90- 120°С fO,l торр). После хроматогра- фирования этого маслянистого продукта на сИликагеле с использованием в качестве элюента смеси этилацетата и петролейного эфира в соотношении 2:3 получают чистый 4-ацетоксиметил-З-ме- тилпиридин (19,0 г; выход 30%) в виде маслянистой жидкости, ИК: 1745 см ,
Полученньй ацетат растворяют затем в 100 мл 10%-ного водного раствора НС1 и кипятят раствор в течение 1 ч с обратньм холодильником. Полученный в результате раствор охлаждают до О С, подщелачивают твердым н трижды подвергают экстракции CHjClj порциями по 100 мл. Органическую фазу промывают рассолом, высушивают () и упаривают, получая в результате 11,0 г беловатого твердого продукта с т.пл. 70-72°С. Этот твердый продукт растворяют с холодным Эфиром, получая 9,5 г (выход 67%) чистого 4-оксиметил-З- метилпиридина в виде твердого белого материала с т.пл. 77-80°С. - Н-ЯКР (CDCl) : 8,27, 7,41 (ABq, Гц, 2Н), 8,18 (с, 1Н), 5,63 (Вг, с, -ОН), 4,67 (с, СН-г), 2,20 (с, СН, ИК (ну- йол): 3160 .
В. Получение 4-(ацетилтиометил)- 3-метилпиридина. К охлаждаемому льдом, механически перекеигяваемому раств ору (17,04 г; 0,065 моль) трифенилфосфина в 250 мл сухого ТГФ добавляют по каплям 12,8 мл (0,065 моль) диизопропил- аэодикарбокснлата и образующуюся суспензию перемешивают при 0°С в течение 1 ч, К этой смеси добавляют по каплям раствор 4,0 г (0,0325 моль) 4-оксиме-тил-З-метилпиридима н 100 мл
сухого ТГФ, а затем 4,6Амл (0,065 моль} .свежеперегнанной тиоуксусной кислоты и перемешивают смесь в течение 1ч при , а затем в течение I ч при , комнатной температуре, получая в результате раствор оранжевого цвета. Этот раствор концентрируют во вращающемся испарителе, а затем разбавляют петролейным эфиром. Образующуюся Ю смесь фильтруют и фильтрат упаривают, получая в результате маслянистую жидкость. После хроматографии этой маслянистой жидкости на силикагеле, используя в качестве элюента сначала и гексан, а затем смесь 10%-ного этил- ацетата и 50%-ного гексана, получают 7,0.г маслянистой жидкости, которую перегоняют в трубке с шаровым расширением. В. результате получают 6,0 г 20 (выход 100%) чистого, я1Родукта в виде желтой маслянистой жидкости с т.кип. (температура воздушной бани) 95-100 G (при 0,1 торр). Н-ЯМР (CDCl,) : 8,40, 7,20 (ABg, Гц, 2Н), 8,37 25 (с, 1Н), 4,08 (с, CH-t), 2,35 (с, СН,), 2,32 (с., CHj), ИК (чистый); 1695 см . С. получение трифторсульфоната 4- (ацетйЛтиометил)-,3-диметилпириди- нина. К охлаждаемому льдом раствору jO 2,95 г (0,016 моль) тиоадетата в 10 мл хлористого метилена добавляют по каплям 4,60 мл (0,04 моль) метил- трифторметансульфоната и смесь пере- мегаивают при ОС в атмосфере азота в течение I ч. После этого реакционную 35 смесь упаривают досуха и остаток растирают с эфиром. Образующийся твердый . продукт отфильтровывают и высушивают в вакууме, получая в результате 4,0 г
льдом раствору 1,45 г (0,0025 коль) енолфосфата в 20 мл ТГФ. Смесь перемешивают в течение 1 ч при О С и переносят в автоклав, добавляют к ней 20 мл эфира, 25 мл 0,1 М фосфатного буфера (рН 7,4) и I,4 г 10%-ного пая- ладия на древесном угле, после чего подвергают гидрированию в течение 1 ч при-давлении 45 фунтов на кв. дюйм. Затем реакционную смесь фильтруют через слой целлита, который дополнительно промывают эфиром и фосфатным буфером с рН 7,4. Водную фазу отделяют и отгоняют растворитель в вакууме. Водимый раствор подвергают об- ратнофазной хроматографии (С-18 Bon- da Pat). Элюированиё осуществляют сначала водой, а затем 10%-ным водным раствором ацетонитрила. После лиофилизации соответствующи: фракций получают 0,9 г твердого продуксз ораи- жевого цвета. Этот продукт подвергают повторной хроматографии, используя в качестве элюента воду, а затем 2%-ный водный раствор ацетонитрила. После лиофилизации получают 0,25 г (выход 57%) чистого конечного продукта ( 10 А) в виде твердого вещества желтого цвета. H-HMP ()5 : 8,55 (с. 1Н), 8,53, 7,96 (ABq, ,8 Гц, 2Н), 4,30-3,99 (м, 2Н), 4,27 (с, 5Н), 3,35 (дд, 1,2,8 Гц, 12-6,0 Гц,, 1НК 3,05 (д, ,8 Гц, 2Н), 2,50 (с, ЗН), 1,23 (д, ,3 Гц, ЗН){ ИК (КВг): 1755, 1590 , УФ (фосфатный буфер, рН 7) 295 им (7180).
Е. Получение 4R,5R,(1,3- димeтилпиpидиний-4-ил)-мeтилlг-тиoi-6-- - - - . .f. |Г1-(к)-оксиэтил -4-метил-7-оксо-1- (выход 72%) белого твердого вещества. азабицикло 3,2,0 гепт-2-ен-2-карбокси- Н-ЯМР (CDCl)S : 8,72 (с, Ш) 8,58, 7,87 (ABq, Гц, 2Н), 4,39 (с, .N-СНз), 4,17 (с, CHi), 2,53 (i, СН,), 2,36 (с, аЦ),ИК(чистый): 1700 .
D. Получение 5R,6s7-3-{(1,3-ди- 45 метилпиридиний-4-ил)-метилЗ -тио 6e(l- (н)-оксоэтилЗ-7-оксо-1-азабицикло 3,2,о гепт-2-ен-2-карбоксилата.
К охлажденному льдом и продутому азотом раствору 0,324 г (0,008 моль) NaOH в 10 мл воды добавляют 1,40 г (0,004 моль} тиоацетата и перемешивают смесь при в атмосфере азота в течение 1 ч. После этого рН раство- ра устанавливают равным 7,2-7,3 с помощью 10%-ного водного раствора од- нозамещенного фосфата калия и по капп. лям добавляют его к охлаждаемому
50
лата.
Охлаждаемый льдом раствор 0,906 г (о,0025 моль) в 10 мл ацетонитрила последовательно обрабатьгоают дифенил- хлорфосфатом (0,544 мл, 0,00263 моль), 0,457 мл (0,00263 моль) диизопропил- этиламина и 0,3 мг 4-диметиламинопи- ридина. Через 50 мин реакционную смесь разбавляют холодным этилацетатом, а затем промывают холодной водой и рассолом. Органическую фазу высушивают () я упаривают при комнатной температуре, получая ц результате енолфосфат в виде грязно-белой пеиы. Эту пену растворяют в 20 мл ТГФ, охлаждают до в атмосфере азота и обрабатывают водным раствором тиолальдом раствору 1,45 г (0,0025 коль) енолфосфата в 20 мл ТГФ. Смесь перемешивают в течение 1 ч при О С и переносят в автоклав, добавляют к ней 20 мл эфира, 25 мл 0,1 М фосфатного буфера (рН 7,4) и I,4 г 10%-ного пая- ладия на древесном угле, после чего подвергают гидрированию в течение 1 ч при-давлении 45 фунтов на кв. дюйм. Затем реакционную смесь фильтруют через слой целлита, который дополнительно промывают эфиром и фосфатным буфером с рН 7,4. Водную фазу отделяют и отгоняют растворитель в вакууме. Водимый раствор подвергают об- ратнофазной хроматографии (С-18 Bon- da Pat). Элюированиё осуществляют сначала водой, а затем 10%-ным водным раствором ацетонитрила. После лиофилизации соответствующи: фракций получают 0,9 г твердого продуксз ораи- жевого цвета. Этот продукт подвергают повторной хроматографии, используя в качестве элюента воду, а затем 2%-ный водный раствор ацетонитрила. После лиофилизации получают 0,25 г (выход 57%) чистого конечного продукта ( 10 А) в виде твердого вещества желтого цвета. H-HMP ()5 : 8,55 (с. 1Н), 8,53, 7,96 (ABq, ,8 Гц, 2Н), 4,30-3,99 (м, 2Н), 4,27 (с, 5Н), 3,35 (дд, 1,2,8 Гц, 12-6,0 Гц,, 1НК 3,05 (д, ,8 Гц, 2Н), 2,50 (с, ЗН), 1,23 (д, ,3 Гц, ЗН){ ИК (КВг): 1755, 1590 , УФ (фосфатный буфер, рН 7) 295 им (7180).
Е. Получение 4R,5R,(1,3- димeтилпиpидиний-4-ил)-мeтилlг-тиoi-6|Г1-(к)-оксиэтил -4-метил-7-оксо-1- азабицикло 3,2,0 гепт-2-ен-2-карбокси-
|Г1-(к)-оксиэтил -4-метил-7-оксо-1- азабицикло 3,2,0 гепт-2-ен-2-карбокси-
лата.
Охлаждаемый льдом раствор 0,906 г (о,0025 моль) в 10 мл ацетонитрила последовательно обрабатьгоают дифенил- хлорфосфатом (0,544 мл, 0,00263 моль), 0,457 мл (0,00263 моль) диизопропил- этиламина и 0,3 мг 4-диметиламинопи- ридина. Через 50 мин реакционную смесь разбавляют холодным этилацетатом, а затем промывают холодной водой и рассолом. Органическую фазу высушивают () я упаривают при комнатной температуре, получая ц результате енолфосфат в виде грязно-белой пеиы. Эту пену растворяют в 20 мл ТГФ, охлаждают до в атмосфере азота и обрабатывают водным раствором тиолата, полученного описанным способом из 0,3 г NaOH (7,5 моль) и 1,3 г (3,76 fмoль) тиоацетата в 10 мл воды. Реакционную смесь перемешивают при в течение 30 мин, а затем при О С в течение 75 мин, после чего переносят в автоклав, содержащий 20 мл,, эфира, 30 мл 0,1 М фосфатного буфера (рН 7,Д) и1,5 г 10%-ного палладия fo на древесном угле. После гидрирования при давлении водорода 45 фунтов на кв. дюйм в течение I ч смесь фильтруют через целлит, водную фазу от- деляюТгИ упаривают в вакууме. Получен-} ный раствор подвергают обратнофазной хроматографии (с-18 Bonda Pak), используя в качестве элтаента воду. После лисфилизации соответствующих фракций получают 1,2 г твердого вещества 20 желтого цвета. Это вещество подверга- ют повторной хроматографии, используя в качестве алюента сначала воду, а затем 4%-ный водный раствор ацётонит- рила. После лиофилизации получают 25 чистый целевой продукт (Ю в) (0,250г, выход 28%) в виде твердого вещества желтого цвета. н-ЯМР () : 8,53 (с, 1Н), 8,49, 7,81 (ABq, ,2 Гц, 2Н), 4,38-3,98 (м, АН), 4,27 (с, ЗН),зо 3,49-3,18 (м, 2Н), 2,51 (с, ЗН), 1,25 .(д, 1-6,7 Гц, ЗН), 1,16 (д, ,6 Гц, ЗН); ИК (КВг): 1750, 1595 УФ (фосфатный буфер, рН 7) 292 нм (67930).
Пример П. Получение 5R,6s3- 3- (1,6- диметилпиридиний-2-ил)-ме- тилтио -6т-( 1Н-оксиэтил)-4Р-метил-7-ок- со- 1-азабицикло 3,2, оЗгепт-2-ен-кар- боксилата.
А, Трифторметансульфонат (1,6-ди- Q метилпиридиний-2-ил)-метилтиола.
Продутьй азотом раствор 1,0 г (5,52 ммоль) (б-метилпиридин-2-ил)- метилтиоацетата в сухом эфире (5 мл) обрабатывают 0,74 мл (6,5 ммоль) ме- , типтрифпата и перемешивают смесь при в течение 4 ч. Эфир отделяют путем декантации, а белый осадок дважды промывают 2 мл эфира и растворяют в 15 кл 6н. раствора (90,0 ммоль) соляной кислоты. Полученный раствор нагре-50 вают в течение 4 ч при 70 С в atмocфe- ре азота и затем упаривают при пониженном давлении до сиропообразной консистенции. Полученный сироп имеет желтый цвет. Остатки соляной кислоты 55 удаляют путем совместной перегонки продукта с водой (2х-10 мл). Очистку сырого продукта осуществляют путем
обрат1(офаз1{ой хроматографии на колонке размерами 2,2X13,0 см (Ргер Pak С-18), используя в качестве элюента воду. Соответствующие фазы собирают и лиофилизуют. В результате получают 1, 3 г белого порошкообразного продута. Выход 85,4%. ИК (КВг)0„а,: 2565 (SH), 1626 (пиридиний), 1585 (пириди ний) см- , УФ () 278 (7355)
Вычислено, %: С 35,64; Н 3,99; N.4,62; S 21 ,14 C,H,,NO,S,F,
Найдено, %: С 35,49; Н 4,05; N 4,56; S 20,99.
В. 5К, (1, 6-диметш1пириди- ний-2-ил)-метилтио -6-(lR-оксаэтил )- 4R-мeтил-7-oкco-l-aзaбициклo З, 2,03 . гепт-2-ен-2-карбоксилат.
- К продутому азотом и охлажденному до 5 С раствору |,11 г (3,06 ммоль, R/S:86/14) 5R,6sJ паранитробензил 3,7-дирксо-б- (1R- оксиэтил) -4RS-MeTl«T- . 1 -азабицикло 3,2, renTaH-2-R-Kap6oK- силата и 90 мл сухого ацетонитрила добавляют одновременно 0,68 мл (3,3 ммоль) дифенилхлорфосфата и 0,57 мл (3,3 имоль) диизопропилэтил- амина в течение 10 мин. Холодную смесь () перемешивают в течение -I ч, охлаждают до и одновре- менно добавляют к ней в.течение 15 мин раствор 1,03 г (3,4 ммоль) трифторметансульфоната (1,6-диметил- пиридиний-2-ил)-метилтиола в 2 мл сухого ацетонитрила и 0,59 мл (3,4 ммоль) диизопропилэтиламина. Образующуюся смесь перемешивают в течение 30 мин при , подогревают до , перемешивают при зтой температуре в течение I ч и разбавляют 35 мл холодной воды. Образующуюся в результате эмульсию выливают в верх нюю часть колонки для обратнофазной хроматографии (Ргер Pak С-18, 2,5 18 см) и осуществляют злюирование смесью 25-50%-ного ацетонитрила в воде. В результате лиофилизации соответствующих фракций получают клейкий твердый материал желтого Ц1вета (1,69 г), который растворяют в 40 мл содержащего воду тетрагидрофурана. К полученному раствору добавляют 70 мл эфира, 50 мл 0,2 М буферного раствора однозамещенного фосфата калия и гидроокиси натрия с рН 7,0 и 10%-ный палладий на древесном угле (1,69 г) и подвергают смесь гидрированию в течение 2 ч при 23 С и дайлеНИИ 42 фунтов на кв. дюйм, после чего фильтруют через слой целлита. Фазы разделяют. Водную фазу дважды промывают эфиром, порциями по 20 мл, и с упаривают в высоком вакууме при температуре ниже 23°С до объема 15 мл. Остаток подвергают обратнофазной хроматографии на колонке (Ргер Pak С-18). Элюирование осуществляют 10 А%-ным водным раствором ацетонитри- ла. После лиофилизации cooTBeTCTpjno- nijix фракций получают 0,23 г целевого соединения в смеси с К, Иа дйфенилфос- фатом (24 мол.%). После повторной |5 обратнофазной хроматографии на колонке (2, см, Ргер Pak С-18) с использопаиием в качестве элюента 400 мл воды и 200 мл 10%-ного водковую (в дальнейшем эта смесь - рас вор А). Раствор 1,23 г (6,8 ммоль) (6-метил-пиридин-2-ил)-метилтиоацет та в 10 мл сухого эфира обрабатьшаю 0,85 мл (7,5 ммоль) метилтрифлата и перемешивают смесь в течение 1,5 ч при 23 С. Эфир отделяют путем декан тации, а белый порошок дважды промы вают эфиром, порциями по 10 мл, и растворяют в воде (20 мл). Полученн водный раствор охлаждают в воде (20 м Полученный раствор охлаждают до CfC в атмосфере, не содержащей кислорода, и обрабатывают 3,4 мл 4н. раство за (3,6 ммоль) гидроокиси на рия. Смесь перемешивают в течение 1 ч при , после чего рН ее довод до 7,6, добавляя к ней одноэамещенного раствора ацетонитрила и последу- 20 ный фосфат калия (в дальнейшем эта
смесь - раствор В). Холодный (5 С) раствор А обрабатывают в течение 30 мин раствором В, поддерживая рН реакционной смеси в пределах 7,25- 7,35 за счет добавления к ней по каплям 4н. раствора гидроокиси натр Смесь перемешивают в течение 30 мин и выливают в колонку обратнофазной хроматогра11)ии размерами 4,018 см (Ргер Pak С-18). ЭАюирование осуществляют 25-50%-ным водным раствором ацетонитрила. После лиофилизации со ответствующих фракций получают цела вое соединение в виде желтого порош ка (2,82 г, 51% (PhO)PO, 49% CF,SO;). Выход 80%. ИК(КВг) : 3700-3000 (он), 1772 ( fb-лактама 1700 ( эфира), 1625 (пиридинии), 1590 (пиридинии) см ; Н-ЯМР (DMCO,
ющей лиофилизации соответртвуютшх фракций получают 0,17 г (выход 15,3%) порошкообразного продукта желтого цвета. ИК (КВг)/лакс: 1750 ( лактама), 1625 (пй{ идиний), 1600 25 ( карбоксилата) см , Н-ЯМР (D ) S: 1,12 (д, ,2 Гц, СН наС-4), 1,24 (д, ,4 Гц, СН,СНОН), 2,80 (с, СН на С-6 пиридинин), 4,41 (центр АВ-квартета), CHj,S (7,5-о,4 Н ЗО на пиридинии); УФ (0,05 М буфер с рН . 7,0) А«„,, : 278 (Е 11504) ,4 (с, 0,2, );Г„1 20,8 (определяется при 37 С в буферном растворе с рН 7,4 при концентрации 10 М). ,,
П р и М ер 12, Получение 5R,6s 3- (1,6-диметш1пиридиний-2-ил)-мётил- тио -б- ( 1 R-рксиэтил )-7-оксо-1-азаби- цикло 3,2,6 гепт-2-ен-2-карбоксила4П
та.
2,84 (с, СН С-б пиридиния), 4,16 (с, СН, на N пиридиния), 4,79 (с, SCH;,),
А. ТрифторметансулЬфонат и дифенил фосфат 5В,.аранитробензил-3-,. .. . . (1,6-диметилпиридиний-2-ил)-метилтиоЗ-/6,6-7,5 (PhO)jPO,, 7,5-8,7 .(Н на пи-.
6-(1Е-оксиэтил)-7-оксо-1-азабициклоридинии и Н на РКБ-эфире).
45
3,2,о1гепт-2-ен-2-карбоксилата.
К продутому азотом холодному (5 С) pdCTBopy 2,14 г (6,14 ммоль) 5B,6S3 1аранитробензил-6-( 1В-оксиэтил)-3,7- диоксо-1-азабицикло 3,2,0 гептан-2Е- карбоксилата в 18 мл сухого ацетонитрила добавляют 1,37 мл (6,6 ммоль) дифенилхлорфосфата, 1,15 мл (6,6 ммоль) диизопропилэтиламина с такой скоростью, чтобы температура смеси была
50
В, 5R,6s -3-t(1,6-Димeтилпиpиди- ний-2-ил)мeтйлтиo |-6-( 1-оксиэтил)-7- оксо-1-азабицикло 3,2,03гепт-3-ен- 3-карбоксилат.
К раствору 0,87 г (1,27 ммоль) смеси трифторметансульфоната и дифе- нилфосфата 5НубВ -паранитробензил- 3-(1,б-диметилпиридиний- -ил)-метил- (1R-оксиэтил)-7-оксо-1-азабицикло 3,2,0 гепт-2-ен-2-карбоксиравна 5 С (.время добавления 7-10 мин) 5дата в соотношении 49:51 в 50 мл сои 6 мг (0,05 ммоль) 4-диметиламинопи-держащего небольшое количество воды
ридина. Смесь перемепшвакуг в течениететрагидрофурана добавляют 50 мл
1,5 ч при 5 С и используют ее как та-эфира, 40 мл 0,1 М буферного раствора
ковую (в дальнейшем эта смесь - раствор А). Раствор 1,23 г (6,8 ммоль) (6-метил-пиридин-2-ил)-метилтиоацета- та в 10 мл сухого эфира обрабатьшают 0,85 мл (7,5 ммоль) метилтрифлата и перемешивают смесь в течение 1,5 ч при 23 С. Эфир отделяют путем декантации, а белый порошок дважды промывают эфиром, порциями по 10 мл, и растворяют в воде (20 мл). Полученный, водный раствор охлаждают в воде (20 мл), Полученный раствор охлаждают до CfC в атмосфере, не содержащей кислорода, и обрабатывают 3,4 мл 4н. раство за (3,6 ммоль) гидроокиси натрия. Смесь перемешивают в течение 1 ч при , после чего рН ее доводят до 7,6, добавляя к ней одноэамещенный фосфат калия (в дальнейшем эта
смесь - раствор В). Холодный (5 С) раствор А обрабатывают в течение 30 мин раствором В, поддерживая рН реакционной смеси в пределах 7,25- 7,35 за счет добавления к ней по каплям 4н. раствора гидроокиси натрия. Смесь перемешивают в течение 30 мин и выливают в колонку обратнофазной хроматогра11)ии размерами 4,018 см (Ргер Pak С-18). ЭАюирование осуществляют 25-50%-ным водным раствором ацетонитрила. После лиофилизации соответствующих фракций получают цела- вое соединение в виде желтого порошка (2,82 г, 51% (PhO)PO, 49% CF,SO;). Выход 80%. ИК(КВг) : 3700-3000 (он), 1772 ( fb-лактама), 1700 ( эфира), 1625 (пиридинии), 1590 (пиридинии) см ; Н-ЯМР (DMCO,
): 1,15 (д, ,2 Гц, СН,СНОН),
2,84 (с, СН С-б пиридиния), 4,16 (с, СН, на N пиридиния), 4,79 (с, SCH;,),
,. .. . . 6,6-7,5 (PhO)jPO,, 7,5-8,7 .(Н на пи-.
ридинии и Н на РКБ-эфире).
5
0
В, 5R,6s -3-t(1,6-Димeтилпиpиди- ний-2-ил)мeтйлтиo |-6-( 1-оксиэтил)-7- оксо-1-азабицикло 3,2,03гепт-3-ен- 3-карбоксилат.
К раствору 0,87 г (1,27 ммоль) смеси трифторметансульфоната и дифе- нилфосфата 5НубВ -паранитробензил- 3-(1,б-диметилпиридиний- -ил)-метил- (1R-оксиэтил)-7-оксо-1-азабицикло 3,2,0 гепт-2-ен-2-карбокси5дата в соотношении 49:51 в 50 мл сооднозамещенного калия и гидроокиси натрия с рН 7,0 и 0,87 г 10%-ного палладия на древесном угле, подвергают смесь гидрированию в течение 2 ч при давлении водорода 36 фунтов на кв. дюйм и температуре , после чего фильтруют ее через слой целлита. Фазы разделяют, водную фазу двалзды промывают эфиром, порциями по 15 мп, упаривают в высоком вакууме до объема 30 МП и выливают в верхнюю часть колонки для обратнофазной хроматографии (Ргер Pak С-18, 2,2X13 см). Элю- ирование осуществляют воДой. Соответствующие фракции объединяют и лиофилизуют, получая в результате 0,179 г (выход 40%) желтого порошка. ИК(КВг).с 755 (С 0/3-лак- тама), 1628 (пиридиний), 1590 (С«0 карбоксилата) Н-ЯМР (D,0)J : 1,25 (д, ,4 Гц, СН -СНОН), 2,82 (с, ШЗ в С-6 пиридиния), 3,12 (дд, 1-9,2 Гц, 1-2,9 Гц, Н-4), 3,39 (дд, 1-6,0 Гц, 1-2,8 Гц, Н-6), 3,7-4,4 ( Н-5, CH,N пиридиния), 4,48 (с, ), 7,6-8,4 (Н пиридиния), УФ (Н40), Амакс 279 (г 9628) с плечом при 296Lei1 55° (с, 0,63, ), r,,5 ч (измерен при в бу35
40
ферном растворе с рН 7,4 для кон- центрации ).
П р и м е р I3. Получение 3-/2- (1,3-диметилимидазолий-метантио}- боб- (1-Сн)-оксиэтш1| -4 -метил-7-ок- со-1-азабицикло ,2,6 гепт-2-еН-2- карбоксилата.
3- f2-(1,3-Диметипимидазолий-ме- t TaHTHo)-6ei- (1-(В)-оксиэтилЗ} -4|5-ме- тил-7-оксо-1-азабицикло 3,2,0 гепг- 2-ен-2-кар боксилат получают в виде желтого порошка с выходом 32% из соединения II таким же способом, как это описано в примере 27. ИК(КВг) у: 3400J 1758 и 1600, см ; УФ;(%0))- гшакс- 294 им ( 7194); ЯМР (J)0}S ; 1,10 (ЗН, д, ,3 Гц, 1,25 (ЗН, д, 1-6,3 Гц), 3,30 (Ш. м), 3,42 (Н, 1-6,0 Гц и 2,2 Гц), 3,85 (6Н, с), 4,2-4,6 (4Н, м) и 7,40 (2Н, с).
П р и м е р 14. Получение 3- (2,4-диметил-1,2,4-триазолий)-мётан- (В)оксиэтил)-4 -метип-7- оксо-1-аз абицикло 3,2,0 гепт-2-е -2- карбоксилата.
з-|2.4-Диметил-,2,4-триазолийТ 5 метантео}-6е4- КЕ)-оксиэтил|-4 -ме- ТИЛ-7-ОКСО-1-азабицикло 3,2,0 гепт- 2-ен-2-карбоксилат получают в виде
20 . : 25 30
45
50
желтого порошка с пыходом 9% из сое- динеьшя II таким же способом, как это описано в примере 27. ИК(КВг)у : 3420, 1756 и 1605 см ; УФ (H,jO)n,: 291 нм (7850); ЯМР () : 1,15 (ЗН, д, 1-6,3 Гц), 1,22 (ЗН, д, I- ,3 Гц), 3,35 (IH, м); 3,48 (1Н, q, ,0 и 1,8 Гц), 3,90 (5Н, с), 4,05 (ЗН, с), 4,2-4,4:(4Н, м) и 8,80
(Ш, с).
Пример 15. Получение 3-f3- (1,2-диметилпиридтио)(В)окси- этил -метил- 7-оксо-1-аз абицикло З, 2,0 гепт-2-ена.
( 1 ,2-Диметилпиридиний)метан- тил -6«{г l-(R)-oкcиэтил -4 -мeтил-7- oкco-l-aзaбициклo 3,2,0 rгeпт-2-etf-2- кapбoкcилaт.
- Указанное соединение получают в ви- де желтого, порошка с выходом 14% из соединения II таким же способом, как описано в примере 27. ИК(ЙВг))Г г 3410, 1750 и 1600 УФ (Hj:0)( макс 296 нм (е 8500); ЯМР () : 1,25 (ЗН, д, ,5 Гц), 1,30 (ЗН, д, ,5 Гц), 2,95 (ЗН, с), 3,40 (1Н, м), 3,50 (IH, д, ,2 и 1,8 Гц), 4,2- 4,4 (4Н, м), 4,35 (ЗН, с), 7,82 (1Н, т, ,5 и 6,3 Гц), 8,40 (Ш, д, I- . -8,5 Гц), 8,72 (1Н, д, 1-6,3 Гц).
П р-и м е р 16, Получение 3-ГЗ-( 1- метилпиридиний)метантио -6Ы- 1-Гн)- оксиэтил -4б-метил-7-оксо-1-азабицйк- ло 3,2,0 -гепт-2-ен-карбоксилата.
. 3-f3-51-Метилпиридикий)метантио - 6et- 1-(Н)-оксиэтил -4Д-метил-7-оксо- 1-азабицикло 3,2,0 гепт-2-ен-2-кар- боксилат.
Указанное соединение получают в виде жёлтого порошка с выходом 27% из соединения 11 таким же способом, как это описано в примере 27.ИК(КВг) jf: 3420, 1750 и 1610 ctT i yit () Л-махе: 295 нм (68750); ЯМР ()5 : 1,10 (ЗН, д, ,9 Гц), 1,25 (ЗН, д, 1-6,9 Гц), 1,27 (1Н, м), 7,43 (IH, q, ,2 и 1,8 Гц), 4, Ь4,35 (4Н, м), 4,39 (ЗН, с), 8,0 (Ш, т, 1-8,5 и 6,2 Гц), 8,45 (1Н, д, ,5 Гц). 8,70 (1Н, д, ,2 Гц) и 8,82 (1Н, с). Вычислено, %: С 51,90; Н 6,36; Я 7,12
C:,7H2oNj04St2, Найдено, %: С 51,92; Н 5,71; И 6,88.
Пример 17. Получение ( -метилпиридинийметантио) -6ti-I-(R)оксиэтил-4р-метил-7-оксо-1 -азабицик-,2,0 гепт--2-ен-2-карбоксилата,
3- А-( 1-Метилпиридинийметантио)3-- 6-et- 1 (R) -оксиэтил -4р-метил- 7-ок- со- 1-азабицикло 3,2,0 гепт-2- ен-2- карбоксилатЦ14).
Указанное соединение получают в виде желтого порошка с выходом I5Z из соединения II так, как это описано в примере 27. ИК(КВг)У : 3410, 1750 и 1650 УФ (Н оНмакс - 293 нм (€7295); ЯМР () : 1,15 (ЗН, д, 1-6,5 Гц), 1,20 (ЗН, д,--Л10
добавляют к раствору 600 мг 10%-ного палладия на древесном угле и подвергают гидрированию при давлении 35 фунтов на кв. дюйм на встряхивающем аппарате в течение 45 мин. Смесь - фильтруют через слой ацетата и промывают катализатор водой (дважды, порциями по 10 мл). Объединенные фильтра и промывные воды подвергают экстракции эфиром (дважды, порциями по 100 мл). После лиофилизации получают желтый порошок, который подвергают очистке на колонке (С-18 Bond Pak)
6,5 Гц), 3,20 (1Н, м), 3,45 (1Н, q, 15 ДОЯобратнофазной хроматографии. ,0 и 2,0 Гц), 4,П (Ш, q, 1-8,0 и 2,0 Гц), 4,20 (Ш, м) и 4,35 (ЗН, с), 7,95 (2Н, д, ,2 Гц) и 8,72 (2Н, д, 1-5,2 Гц)..
П -р и м е р 18. Получение (1,4-диметилпиридиний)метантиоЗ-6о6- { |-(Н)-оксиэтил -4 метидг-7-оксо-1 - азабицикло з,2,о гепт-2-ен-2-карбок- силата.
( 1,4-Диметилпиридиний)метан- 25 И) целевого соединения в виде бледг- тио -6в6-.1-(Н)-оксиэтилП-4 -метил-7- но-желтого порошка. ИК(КВг)у : 3400, оксо-1-азабицикло 3,2,0 -гепт-2-ен-2- .-«« .r-i. „ /,. rt
карбоксилат}13).
Указанное соединение получают в виде желтого порошка с выходом I7% из соединения II таким же образом, как это описано в примере 27. ИКf : 3400, 1755 и 1600 , УФ (Н40)ма«Г
ирование осуществляют 5Х-ным водным раствором CH|CN при давлении 8 фунтов на кв. jcuoAM, 15 мл из каждой фракция подвергают высокопроизводительной 20 жидкостной хроматографии. Отбирают фракции с максимумом поглощения в ультрафиолетовой области при 300 нм и подвергают их лиофилизации, в результате чего получают 58 мг (выход
30
300 нм (е 7600); ЯМР (DjiO) : 1,20 (ЗН, д, 1-6,7 Гц), 1,28 (ЗН, д, I1750 и 1590 УФ (Н.40)мам : 292 им (е7081); ЯМР (): 1,13 (ЗН, д, 1-6,5 Гц), ,23 (.1Н, д, I- -6,5 Гц), 3,18 (1Н, м), 3,45 (1Н, q, ,0 и 2,1 Гц), 4,0-4,4 (4Н, м), 4,65 (ЗН, с), 7,79 (2Н, м), 8,30 (IH, м), 8,60 (1Н, м).
G. п-Нитробензил-3- пиридин-2-ил- метантисГ -6bL- l - (Н)-оксиэтил -4/3-ме6,7-Гц), 2,60 СЗН. с), 3,4-3,5 (2Н, тил-7-оксо-1-азабицикло 3,2,о Еепт-2м), 4,2-4,4 (4Н, м), 4,52 (ЗН, с), 7,82 (Ш, т, 1-6,5 и 4,2 Гц), 8,32 (Ш, д, ,5 Гц) и 8,60 (Ш, д, I- -4,2 Гц).
И. 3- 2- (N-Метилпиридиний) метан- THo -6ot- {1-(К)-оксиэтш{1-4р-метил-7- оксо-1-азабицикло 3,2,0 гепт-2-е -2- карбоксилат, защищенный по карбоксильной группе.
К раствору 1,0 г (2 ммоль) соединения I в 10 мл добавляют 450 мг (3,3 ммоль) метилтрифторметан- сульфонс«та и перемешивают смесь при 23°С в течение 90 мин. После отгонки
40
4S
50
ен-2-карбоксилат.
К охлажденному до раствору
(Г,2 г; 2 ммоль) фосфоната- 0 в 10 мл CHjGN добавляют в атмосфере азота 390 мг (3 ммоль) диизопропкпэтилами- на и затем 370 мг (3 ммоль) 2-мер- каптометилпиридина. Смесь перемешивают в течение 60 мин при 15 С и затем еще 60 мин при ( после чего разбавляют EtOAc, промывают ледяной водой, рассолом и высушивают (MgSO). После отгонки растворителя в вакууме получают желтую маслянистую жидкость, которую подвергают очистке с помощью хроматографии на колонке, заполнен
добавляют к раствору 600 мг 10%-ного палладия на древесном угле и подвергают гидрированию при давлении 35 фунтов на кв. дюйм на встряхивающем аппарате в течение 45 мин. Смесь - фильтруют через слой ацетата и промывают катализатор водой (дважды, порциями по 10 мл). Объединенные фильтра и промывные воды подвергают экстракции эфиром (дважды, порциями по 100 мл). После лиофилизации получают желтый порошок, который подвергают очистке на колонке (С-18 Bond Pak)
ДОЯобратнофазной хроматографии.
ирование осуществляют 5Х-ным водным раствором CH|CN при давлении 8 фунтов на кв. jcuoAM, 15 мл из каждой фракция подвергают высокопроизводительной жидкостной хроматографии. Отбирают фракции с максимумом поглощения в ультрафиолетовой области при 300 нм и подвергают их лиофилизации, в результате чего получают 58 мг (выход
И) целевого соединения в виде бледг- но-желтого порошка. ИК(КВг)у : 3400, .-«« .r-i. „ /,. rt
1750 и 1590 УФ (Н.40)мам : 292 им (е7081); ЯМР (): 1,13 (ЗН, д, 1-6,5 Гц), ,23 (.1Н, д, I- -6,5 Гц), 3,18 (1Н, м), 3,45 (1Н, q, ,0 и 2,1 Гц), 4,0-4,4 (4Н, м), 4,65 (ЗН, с), 7,79 (2Н, м), 8,30 (IH, м), 8,60 (1Н, м).
G. п-Нитробензил-3- пиридин-2-ил- метантисГ -6bL- l - (Н)-оксиэтил -4/3-метил-7-оксо-1-азабицикло 3,2,о Еепт-2тил-7-оксо-1-азабицикло 3,2,о Еепт-2
ен-2-карбоксилат.
К охлажденному до раствору
(Г,2 г; 2 ммоль) фосфоната- 0 в 10 мл CHjGN добавляют в атмосфере азота 390 мг (3 ммоль) диизопропкпэтилами- на и затем 370 мг (3 ммоль) 2-мер- каптометилпиридина. Смесь перемешивают в течение 60 мин при 15 С и затем еще 60 мин при ( после чего разбавляют EtOAc, промывают ледяной водой, рассолом и высушивают (MgSO). После отгонки растворителя в вакууме получают желтую маслянистую жидкость, которую подвергают очистке с помощью хроматографии на колонке, заполнен

















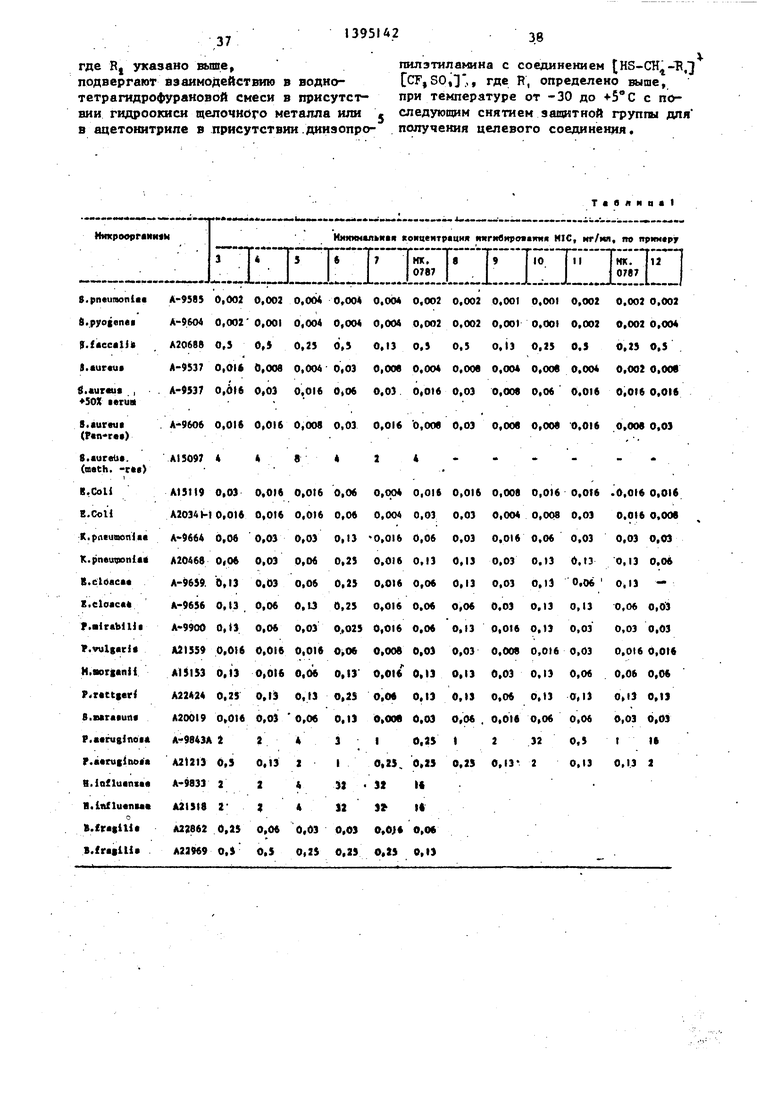


Изобретение относится к способу получения карбапенемов формулы ОН Щг + .СН. гУ S-CH2-RI , / где R - присоединенный через кольцевой атом углерода катион 2-(Н-метил- пиридиний), или 2-(I,6-диметилпириди- ний)« или 4-(1 3-диметилпиридиний), или Н,Н-диметил-1,2,3-триазолий, или 2-метил-1,2,3-тиодиазолий, или калиевая соль 4-Ш1и-5-(l-кapбoкcимeтйл3-мeтил-l,2,3-тpиaзoлия) или 5-(1,4- диметилтетразолия); 3-(I 3-диметил- пиридин-4-ил), 3-(1,6-диметилпири- дин-2-ил)-б е та-метил, 3-(1,6-диметял- пиридин-2-ил), (1 ,3-диметилимнг дазолкл )|, 3- З- (, 2-диметилпирндил) .(1-метиппиридил) , Нметил- пиридил) , 3- 2- (1,4-диметилпиридял J, 3- 2-(Н-метилпиридил) , 3,5-(1,4-ди- метил-1,2,4-триазолил), 3-(2,4-диме- тил-1,2,4-триазолил), 3-(2,4-диметил- 1,2,4-триазолил), 2-метил-1,2,3-тио- диазолил; 5-(,4-диметил-1,2,4-триазолил), 3-(Г;3-диметш1-5-тетразолил); Rj - Н или метильная группа, котр1 1е используются в медицине. Получение . целевых соединений ведут из соответствующего енолфосфата и соединения (HS-CHj.-R,)+(CF,SO,), где R указано вьше, в водно-тетрагидрофурановой смеси в присутствии гидроокиси щелочного металла или в ацетонитриле в присутствии диизопропилэтиламина. Процесс ведут при (-30)-(+5) с последующим снятием защитной группы для получения целевого соединения, 3 табл.
в вакууме получают четвертич- ной SiO. Элюирование осуществляют ное основание пиридина в виде пены, которое непосредственно, без дополнительной очистки, подвергают гидрированию. Сырую cojTfy пиридина растворяют в смеси ТГФ, эфира и буферного раствора с рН 7 в соотношении 1:1:1 (по 100 мл каждого из компонентов).
55
1
20%-ным раствором EtOAc. в CHjClj., В результате получают 375 мг (выход 4 соединения в виде белой аморфной ne ны. ИК(КВг)у : 3400, 1775 и 1710 см ЯМР: (IH, q, Т-6,2 и 2,0 Гц), 3,40 (Н, м), 4,0 (1Н, д, 1«7,7 nl), 4, (1Н, д, ,6 Гц), 4,18 (1Н, q, SiO. Элюирование осуществляют
5
1
20%-ным раствором EtOAc. в CHjClj., В результате получают 375 мг (выход 40Х) соединения в виде белой аморфной ne-j ны. ИК(КВг)у : 3400, 1775 и 1710 см ; ЯМР: (IH, q, Т-6,2 и 2,0 Гц), 3,40 (Н, м), 4,0 (1Н, д, 1«7,7 nl), 4,12 (1Н, д, ,6 Гц), 4,18 (1Н, q, I-6,7 и 2,0 Гц), 4,25 (1Н, м), 5,25 (1Н, д, ,3 Гц), 7,15-8,2 (4Н,м).
F. п-Нитробензил-3-дифеноксифосфи нил-бсС- |-(к)-оксиэтил -4/ -метил-7- оксо- 1-аэабицикло 3,2,.0 гепт-2-ен-2- карбоксилат.
К охлажденноьсу до раствору (20,0 г; 55,2 ммоль) 3-кетосоединени в 150 мл CHjCN добавляют 7,18 г (55 ммоль) диизопропилэтиламина, а затем 14,85 г (55 ммоль) дифенилхлор фосфоната в 20.мп в течение 5 мин. Полученный раствор перемешивают в течение 60 мин при , затем разбавляют 600 мл Etc Ас , промывают .охлажденным льдом 10%-ным раствором и рассолом. После высушивания (MgSO) и отгонки растворителя получают сырую маслянистую жидкость, ко- торую подвергают очистке на колонке до твердого продукта. Т.пл. 54-55 С, ИК(КВг): 2080 и 1695 .
Пример 19. Получение (N- метилпиридиний)метантио -6в&- 1- (н)-ок- сиэтил1-4Б-метил-7-окс i-азабицикло 3,2 ,.01 гепт- 2- ен-2-карбоксил ата,
Раствор 512 мг (12,8 ммоль) гидроокиси натрия в 5 мл воды добавляют к охлажденному льдом раствору 1,45 г (4,35 ммоль) соли триазолия в 5 мп воды. Через 45 мин его .--азбавляют водой до 25 мл и с помощью однозаме- щенного фосфата калия устанавливают его рН равным 7,6. Полученный таким образом раствор добавляют к охлажденному льдом, перемешиваемому раствору 2,00 г (3,45 ммоль) енолфосфата в 25 мл ТГФ. Через 30 мин реакционную смесь переносят в автоклав, содержа- щий 40 мл зфира и 2,0 г Г0%-ного-палладия на древесном угле, и подвергают смесь гидрированию в течение 1,25 ч при давлении 45 фунтов на кв. дюйм. После этого реакционную смесь разбавляют 25 мп эфира и фильтруют. Органическую фазу отделяют и двалады промывают водой порциями по 25 мл. Объединенные водные фазы трижды пропивают зфиром, порциями по 25 мл, и упаривают в вакууме. После колоночной обратнофазной хроматографии (колонка размерами 45x130 мм) с использо ванием воды в качестве элюента и последующей лиофилизации карбапенемсо- держащих фракций получают 650 мг сы- рого продукта, который подвергают по вторной хроматографии. В результате получают 450 мг (выход 39%) чистого
целевого продукта. Н-ЯМР () : 1,24 (ЗН, д, ,4 r:i), 3,19 (2Н, q, ,6 и 9,2 Гц), 3,45 (1Н, q, ,8 и 6,0 Пд), 3,91 (ЗН, с), 4,06 (ЗН, с), 4,08-4,36 (2Н, м), 4,54 (2Н, д, ,8 Гц), 8,71 (1Н, с), ИК (нуйол): 1755 , УФ (фосфатный буферный раствор с рН 7,4),: 294 им. (б 8,202); T,,l ч (фосфатный буферный раствор с рН 7,4, ,067, «37 С); 3,1 (2Н, д, Гц), 3,37 (1Н, q, ,1 Гц), 4,02 (7Н м), 5,18 (2Н, с), 8,53 (1Н, с), ИК (нуйол): 1750 , УФ (фосфатный буферный раствор с рН 7,4) Л д, : 205 им ().
Пример 20. (1,4-Дикетил 1,2,4-триазрлий)метантиол | 6et-l-(R)- оксизтил-7-оксо-1-азабицикло 3,2,0 гепт-2-ен-2-карбоксилат.
A.1-Метил-5-метантиолацетат-1,2, 4-триазол.
мл (6,0 ммоль) метансульфо- нилхлорида добавляют по каплям к охлаждаемому льдом, перемешиваемому раствору 565 мг (5,0 ммоль) f-метил- 5-оксиметил-1,2,4-триазопа и 0,91 мп (6,5 ммоль) триэтиламина в 5 мп хлористого метилена. Через 20 мин добавляют еще 1,05 мл (7,5 ммоль) тризтиламина и затем 0,53 мгг,-. (7,5 ммоль) уксусной кислоты и смесь перемешивают в течение 45 мин. После этого реакционную смесь разбавляют хлористым метиленом и промывают водой. Водную фазу подвергают экстракции хлористым метиленом (трижды, порциями по 5 мл). Объединенные органические вытяжки высушивают (MgSO) и отгоняют из них растворитель. После колоночной хроматографии на силнка- геле получают чистый 1-метил-5-мётан- тиолацетат-1,2,4-триазол (570 мп) в виде желтой маслянистой жидкости. Содержащую примеси фракцию (200 мг) подвергают повторной хроматографии (препаративная тонкослойная хроматография на силикагеле), в результате чего получают дополнительно 100 мг чистого целевого продукта. Общий выход 85%. Н-ЯМР (CDC1,)S : 2,38 (ЗН, с), 3,90 (ЗН, с), 4,25 (ЗН, с), 7,80 (1Н, с).
B.(I,4-Лиметил-I,2,4-трй- азолий) метантнп -6й;- 1-(к)-оксиэтил 7-оксо- 1-азабицикло f3,2,Ojгепт-2- ен- 2-карбоксилат.
1,20 мл (10,7 ммоль) трифторметансульфояата добавляют по каплям к охлажденному льдом раствору 730 мг (А,27 ммоль) 1-метил-5-метантиолаце- тат-1,2 2 триазола в 7 мя хлористого метилена. Реакционную скесь ме ленно (в течение 3 ч) нагревают до комнатной температурм. после чего концентрируют Остаток в виде масля- нистой жидкости растирают с эфиром, в результате чего получают 1,4б г сырого 1,4-димeтил-5-мeтaнтиoлau eтaт- l, 2, 4-триа9 олий-трифторметансульфо- ната, который используют без допол- нительной очисткн,
П.р и м е р 21, Получение (N- мвтилпиридиний)метантяоЗ-6в6- tl-(R)- оксиэтилТ -4(1 метш1-7-оксо-1 - аз абицик- по 5,2,0j| гепт 2-вяе-2-карбоксилата,
А. п-Нитробензил-З-дифеноксифос- финил-беС-р - (R)-оксиэтил - 4й-метил- 7-оксо- 1-азабицикло 3,2, oj гепт-2-ен- 2-карбоксилат (II).
К охпаященному () раствору 20,0 г-(55,2 ииоль) 3-кетопромежуточного соединения в 150 мл ацетонитри- ла добавляют 7,18 г (55 моль) ди - нзопропилэтиламина, затем 14,85 г (55 ммоль) диизопропилэтиламииа, затем 14,85 г (55 ммоль) дифенилхлор- фосфоната в 20 мл ацетоиитрила в течение 5 мин. Полученный раствор перемешивают 60 мин при , затем раз- бавляют 600 мл этилацбтата, промыва- ют ледяным 10%-ным раствором фосфорной кислоты и рассолом. Выпаривание высушенного (сульфат магния) растворителя дает сьфое масло, которое очи- щают на колонке с силикагелем} )в- ка из адсорбента в колонке lOZ-ным этидацбтатом в метилёнхлориде дает 3,7 г (lt,5Z) фосфоната-10 в форме белою вещества. ИК (СНС1,) Ir : 3400, 1790 и 1720 см- .
ЯМР ( дейтерированный хлороформ) (У: 1,20 (ЗН, дуплет, На), 1,3В (ЗН,- дуплет, ,3 Hz), 3,35 (1H, 1, ,7 ,0 Hz), 3,50 (IH, мульти-. плет), 4,2-4,25 (2Н, льтипле), 5,20 (IH, дуплет, J-10,5 Hz), 5,37 (IH, дугшёт, J-10,5 Hz), 7,1-7,4 . (lOH, 1мультиплеТ), 7,56 (IH, дуплет, J«9,0 Hz) и 8,10 (IH, дуплет, J 9,0 Hz).-
Ядерные эффекты Оверхаузера применяют для определения конфигурации 4метила у соединения 10. В случае облучения И, для 4-метила не наблюдают увеличения сигнала, что указывает на трансзависимость Н и 4-метила.
B.п-Нитробенэил-3 пиридин-2-ил- метантио -бо р-(н)-оксиэтил -4 ме- тил-7-оксо-1-азабицикло 3,2,0 гепт- 2-ен-2-карбоксилат. .
К охлажденному (-15 CJраствору 1,2 г (2 ммоль) фосфоната-10 в 10 мл ацетонитрила добавляют 390 мг (З ммоль} диизопропилэтиламина, зате 370 мг (3 ммолв) 2-меркаптометилпиридина в атмосфере азота. Реакционную смесь перемешивают 60 мин при -15 С затем дополнительно 60 мин при О С. Реакционную смесь разбавляют этилацетатом, промывают ледяной водой, рассолом и высушивают (сульфат магния). Выпаривание растворителя в вакуум дает желтое масло, которое очищают на колонке с силикагелем; отмывка из адсорбента в колонке 20%-ны зтилацетатом в метилёнхлориде дает 375 мг (40%-кый выход) соединения II в виде белой аморфной губчатой массы ИК(КВг)5 : 3400, 1775 и 1710 ЯМР (лейтерированный хлороформ)5 : 2,14 (ЗН, дуплет, ,7 Hz), 2,19 (ЗН, дуплет, ,7 Hz), 3,14 (IH, . квартет, J,2 и 2,0 Hz), 3,40 (IH, мультиплет), 4,0 (IH, дуплет, J« 7,6 Hz), 4,12 ClH, дуплет, ,6 Hz 4,18 (IH, квартет, ,7 и 2,0 Hz), 4,25 (IH, мультиплет), 5,25 (IH, дуплет, ,3 Hz), 5,40 (Ш, дуплет, J «n,3 Hz), 7,15-8,2 (4Н, мульти- , плет). .
C,(N-метилпиридиний)метан- тис -6ot- 1-(R)-оксиэтилТ-4 -мeтил-7- oкco-l-aзaбициклo 3,2,0 -2-ен-2-кар- боксилат.
К раствору 1,0 г (2 ммоль) соединения, полученного по В, в 10 мл метиленхлорида добавляют 450 мл (3,5 ммоль) метилтрифторметансуль- фоната и перемешивают при 23 с ЭОыяя Выпаривание метиленхлорида в вакууме дает очетверённый пиридин в виде губки, которую точас же гидрируют без предварительной очистки. Сырую соль пиридиния растворяют в смеси ТГФ - простой зфир - буферный раствор с рН 7 в соотнощении 1:1:1 (по 100 мл каждого), затем добавляют 600 мг 10%-ного палладия на активированном угле. Смесь гидрируют при 2,46 кг/см
(35 фунтов на кв. дюйм) в аппарате для перемешивания встряхиванием 45 мин. Смесь фильтруют через слой целлита и катализатор промывают во- дои (2x10). Объединенные фильтрат и промывные жидкости экстрагируют эфиром ( мл) и лиофилизируют, что дает желтый порошок, который очищают на колонке с обратимой фазой С-18 Bond Pak (10 г) и откывают из адсорбента 5%-ным ацетонитрилом в воде под давлением 0,56 кг/см (8 фунтов на кв. дюйм). Каждую фракцию (15 мп) оценивают.на чистоту методом жидкостной хроматографии, проводимой под высоким давлением, причем фракцию, имеющую ультрафиолетовую абсорбцию при
маис 300 нм, собирают и лиофилизи20
25
руют, что дает 58 мг (ПХ-ный выход) указанного в заголовке соединения в виде светло-желтого порошка. ИК(КВг) Г: ЗАОО, 1750 и 1590 см ,,(с (Н,0) 92 вм (с«7081); ЯМР (окись дейтерия) S: 1,13 (ЗН, дуплет, ,5 Hz),.1,23 (ЗН, дуплет, 1-6,5 Hz), 3,18 (Н, мультиплет), 3,45 (1Н, квартет ,0 и 2, Hz), 4,0-4,А (4Н, мультиплет), 4,65 (ЗН, синглет), 7,79 (2Н, мультиплет), 8,30 (1Н, мультиплет) и 8,60 3i, (IH, мультиплет).
Данные по биологической активности приведены в табл. 1-3..
В табл. I приведена активность in vitro, пробы карбапенемов (обозначенных номером соответствующего примера) после растворения в воде и разведения питательным бульоном (инкубацию осуществляли при 37 С в течение ночи при разведении в пробирке; для 0 сравнения использовали N-формамидоил- тиенамицин - МК.0787).
В табд. 2 представлены данные по терапевтической активности in vivo некоторых предлагаемых соединений д и R-формамидоил-тиенамицина (МК.0787), вводившихся внутримышечно мышам, искус- ственнс) зараженным различными микроорганизмами.
35
В табл. 2 для каждого соединения указана доза (PDjo, мкг/кг), обеспечивающая защиту 50% зараженных мышей.
Пример 5
Концентрация исследуемых соедине-м НИИ в крови мышей после внутримышеч ного введения.
В табл. 3 приведены концентрации некоторых предлагаемых соединений в крови мышей и период их пулараспада после внутримышечного введения в количестве 20 мг/кг (соединен{1я раство- .рялись в 0,1 М фосфатном буферном растворе с рН 7, причем каждое из соединений испытывалось на 4 мышах)..
Формула изобретения
Способ получения карбапен емов общей формулы
20
3i,
где н, - лрисоединенный через кольцевой атом углерода катион 2- (N-метилпирндиний), или IJ- (I,6-диметш1пиридиний),или 4-(I,3-диметилпиридинйй), или N,N-диметил-1,2,3-три- азолий, или 2-метил-1,2,3- тиодиаэолий, или калиевая соль 4- или 5-(1-карбоксиме- тил-З-метил- 1 ,2 ,3-триазолия) или 5-(I,4-диметш1тетразо ЛИЯ); 3-( 1,3- диметилпири- ДИН-4-ИЛ), 3-(I,6-диметилпи- ридин-2-ил)-бета-метил, 3 (I,6-диметилгшридин-2-ил), (1,3-диметилимидазолил Я; 3-Сз-( 1,2-диметилпиридил) , 3- ГЗ-( 1- нетилгиридил) , 3- С4-( 1-метилпиридил)} , (1,4-диметилпиридил) , 3-С2 (Н-метилпиридил) , 3,5-(1,4- диметил-1,2,4-триазолил}, 3-(2,4-диметил-1,2,4-трилзо- лил), 3-(2,4-диметил-1,2,4- триазолкп), 2-метил-1,2,3- тиодиазолил; 5--( 1,4-димeтил- l ,2,4-триазолил), 3-(1,3-ди- метил-5-тетразолил); R - водород или метильная группа;отличающийся
динение общей формулы 2
тем, что сое «--r-rV°- -i° .
где Rj указано вьше, пилэтиламина с соединением HS-CH -B,
подвергают взаимодействию в водно-CCF,SO,, где R, определено выше,
тетрагидрофурановой снеси в присутст-при температуре от -30 до 5 С с пегвин гидроокиси щелочного металла или jследующим снятием защитной групгы для
в ацетоннтриле в присутствии.диизопро-получения целевого соединения.
т о л и п I
19
Микрворпяиэм
Н«иннй.1М1«я концгнтрянйя ингиСиропякпя HIC, НГ/Н11, по примеру
и
114 Г мк. 1 If 17 ПК. Т 18 Т|9 Тго Тмк. .LJiiiLJ....i-...L...i.....
.S.pncuffloniat З.руоцсп S.r cc«ll S.iurtui
«.urcui )OZ crum
S.iureu (P n-r««)
S.aurcui . (etti. -rci)
E.Coii
e.CoK
K.pneuooni
K.poetinoni
B.clocca
B.etoacM
P.«lr«b lli
P.«ul|«r(«
Н.«юг(пН
P.r«tt|cri
S.B raiun«
T,««nitino «
P.««ru|ino(«
N
BtinfluttiM B.influ«ni«« S.fratUii B.«|iUi
A-958JO.OOOS0,000}0,O020,O020,016 O.OOII
A-96O d,OOOS0,000$0,0020,0020,016 0,001. 2
А206в8I.IJ0,130,25I4 0,25«3
A-95J70,00eО.ООЯ0,0020,0080,25 0,
A-953T0,0160,03. 0,0080,03I 0,
0,0020,060,001
0,020,13- 0,002
0,5160,25
0,00«0,50,002
0,008г0,004
A-9606 0,008 0,0160,008 0,016 0,5 0,002 I25 0,016 2J O .OO
A15097 At5U9
A2034I-I
A-9664
А2046в
A-9659
A-9656
A-9900
A2I559
AI3I53
A22424
A200t9
А-9в43А
A2I2I3
A2l5tB
А22в62
A2Z969
0,0040,00e
O,0080,000
0,030,03
0,00,03
0,0)6 0,016 0,03 0,06
0,03 , 0,030,06
0,0160,0160,016
0,0080,0160,016
0,0160,030,06
0,230,06OJ3
0,0160,0160,03
328I
r0,50,13
0,016 0,016 0,06 0,06 0,06 0,0« 0,06 0,03 0,13 0,25 0,06 0,25 0,13
0,60,008
0,60,008
0,130,03
0,50,06
20,06
20,06 0,130,016 0,130,008 0,50,06 .
30,06 0,130,
630,5
160,13
16
16
32
63
63
125
32
32
32
32 ;
32
63
63
O,OCM I
0,0082
0,034
. 0,064
0,03e
0,0316
0,034
0,0164
0,06,e
0,t38
0,034
0,53J
0,06Г6
0,01 0,016 0,OJ 0,06
0,06
0,06
0,016
0,016
0,0
0,13
0,03
0,J
0,13
Мафоорг«м м
.r:i
Л9606 0,07 0,1.
FDff , мсг/кг, для соедикежк по примеру
7 ТнК.0787Тб Т 9 J 12 Tit Т ,15А Т 15
8, «urcUtА9606 0,070,1.0,20,07
E.eoliAI5II9 I0,40,23.
К. potoMmiMА9664 З .3IЗ
Р, «{гвЫИ А9900. 242«49
Р, eerueinoeaА9в43А 0,50,20,20,5
Р. «eruelnoMA2408I 0,90,20,10,4
0,21 0,86 1,8
I;A
0,89 1,2 - 1,8 - Л 0,07 0,4 0,19 0,19 1,8 0,33 0,t9 -
-
0,45 0,39 J,89
I395U2
40
Продолжение табл.1
0,0020,060,001
0,020,13- 0,002
0,5160,25
0,00«0,50,002
0,008г0,004
,016 ,016 ,06 ,06 0,06 0,0« ,06 ,03 ,13 ,25 ,06 ,25 ,13
0,60,008
0,60,008
0,130,03
0,50,06
20,06
20,06 0,130,016 0,130,008 0,50,06 .
30,06 0,130,
630,5
160,13
16
16
32
63
63
125
32
32
32
32 ;
32
63
63
O,OCM I
0,0082
0,034
. 0,064
0,03e
0,0316
0,034
0,0164
0,06,e
0,t38
0,034
0,53J
0,06Г6
0,01 0,016 0,OJ 0,06
0,06
0,06
0,016
0,016
0,0
0,13
0,03
0,J
0,13
т б я ц « 2
МК.0787
0,21 0,86 1,8
I;A
0,89 1,2 - 1,8 - Л 0,07 ,4 0,19 0,19 1,8 0,33 0,t9 - 0,07 3
3
- t
0,45 0,39 J,89 I
0,4
4t
г
Таблица 3
iZ
| Патент ClOA if 4350631, кл | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
