натом серебра при кипячении, а деми- тилирование ведут с помощью А1С1s в нитробензоле. Полученное соединение обрабатывают этиленгликолем в присутствии каталитических количеств п-то- луолсульфокислоты с последующим бронированием бромом в присутствии 2,2 азо-бис(изобутиронитрила)гидролизом и обработкой кислотой (для удаления кетальной группы). Эти условия обеспечивают получение только 6-диокси- производных в противоположность в известном случае смеси изомеров 6-диок- си- и 11-диоксипроизводных.




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения антрациклиновых гликозидов | 1986 |
|
SU1553015A3 |
| Способ получения 6-деоксиантрациклингликозидов | 1984 |
|
SU1429935A3 |
| Способ получения рацемических аглюконов | 1983 |
|
SU1311616A3 |
| Способ получения сконденсированных производных пиразола или их фармацевтически приемлемых солей | 1989 |
|
SU1731059A3 |
| Способ получения производных 7-оксо-4-тиа-1-азабицикло(3,2,0) гептана и его варианты | 1979 |
|
SU942598A3 |
| Способ получения гликозида | 1987 |
|
SU1590045A3 |
| Способ получения замещенных производных карбокситиазоло[3,2-а]пиримидина или их фармацевтически приемлемых солей | 1984 |
|
SU1355131A3 |
| Способ получения хлоргидратов замещенных антрациклинов | 1979 |
|
SU867315A3 |
| Способ получения производных бета-лактама | 1989 |
|
SU1750430A3 |
| Способ получения пенемовых соединений или их фармацевтически приемлемых солей щелочных металлов | 1986 |
|
SU1586516A3 |
Изобретение касается 6-дезоксиантрациклинов, в частности получения соединений общей формулы @ , где R, H, OH, OCH3 - полупродукты для синтеза гликозидов антрациклина, используемые в фармакологии. Цель - повышение селективности процесса. Последний ведут последовательной обработкой диметилового эфира 1,2,3,6-тетрагидрофталевой кислоты: а) уксусным ангидридом в присутствии SPCL4
б) слабым основанием или слабой кислотой
в) тозилгидразином
г) бораном кахетина (восстановление)
д) ацетатом натрия при кипячении реакционной смеси (перегруппировка двойной связи из эндо- в экзоположение)
е) KMNO4 при комнатной температуре (окисление и разрушение избытка окислителя), ж) этилацетатом (экстракция с отделением органической фазы, промыванием ее водой, высушиванием и упариванием до сухого остатка). Последний растворяют в бензоле и кипятят с этиленгликолем в присутствии каталитических количеств N-толуолсульфокислоты с последующей конденсацией с н-бутиллитием и с 5R-1,4-диметокси-3-бромнафталином при -78°С в среде безводного тетрагидрофурана (1 ч). Полученный при этом лактон подвергают метанолизу (для размыкания лактонного кольца) с одновременной обработкой кислотой (для удаления диоксалановой группы). Далее ведут восстановление оксогруппы с помощью пиридинборанового комплекса в присутствии CF3-C(O)-OH с последующей обработкой реакционной смеси фенилдиазометаном (превращение метоксикарбонильной группы в бензилоксикарбонильную), а затем уксусным ангидридом и пиридином в присутствии 4-диметиламинопиридина (удаление бензильной группы). После этого смесь кипятят с циклогексеном в присутствии палладия на угле и полученный продукт циклизуют действием трифторуксусного ангидрида с трифторуксусной кислотой с дальнейшим гидролизом ацетоксигрупп. 1-Оксиэтильную группу окисляют карбонатом серебра при кипячении, а демитилирование ведут с помощью ALCL3 в нитробензоле. Полученное соединение обрабатывают этиленгликолем в присутствии каталитических количеств п-толуолсульфокислоты с последующим бромированием бромом в присуствии 2,2Ъ-аза-бис(изобутиронитрила), гидролизом и обработкой кислотой (для удаления кетальной группы). Эти условия обеспечивают получение только 6-диоксипроизводных в противоположность в известном случае смеси изомеров 6-диокси- и 11-диоксипроизводных.
Изобретение относится к способу получения 6-дезоксиантрациклинов общей Аормулы
(I)
но он
R - является водородом, гидроксилом
или метоксигруппой,
которые являются промежуточными соединениями при получении гликозидов антрациклина, которые в свою очередь используются в фармакологии.
Цель изобретения - повышение избирательности процесса.
Изобретение иллюстрируется следующими примерами.
Пример 1. Диметиловый эфир 1,2,3,6-тетрагидро-4 -ацетилфталевой кислоты (ill).
10 г диметилового эфира 1,2,3,6- тетрагидрофталевой кислоты (II) обрабатывают при -5 С 25 мл уксусного ангидрида в присутствии 9 мл четырех- хлористого олова. Затем реакционную смесь переносят в ледяную воду и экст рагируют диэтиловым эфиром. Органический слой промывают насыщенным водным раствором бикарбоната натрия и затем водой, после чего раствор упаривают досуха в вакууме-. Полученное масло растворяют в бензоле и обрабатывают метанольным раствором хлористого водорода. Раствор испаряют досуха, а остаток очищают колоночной хроматографией на силикагеле с получением 9 г указанного соединения с общим выходом 75%.
Масс-спектр: m/Z 240 (Н); ИК(КВг) :. 1720 ( сложного эфира); 1660 см ( d,р ненасыщенного кето- на). ПМГ (CDC13) inter alia Ј, (2,33 синглет, COCIi,), 3,70 (синглет, -COOCHj) и 6,91 (мультиплет, ).
5
0
5
0
5
0
45
0
5
П р и м е р 2. 1,2-Ди(метоксикарбо нил)-4-этилиденциклогексан (V) 17 г диметилового эфира 1,2,3,6-тетрагидро- 4-ацетилфталевой кислоты (III), приготовленного, как указано в примере 1, кипятят в безводном этаноле с 14,6 г тозилгидразина. После удаления растворителя перекристаллизацией из воды получают 24 г диметилового эфира 1,2,3,6-тетрагидро-4-(1-тозилгидразо- ноэтил)фталевой кислоты (IV) с т.пл. 162-163°С, m/Z 408 (М+). Полученное соединение растворяют в хлороформе и-обрабатывают при О С 14 мл борана катехина. Затем к реакционной смеси добавляют ацетат натрия и полученную смесь кипятят. После промывания водой растворитель испаряют, а остаток очищают колоночной хроматографией на си- ликагеле с получением 10 г указанного соединения (выход 80%) m/Z 226 (); ПМР (CDC1): inter alia 8 , 1,6 (дублет, Гц, СН3СН), 5,3 (квартет, Гц, CHSCH).
П р и м е р 3. 2-Метоксикарбонил- 5-(2-метилдиоксалан-2-ил)-6-оксабицик- ло j3,2,1 октан-7-он (VI).
8 г 1, (метоксикарбонил)-4 этилиденциклогексана, синтезированного, как указано в примере 2, растворяют в водном ацетоне, содержащем 4,8 мл уксусной кислоты. Затем прибавляют водный раствор перманганата калия и полученную смесь выдерживают при комнатной температуре 60 мин с получением соответствующего -гидрокси- кетона, Избыток окислителя разрушают, реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают водой и высушивают над безводным сульфатом натрия, после чего испаряют в вакууме. Остаток растт воряют в бензоле и кипятят 60 мин в присутствии каталитического количества п-толуолсульфокислоты. Затем добавляют 4 мл этиленгликоля и реакционную смесь кипятят еще 2 ч. После обыч-
515618216
ной обработки остаток, полученный ис- ственным выходом 1,5 г 1( 1,4,5-три;- парением растворителя, очищают коло- метокск-3 нафтилкарбонил)2-метокси ночной хроматографией на силикагеле карбонил-4-ацетил-4-гидроксициклогек с применением в качестве элюента смег- си толуола с ацетоном (15:1 по объему) . Выделено 3 г заглавного соедине5 сана (VIII ). m/Z 444/М2/; ИК (пленка): 3460 (ОН) , 1730
(
(С-0 сложного эфира), 1710 см кетона), 1665 ( бензильного кетона); ПМР (CDC13) inter alia & ,
ния (выход 33%), т.пл. 69-71 С; m/Z 271 (Ш4-). ИК (КВг): 1790 (СО
пятичленного кольца лактона); 1735 см ю 2,3 (синглет, СН3СО), 3,75-4,05 (син( сложного эфира); 1720 ( кетона), ПМГ (CDC1,): inter alia &, 1,25 (синглет, СН3), 3,65 Тсинглет, COOCHj), 3,9 (синглет, гС-).
глет, четыре 0(Ш3), 6,8 (синглет, ароматические Н), 6,85-8,0 (мульти- плет, три атоматических Н). 1,5 г полученного соединения растворяют 15 в 1-5 мл трифторуксз сной кислоты и кипятят с 1,4 мл пиридин-боранового комплекса. После удаления растворителя остаток обрабатывают 10%-ным водным раствором гидроокиси натрия и
глет, четыре 0(Ш3), 6,8 (синглет, ароматические Н), 6,85-8,0 (мульти- плет, три атоматических Н). 1,5 г полученного соединения растворяют 15 в 1-5 мл трифторуксз сной кислоты и кипятят с 1,4 мл пиридин-боранового комплекса. После удаления растворителя остаток обрабатывают 10%-ным водным раствором гидроокиси натрия и
П р и м е р 4. 2-0 ,4,5-Tpимeтoкcи- 3 нaфтилкapбoнил)5-(2 мeтилдиoкcaлин 2-ил)6-оксабицикло 3,2,1 октан-7-он (VII, )„
В 30 мл безводного тетрагидрофурана20 после подкисления слабой кислотой растворяют 7 мл 1,65 М гексанового полученного свободную кислоту экстрараствора н-бутиллития и к полученному гируют этилацетатом. Растворитель раствору при -78°С добавляют раствор испаряют, а остаток непосредственно 3,3 г 1,4,5-триметокси-З-бромнафтали- обрабатывают эфирным раствором фенил-. (на () в 30 мл безводного тетра- 25 диазометана с получением заглавного гидрофурана. Затем к реакционной сме- соединения, выход 74%, который очища- си прибавляют раствор 2,5 г 2-метокси- ют хроматографированием. m/Z 508(М+). карбонил-5-(2-метилдиоксалан-2-ил)-6™ IM (CUC13): inter alia , 1,25 (дуб- оксабицикло 3,2,1 онтан-7 она, синте- лет, СН3СК), 3,70-3,95 (синглет, три зированного, как указано в примере 3, 30 ОС113), 5,15 (дублет, CH2Ph), 6,4-8,1
(мультиплет, девять ароматических Н).
II р и м е р 6. 1,2,3,4,4а,5,12, 12а-октагидро-2- (1 -гидроксиэтил) -2- гидрокси-6,7-11-триметокси-12-оксо- 35 нафтацен (XI, ).
0,48 г 1-(1,4,5-триметокси-З-нафв 50 мл безводного тетрагидрофурана. Реакционную смесь выдерживают 1 ч при -78 С к затем промывают уксусной кислотой. Растворитель удаляют в вакууме. Остаток очищают колоночной хроматографией на силикагеле с получением 3 г (выход 73%) заглавного сое- тилметил)-2-бензилоксикарбонил-4-(1- динения. m/Z 456 (М+). ИК (КБг): 1775 см ( пятичленного кольца лактона), 1680 см ( бензильного кетона), ПМР (CDC13) inter alia $ 1,3 (синглет, СН3), 3,75 (синглет, ОСН3), 3,95-4,05 (мультиплет, две ОСН3 и ), 6,8 (синглет, ароматические СН), 6,8-8,1 (мультиплет, три Н).
П р и м е р 5. 1-1,4,5-Триметокси- З-нафтилметил-2-бензилоксикарбонил- 4-(1-гидроксиэтил)-4-гидроксицикло- гексан (IX, ).50
Метанольный раствор 1,6 г 2(1,4,5- триметокси-2-нафтилкарбонил)5-(2- метилоксалан-2-ил)-6-оксабицикло Ј3,2, 1 октан-7-она, полученного, как указано в примере 4, обрабатывают в течение 1 ч при комнатной температуре 1 н, раствором хлористого водорода в безводном метаноле. После испарения растворителя получают почти с количе-
гидроксиэтил)-4-гидроксициклогексана, синтезированного, как указано в при-
40 мере 5, обрабатывают уксусным ангидридом и пиридином в присутствии 4-ме- тиламинопиридина. После выдерживания реакционной смеси в течение суток при комнатной температуре ее переносят
45 в ледяную воду и экстрагируют этил- ацетатом. Органический слой промывают водой и концентрируют. Сырой продукт растворяют в метаноле и кипятят с цик- логексаноном в присутствии 10%-кого (по массе) палладия на угле. Затем катализатор отфильтровывают, а раствор концентрируют до небольшого объема, после чего обрабатывают при О в течение 60 мин смесью трифторуксус-
55 ного ангидрида с трифторуксусной
кислотой. Полученной раствор разбавляют этилацетатом, промывают водным насыщенным раствором бикарбоната нат- рия и водой и концентрируют досуха
ственным выходом 1,5 г 1( 1,4,5-триметокск-3 нафтилкарбонил)2-метокси карбонил-4-ацетил-4-гидроксициклогек
сана (VIII ). m/Z 444/М2/; ИК (пленка): 3460 (ОН) , 1730
(
(С-0 сложного эфира), 1710 см кетона), 1665 ( бензильного кетона); ПМР (CDC13) inter alia & ,
2,3 (синглет, СН3СО), 3,75-4,05 (синглет, четыре 0(Ш3), 6,8 (синглет, ароматические Н), 6,85-8,0 (мульти- плет, три атоматических Н). 1,5 г полученного соединения растворяют в 1-5 мл трифторуксз сной кислоты и кипятят с 1,4 мл пиридин-боранового комплекса. После удаления растворителя остаток обрабатывают 10%-ным водным раствором гидроокиси натрия и
тилметил)-2-бензилоксикарбонил-4-(1-
гидроксиэтил)-4-гидроксициклогексана, синтезированного, как указано в при-
мере 5, обрабатывают уксусным ангидридом и пиридином в присутствии 4-ме- тиламинопиридина. После выдерживания реакционной смеси в течение суток при комнатной температуре ее переносят
в ледяную воду и экстрагируют этил- ацетатом. Органический слой промывают водой и концентрируют. Сырой продукт растворяют в метаноле и кипятят с цик- логексаноном в присутствии 10%-кого (по массе) палладия на угле. Затем катализатор отфильтровывают, а раствор концентрируют до небольшого объема, после чего обрабатывают при О в течение 60 мин смесью трифторуксус-
ного ангидрида с трифторуксусной
кислотой. Полученной раствор разбавляют этилацетатом, промывают водным насыщенным раствором бикарбоната нат- рия и водой и концентрируют досуха
в вакууме. Остаток растворяют в метаноле, содержащим каталитическое количество метилата натрия. После обычной обработки и очистки хроматографией получают 0,18 г (выход 49%) заглавного соединения. m/Z 400 (М ); ПК (КБг): 3450 (ОН), 1675 ( бензильного кетона); IMP (CDC13) inter alia Ј, 1,2 (дублет, СНЭСН), 3,7-3,9 (синглет, три ОСНЭ), 6,4-8,0 (мультиплет, три ароматических водорода) .
Пример. 1,2,3,4,4а,5,12, 12а-Октагидро-2 ацетил-2-гидрокси- 6;7,11-триметокси-12-оксонафтацен.
К бензольному раствору 0,09 т 1,2, 3,4,4а,5,12,12а-октагидро-2-(1-гид- роксиэтил)-6,7,11-триметокси-12- оксонафтацена, приготовленного, как указано в примере 6, добавляют 0,8 г карбоната серебра, после чего полученную смесь кипятят. После отфильт- ровывания твердого вещества и испаре- ния в вакууме растворителя получают 0,08 г (выход 90%) указанного соединения. ИК (kBr): 3360 ( (ОН), 1705 ( кетона), 1 680 см ( бензильного кетона); ПМР (CUC13) inter aliaS, 2,2 (синглет, ) 3,65-3,80 (синглет, три ОСИ 3).
Нримерв. Ь,7-Дидезоксикарми- номицинов (XII. ).
Нитробензольный раствор 0,06 г 1,2,3,4,4а,5,12,12а октагидро-2-аце- тил 2-гидрокси-6,7,11-тримётокси-12- оксонафтадена, полученного, как описано в примере 7, обрабатывают 0,12 г треххлористого алюминия, после чего полученную смесь выдерживают при 70°С до момента, пока не перестает обнаруживаться исходное вещество. Затем реакционную смесь переносят в водный насыщенный раствер щавелевой кислоты и экстрагируют этилацетатом. Органический слой отделяют, промывают водой, высушивают и испаряют досуха. Остаток очищают колоночной хроматографией на силикагеле с получением чистого 6,7-дидезоксикарминомицинона (выход 40%). m/Z 352 () ИК (КВг): 3420 см (ОК), 1705 см ( кетона), 1625 ( хелатного хинона) ; ПМР (GDC13): inter aliaS , 1,7-2,2 (мультиплет, СК2), 2,3 (синглет, С1ЦСО), 2,8-3,2 (мультиплет, две бен- зильных CHj), 7,0-7,8 (мультиплет, четыре ароматических Н) 12,6 (син
5
0
5
0
5
0
5
0
5
глет, фенольный ОН), 12,9 (синглет, фенольный ОН).
II р и м е р 9. 6-Дезоксикармино- мицинон (1, ).
Раствор 6,7-дидезоксикарминомице- нона, синтезированного, как указано в примере 8, в бензоле обрабатывают при кипячении в течение 4 ч 1,2 мл этиленгликоля в присутствии каталитического количества п-толуолсульфокис- лоты с получением соответствующего 13-кетального производного, которое растворяют в четыреххлористом углероде и обрабатывают 2 мл раствора, приготовленного растворением 3,2 г брома в 32 мл четыреххлористого углерода, при 45°С в течение 6 ч, в присутствии 2,2-аэо-бис (изобутиронит- рила). Охлажденную реакционную смесь экстрагируют 1 н.водным раствором гидроокиси натрия, окрашенный водный слой доводят до рК 8,5 и экстрагируют хлороформом. Органические экстракты испаряют досуха с получением 6-дезок- си-13-кеталькарминомицинона, который растворяют в ацетоне, содержащем хлористый водород (300 мл 0,25 н. раствора), и выдерживают 3 ч при комнатной температуре с целью гидролиза ке- тальной группы, в результате чего получают целевой 6-дезоксикармииомици- нон, выход 58%.
ПримерЮ, 6,7-Дидеоксидауно- мицинон,
2,77 г 1,2,3,4,4а,5,12,12а-0кта- гидро-2-(1-оксиэтил)-2-ок си-6,7-11- триметокси 12-оксонафтацена (XI, R ОСН3), полученного, как описано в примере 6, растворяют в диоксане (160 мл) и обрабатывают 2,2 -диметок- синропаном (8,47 г) в присутствии ФТСК (0,07 г) при комнатной температуре в течение 3 часов. Этот раствор разводят водой и экстрагируют мети- ленхлоридом, после выпаривания растворителя получают 2,93 г твердого вещества.
Остаток растворяют в ацетонитриле (100 мл), охлаждают до ОаС и обрабатывают раствором церий аммоний нитрата ОНО (9,87 г) в воде (30 мл). После выдержки в течение 30 мин добавляют воду (200 мл) и тщательно экстрагируют . Органический слой промывают раствором , водой, высушивают над NajSO, растворитель отгоняют и получают 2,57 г
твердого вещества. Остаток растворяют с СНгС1г (275 мл), охлаждают при 0°С, обрабатывают комплексом РуНВг Вг2 (2,01 г). Через 1 ч добавляют при OffC ТЭА (1,75 мл), смесь перемешивают в течение 1,5 ч и затем экстрагируют СНгС1а. Органический слой промывают 5%-ным Na2S205 водным раствором, водой, высушивают над , к концентрируют. Твердое вещество подвергают хроматографии на диатомите и получают (1,08, общий выход 36%) 6,7-дидеокси-1З-дигидро-8,13-изопро- пилидендауномицинон. Т.пл. 13Ь-138 С (разлагается) m/Z 408 (М4); РМР (СВСЦ)Ј : 1,45, 1,39, 1,24 (с, ЬН), 4,04 (с, ЗН, ОСИ;}), 4,07, 4,16 (к, ,4 Гц, 1Н, СН-СН3), 7,54-7,57 (с, 1Н, 6-Н), 7,35-/,79 (м, ЗН), 12,92 (с, 1Н, 11-ОН).
Очищенный продукт обрабатывают при 0°С 90% CF3COOH (50 мл) в тече- , ние 2 ч. Раствор нейтрализуют твердым NaHCO з и экстрагируют метиленхлори- дом. Растворитель отгоняют в вакууме, остаток растворяют в диметилсульфо- ксиде ДМСО (20 мл). Добавляют ТЭА (4 мл) и раствор комплекса ТЭА. Оз (1,5 г) в ДМСО (5 мл). Смесь перемешивают в течение 40 мин при комнатной температуре, вливают в 1 н. НС1 и экстрагируют . Органическую фазу промывают NaKCO насыщенным водным раствором, водой, высушивают над . и растворитель отгоняют в вакууме. Сырое вещество очищают с помощью хроматографии на диатомите, получают вещество (XIV), (0,72 г, выход 80%). Т.пл. 149-151 С (с разложением): m/Z 366 (М+)8 : 1,8-2,2 (м, 2Н, 8-CHz), 2,37 (с, 311, СОСН,), 2,8-3,4 (м, 4Н, 10-СН4, 7-СН4), 4,04 (с, ЗН, ОСНЭ), 7,35 (дд, ,0, 8 Гц, 1Н, 3-Н), 7,58 (с, 1Н, 6-Н), 7,72 (т, Гц, 1HS 2-H), 7,97 (дд, .,0, 8 Гц, 1Н, 1-Н) , 12-9 (с, 1Н, 11-ОН);
УФи видимый спектр: 228, 261, 394, |412 нм.
Приме р 11. 6-Деоксидауноми- цинон (1, 3).
Продукт (XII) () (0,6 г) по
10
1Н, 10-Нах), 3,15 (д, Гц, 1Н, Ю-Н9К), 4,06 с, ЗН, ОСН), 4,96 1Н, 7-Н), 7,40 (д, Гц,1Н, 3-Н)
II р и м е р 12. 2-(1,4-Диметокс 3-нафтилкарбонил)-5-(2-метил-диокс ) -6-оксабицикло JJJ, 2, Г{ окта 7-он ().
Но методике, приведенной в прим ре 4, раствор 3,2 г 1,4 -диметокси бромнафталина в безводном тетрагид фуране обрабатывают при -78 С н-бу
15 тиллитием и затем прибавляют к рас вору 2,7 г соединения, полученного в примере 3, в безводном тетрагидр фуране. После очистки на силикагел получают 2,8 г указанного соединен
2о (выход 65%), m/Z 426 (М2); ИК (пле ка): 1780 ( пятичленного ко ца лактона), 1670 см ( бензильн го кетона); 1ШР (CDC13): inter ali о, 1,4 (синглет, СИ§, 3,85 (сингле
25 две ОСИ,), 3,9 (сикглет, -ОСНаСН4(Ь
Пример 13. 1-(1,4-Диметокс 30 3-нафтилметил)-2-бензилоксикарбони 4-(1-гидроксиэтил)-4 гидроксицикло гексан (IX, ).
По методике примера 5 обработко 2-.(1 ,4-диметокси-3-нафтилкарбонил) 5-(-метилдиоксалан-2-ил)-6-оксаби цикло 3,2,1 октан-7-она, синтезиро ванного, как указано в примере 12, раствором хлористого водорода в ме ноле получают с почти количественн 40 выходом 1-(1,4-диметокси-З-нафтил- карбонил)-2-метоксикарбонил-4-ацет 4-гидроксициклогексан (), m/Z 414 (М+-); ИК (пленка): 3460 (О 1730 ( сложного эфира),
35
45 1710 см ( кетона), 1670 см( (С бензильного кетона); НИР (CDCl): inter alia $ , 2,3 (синглет, СН3СО) 2,9-3,6 (мультиплет, два Н), 3,7-3 (синглет, три ОСИ}), 6,9 (синглет,
50 ароматические Н), 7,4-8,4(мультипл четыре ароматических Н).
1 г полученного соединения восс новлением пиридин-борановым компле сом, действием основания и этерифик
методике примера 9 превращают в про- 55 Чией превращены в 0,7 г заглавного
ДУкт (I) () (0,210 г, выход 33%). Т.пл. 268-27Q°C; ra/Z 382 М+); РМР (CDC1,; 8 : 2,3 (м, 2Н, 8-СНг., 2,42 (с, ЗН, СОСНз), 3,0 (д, Гц,
соединения (выход 63%), m/Z 478 (М ИК (пленка): 3450 (ОН), 1725 см ( сложного эфира); ПМР (CDC13): inter aliaS , 1,3 (дублет, Гц,
0
1Н, 10-Нах), 3,15 (д, Гц, 1Н, Ю-Н9К), 4,06 с, ЗН, ОСН), 4,96 (м, 1Н, 7-Н), 7,40 (д, Гц,1Н, 3-Н),
II р и м е р 12. 2-(1,4-Диметокси- 3-нафтилкарбонил)-5-(2-метил-диокса- ) -6-оксабицикло JJJ, 2, Г{ октан- 7-он ().
Но методике, приведенной в примере 4, раствор 3,2 г 1,4 -диметокси-З- бромнафталина в безводном тетрагидро- фуране обрабатывают при -78 С н-бу-, о
5 тиллитием и затем прибавляют к раствору 2,7 г соединения, полученного в примере 3, в безводном тетрагидро- фуране. После очистки на силикагеле получают 2,8 г указанного соединения
о (выход 65%), m/Z 426 (М2); ИК (пленка): 1780 ( пятичленного кольца лактона), 1670 см ( бензильно- го кетона); 1ШР (CDC13): inter alia о, 1,4 (синглет, СИ§, 3,85 (синглет,
5 две ОСИ,), 3,9 (сикглет, -ОСНаСН4(Ь),
Пример 13. 1-(1,4-Диметокси- 0 3-нафтилметил)-2-бензилоксикарбонил- 4-(1-гидроксиэтил)-4 гидроксицикло- гексан (IX, ).
По методике примера 5 обработкой 2-.(1 ,4-диметокси-3-нафтилкарбонил)- 5-(-метилдиоксалан-2-ил)-6-оксаби- цикло 3,2,1 октан-7-она, синтезированного, как указано в примере 12, раствором хлористого водорода в метаноле получают с почти количественным 0 выходом 1-(1,4-диметокси-З-нафтил- карбонил)-2-метоксикарбонил-4-ацетил- 4-гидроксициклогексан (), m/Z 414 (М+-); ИК (пленка): 3460 (ОН) , 1730 ( сложного эфира),
5
5 1710 см ( кетона), 1670 см( ( бензильного кетона); НИР (CDCl): inter alia $ , 2,3 (синглет, СН3СО), 2,9-3,6 (мультиплет, два Н), 3,7-3,9 (синглет, три ОСИ}), 6,9 (синглет,
0 ароматические Н), 7,4-8,4(мультиплет, четыре ароматических Н).
1 г полученного соединения восстановлением пиридин-борановым комплексом, действием основания и этерификаЧией превращены в 0,7 г заглавного
соединения (выход 63%), m/Z 478 (М) ИК (пленка): 3450 (ОН), 1725 см ( сложного эфира); ПМР (CDC13): inter aliaS , 1,3 (дублет, Гц,
11156182112
СН5СН), 3,85, 3,9 (синглет, две ОСИ,), Но методике примера 8 0,1 г 1,2,3; 5,1 (синглет, бензильная СН2-группа), 4,43,5,12,12а-октагидро-2-ацетил-2 6,6 (синглет, ароматические К), 7,2- 8,4 мультиплет, девять ароматических водородов).
И р и м е р 14. 1,2,3,4,4а,5,12, 12а Октагидро 2-(1-гидроксиэтшг-2- гидрокси-6,11-диметокси-12 оксонафта- цен (XI, ).
Но методике примера 6 0,44 г 1- (1,4 димeтoкcи-3-нaфтилмeтил)2-бeн- зилoкcикapбoнил-4- (1-гидроксиэтил)- 4-гидроксициклогексана, приготовленного, как указано в примере ТЗ, обра- 15 оксидауномицинона ( 1). батывают уксусным ангидридом в присутствии 4-диметиламинопиридина и пиридина. Полученный ацетат восставав - ливают в циклогексане на 10%-ном палладии на угле с целью удаления бен-. 20 вают при температуре дефлегмации в зильной группы. Затем полученную кис- течение 4ч 1,2 мл этиленгликоля в лоту (X) циклизуют обработкой смесью присутствии 0,045 г п-толуолсульфо- трифторуксусного ангидрида и трифтор- уксусной кислоты при 0°С. И наконец проводят удаление ацетил О-защитных групп действием метилата натрия и очистку колоночной хроматографией на
гидрокси-6,11-диметокси-12 оксонафте цена, пригот9вленного, как указано в примере 15, в 3 мл нитробензола обрабатывают 0,25 г треххлористого алюминия, после чего выдерживают в течение суток при комнатной температуре. Ю Очисткой колоночной хроматографией на силикагеле получено 0,055 г (выход 63%) заглавного соединения с т.пл. 203-204°С.
II р и м е р 17. 4 Деметокси 6-де
Раствор 0,5 г 4-деметокси-6,7-ди- деоксидауномицинона, полученные по примеру 16 в 50 мл бензола обрабаты-
кислоты и получают соответствующее 13-кетальпроизводное (0,4 г), которое 25 кристаллизовалось непосредственно из охлажденной реакционной смеси. Это соединение растворяют в 250 мл четы-
силикагеле с получением 0,225 г указанного соединения (общий выход 66%).
ИК (пленка): 3450 (ОН), 1675 ( бензильного кетона), IMP (CDC1,) inter aliaS , 1,3 (дублет, . Гц, СК-эСН), 1,6-3,5 (мультиплет, ЗН), 3,85 (синглет,
реххлористого углерода и обрабатывают 2 мл раствора 3,2 г брома в 32 мл 30 четыреххлористого углерода при 45 в
течение 6 ч в присутствии 0,46 г 2,2 азобис-изо-бутиронитрила. Охлажденную реакционную смесь экстрагируют 1 н. водным раствором гидроокиси натрия и
40
-ОСНЭ), 3,90 (синглет, ОСН3), 7,2-8,4 35 окрашенную водную фазу подрабатывают (мультиплет, четыре ароматических Н).
Пример 15. 1,2,3,4,4а,, 12а-0ктагидро 2 ацетил 2-гидрокси- 6,11-диметокси-12-оксонафтацен.
Бензольный раствор 0,1 г 1,2,3,4, 4а,5,12,12а-Октагидро-2-(1-гидроксиэтил) -2-гидрокси-6, 11-диметокси-12- оксонафтацена, синтезированного, как указано в примере 14, обрабатывают 1 г карбоната серебра при температуре кипения раствора. После отфильтро- вывания неорганического твердого вещества и удаления растворителя получают 0,1 г заглавного соединения..,
ИК (пленка): 3460 (ОН), 1710 ( кетона), 1680 ( бензилового кетона); ПМР (CDC1): inter alia 8 , 2,4 (синглет, ), 3,85 (синглет, ОСИ), 3,90 (синглет, ООН), 7,2-8,4 (мультиплет, четыре ароматических К).
до рН 835 и экстрагируют хлороформом В органических экстрактах, выпаренных до малого объема, было 0,11 г кристаллического 4-диметокси-6-деок- си- 13- кетальдауномицинона. ТСХ на диатомитовых пластинах (Мерк Г254) система растворителей СНС13-(СН3)2СО (9:1 по объему): Rf 0,21 Е1-М m/Z 396 (М4) РМР (CDC13)Ј : 1,47
454с, ЗН, 14-СН), 1,53 (с, 1Н, ОН-9), 2,27 (ддд, 2Н, Н-8) ,5 Гц, 4,5 Гц 6.0 Гц, 3,02 (дд, 2Н, Н-10) ,5T4 3,90 (д, 1Н, ОН-7) ,5 Гц, 4,09 (с, 4Н, ), 4,90 (дд, 1Н, Н-7 ,5; 6,0 Гц, 7,85, 8,26 (м, 4Н аро матические), 7,98 (с, 1К, Н-6), 13,1 (сл JH, ОН-11) ИК (КВг): 1620 связанный хинон, 1670 свобод ный хинон.
55 В конце проводят гидролиз кеталь- ной группы путем обработки водным раствором хлористого водорода в ацетоне (300 мл 0,25 н. раствора) при комнатной температуре в течение 3 ч.
50
Пример 16. 4-Деметокси-6,7 дидеэоксидауномицинон (XII;, .
Но методике примера 8 0,1 г 1,2,3; 4,43,5,12,12а-октагидро-2-ацетил-2
оксидауномицинона ( 1). вают при температуре дефлегмации в течение 4ч 1,2 мл этиленгликоля в присутствии 0,045 г п-толуолсульфо-
гидрокси-6,11-диметокси-12 оксонафте цена, пригот9вленного, как указано в примере 15, в 3 мл нитробензола обрабатывают 0,25 г треххлористого алюминия, после чего выдерживают в течение суток при комнатной температуре. Очисткой колоночной хроматографией на силикагеле получено 0,055 г (выход 63%) заглавного соединения с т.пл. 203-204°С.
II р и м е р 17. 4 Деметокси 6-де
оксидауномицинона ( 1). вают при температуре дефлегмации в течение 4ч 1,2 мл этиленгликоля в присутствии 0,045 г п-толуолсульфо-
Раствор 0,5 г 4-деметокси-6,7-ди- деоксидауномицинона, полученные по примеру 16 в 50 мл бензола обрабаты-
оксидауномицинона ( 1). вают при температуре дефлегмации в течение 4ч 1,2 мл этиленгликоля в присутствии 0,045 г п-толуолсульфо-
кислоты и получают соответствующее 13-кетальпроизводное (0,4 г), которое кристаллизовалось непосредственно из охлажденной реакционной смеси. Это соединение растворяют в 250 мл четы-
реххлористого углерода и обрабатывают 2 мл раствора 3,2 г брома в 32 мл 30 четыреххлористого углерода при 45 в
течение 6 ч в присутствии 0,46 г 2,2 - азобис-изо-бутиронитрила. Охлажденную реакционную смесь экстрагируют 1 н. водным раствором гидроокиси натрия и
окрашенную водную фазу подрабатывают
до рН 835 и экстрагируют хлороформом. В органических экстрактах, выпаренных до малого объема, было 0,11 г кристаллического 4-диметокси-6-деок- си- 13- кетальдауномицинона. ТСХ на диатомитовых пластинах (Мерк Г254) система растворителей СНС13-(СН3)2СО (9:1 по объему): Rf 0,21 Е1-М m/Z 396 (М4) РМР (CDC13)Ј : 1,47
с, ЗН, 14-СН), 1,53 (с, 1Н, ОН-9), 2,27 (ддд, 2Н, Н-8) ,5 Гц, 4,5 Гц, 6.0 Гц, 3,02 (дд, 2Н, Н-10) ,5T4, 3,90 (д, 1Н, ОН-7) ,5 Гц, 4,09 (с, 4Н, ), 4,90 (дд, 1Н, Н-7) ,5; 6,0 Гц, 7,85, 8,26 (м, 4Н ароматические), 7,98 (с, 1К, Н-6), 13,11 (сл JH, ОН-11) ИК (КВг): 1620 связанный хинон, 1670 свободный хинон.
В конце проводят гидролиз кеталь- ной группы путем обработки водным раствором хлористого водорода в ацетоне (300 мл 0,25 н. раствора) при комнатной температуре в течение 3 ч.
который обрабатывают уксусным ангидридом в присутствии четыреххлористого олова и затем слабым основанием или слабой кислотой, полученный при этом диметиловый эфир 1,2,3,6-тетрагидрот 4-ацетилфталевой кислоты формулы
О
I)
нзсоос -пГ,СНз
Н3СООС
растворяют в безводном тетрагидрофу- ране и конденсируют с н-бутиллитием и 5R 1,4-диметокси-3 бромнафталином при (-78) С 1 ч, с получением лак- тона общей формулы
R ОСН30
R имеет указанные значения, который подвергают метанолизу для размыкания лактонного цикла с одновременной обработкой кислотой для ; удаления диоксалановой группы, в полученном соединении общей формулы
ОСН3 ОСН30
V
СН3 ОН
R OCH3
R имеет указанные значения, восстанавливают пиридин-борановым комплексом в присутствии трифторук -,, сусной кислоты, с последующей обработкой реакционной смеси фенилдиазо- метаном, для превращения метоксикар- бонильной группы в бензолоксикарбо- нильную с получением соединения общей фирмулы
ОСНзОШ2СбН5 0Н
ОСН3
R имеет указанные значения, которое для удаления бензильной группы обрабатывают уксусным ангидридом и пиридином в присутствии 4-диметил- аминопиридина, после чего реакционную смесь кипятят с циклогексеном в присутствии в качестве катализатора палладия на угле, получая при этом соединение общей формулы
ОСН30
ОСОСНз
СН3 ОСОСНз
где R.-K, ацетоксигруппа или метоксигруппа,
которое подвергают циклизации действием смеси трифторуксусного ангидрида с трифторуксусной кислотой, и с последующим гидролизом ацетоксигрупп в присутствии метилата натрия, в полученном при этом соединении общей фор-
МЛ ЛЫ
ОСНзО ОН
/
ОСН3
где R имеет указанные значения, окисляют карбонатом серебра при кипячении и деметиллируют треххлористым алюминием в нитробензоле, получая соединение общей формулы
о
30
R О
где R имеет указанные значения,
которое обрабатывают этиленгликолем в присутствии каталитических количеств п-толуолсульфокислоты и последующим бромированием бромом в присутствии 2,2 -азо-бис(изобутиронитрила) гидролизом 7-бромпроизводного с последующей обработкой кислотой для удаления кетальной группы.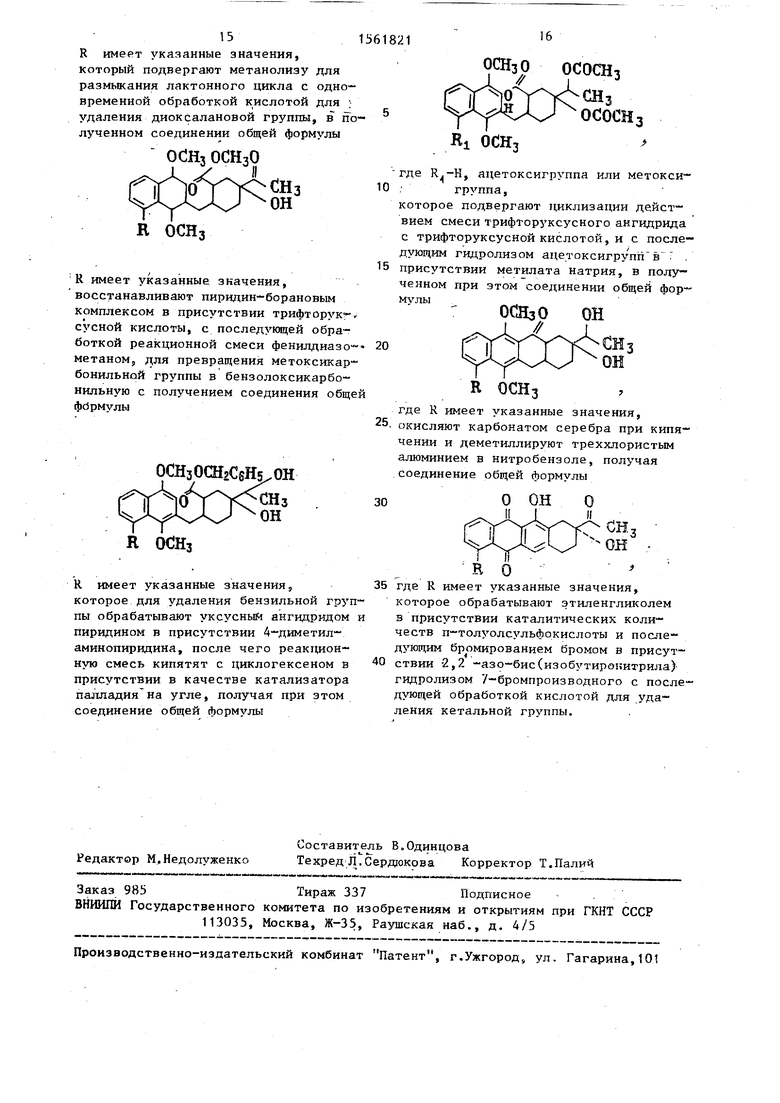
| J | |||
| Am | |||
| Chem | |||
| Парный автоматический сцепной прибор для железнодорожных вагонов | 0 |
|
SU78A1 |
| J | |||
| Am | |||
| Chem | |||
| Цилиндрический сушильный шкаф с двойными стенками | 0 |
|
SU79A1 |
| САМОЛЕТ-ЗАПРАВЩИК | 1994 |
|
RU2100257C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
| СИСТЕМА АМОРТИЗАЦИИ ДЛЯ ОБУВИ | 2007 |
|
RU2429770C2 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
| Способ получения рацемических аглюконов | 1983 |
|
SU1311616A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
