Изобретение относится к микробиологической промышленности, генетической инженерии и биотехнологии. Предлагаются ген, кодирующий гормон роста (ГР) овцы, рекомбинантная плазмида. содержащая этот ген, и штамм бактерий - продуцент ГР овцы.
ГР животных - полипептидные гормоны, состоящие из одной цепи длиной около 190 остатков. Особенностью ГР является видовая специфичность. Указанные соединения представляют интерес для животноводства, поскольку они необходимы для сбалансированного роста молодых животных. Их действие приводит к увеличению транспорта аминокислот в клетки, стимуляции синтеза белка на рибосомах, увеличению митотической активности клеток. Введение ГР вызывает увеличение веса животных, в связи с этим они рассматриваются
в качестве препаратов, перспективных для повышения продуктивности животноводства.
Существующие методы выделения ГР из гипофизов животных не позволяют получать их в достаточных количествах. Альтернативным подходом является синтез ГР в клетках микроорганизмов на основе методов генетической инженерии. Известны клонированный ген (кДНК) предшественника ГР овцы и плазмида, несущая этот ген. однако нет каких-либо сведений о получении штамма-продуцета ГР овцы.
ГР овцы отличается от ГР крупного рогатого скота (КРС)заменой глицина на валин в положении 129 аминокислотной последовательности. При использовании ГР КРС в качестве стимулятора роста в овцеводстве нельзя исключить возможности выработки нейтрализующих антител при длительном
W
о
СП
о
введении препарата, что приведет к снижению его эффективности. Для применения в овцеводстве предпочтительным представляется препарат ГР овцы. Этим определяется практическое значение ГР овцы.
Целью изобретения является создание штамма - продуцента ГР овцы,
Для достижения поставленной цели сконструированы ген, кодирующий зрелый ГР овцы, рекомбинантная плазмидная ДНК, poGHtrpH, содержащая данный ген под контролем промотора trp-оперона Е. coli, и подобран штамм-реципиент, трансформация которого плазмидой poGHtrpH приводит к эффективному и стабильному синтезу ГР овцы.
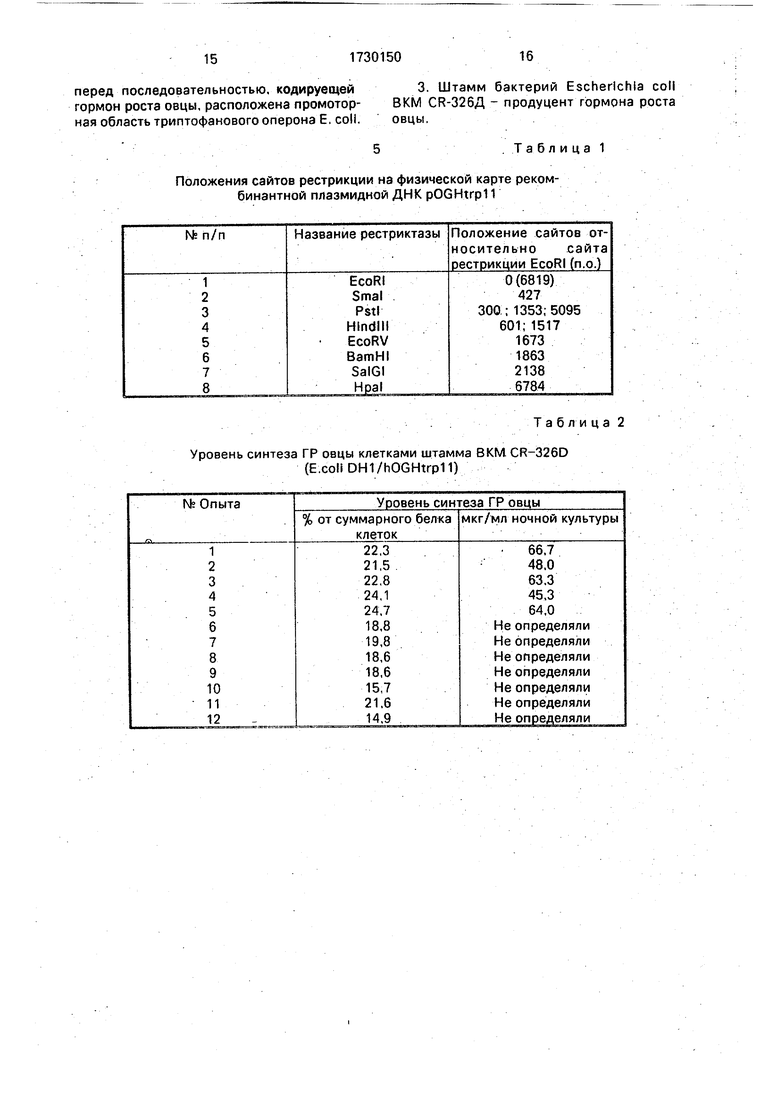
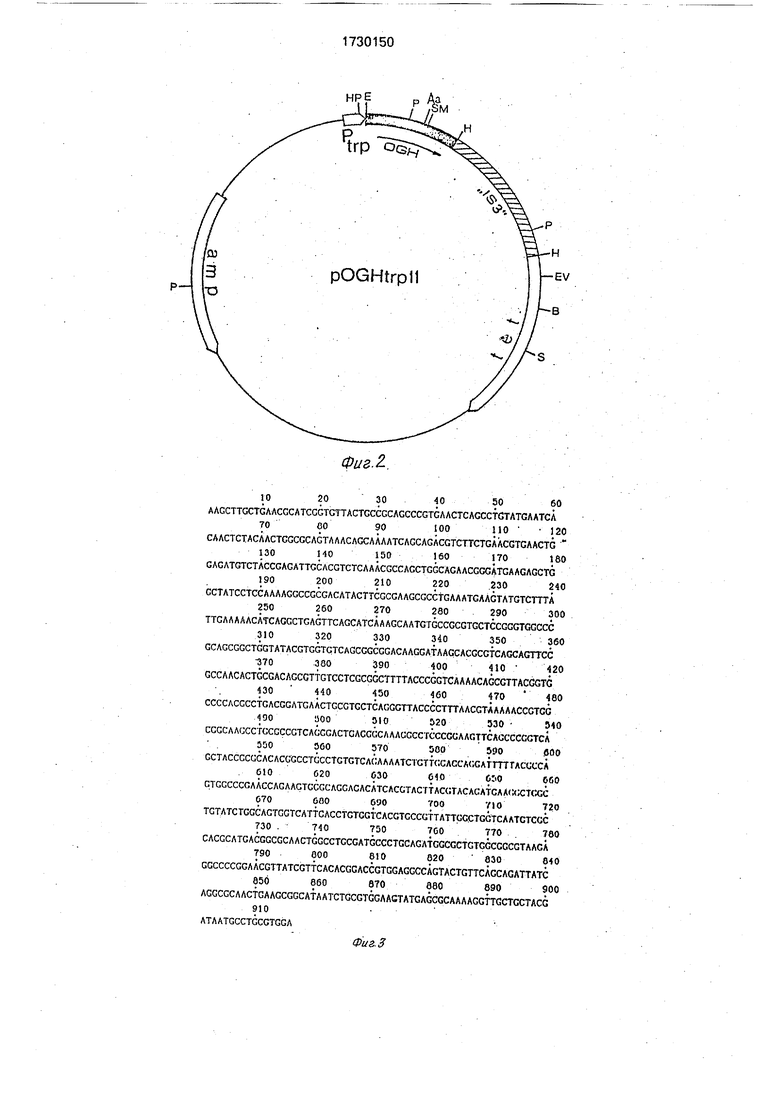
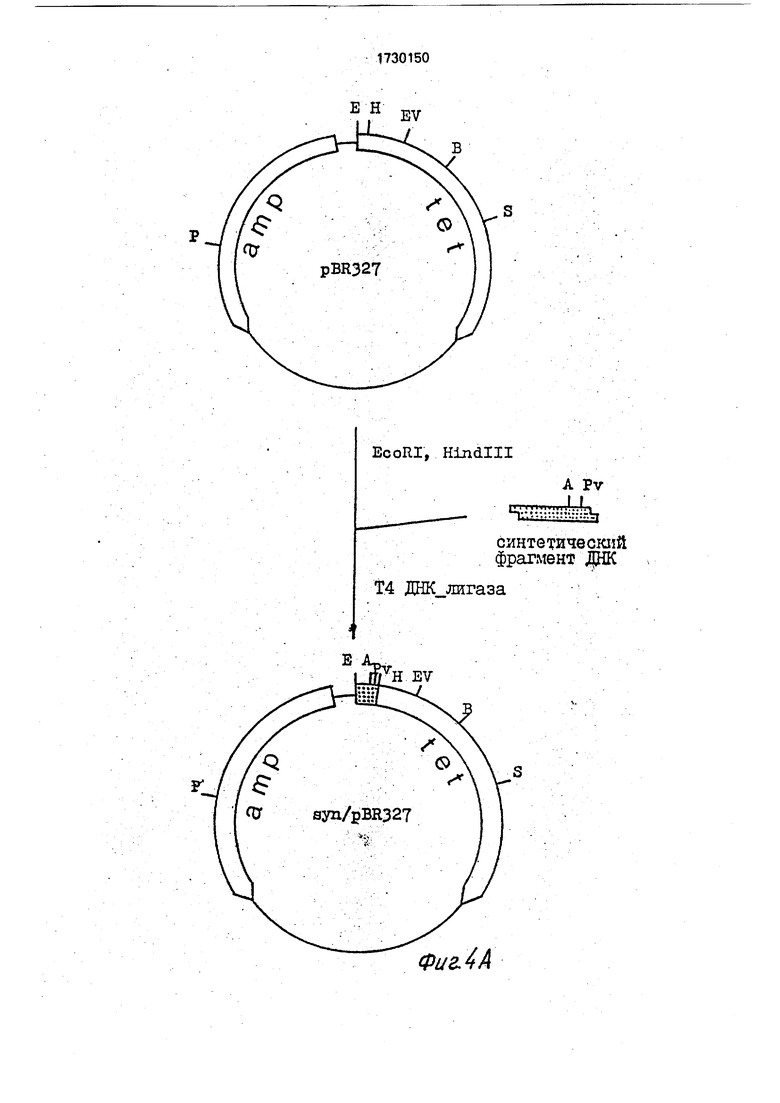
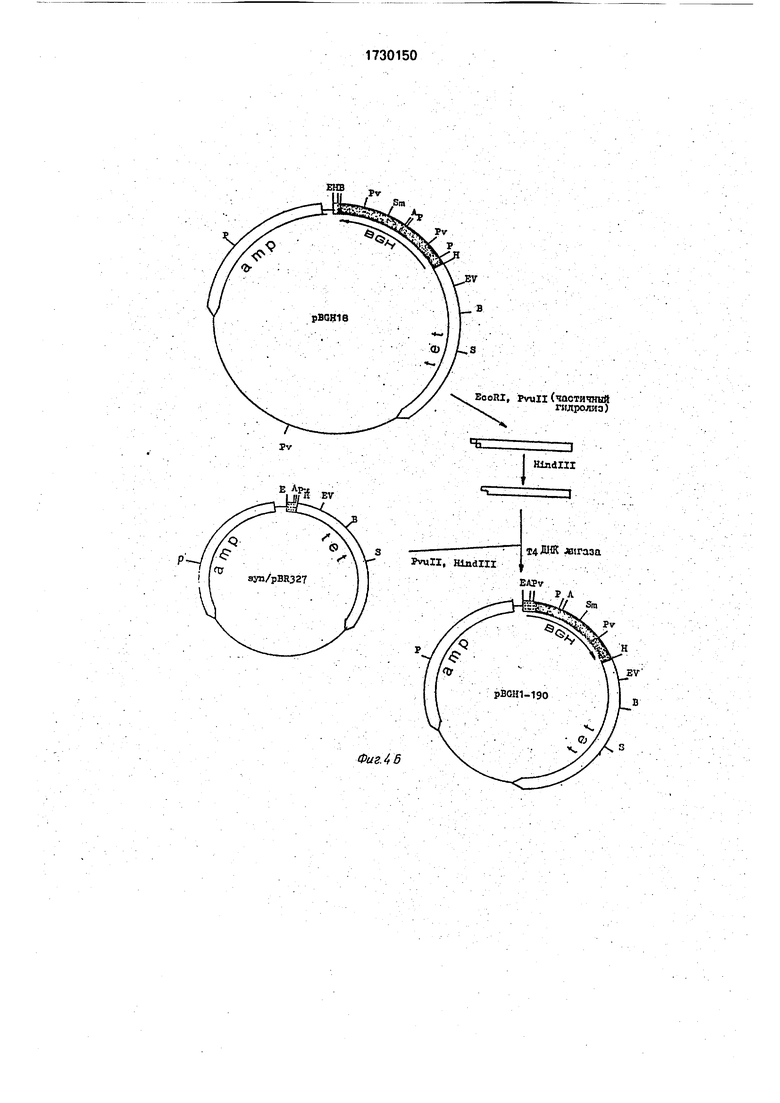
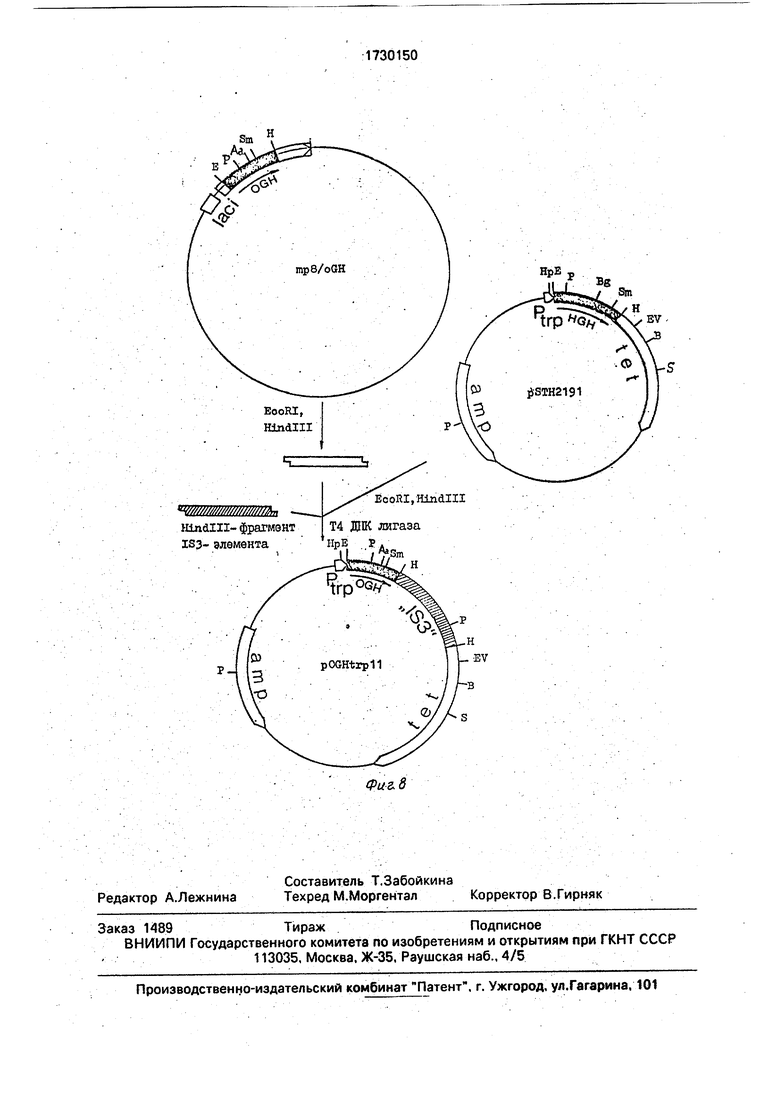
На фиг. 1 изображена нуклеотидная последовательность EcoRI-Hind Ill-фрагмента, кодирующего ГР овцы, и аминокислотная последовательность целевого продукта (подчеркнут участок расщепления рестрик- тазы Aatll, введенный с помощью мутагенеза); на фиг. 2 - схема рекомбинантной плазмидной ДНК pOGHtrpH, amp, tet-гены устойчивости к ампициллину и тетрацикли- ну, Ptrp-npoMOTop триптофанового оперона, OGH-фрагмент, кодирующий гормон роста овцы, IS3 - фрагмент 133-элемента (сайты рестрикции обозначены Е - EcoRI, EV - EcoRV, В - BamHt, H - Hlndlll, HP - Hpal, P - Pstl, S-Sal 3l, SM-Smal, Aa- Aatll); на фиг. 3 - нуклеотидная последовательность фрагмента 153-элемента; на фиг. 4 - схема конструирования плазмиды pBGHBal12 (сайты рестрикции обозначены: А - Apal, Pv - Pvull, остальные обозначения, как на фиг. 2; на фиг. 4А - схема конструирования плазмиды syn/pBR327; на фиг. 4Б - схема конструирования плазмиды pBGH1-190; на фиг. 4В - схема конструирования плазмиды pBGHBa 12; на фиг. 5 - структура синтетических олигонуклеотидов, использованных для сборки фрагмента, кодирующего N-кон- цевую часть гормона роста крупного рогатого скота; на фиг. 6 - схема конструирования рекомбинантного бактериофага mp8/bGH; на фиг. 7 - схема конструирования рекомбинантного бактериофага mp8/oGH; на фиг. 8 - схема конструирования рекомбинантной плазмиды pOGHtrpH.
Фрагмент ДНК, кодирующий ГР овцы, имеет длину 601 п.о., получен путем олиго- нуклеотид-направленного мутагенеза фрагмента кДНК ГР крупного рогатого скота (КРС) и имеет последовательность нуклео- тидов, представленную на фиг. 1.
Сущность рекомбинантной плазмиды poGHtpr11 (фиг. 2) состоит в том, что она содержит репликон и гены устойчивости к ампициллину и тетрациклину плазмиды
pBR322, промотор trp-оперона Е; coll, фрагмент, кодирующий ГР овцы, и фрагмент IS3- элемента бактерий, а именно состоит из следующих элементов:
EcoRI-Hindlll-фрагмента плазмиды pSTH2191 размером 5300 п.о., включающего участок начала репликации, гены устойчивости к ампициллину (Ыа) и тетрациклину (tet) и промотор trp-оперона E. coli;
EcoRI-Hindlll-фрагмента, кодирующего ГР овцы, размером 601 п.о., полученного с помощью мутагенеза фрагмента ДНК, кодирующего ГР КРС;
Hind Ill-фрагмента 83-элемента размером 916 п.о..
Размер плазмиды pOGHtrpH составляет 6819 п.о. Копийность плазмиды около 20 молекул на клетку.
ДНК плазмиды pOGHtpr11 содержит уникальные сайты рестрикции EcoRI, Hpal, Smal, BamHI, SalGI, EcoRV, 2 сайта рестрикции Hindlll и З сайта рестрикции Pstl.
Положения всех перечисленных сайтов рестрикции относительно уникального EcoRI-сайта показаны в табл. 1.
Синтез ГР овцы осуществляется под контролем trp-промотора. Наличие в плаз- миде фрагмента 153-элементэ стабилизирует синтез целевого продукта.
Полная первичная структура всех элементов, из которых построена плазмида pOGHtrpH, известна.
Первичные структуры фрагмента, кодирующего ГР овцы, и фрагмента 153-элемен- та представлены на фиг. 1 и 3 соответственно.
Штамм-продуцентр ГР овцы получен трансформацией клеток Е. coli DHT плазмидной pOGHtrpH. Уровень синтеза ГР по данным-иммуноферментного анализа составляет 50-60 мкг на 1 мл ночной культуры. Штамм депонирован во Всесоюзной коллекции микроорганизмов при ИБФМ АН СССР под номером ВКМ СР-326Д. Штамм Е. coll DH1, содержащий плазмиду pOGHtrpH, характеризуется следующими признаками.
Морфологические признаки: клетки прямые, палочковидной формы, грамотри- цательные, неспороносные.
Культуральные признаки: клетки хорошо растут на обычно используемых питательных средах. При росте на питательном агане Дифко колонии гладкие, круглые, блестящие, серые. Края колоний ровные. При росте в жидких средах YT.LB.M9 образуют ровную интенсивную муть.
Физиолого-биохимические признаки: оптимальная температура культивирования 37°С, оптимум рН 6,8-7,5. В качестве источника углерода используют многие углеводы.
спирты, органические кислоты. В качестве источника азота используют как минеральные соли в аммонийной или нитратной форме, так и органические соединения - пептон, триптон, аминокислоты.
Устойчивость к антибиотикам: проявляют устойчивость к ампициллину, обусловленную наличием плазмиды. Проявляют устойчивость к тетрациклину при концентрации последнего не более 5-7 мг/л.
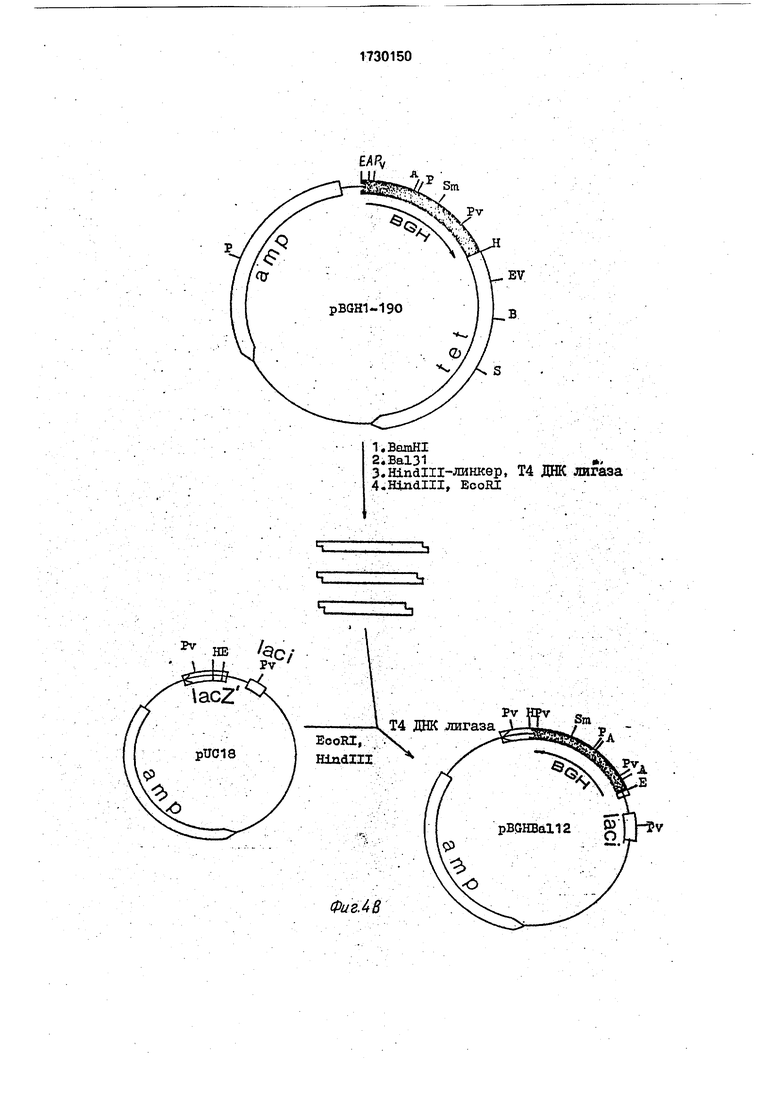
П р и м е р 1. Конструирование реком: бинантной. плазмидной ДНК pBGHBal 12.
Схема конструирования плазмиды pBGHBa12 представлена на фиг. 4 (А-В).
А) Конструирование плазмиды зуп/рВР327(фиг. 4А).
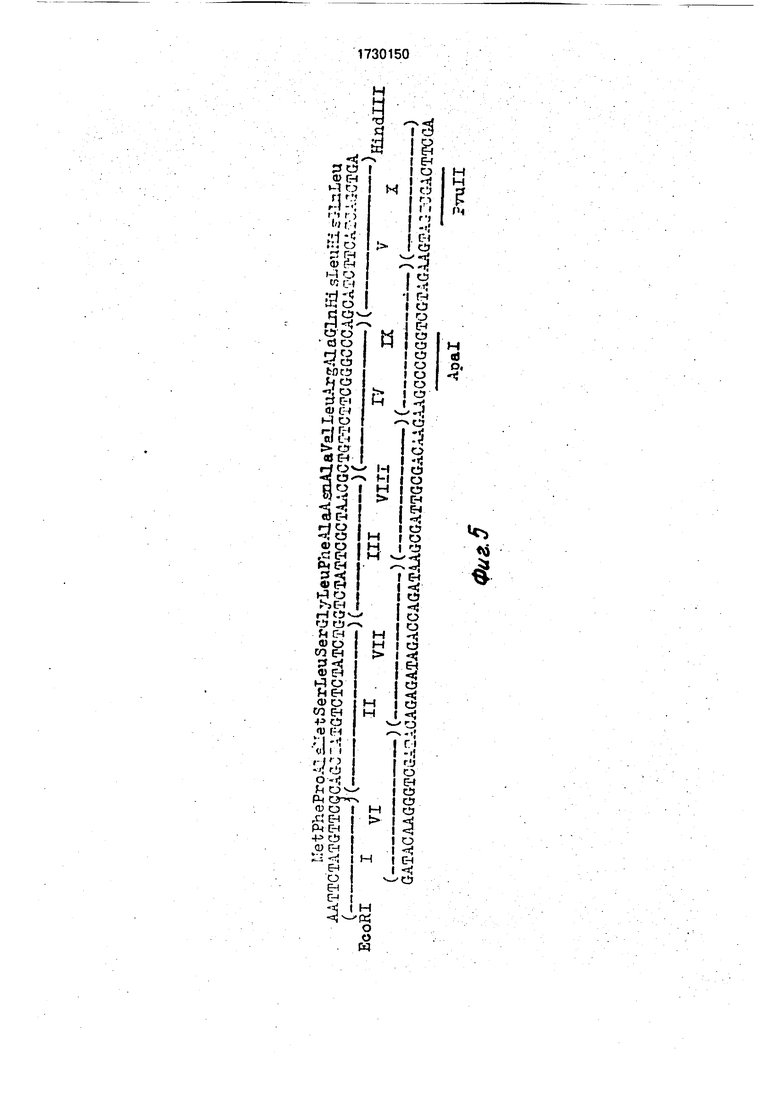
Для получения фрагмента ДНК. кодирующего N-концевую часть ГР КРС (аминокислоты 1-23), использовали синтетические олигонуклеотиды, структура которых представлена на фиг. 5. Структура олигонуклео- тидов выбрана с таким расчетом, чтобы синтетические фрагмент ДНК.имел липкие концы для рестриктаз EcoRI и Hindlll. Инициирующий ATG-кодон расположен перед кодоном 1-й аминокислоты зрелого ГР КРС- фенилала нина. В участке, соответствующем кодонам Glu2 -Leu23, предусмотрен сайт рестриктазы Pvull, позволяющий сшить синтетический фрагмент ДНК с фрагментом, кодирующим остальную часть молекулы ТР (остатки 24-190).
Фосфорилирование и сшивку олигонук- леотидов проводили следующим образом.
5 мкг каждого из 10 олигонуклеотидов инкубировали при 37°С 15 мин в 20 мкл инкубационной смеси, содержащей 50 тМ трисНС, рН 7,5, 5 mM MgCt. 5 тМ дитиот- рейтола (ДТТ), 5 mkCi y-32P-ATP (800 Ci/mmol), 4 ед. полинуклеотидкиназы фага ТА. Затем добавляли 1 мкл 10 тМ раствора немеченного АЕР и инкубировали еще 30 мин при 37°С. Эффективность фосфорили- рования проверяли с помощью электрофореза .1 мкл каждой инкубационной см.еси в 20% денатурирующем полиакриламидном геле с последующей авторадиографией. Инкубационные смеси объединяли, добавляли 50 ед. ДНК-лигазы фага Т4 и инкубировали в течение 12 ч при 4°С. Смесь прогревали 10 мин при 70°С, добавляли 5 М раствора NaCI до концентрации 100 тМ, 50 ед. рестрикта- зы EcoRI, 50 ед. рестриктазы Hindlll и инкубировали при 37°С 5 ч. Инкубационную смесь пропускали через колонку с сефадек- сом G-50 и фракционировали с помощью электрофореза в 8% полиакриламидном геле. После авторадиографии вырезали из геля полосу, соответствующую фрагменту размером около 75 п.о., и элюировали ДНК
:
из геля стандартным методом. ДНК после переосаждения спиртом растворяли в 20 мкл воды.
5 мкг плазмиды pBR327{Soberbn X. et al. 5 Gene, 1980,9,287) гидролизовали рестрикта- . зами EcoRI и Hindlll в инкубированной смеси следующего состава: 50 тМ трисНС, рН 7,5, 100 mM NaCI, 10 тМ MgCl2, 1 тМ ДТТ, 10 ед. EcoRI и 10 ед. Hindlll в течение 2 ч при
10 37°С. Смесь фракционировали в 1 % агароз- ном геле и элюировали линейную форму плазмиды, используя легкоплавкую агарозу фирмы BRL ДНК переосаждали спиртом и растворяли в 50 мкл воды.
15 Лигирование фрагмента ДНК и плазмиды проводили в смеси объемом 20 мкл, содержащей 1 мкл раствора фрагмента, 1 мкл раствора плазмиды/ 20 тМ трисНСЦ рН . 7.5, 10 тМ ДТТ, 10 тМ MgCl2. 1 тМ АТР, 5
20 ед. ДНК-лигазы фага Т4, при 4°С в течение 12 ч. К 2 мкл инкубационной смеси добавля ли 48 мкл ТЕ-буфера (50 тМ трисНС, рН 8,0, 1 плМ ЭДТА). Раствором ДНК трансформировали клетки штамма Е. coll HB101, ис5 пользуя стандартную методику. Отбирали клоны с фенотипом Ар Тс . Наличие вставки с нужной ориентацией проверяли секвенированием по методу Максама- Гилберта. В результате была отобрана
0 плазмида. обозначенная syn/pBR327.
Б) Конструирование плазмиды.pBGH 1- 190 (фиг. 4Б).;
10 мкг ДНК плазмиды pJbGH18 гидролизовали в инкубационной смеси объемом 50
5 мкл 30 ед. рестриктазы EcoRJ в течение 1,5 ч при 37°С, как описано ранее, ДНК из смеси осаждали спиртом, высушивали и растворяли в воде. Гидролиз линеаризованной плазмиды pbGH18 рестрихтазой Pvull
0 проводили в 100 мкл инкубационной смеси в среднесолевом буфере в течение 10 мин при 37°С. На .10 мкг ДНК брали 30 ед. фермента. Реакцию останавливали добавлением раствора ЭДТА до концентрации 20 тМ.
5 Реакционную смес.ь экстрагировали равным объемом смеси фенол - хлороформ. ДНК из водной фазы осаждали спиртом, высушивали, растворяли в воде и фракционировали в 1% агарозном геле. Нужный
0 фрагмент ДНК (Pvull-EcoRI-фрагмент раз- .. мером 680 п.о,) элюировали из легкоплавкой агарозы с помощью стандартной процедуг ры, осаждали спиртом, высушивали и рас. творяли в воде.
5 -.-.- :..
0,5 мкг Pvull-EcoRI-фрагмента инкубировали в 10 мкл среднесолевого буфера с 5 ед. рестриктазы Hindlll в течение 1ч при 37°С. ДНК из реакционной смеси осаждали спиртом, высушивали и растворяли в воде.
Для получения вектора из плазмиды syn/pBR327 4 мкг ДНК гидролизовали в 20 мкл инкубационной смеси на основе сред- несолевого буфера, содержащей 15 ед. Pvull и 20 ед. Hind ИI-, в течение 1,5 ч при ,- 37°С. Реакционную смесь фракционировали в 1 % агарозном геле и вектор элюирова- ли, используя легкоплавкую агарозу. ДНК осаждали спиртом, высушивали и растворяли в воде..
Лигирование Pvull-Hindlll-Фрагмента к ДНК ГР КРС с Pvull-Hlndlll-вектором (syn/pBR327)осуществляли в 20 мкл инкубационной смеси следующего состава: 50 тМ трисНС. рН 7.6.10 mM MgCIa, 5 тМ ДТТ. 0,1 тМ спермидина, 0,05 тМ ЭДТА, 1 тМ АТР. Соотношение вектор-вставка (по молям) составляло 1:2. Реакционную смесь инкубировали с 10 ед. ДНК-лигазы фага Т4 в течение ночи при 8°С. Лигазной смесью трансформировали клетки штамм E.coli НВ 14)1. Плазмиды из полученных клонов анализировали с помощью гидролиза ре- стриктазами. Правильность структуры подтверждали секвенированием методом Максама-Гилберта. В результате была отобрана плазмида нужной структуры, обозначенная pBGH 1-190.
В) Конструирование плазмиды рВОНВа112(фиг. 4В)
Для удаления З -нетранслируемого участка кДНК ГР КРС 10 мкг плазмиды pBGH1- 190 гидролизовали рестриктэзой Bam HI в 20 мкл инкубационной смеси на основе среднесолевого буфера в течение 1 ч при 37°С. Линейную форму плазмиды обрабатывали нуклеазой Ва131. Далее проводили обработку полученных ДНК фрагментом Кленова ДНК полимеразы I в присутствии dNTP и пришивку синтетического Hindllf- линкера 5 -CCAAGCTTGG-3 . Смесь прогревали 5 мин при 70°С, добавляли 10 ед. рестриктазы EcoRI и 30 ед. рестриктазы HlndHI и инкубировали в течение 3 ч при 37°С. Смесь полученных фрагментов фракционировали в 1% агарозном геле и выделяли фрагменты длиной 580-650 п.о. После переосаждения этанолом ДНК растворяли в 20 мкя воды. 5 мкг плазмиды pUC18 гидроли- эовали рестриктазами EcoRI и Hindlli, как описано ранее. К 100 нг pUC18, расщепленной EcoRI и Hlndlll добавляли 5 мкл раствора фрагментов ДНК. выделенных из агарозы, и проводили инкубацию с ДНК-ли- газой фага Т4. как описано выше. Лигазной смесью трансформировали штамм TG1. Клетки высевали на индикаторые чашки с X-gal и ИПТГ, содержащие ампициллин. Проводили отбор белых колоний. Из полученных клонов выделяли плазмиды и определяли нуклеотидную последовательность 3- -концевой части кДНК ГР КРС методом Сэн- гера. Для дальнейшей работы- отобрали плазмйду, содержащую EcoRI-Hindlll-фрагмент с геном ГР КРС размером 601 и.о.. обозначенную pBGHBal12.
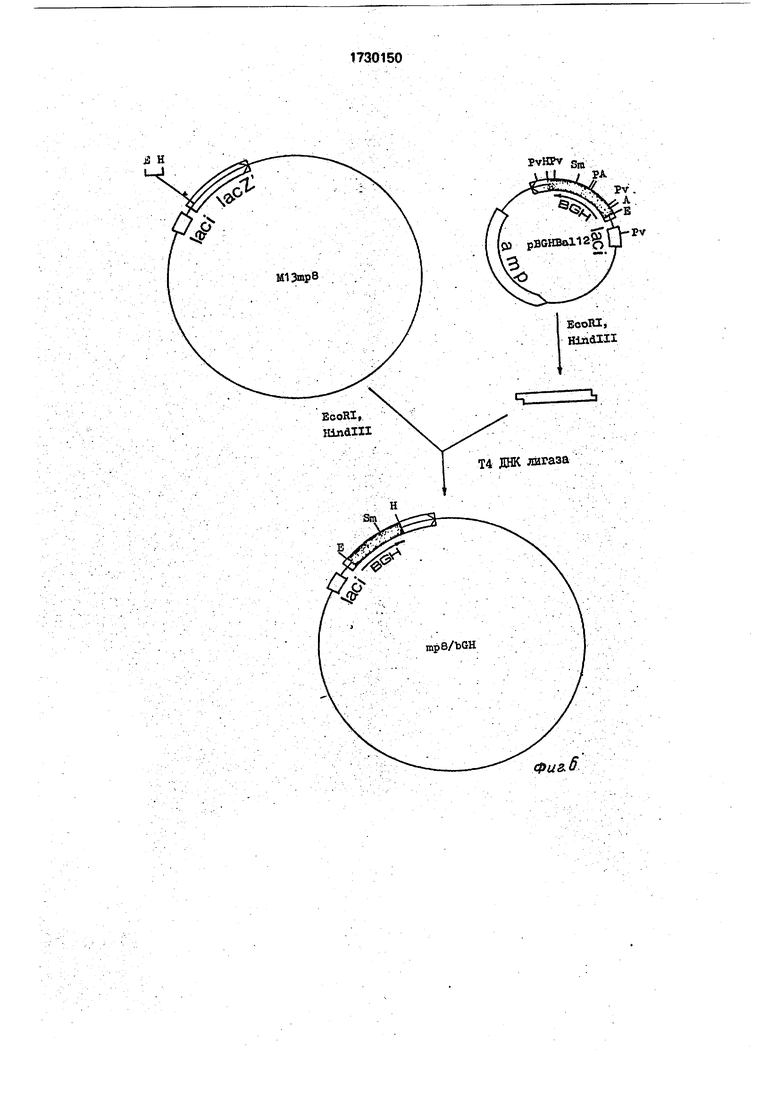
П рим ер2. Получение фрагмента ДНК, кодирующего ГР овцы. А) Клонирование фрагмента ДНК, кодирующего ГР КРС, в векторе М13 (фиг. 6).
10 мкг плазмидной ДНК pBGHBal12 гидролизовали рестриктазами EcoRI и Hindfll в 50 мкл инкубационной смеси следующего состава: SO mM NaC|, 10 тМ
5 трисНС. рН 7,5,10 mM MgCl2, 1 тМ дитиот- рейтола(ДТТ),10ед. рестриктазы EcoRI и 10 ед. рестриктазы Hlndlll. в течение 2 ч при 37°С. Проводили разделение фрагментов ДН К в 1 % ага- розном геле и выделяли из геля фрагмент раз® мером 601 п.о.. используя стандартный метод. 5 мкг ДНК репликативной формы бактериофага М13тр8 гидролизовали рестриктазами EcoRI и Hindlli, как описано выше. Продукты гидролиза разделяли в 1 % агароз5 ном геле и выделяли векторную ДНК размером около 7 тыс. п.о .. Растворы фрагмента и вектора смешивали в молярном соотношении 2:1, добавляли лигазный буфер, смесь инкубировали в течение 10 мин при 65°С. в
Q теченине 5 мин при комнатной температуре и помещали в лед. К смеси добавляли АТР до кон центрации 0.5 тМ и ДНК-лигазу фага Т4 (1 ед.) и инкубацию продолжали в течение ночи. Проводили трансформацию компетентных клеток Е. coll TG1, полученных кальциевым методом.
5 Для трансформации брали 10 мкл расг твора, содержащего 100 нг ДНК (в пересчете на репликативную форму ДНК фага М13тр8). смешивали раствор ДНК со 100 мкл суспензии компетентных клеток и инкубировали 30
0 мин при 0°С. 1.5 мин при 42°С и охлаждали смесь во льду. Добавляли 2 мл среды YT (5 г/л бактотриптона, 8 г/л дрожжевого экс- трактра, 5 г/л NaCI) и инкубировали 1 ч при 37°С. В стерильные пробирки разливали по
5 2 мл верхнего агара и выдерживали их в водяной бане с температурой 45°С. Добавляли в пробирки по 40 мкл 100 тМ раствора иэопропилтиогалактозида (ИПТГ) и 2% раствора X-gal, 100 мкл культуры Е. coli с плотQ ностью 0,3 о.е./мл и 100 мкл суспензии трансформированных клеток. Смесь тщательно перемешивали, выливали на поверхность чашки с нижним агаром. Чашки инкубировали в течение ночи при 37°С. С
бесцветных фаговых бляшек, образовав-/. шихся на бактериальном газоне, проводили заражение 2 мл ночной культуры штамма TG1, разбавленной в 100 раз двукратной средовй YT. Культуру выращивали на качалке
9 173015010
при 37°С в течение 6ч. Затем культуру4,5 мкл НгО.
центрифугировали и отбирали 1 мл суперна-Смесь прогревали при 80°С, медленно в
такта, содержащего фаговые частицы. До-течение 1 ч снижали температуру до 30°С,
бавляли к супернатанту 200 мкл 20%затем охлаждали во льду
раствора полиэтиленгликоля (М.в. 6000) в5 Реакцию пролонгирования праймеров
2,5 М растворе NaCt и оставляли на 15 минпроводили в инкубационной смеси следуюпри комнатной температуре. После центри-щего состава: .
фугирования в течение 5 мин супернатант1 мкл 10 х ТМ-буфера:
удаляли, а осадок растворяли в 100 мкл ТЕ-1 мкл смеси dNTP с концентрацией 5
буфера(50тМтрисНС, рН 8.0,1 тМЭДТА).10 тМ;
Раствор экстрагировали равным объемом1 мкл 100 тМ ДТТ;
фенола, насыщенного ТЕ-буфером и 5 объе-1 мкл 10 mM ATP;
мами эфира, насыщенного ТЕ-буфером.6 мкл Н2О;
ДНК из раствора осаждали этанолом и ис-. 10 мкл смеси, содержащей матрицу и
пользовали для секвенирования вставки ме-15 праймеры.
тодом Сэнгера и для проведенияДобавляли 1 мкл раствора фрагмента
мутагенеза.Кленова ДНК полимераэы 1 (5 ед.) и 1 мкл
Секвеннрование 6 полученных такимраствора ДНК-лигазы фага Т4(2 ед.) и инкуспособом препаратов ДНК показало, что 3бировали в течение ночи при 12°С. Смесь
из них содержат вставку фрагмента, кодиру-20 разбавляли в 10 раз раствором 10 тМ
ющего ГР КРС. Один из них, обозначенныйтрисНС. рН 8,0,10 тМ ЭДТА итрансформиmp8/bGH. использовали в дальнейшей рабо-ровали клетки штамма ВМН 71-18 mutL. Для
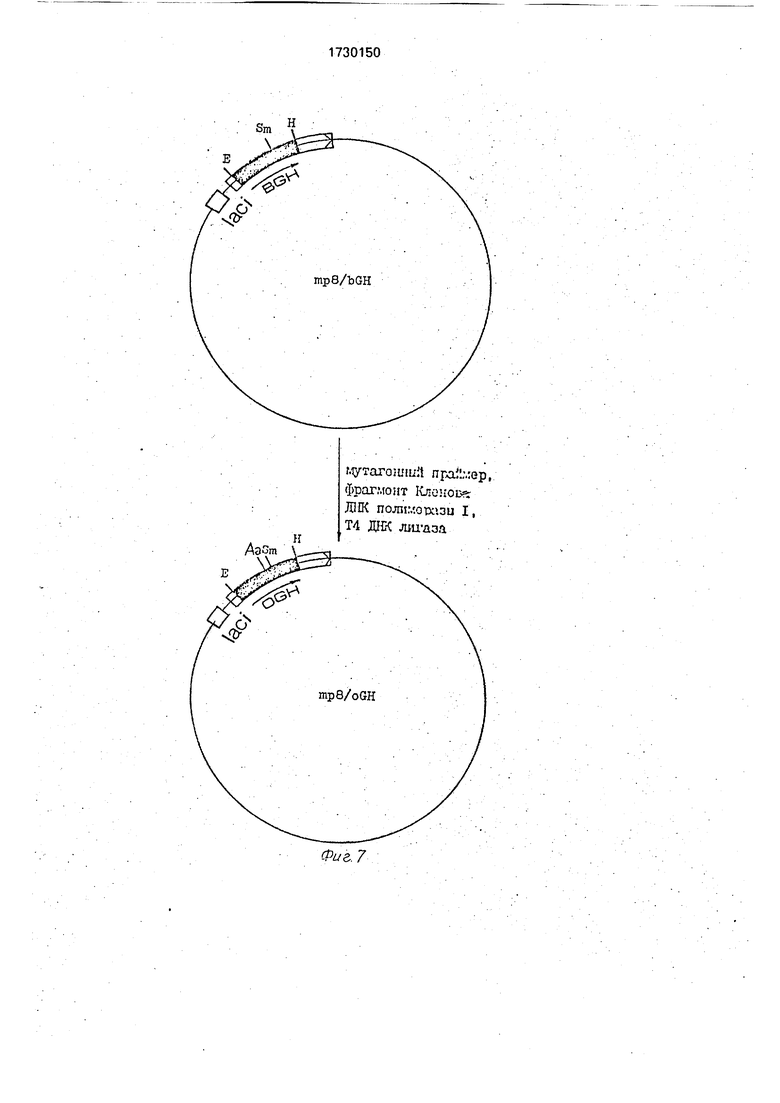
те.получения бактериального газона использоБ) Олигонуклеотид-направленный мута-вали штамм ВМН 71-18. ..
генез фрагмента, кодирующего ГР КРС (фиг.25 200 бляшек перекалывали на нитро7).целлюлозный фильтр и чашку-реплику, инДля проведения мутагенеза испольэо-кубировали 8 течение ночи при 37°С. вали синтетически олигонуклеотид (мутаген-Нитроцеллюлозный фильтр с колониями об- ный праймер), комплементарный участкурабатывали по известному методу. Фильтр гена ГР КРС. содержащий 2 точечные заме-30 инкубировали в 10мл раствора для предгиб- ны, одна из которых приводит к замене ко-ридизации{6х SSC, 10X раствор Денхардта, донаСОС(С1у 29) на GTC(Vat), т.е. позволяет0.2% SDS) в течение 1 ч при 65°С. Далее, превратить ген ГР КРС в ГР овцы. Втораяфильтр гибридизовали с меченым мутаген- замена приводит к образованию в нуклео-ным праймером в 10мл раствора 6XSSC. 10 тидной последовательноти фрагмента уча-35 X раствор Денхардта в течение 2 ч при ком- стка узнавания рестриктазы Aatflнатной температуре. Затем фильтр отмыва- (б -САССТС-З1). Структура .мутагенноголи 3 раза по 1 мин в 100 мл 6XSSC и праймера: 5-GGTGGTTGaCGTCTTCCAG-3 .авторадиографировали в течение 30 мин. Фосфорилирование праймера проводили вДалее проводили отмывку фильтра в том же инкубационной смеси следующего состава:40 растворе при 59°С и снова авторадиографи2 мкл 10 х буфера для полинуклеотидки-ровали. Для дальнейшего анализа отбирали
назы;клоны, гибридизующиеся с мутагенным
10 мкл раствора мутагенного праймерапраймером. Выделяли одноцепочную ДНК и
20 пмолей/мкл):определяли нуклеотидную последоватеяь2мкл 10 тМ раствора АТР,45 ность в участке, подвергавшемся мутагене- 6 мкл НаО;зу. В результате был получен бактериофаг 1 мкл полинуклеотидкиназы фага Т4 (5mp8/oGH со вставкой, кодирующей ГР овцы
ед.). . .и содержащей новый сайт расщепления ре- Смесь инкубировали 1 ч при 37°С и про-стриктазы Aatll на расстоянии 391 п.о. от
гревали 10 мин при 70°С. Отжиг мутагенного50 EcoRI-сайта.
и секвёнирующего праймеров на матрицеП р и м е р 3. Получение рекомбинантной
mp8/bGH проводили в инкубационной сме-плазмидной ДНК pOGHtrpH (фиг. 8).
си следующего состава:5 мкг репликативной формы фага
1 мкл раствора фосфорилированнргрmp8/oGH гидролизовали pectpHKTaaaMH
мутагенного праймера (10 пикомолей);.55 EcoRi и HindiII. как описано ранее, и выде0.5 мкл раствора универсального секве-ляли после разделения в 1 % агарозном геле
пирующего праймера (5 пикомолей);фрагмент, кодирующий ГР овцы, размером
3мкл матричной ДНК mp8/bGH (1 мкг);601-п.о..
1 мкл 10 х ТМ-буфера (100 тМ трисНС.10 мкг плазмидной ДНК pSTH2191 гидрН 7,5. 100 mM MgCl2);ролизовали реетриктазами EcoRI и Hindlll,
продукты гидролиза фракционировали в геле и выделяли больший фрагмент размером 5300 п.о..
Фрагмент и вектор сшивали ДНК-лига- зой фага Т4, как описано выше. Смесью трансформировали клетки штамма Е. coli DH1. Транформированные клетки высевали на чашки с агаризованной средой YT. содержащей 20 мг/л ампициллина и 5 мг/л тетрациклина, Полученные клоны выращивали в 3 мл среды YT. содержащей 20 мг/л ампициллина, в течение ночи. Клетки собирали центрифугированием, суспендировали в растворе, содержащем 50 тМ трисНС, рН 8,0, 1 тМ ЭДТА, 1% SDS и инкубировали в течение 10 мин на кипящей водяной бане. После охлаждения до комнатной температуры лизаты центрифугировали. Аликво ты ли- затов (10 мкл) наносили на полиакриламидный гель с SDS и проводили электрофорез по Лэммли. После электрофореза гель окрашивали Кумасси R-250 по стандартной методике. В результате анализа лизатов более чем 100 клонов был выявлен один клон, направляющий синтез полипептида с мол. массой около 22 KD, представляющего собой ГР овцы. Уровень синтеза этого белка составлял около 20% от суммарного белка клетки. Исследование плазмиды, выделенной из этого клона и обозначенной pOGHtrp11, показало, что она содержит кроме векторной части и фрагмента, кодирующего ГР овцы, дополнительный Hlndlll-фрагмент ДНК размером 916 п.о.. Этот фрагмент представляет собой часть 153-элемента Е. coli и имеет нуклеотидную последовательность, представленную на фиг. 3.
П р и м е р 4. Получение штамма Е. coli DH1 - продуцентра ГР овцы.
Плазмидой pOGHtrpU трансформировали штамм Е. coli DH1 по стандартной методике и получали штамм-продуцент ГР овцы. Штамм депонирован во Всесоюзной коллекции микроорганизмов при 1/1БФ АН СССР под номером ВКМ СР-326Д.
Содержание гормона е биомассе штамма определяли путем лизиса бактерий и измерения гормона в лизате методом иммуноферментного анализа. Накопление
ГР овцы в биомассе штамма ВКМ СР-326Д, содержащем плазмиду pOGHtrpll, составляло 50-60 мг на 1 л ночной культуры.
П р и м е р 5. Анализ накопления ГР овцы
в биомассе штамма ВКМ СР -326Д.
Штамм ВКМ СН-3-26Д выращивали в течение ночи в 3 мл среды YT. содержащей 25 мг/л ампициллина. Клетки осаждали центрифугированием и суспендировали в 100
мкл буфера, содержащего 100 тМ трисНС, рН 8,0. 1 тМ ЭДТА и -1% SDS. Суспензию прогревали в течение 10 мин на кипящей водяной бане, охлаждали до комнатной температуры и центрифугировали на центрифуге Эппендорф в течение 10 мин при 10000 об/мин. Отбирали 10 мкл супернатанта, добавляли 10 мкл буфера, содержащего 20% глицерина, 40% /3-меркаптоэтанола, 0,2 М трисНС, рН 7,0,8% SDS. 0,025% бромфенолового синего. Образец прогревали 5 мин при 100°С и подвергали электрофорезу в полиакриламидном геле с SDS. Электрофо- реграмма белков штамма ВКМ CR 326Д представлена на фиг. 9. Гель сканирования
на лазерном денситометре UltroScan XL фирмы LKB.
Результаты обработки данных представлены в табл. 2.
Как следует из табл. 2, содержание ГР
овцы в клетках штамма ВКМ СН-326Д достигает 20-25% от суммарного белка клеток.
Замена глицина на валин в аминокислотной последовательности приводит к увеличению молекулярной массы ГР овцы по
сравнению с молекулярной массой ГР КРС. Небольшое различие в молекулярных массах удается выявить при фракционировании соответствующих препаратов с помощью электрофореза в полиакриламидном геле.
ГР овцы мигрирует при электрофорезе в геле медленнее, чем ГР КРС.
Ф ор м у л а и з о б р е т е н и я 1. Фрагмент ДНК, кодирующий гормон роста овцы, полученный с помощью сайт- специфического мутагенеза фрагмента ДНК, кодирующего гормон роста крупного рогатого скота, размером 601 п.о., имеющий следующую последовательность нуклеоти- дов:
.:.-.- АЛТ
-1+1+10
МЕТ ЭД|Ј PRO ALA MEJ0SER LEU SER3GLY LEU PHE ALA ASM ALA
TCT ATG TTC CCA GCT ATG T.CT СТА TCT GGT СТА TTC GCT ЛАС GCT GTT CTC
+20 .-4-30
AfiG ALA6.gLN HIS LEU HJS, GLN LEU ALAQALA ASP THRgPHE LYS GLU PHE0GLU
CGG GCC CAG CAT CTT CAT CAG CTG GCT GCT GAC ACC TTC АЛЛ GAG TTT GAG
+ 40
ARC THpjJYR ILE GLY GLN ARGQTYR SER ILE4gLN ASN VAL
CGC ACC TAC ATC CCG GAG GGA CAG AGA TAG ТСС АТС CAG AAC ACC CAG GTT
+50:+60
ALA PH|0CYS PHE THR ILEjggO ALA PRO THg0GLY LYS ALA
GCC TTC TGC TTC TCT GAA ACC ЛТС CCG GCC CCC ACG GGC AAG A AT GAG GCC
+70 : . +80
LYS SER A§P0LH:U GLU LEy.3bEUARG LEU LEU М.Уо11- GLN
AAA TCA GAC TTG GAG CTG CTTCGC ЛТс тСА CTG CTC CTC АТС CAG
+90+100
LEU GLY2| gO LEU GLN Py|QLEUSF.R PHE SER LEU
TCG TGG CTT GGG CCC CTG CAG TTC CTC AGC AGA GTC TTC ACC AAC AGC TTG
. +110
VAJ.0PHE GLY THg2§ER ASP TYR GLU LYS GLU GLU
GTG TTT GGC GAC CGT GTC TAT GAG AAG CTG AAG GAC CTG GAG GAA
+ 120- . +130
ILE LEU MET LEU GLU3AgP VAL THR ALA GLY
GGC АТС CTG.GCC CTG ATG CGG GAG CTG GAA GAC GTC ACC CCC CGG GCT GGG
+140+150
GLN ILE GLH THR TYJ0A5P LYS PilE4 }SP TilR ASN4|1ET ARG SER Aj|u
CAG АТС-CTC AAG CAG ACC TAT GAC AAA TTT GAC АСА ЛАС ATG CGC ACT GAC - .-. .-+160.-.-
ASP ALA LEy7J,EU LYS ASN4JJfR .GLY LEU LEASER CYS PH|OARG LYS ASP,.LgU
GAC GCG CTG CTC AAG AAC TAC GGT CTG CTC TCC TGC TTC CGG AAG GAC CTG
+ 170:+160:
HIS LYS Tyg0GLU THR .U ARG LYS CYS PHE GLYegLU
CAT AAG ACG GAG ACG ..TAG CTG AGG GTC ATG AAG TGC CGC CGC TTC GGG GAG
+Г90:; .
ALA SEU5Cp ALA PHE .
GCC. AGC TGT GCC TTC TAG TTGCCAGCCATCTGTTGTT






Изобретение относится к микробиологической промышленности, генетической инженерии и биотехнологии. Цель изобретения - создание штамма-продуцента гормона роста овцы. Сконструированы ген, кодирующий зрелый гормон роста овцы, ре- комбинантная плазмидная ДНК, pOGHtrp 11. содержащая данный ген под контролем промотора trp-оперона Е. coil. Получен штамм, трансформация которого плазмид- ной pOGHtrp 11 приводит к эффективному и стабильному синтезу гормона роста овцы. 3 „. с.п.ф-лы, 8 ил., 2 табл.5 со С
EcoRI-Hind III - фрагмент плазмиды pSt.H2.191 размером 5300 п.о.:
EcoRI-Hind III - фрагмент, кодирующий гормон роста овцы, размером 601 п.о.;
Eco.RI-H.lnd III - фрагмент 153-элемента размером 916 п.о.;
1 участок расщепления рестриктазы EcoRI;
1участок расщепления рестриктазы Sma1, расположенный на расстоянии 427 п.о. от ЕсоЯ1гсайта;
3 участка расщепления рестриктазы Pstl, расположенные на расстоянии 300, 1353 и 5095 п.о. от EcoRI-сайта;
2участка расщепления рестриктазы Hind III, расположенные на расстоянии 601 п.о. и 1517 п.о. от EcoRT-сайта;
Т участок расщепления рестриктазы EcoRV, расположенный на расстоянии 1673 п.о. от EcoRt-сайта;
1 участок расщепления рестриктазы
ВатН1, расположенный на расстоянии 1863 п.о. от EcoRI-сайта;
1 участок расщепления рестриктазы SalGt, расположенный на расстоянии 2138 п.о. от EcoRI-сайта;
1 участок расщепления рестриктазы Hpal, расположенный на расстоянии 6784 п.о. от EcoRI-сайта;
генетические маркеры:
Ыа-ген, обеспечивающий синтез fi-пак- тамазы - устойчивость к ампициллину, расположенный в участке от 1200 до 2100 п.о. против часовой стрелки от Hpal-сайта;
tet-ген, обеспечивающий устойчивость к тетрациклину, расположенный в участке от 1500 до 2500 п.о. по часовой стрелке от EcoRI-сайта:.
перед последовательностью, кодируещей гормон роста овцы, расположена промотор- ная область триптофанового оперона Е. coll.
Положения сайтов рестрикции на физической карте реком- бинантной плазмидной ДНК pOGHtrp Г
Уровень синтеза ГР овцы клетками штамма ВКМ. CR-326D (E.coliDH1/hOGHtrp11)
.Таблица
Таблица 2
. АЛТ
-1 +1 +10
МЕТ РЭД PRO ALA MEJ0SEn LEU LEU PHE Ay ASN ALA
TCT ATG TTC CCA GCT ATG ТОТ СТА TCT GGT СТА TTC GCT AAC GCT GTT CTC
+20+30
ARG A.LAgGJLN HIS LEU ,RJ§ GLN LEU ASP THRgF,HE LYS GLU PHg0GLU
CGG GCC CAG CAT CTT CAT CAG CTG GCT GCT GAC ACC TTC.AAA GAG TTT GAG
. - : +40 . : : -.: :
ARG THHjTYR ILE-PROjC U GLY GLN ARG0TYR SER ILE4gLN ASN TH Rt.gЈN VAL
CGC ACC ТЛС АТС CCG GAG GGA CAG AGA TAG ТСС АТС CAG AAC ACC CAG GTT
ALA .CYS РИЕ THR ILEjgRO ALA PRO LYS ALA
GCC TTC TGC TTC TCT GAA ЛСС АТС CCG GCC CCC ACG GGC AAG AAT GAG GCC
. +70+80
LYf) SER A5C0I.KU GLU LEU.3L,EU ARG LEU LEU GLN
CAG CAG AAA TCA GAC TTG GAG CTG CTT CGC CTG стс стс АТС CAG
. , +90:+100
SEggJHP LEU . LEU GLN PggeLEU SER РЫЕ SEB LEU
TCG TGG CTT GGG CCC C1G CAG TTC CTC AGO AGA GTC TTC ACC AAC AGC TTG
+110 :V§L0PHE GLY ASP TYR GLU LYS GLU GLU
GTG. TTT GGC ACC TCG GAC CGT GTC TAT GAG AAG CTG AAG GAC CTG GAG GAA
+120 +130
, ILE LEU MET ARg8GLU LEU VAL THR .RG ALA GLY
GGC GCC CTG ATG CGG GAG CTG GAA GAC GTC ACC CCC CGG GCT GGG
Ч-10 У .:-.: .+150
GLN ILE GLH THR LYS THR ASN4| gt ABG SER
CAG AJЈ CTC AAG CAG ACC TAT GAG AAA TTT GAC АСА ЛАС ATG CGC ACT GAC
+160 ASP ALA LYS GLY LEU LgyoSER CYS LYS ASPgL§U
GAC GCG CTG CIC AAG AAC TAG GGT CTG CTC TCC TGC TTC CGG AAG GAC CTG
+ 170..:+180
HIS LYS TtJK0GLU THR AHG VALg.gT LYS CYS PH.E
CAT AAG ACG GAG ACG TAG-CTG AGG GTC ATG AAG TGC CGC CGC TTC GGG GAG
;490
ALASER p ALA P1IE S
GCC AGC TGT GCC TTC TAG TTGCCAGCCATCTGTTGTT
Фиг.1
102030-405060
AAGCTTGCTGAACGCATCGGTGTTACTGCCGCAGCCCGTGAACTCACCCTGTATGAATCA
700090100ПО 120
CAACTCTACAACTCGCGCAGTAAACAGCAAAATCAGCAGACGTCTTCTGAACGTGAACTG
130HO150160170160
GAGATGTCTACCGAGATTGCACGTCTCAAACGCCAGCTGGCAGAACGGGATGAAGAGCTG
190200210220.230240
GCTATCCTCCAAAAGGCCGCGACATACTTCGCGAAGCGCCfGAAATGAAGTATGTCTTTA
250260270280290300
TTGAAAAACATCACGCTGAGTTCAGCATCAAAGCAATGTGCCGCGTGCTCCGCGTGGCCC
310320330340350360
GCAGCGGCTGGTATACGTGGTGTCAGCGGCGGACAAGGATAAGCACGCGTCAGCAGTTCC
370300390400410 420
GCCAACACTGCGACAGCGTTGTCCTCGCGCCmTACCCGGTCAAAACAGCCTTACGGTG
430440450460470 480
ССССЛССССТСЛСССЛТСЛЛСТСССТССТСАСССТТЛССССТТТЛЛССТЛАЛАЛСССТСС
4901500010520330-540
CCGCAACCCTGCCCCGTCACGGACTGACCGC/iAAaGCCTCCCCGAAGTTCAGCCCCCTCA
550360570500500600
GCTACCCCGCACACGGCCTGCCTCTGTCAGAAAATCTGTTCGACCACJCATmTACCCCA
. сюего630еюс: ообо
CTGGCCCCAACCAGAAGTCCGCAGGACACATCACGTACTTACGTACACATGA/l(UX:T(X;C
670 600 690 700 710 720 ТСТЛТСТСССЛСТССТСЛТТСЛССТСТССГСЛССТСССаТТЛТТСССТССТСААТСТССС
790 COO 610 820 830 640 GGCCCCGGAACGTTATCGTTCACACGGACCGTGGAGGCCAGTACTGTTCACCAGATTATC
850 860 670 880 890 900 AGGCGCAACTGAAGCGGCATAATCTGCGTGGAAGTATGAGCGCAAAAGGTTGCTGCTACG
910
ATAATGCCTGCGTGGA
Фиг.З
Фиг.2-.
A Pv I 1
синтетический фрагмент ДНК
T4 ДЩ лигаза
T.BamHI
«tf
4
г. 7
цггагошшй .:ep, фрагмент Клоно й Д11К полиморази Г, Т4 ЛЦК лш-азл
| ЕПВ, 0125818 | |||
| кл | |||
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
