Изобретение относится к способам получения биологически активных соединений, меченных радиоактивными изотопами.
Простагландины (ПГ) являются низкомолекулярными биорегуляторами липидной природы. Механизм действия простагландинов и их роль в регуляторных процессах организма изучены недостаточно. В то время известно, что простагландин F2α участвует в регуляции дыхательной, репродуктивной и других функций, роль ПГF3α в живых системах до сих пор не выяснена. Меченный тритием ПГF3α может быть использован для изучения метаболизма, механизма действия, а также для определения уровня эндогенного ПГF3α в тканях человека и животных методом радиоиммунного анализа. Препараты [3H]ПГF3α не выпускаются ни в СССР, ни за рубежом. Таким образом, синтез меченного тритием простагландина F3α является актуальной задачей.
Известен способ получения [3H]ПГF3α, в котором [3H]ПГF3α получают в две стадии.
1. К раствору 2,3 ГБк [5, 6, 8, 9, 11, 12, 14, 15, 17, 18-3Н] тимнодоновой кислоты (ТК) с молярной радиоактивностью 9,25 ПБк/моль и 150 мкл этилового спирта добавляют 3,63 мл воды, 250 мкл 200 мМ К-фосфатного буфера (рН7,4), содержащего 0,5% Lubrol PX, 100 мкл 100 мМ раствора L-адреналина, 50 мкл 200 мкМ раствора гемина, 88 мкл 100 мМ раствора восстановленного глутатиона. Предынкубируют реакционную смесь 5 мин при 23оС и добавляют предварительно проинкубированную при 23оС смесь 500 мкл препарата ПГН-синтетазы (активность 7,5 мкмоль арахидоновой кислоты˙мин-1˙мл-1 при 25оС, рН8) и 250 мкл ПГН-ПГЕ изомеразы (активность 0,6 мкмоль ПГЕ2 за 30 мин на 1 мл ферментного препарата при 32оС, рН 7,4). Выдерживают инкубационную смесь 5 ч при 23оС, затем добавляют 86 мкл 2М раствора лимонной кислоты и экстрагируют этилацетатом (5х20 мл). Экстракт сушат безводным сульфатом натрия, фильтруют, растворяют в 5 мл метанола. Выход [3H]ПГЕ3 47-50%. Очистку [3H] ПГЕ3 осуществляют методом ВЭЖХ на колонке Separon 5С 18, 3,3х150 мм (ЧССР). Получают 0,93 ГБк [3H] ПГЕ3 с молярной радиоактивностью 7,4 ПБк/моль.
II. Полученный [3H]ПГЕ3 восстанавливают до [3H]ПГF3α, добавляя к 0,42 ГБк [3H]ПГЕ3 2,5 мкг боргидрида натрия в 0,3 мл метанола и выдерживая смесь 5-7 мин при комнатной температуре. Затем реакционную смесь подкисляют 1 н. НСl до рН 3, упаривают метанол, разбавляют водой и экстрагируют этилацетатом (3х1 мл). Экстракты объединяют, промывают водой (2х1 мл), упаривают, растворяют в 0,1 мл метанола и очищают с помощью ТСХ. При этом [3H]ПГF3α отделяют от получающегося при восстановлении β-изомера. Получают [3H]ПГF3α с выходом 34% (в расчете на [3H] ПГЕ3 и молярной радиоактивностью 7,3 ПБк/моль. Выход [3H]ПГF3α в расчете на исходную ТК равен 16-17%.
Целью изобретения является увеличение выхода и молярной радиоактивности [3H]ПГF3α, а также упрощение процесса синтеза.
Поставленная цель достигается инкубацией [3H]ТК с концентрацией 25-500 мкМ с частично очищенным препаратом ПГН-синтетазы из везикулярных желез барана при соотношении 0,5-0,6 мкмоль [3H]ТК на 1 мг ПГН-синтетазы (или 60-70 нмоль [3H]ТК на единицу активности ПГН-синтетазы) при 15-50оС в течение 1-7 мин, после чего добавляют раствор SnCl2 в метаноле до концентрации 35-50 мМ.
Сущность изобретения заключается в быстром превращении исходной [3H]ТК под действием очищенного препарата ПГН-синтетазы из везикулярных желез барана за счет использования низких значений соотношения [3H]ТК и фермента ПГН-синтетазы и в последующем добавлении избыточного количества SnCl2.
Оптимизацию синтеза [3H]ПГF3α проводят относительно выхода [3H]ПГF3α, рассчитанного, исходя из количества кратномеченной тритием тимнодоновой кислоты, так как она является дорогостоящим продуктом трудоемкого многостадийного химического синтеза.
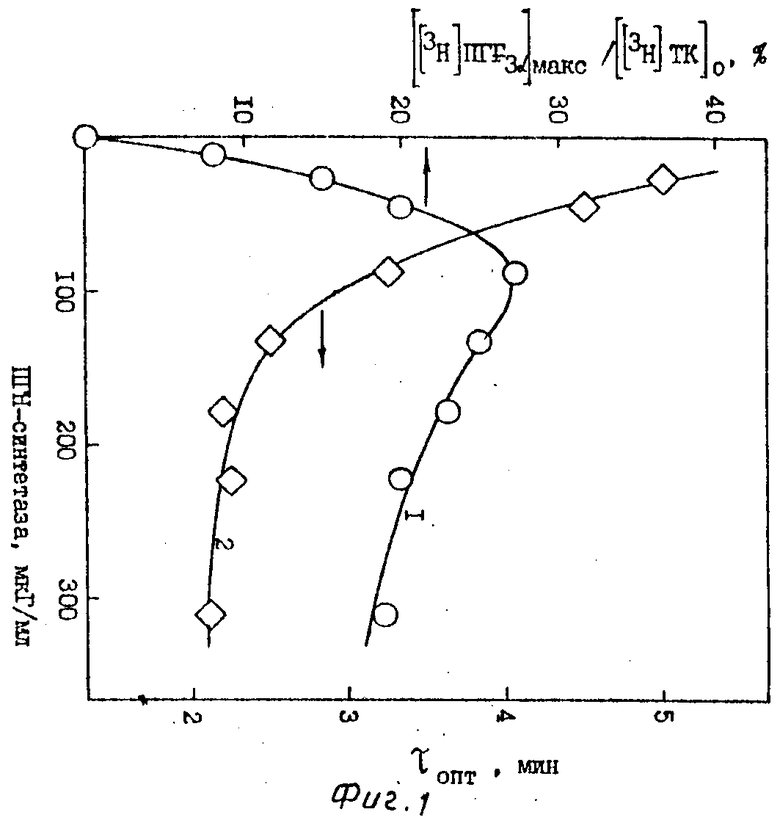
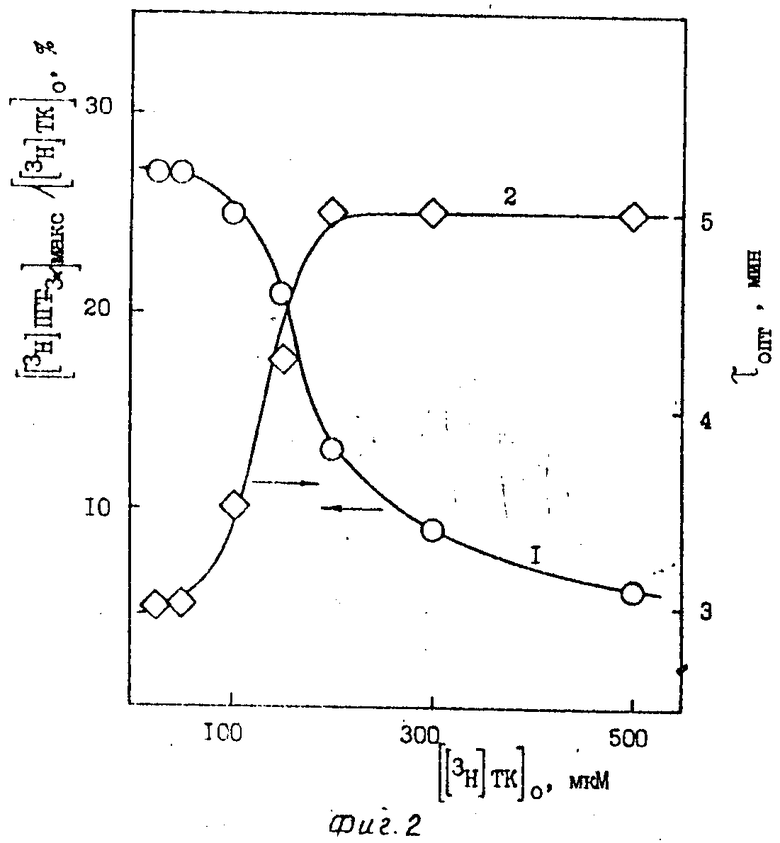
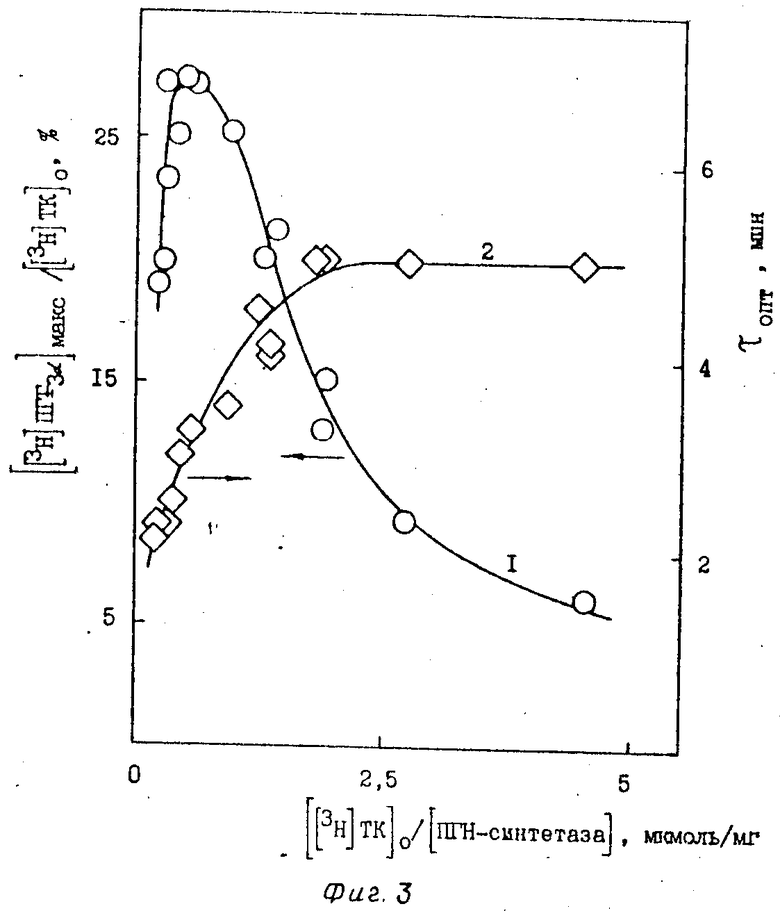
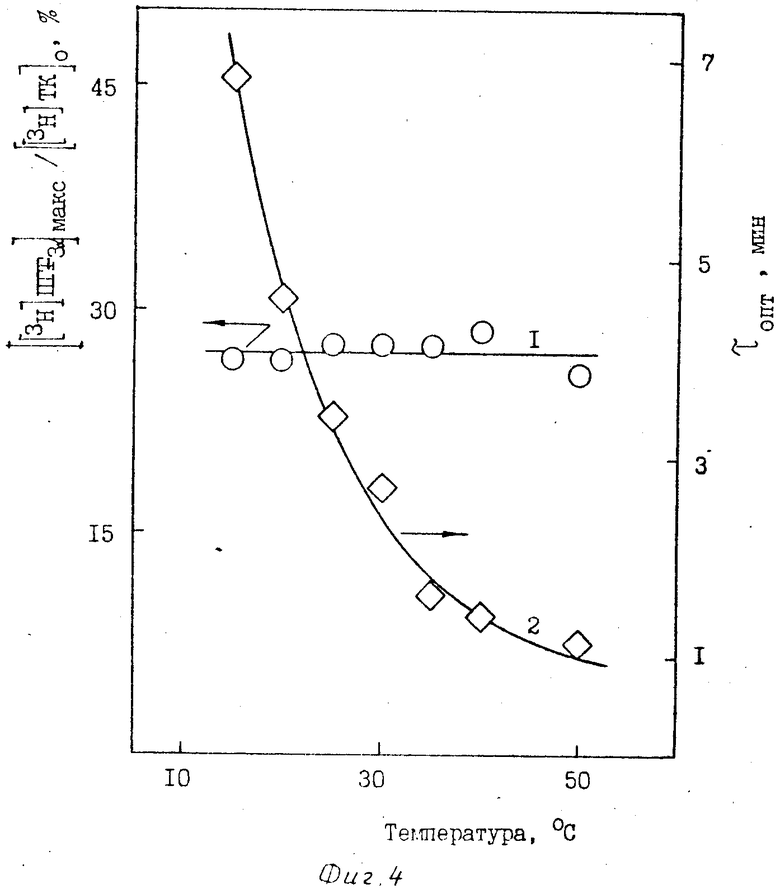
Для определения оптимальных условий синтеза [3H]ПГF3α исследуют зависимость выхода [3H]ПГF3α от концентрации ферментного препарата (фиг. 1), субстрата - [3H]ТК (фиг. 2), соотношения между количествами [3H]ТК и препарата ПГН-синтетазы (фиг. 3), температуры (фиг. 4), концентрации SnCl2 (фиг. 5) и времени инкубации.
Во всех экспериментах реакцию начинают добавлением препарата ПГН-синтетазы в реакционную смесь, содержащую [3H]ТК, а останавливают добавлением раствора SnCl2 в метаноле (кроме зависимости на фиг. 5). Время, за которое концентрация [3H]ПГF3α (после добавления SnCl2 к инкубационной смеси) достигает максимального значения, обозначают как τопт.
Выход [3H]ПГF3α достигает максимума при концентрации препарата ПГН-синтетазы 105 мкг/мл при начальной концентрации [3H]ТК 50 мкМ (фиг. 1). Изменение выхода [3H]ПГF3α в зависимости от начальной концентрации [3H]ТК (фиг. 2) определяют при концентрации ПГН-синтетазы, равной 105 мкг/мл. Максимальный выход [3H]ПГF3α (27-30%) достигается при использовании низких концентраций исходной [3H]ТК - 25-50 мкМ. На фиг. 3 представлены результаты, показывающие, что при варьировании концентрацией [3H]ТК (от 25 до 500 мкМ) и ПГН-синтетазы (от 10 до 350 мкг/мл) выход [3H]ПГF3α определяется только соотношением этих параметров. Максимальный выход [3H]ПГF3α наблюдается в узком диапазоне значений соотношением [3H] ТК и ПГН-синтетазы - 0,5-0,6 мкмоль [3H]ТК на 1 мг ПГН-синтетазы (или 60-70 нмоль [3H]ТК на единицу активности ПГН-синтетазы); τопт. при этом равно 3,4-3,6 мин. Наблюдается резкое снижение выхода [3H]ПГF3α при уменьшении этого соотношения, что связано с сильной сорбцией [3H]ТК на балластных белках, присутствующих в препарате ПГН-синтетазы. Выход [3H]ПГF3α не зависит от температуры инкубации (в интервале 15-50оС) при соответствующих τопт. (фиг. 4).
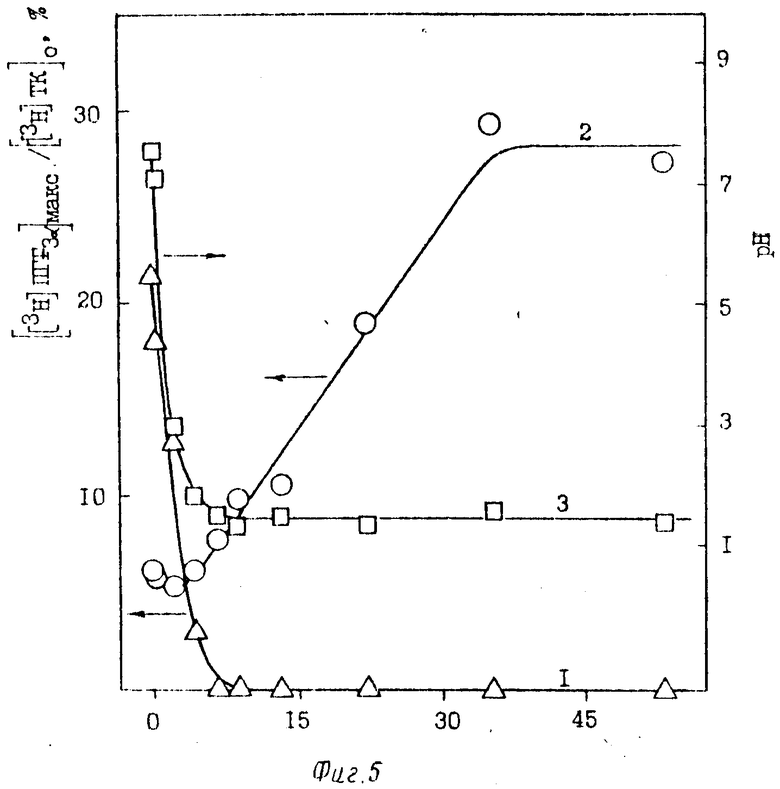
Максимальный выход [3H]ПГF3α (при τ= 3,5 мин) достигается при концентрациях SnCl2, равных 35-50 мМ (фиг. 5). При этих концентрациях добавляемый в инкубационную смесь SnCl2 приводит к снижению рН среды инкубации до 1,4-1,5 (фиг. 5, кривая 3). Изменение рН среды инкубации, вызванное добавлением SnCl2, приводит к снижению активности ПГН-синтетазы, и, как следствие, - выхода [3H]ПГF3α при τ= 0 мин.
Из представленных данных видно, что значение τопт. снижается при увеличении концентрации ПГН-синтетазы, при снижении концентрации исходной [3H] ТК и увеличении температуры инкубации.
При добавлении SnCl2 в инкубационную смесь (конечная концентрация 10 мг/мл или 44 мМ) происходит изменение рН среды с 7,9 до 1,4-1,5 в связи с чем не требуется обычное подкисление инкубационной смеси лимонной кислотой перед экстракцией. Кроме того, при добавлении SnCl2 обнаруживается заметное увеличение сорбции продуктов реакции и тимнодоновой кислоты на денатурированных белках по сравнению с сорбцией в присутствии лимонной кислоты. В связи с этим требуется тщательная экстракция продуктов из реакционной смеси - не менее 5 экстракций 3-4 объемами этилацетата.
Было обнаружено, что тимнодоновая кислота в отличие от арахидоновой и дигомо- γ-линоленовой значительно более интенсивно сорбируется на белках, присутствующих в препарате фермента. Поэтому при синтезе [3H]ПГF3α очень важно использовать препарат ПГН-синтетазы с высокой специфической активностью. Препарат ПГН-синтетазы из везикулярных желез барана очищают с помощью хроматографии на ДЕАЕ-целлюлозе. Специфическая активность ПГН-синтетазы увеличивается в 7,5-10 раз по сравнению с активностью микросомальной фракции.
Таким образом, инкубация [3H]ТК с концентрацией 25-500 мкМ с очищенным препаратом ПГН-синтетазы при их соотношении 0,5-0,6 мкмоль [3H]ТК на 1 мг ПГН-синтетазы (или 60-70 нмоль [3H]ТК на единицу активности ПГН-синтетазы) при 15-50оС в течение 1-7 мин с последующим добавлением раствора SnCl2 в метаноле позволяет получать [3H]ПГF3α с выходом по радиоактивности относительно исходной [3H]ТК 27-30% и молярной радиоактивностью 8,1 ПБк/моль.
П р и м е р. Получение [3H] простагландина F3α.
1. Получение ПГН-синтетазы.
100 г везикулярных желез барана в 200 мл 500 мМ трис-НCl буфера, рН8, содержащего 0,5 мМ ЕДТА 0,1 мМ диэтилдитиокарбамата, 0,1% неионного детерганта Lubrol PX, (буфер А), охлажденного до 4оС, гомогенизируют в Waring Comm. Blender (8 раз по 0,5 мин) и центрифугируют 15 мин при 12х х103 g. Супернатант (200 мл) фильтруют через 4 слоя марли и центрифугируют 90 мин при 100х103g. Осадок гомогенизируют в 180 мл буфера А и центрифугируют 90 мин при 100х103g. Осадок (микросомальная фракция) ресуспендируют в 42 мл буфера А и солюбилизируют мембранные белки добавлением 4,2 мл 10% Tween 20. Смесь выдерживают 30 мин при 4оС, затем центрифугируют 90 мин при 100х103 g 42 мл солюбилизированного ферментного препарата наносят на колонку (2,5х30 см) с ДЕАЕ-целлюлозой, предварительно уравновешенной 20 мМ К-фосфатным буфером рН 7,4, содержащим 0,5 мМ ЕДТА 0,1 мМ диэтилдитиокарбамата и 0,05% Lubrol PX. ПГН-синтетазная активность элюируется этим же буфером.
Получают 60 мл ферментного препарата ПГН-синтетазы (концентрация белка 0,89 мг/мл; специфическая активность 7,4 мкмоль арахидоновой кислоты˙мин-1˙мг-1 при 25оС в трис-буфере, рН8).
II. Получение [3H]ПГF3α.
К раствору 2,3 ГБк (0,25 мкмоль) [5, 6, 8, 9, 11, 12, 14, 15, 17, 18 (n)-3Н] тимнодоновой кислоты с молярной радиоактивностью 9,25 ПБк/моль в 0,15 мл этилового спирта добавляют 3,61 мл воды, 0,5 мл 200 мМ калийфосфатного буфера, рН 7,8, содержащего 0,5% Lubrol PX, 0,1 мл 100 мМ раствора L-адреналина, 0,05 мл 200 мкМ раствора гемина. Прединкубируют реакционную смесь 5 мин при 25оС и добавляют 0,59 мл предварительно проинкубированного в течение 5 мин при 25оС препарата ПГН-синтетазы.
Выдерживают реакционную смесь 3,5 мин при 25оС при интенсивном перемешивании, затем добавляют 0,5 мл раствора SnCl2 в метаноле (100 мг/мл) и экстрагируют этилацетатом (5х25 мл). При этом в экстракте оказывается не менее 90% всей радиоактивности. Экстракт сушат безводным сульфатом натрия, фильтруют, упаривают, растворяют в 5 мл метанола. Выход [3H]ПГF3α определяют с помощью ТСХ продуктов реакции на пластинках Силуфор (ЧССР) в системе растворителей хлороформ- метанол-уксусная кислота (85:14:1, v/v/v). Распределение радиоактивности по пластинке определяют с помощью сканирования. Выход [3H]ПГF3α 27-30%. Очистку [3H]ПГF3α осуществляют методом ВЭЖХ следующим образом. Метанольный раствор продуктов синтеза упаривают, остаток растворяют в 0,15 мл метанола, фильтруют через патрон с обращенной фазой Servachrom Si100 = Polyol - RP 18-30 мкм ("Serva"), промывают 2 раза 0,15 мл метанола, упаривают, растворяют в 0,1 мл 20%-ного раствора ацетонитрила в воде, содержащего 0,1% уксусной кислоты, и вводят в колонку (Separon 5C 18,33 x 150 мм, ЧССР). Элюацию проводят 33-ным раствором ацетонитрила в воде, содержащим 0,1% уксусной кислоты, со скоростью 0,5 мл/мин. Получают 0,64 ГБк [3H] ПГF3α с молярной радиоактивностью 8,1 ПБк/моль и радиохимической чистотой 98%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КРАТНОМЕЧЕННЫХ ТРИТИЕМ ПРОСТАГЛАНДИНОВ ТИПА E ИЛИ ИХ ПРОИЗВОДНЫХ ТИПА A, B, F, ИЛИ МЕТИЛОВЫХ ЭФИРОВ ПРОСТАГЛАНДИНОВ ТИПА J | 1988 |
|
SU1646252A3 |
| СПОСОБ ПОЛУЧЕНИЯ МЕЧЕННОГО ТРИТИЕМ ТРОМБОКСАНА B | 1990 |
|
SU1732513A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОСТАГЛАНДИНА F | 1990 |
|
RU1774659C |
| СПОСОБ ПОЛУЧЕНИЯ МЕЧЕННОГО ТРИТИЕМ ТРОМБОКСАНА B | 1990 |
|
SU1732514A1 |
| КРАТНОМЕЧЕННАЯ ТРИТИЕМ ПО ДВОЙНЫМ СВЯЗЯМ 15-ГИДРОКСИ-5Z,-8Z,11Z, 13Е-ЭЙКОЗАТЕТРАЕНОВАЯ КИСЛОТА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1993 |
|
RU2073665C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ ЧЕТВЕРТИЧНЫХ АМИНОВ, СОДЕРЖАЩИХ МЕЧЕНЫЙ ТРИТИЕМ АРОМАТИЧЕСКИЙ РАДИКАЛ ПРИ АТОМЕ АЗОТА | 1995 |
|
RU2089532C1 |
| СПОСОБ ПОЛУЧЕНИЯ КРАТНОМЕЧЕННОГО ТРИТИЕМ ПО α-ПОЛОЖЕНИЮ АМИНОКИСЛОТНЫХ ФРАГМЕНТОВ ГЕКСАПЕПТИДА | 1988 |
|
SU1736126A3 |
| СПОСОБ ПОЛУЧЕНИЯ КРАТНОМЕЧЕННЫХ ТРИТИЕМ ПО ДВОЙНЫМ СВЯЗЯМ НЕНАСЫЩЕННЫХ ПРОСТЫХ ЛИПИДОВ | 1988 |
|
SU1582553A3 |
| Способ получения [4,5- @ Н] докозагексаеновой и [5,6- @ Н] тимнодоновой кислот | 1988 |
|
SU1631942A1 |
| ВЫСОКОМЕЧЕННЫЙ ТРИТИЕМ [H]-7-ХЛОРО-1,3-ДИГИДРО-1-МЕТИЛ-5-ФЕНИЛ-2H-1,4- БЕНЗДИАЗЕПИН-2-ОН И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2003 |
|
RU2247116C2 |
Использование: получение биологически активных соединений, в том числе меченных радиоактивными изотопами. Сущность изобретения: для получения меченного тритием простагландина F3α используют очищенный препарат ПГН-синтетазы, который инкубируют в меченной тимиодоновой кислотой при соотношении 60 - 70 нмоль на единицу активности ПГН-синтетазы при 15 - 50°С в течение 1 - 7 мин, с последующим добавлением SnCl2 в метаноле до концентрации 35 - 50 мМ. Способ позволяет увеличить выход меченого простагландинсинтетазы F3α в 1,6 - 1,9 раза, молярную радиоактивность на 11%. 5 ил.
СПОСОБ ПОЛУЧЕНИЯ МЕЧЕННОГО ТРИТИЕМ ПРОСТАГЛАНДИНА F3α путем инкубации фермента с меченной тритием тимнодоновой кислотой в присутствии этанола, гемина, адреналина, отличающийся тем, что, с целью увеличения выхода и молярной радиоактивности простагландина F3α и упрощения процесса, в качестве фермента используют препарат ПГН-синтетазы, инкубацию его с меченной тритием тимнодоновой кислотой проводят при соотношении 60 - 70 нмоль меченой тимнодоновой кислоты на единицу активности ПГН-синтетазы в течение 1 - 7 мин с последующим добавлением SnCl2 в метаноле до концентрации 35 - 50 мМ.
| СПОСОБ ПОЛУЧЕНИЯ КРАТНОМЕЧЕННЫХ ТРИТИЕМ ПРОСТАГЛАНДИНОВ ТИПА E ИЛИ ИХ ПРОИЗВОДНЫХ ТИПА A, B, F, ИЛИ МЕТИЛОВЫХ ЭФИРОВ ПРОСТАГЛАНДИНОВ ТИПА J | 1988 |
|
SU1646252A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
