Изобретение относится к химической технологии, конкретно к способу получения -хлормолочной кислоты (ХМК) - исходного продукта для получения мономеров, служащих основой для получения негорючих органических стекол.
Известен способ получения ХМК окислением эпихлоргидрина 70%-ной азотной кислотой при температуре 46-68°С и мольном соотношении эпихлоргидрина и азотной кислоты 1:(2 5-4,0) с последующим разложением избытка азотной кислоты 37%-ным раствором формальдегида при 65-68°С. Получают целевой продукт с выходом 84,6%.
Недостатком известного процесса является его сложность чз-за применения формальдегида.
Известен способ получения ХМК, согласно которому гроцесс окисления эпихлоргидрина азотной кислотой проводят
при температуре 55°С и мольном роотноше- нии компонентов ЭПХГ и НМОз 1:2,65, с последующим разложением избытка азотной кислоты формальдегидом. Выход целевого продукта 86,1 %.
Недостатком описанной технологии яв ляется низкое качество целевого продукта, большой расход формальдегида, который разлагается деструктивно, а также высокая опасность процесса разложения и избыточной азотной кислоты формальдегидом вследствие большого теплового эффекта реакции.
Наиболее близкие по технической сущности и достигаемым результатом явлчется способ получения / -хлормолочной кислоты окислением эпихпоргидрина 70%-ной частной кислотой при их вольном соотношении 1:2,5-4,0 при 4б-68°С. После окисления эпихлоргидрина проводят частичное упаривание реакционной массы при температуре
VI
00
ю чэ
VI Ы
55-57°С и остаточном давлении 130-150 мм рт.ст,, охлаждают до 15-18°С, отделяют фильтрацией выпавшие кристаллы побочного продукта - щавелевой кислоты, фильтрат повторно упаривают при 10-30 мм рт.ст. и температуре 55-57°С. Получают целевой продукт в виде плава с массовой долей основного вещества 84% и выходом 64,5% в расчете на опихлоргидрин (ЭПХГ) 1.
Недостатком данного способа является высокий расход концентрированной азотной кислоты, низкий выход и, как следствие, низкая экономичность процесса.
Целью изобретения является повышение экономичности процесса за счет более полного и эффективного использования сырья, а также повышение выхода и качества целевого продукта.
Поставленная цель достигается тем, что процесс окисления эпихлоргидрина водным раствором азотной кислоты проводят в две стадии: на первой стадии смешивают элихлоргидрин с 60-80%-ной азотной кислотой при температуре 8-20°С в мольном соотношении 1:(2,0-2,8) На второй стадии смесь выдерживают под давлением 0,3- 0,6 МПа и температуре 60-80оС. В реакционную массу на второй стадии добавляют 10-40%-ную азотную кислоту, предварительно обработанную отходящими окислами азота. Мольное соотношение ЭПХГ и разбавленной азотной кислоты 1:(0,2-0,8). Температура обработки разбавленной азотной кислоты отходящими окислами азота 20-30°С. На заключительной стадии процесса в реакционную массу подают воздух в количестве 0,1-1,0 мас.ч на 1 мае.ч. введенного в процессе ЭПХГ.
Массу упаривают в вакууме, постепенно углубляя его от 130 до 100 мм рт.ст., отгоняют 10%-ную азотную кислоту в количестве 1/10 мас.ч. от общей массы реакционной массы. После обработки отходящими окислами азота разбавленная азотная кислота укрепляется и вновь возвращается в процесс на вторую стадию.
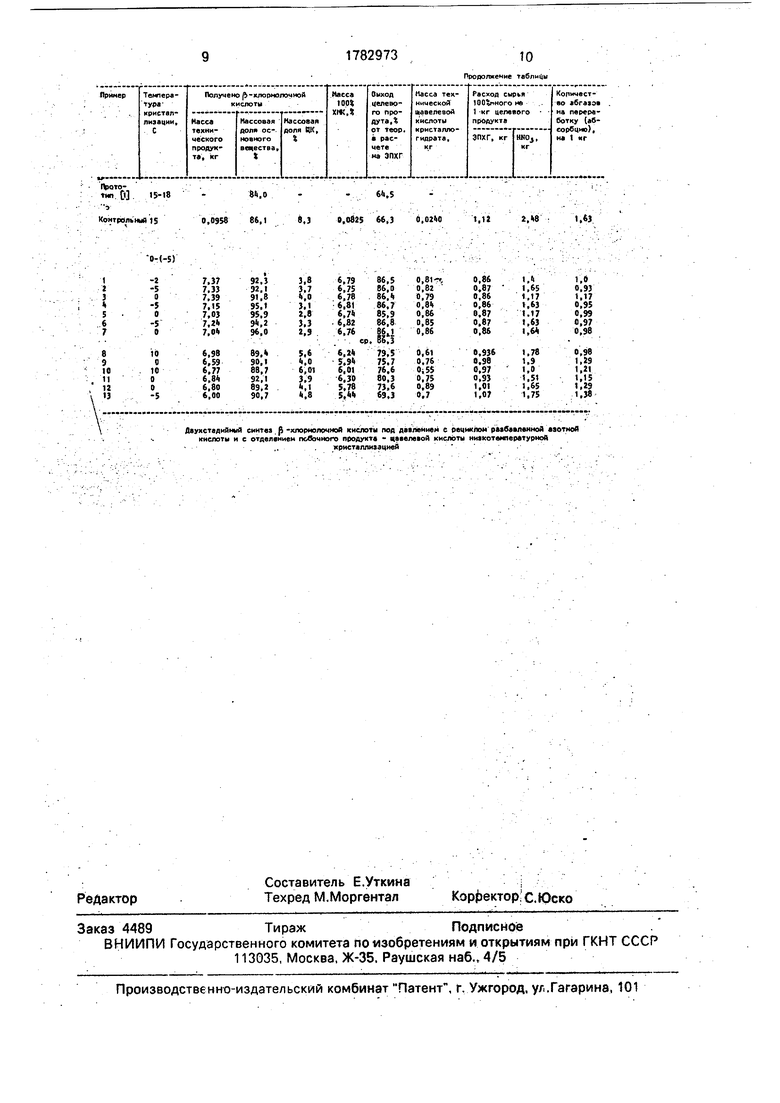
Массу после отгонки слабой кислоты охлаждают до 0-(-5)°С, отделяют фильтрацией выпавшие кристаллы щавелевой кислоты, фильтрат упаривают в вакууме, при температуре в массе 55 ± ёиС постепенно углубляя его от 100 мм до 40 мм, так как состав массы меняется в процессе отгонки. Получают целевой продукт в виде обезвоженного плава с массовой долей основного вещества 90- 92%, выходом 84-86,5% в расчете на ЭПХГ.
Отличительными признаками процесса является проведение его в две стадии: на первой стадии смешивают эпихлоргидрин и концентрированную азотную кислоту с массовой долей 60-80% в мольном соотношении эпихлоргидрина и азотной кислоты 1:2,0-2,8 при температуре 8-20°С, на второй стадии процесс ведут при температуре
в массе 60-80°С и давлении 0,3-0,6 МПа и вводят разбавленную 10-40%-ную азотную кислоту в количестве 0,2-0,8 моль на 1.моль эпихлоргидрина, предварительно обработанную отходящими газами при температу0 ре 20-30°С, на заключительной стадии процесса окисления в реакционную массу подают воздух в количестве 0,1 -1.0 мас.ч. на 1 мас.ч. введенного в процесс эпихлоргидрина, при этом частичное упаривание про5 водят при углублении вакуума от 130 до 100 мм рт.ст., охлаждение остатка проводят до 0 - -5°С, а повторное упаривание - при вакууме от 100 мм рт.ст. до 40 мм рг.ст ., что позволяет повысить эффективность про0 цесса, а также его выход и качество.
Разделение процесса окисления на две стадии - низкотемпературную и высокотемпературную, позволяет существенно упростить процесс с точки зрения регулирования
5 теплового режима. В-результате введения низкотемпературной стадии снимается 30-35% общего количества тепла целевого процесса. Смешение реагентов при температуре 8-20°С обуславливает мягкий гидро0 лиз ЭПХГ (раскрытие эпоксидного кольца) и перевод его в нитроэфир. Получение нитро- эфира с высоким выходом протекает при небольшом избытке азотной кислоты, что позволяет проводить эту стадию с количест5 вом азотной кислоты, близкой к теоретическому расходу на окисление 2,0-2,8 моль на 1 моль ЭПХГ.
Выбор верхнего предела смешения ЭПХГ с азотной кислотой 20°С определяет0 ся устойчивостью реакционной массы. При температуре ниже 20°С исключается саморазгон окислительной реакции, что важно для проведения процесса окисления з непрерывном режиме. Температуру ниже 8°С
5 поддерживать нецелесообразно ввиду непроизводительного расхода хладоагента без достижения положительного эффекта,
Применение на второй высокотемпературной стадии давления 0,3-0,6 МПа
0 позволяет повысить эффективность использования разбавленной азотной кислоты за счет увеличения растворимости окислов азота в жидкой фазе Нижний предел давления 0,3 МПа обусловлен возмож5 ностью конденсации четырехокиси азота при температуре реакции. Верхний предел определяется технико-экономическими соображениями, давление 0,6 МПа является одной из ступеней, давления для создания и применения типовой аппаратуры. В то же
время более высокое давление не дает существенного положитепьного эффекта в пределах выбранного температурного режима окисления.
Температурный режим второй стадии 60-80°С обуславливает достижение максимальной селективности процесса: ниже 60°С не обеспечивается достаточно глубокий уровень конверсии при приемлемой продолжительности процесса (4-5 ч). Выше 80°С селективность быстро падает из-за протекания побочных процессов деструктивного окисления.
Эффект применения на второй ступени разбавленной азотной кислоты достигается путем обработки этой кислоты отходящими газами (окислами азота) при температуре 20-30°С. При температуре выше 30°С снижается степень насыщения разбавленного раствора азотной кислоты окислами азота, температуру ниже 20°С держать нецелесообразно ввиду отсутствия положительного эффекта,
Массовая доля применяемой разбавленной кислоты на второй стадии обуславливается тем, что получаемая на стадии выделения целевого продукта упариванием азотная кислота имеет минимальное содержание основного вещества 10% (стадия предварительной отгонки) и 40% - после второй окончательной отгонки. Такая же 40%-ная азотная кислота получается со стадии абсорбции отходящих газов. Часть этой кислоты без дополнительной очистки и концентрирования может быть снова введена в процессе.
Введение воздуха в реакционную массу повышает концентрацию окисляющего агента - четырехокиси азота в жидкой и газовой фазе на заключительной стадии процесса окисления, что способствует более глубокой конверсии и эффективности использования разбавленной азотной кислоты. Количество воздуха 0,1-1 мае.ч. на 1 мае.ч, ЭПХГ. Нижний предел расхода воздуха 0,1 мае.ч. на 1 мае.ч, ЭПХГ обуславливается тем, что это самое минимальное количество, когда в реакционной массе ощущается избыток воздуха. Количество воздуха свыше 1,0 мае ч на 1 мае.ч. ЭПХГ не приводит к дополнительному положительному эффекту
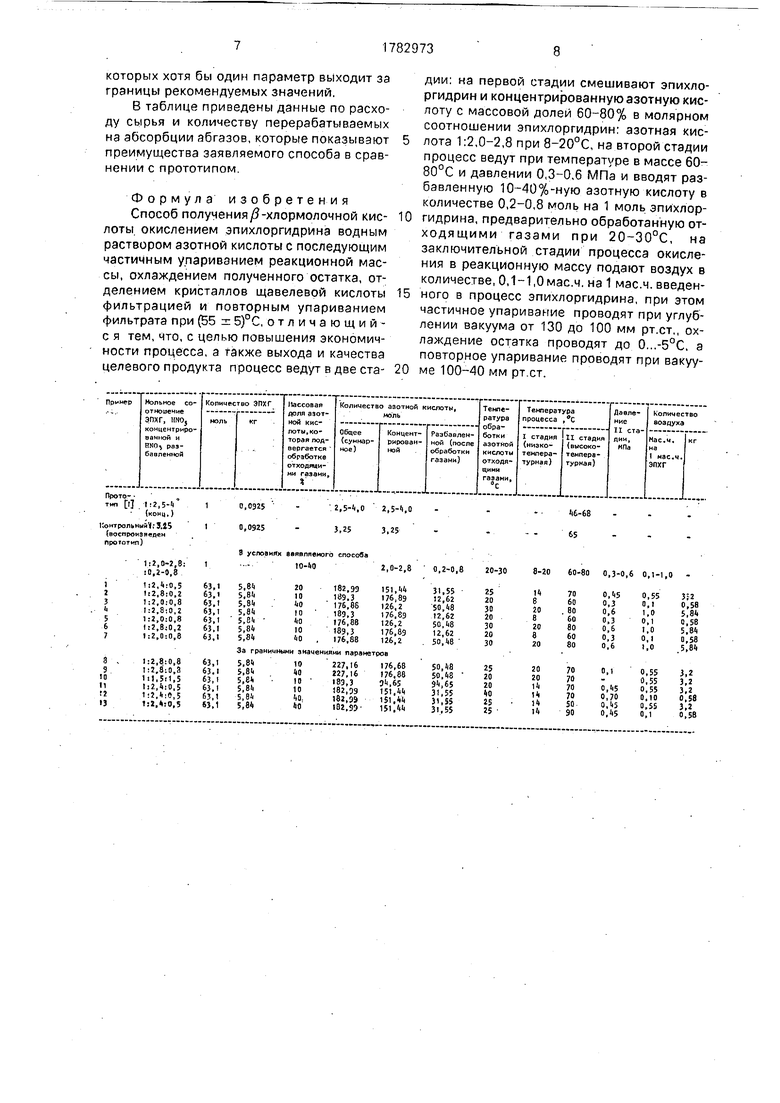
Пример 1. В реакторе вместимостью 20 дм3 смешивают при температуре 14 ± 2°С 5,84 кг(63.1 моль) 100%-ного эпихлоргидри- на и 13,63 кг 70%-ной азотной кислоты (151,4 моль). Полученная смесь со скоростью 3,91 кг/ч (3,0 л/ч) поступает в теплообменник, где подогревается до температуры 70°С, а далее последовательно проходит через реакторы-окислители (3 неактора), в которых поддерживается такая же температура.
Отходящие газы (окислы азота) проходят через промывной аппарат, заполненный 20%-ной азотной кислотой. В промывном аппарате при температуре 25°С происходит укрепление азотной кислоты до концентрации 50%. Такая разбавленная кислота по0 ступает во второй реактор-окислитель. Количество подаваемой разбавленной азотной кислоты 31,55 моль или 1,98 кг в 100%- ной массе, что составляет 3,97 кг в расчете на 50%-ную кислоту.
5 Скорость подачи 0,79 кг/ч.
Газы, выходящие после промывного аппарата, поступают в абсорбционные колонны. Вся система (реакторы-окислители, промывной аппарат, абсорберы) находится
0 под давлением 0,45 МПа, создаваемым выделяющимися газами. Избыточное давление сбрасывается дросселированием после узла абсорбции.
В третий реактор-окислитель подается
5 воздух в количестве 3,2 кг(0,64 кг/ч). Общее время пребывания реакционной массы в реакторах-окислителях 4 ч.
Реакционную массу по окончании процесса окисления упаривают на пленочном
0 испарителе под вакуумом 100-130 мм рт.ст., отгоняя 10%-ную азотную кислоту в количестве 1,58 кг (ее направляют на следующий синтез в промывной аппарат и после обработки (укрепления) отходящими газами воз5 вращают в процесс. Упаренную массу охлаждают до (-2)°С, отфильтровывают 0,81 кг технической щавелевой кислоты - кристаллогидрата, после чего при температуре в массе не выше 60°С и остаточном
0 давлении 100-40 мм рт.ст. на роторноп- леночном испарителе отгоняют воду и азотную кислоту. Получают 7,36 кг обезвоженной кристаллической в-хлормолочной кислоты с массовой долей основного веще5 ства 92,3%, что составляет 6,79 кг в 100%ной массе (86,5% от теории в расчете на
ЭПХГ). Количество отгона 40%-ной HNOa
9,8 кг. Кислота также может быть возвраще, на в промывной аппарат и послеукрепления
0 отходящими окислами азога используется на второй высокотемпературной стадии ,в следующем синтезе.
Другие примеры получения ХМК приведены в таблице. Примэры 2 и 3 - на нижние
5 граничные значения гараметров и на верхние граничные значения. Примеры 4-7 - смешанные значения параметров внутри рекомендуемых границ. Кроме того, приведен контрольный пример в условиях прототипа и серия примеров (примерм 8-13) с
которых хотя бы один параметр выходит за границы рекомендуемых значений.
В таблице приведены данные по расходу сырья и количеству перерабатываемых на абсорбции абгазов, которые показывают преимущества заявляемого способа в сравнении с прототипом
Формула изобретения Способ получения/ -хлормолочной кислоты окислением эпихлоргидринэ водным раствором азотной кислоты с последующим частичным упариванием реакционной массы, охлаждением полученного остатка, отделением кристаллов щавелевой кислоты фильтрацией и повторным упариванием фильтрата при (55 и 5)°С, отличающий- с я тем, что, с целью повышения экономичности процесса, а также выхода и качества целевого продукта процесс ведут в две стадии: на первой стадии смешивают эпихло- ргидрин и концентрированную азотную кислоту с массовой долей 60-80% в молярном соотношении эпихлоргидрин: азотная кислота 1:2,0-2,8 при 8-20°С, на второй стадии процесс ведут при температуре в массе 60- 80°С и давлении 0,3-0,6 МПа и вводят разбавленную 10-40%-ную азотную кислоту в количестве 0,2-0,8 моль на 1 моль эпихлоргидрина, предварительно обработанную отходящими газами при 20-30°С, на заключительной стадии процесса окисления в реакционную массу подают воздух в количестве, 0,1-1,0 мае.ч. на 1 мае.ч. введенного в процесс эпихлоргидрина, при этом частичное упаривание проводят при углублении вакуума от 130 до 100 мм рт.ст,, охлаждение остатка проводят до 0...-5°С. а повторное упаривание проводят при вакууме 100-40 мм рт ст.
Продолжение таблицы


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 5-хлорметил-1,3-диоксолан-4-она | 1976 |
|
SU609290A1 |
| Способ получения метилового эфира @ -хлормолочной кислоты | 1989 |
|
SU1778111A1 |
| Способ получения алифатических @ -оксикарбоновых кислот | 1988 |
|
SU1512964A1 |
| Способ получения замещенных 5-метилен-1,3-диоксолан-4-онов | 1970 |
|
SU606313A1 |
| СПОСОБ ПОЛУЧЕНИЯ р ХЛОРМОЛОЧНОЙ КИСЛОТЫ | 1972 |
|
SU426999A1 |
| СПОСОБ ПОЛУЧЕНИЯ АДИПИНОВОЙ КИСЛОТЫ | 1994 |
|
RU2069654C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-НИТРО-5-АЗИДОМЕТИЛОКСАЗОЛИДИНА-1,3 | 2009 |
|
RU2404168C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-НИТРО-5-АЗИДОМЕТИЛОКСАЗОЛИДИНА-1,3 | 2011 |
|
RU2461548C1 |
| Способ получения лимонной кислоты | 1981 |
|
SU1065403A1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕРМИЧЕСКОЙ ОРТОФОСФОРНОЙ КИСЛОТЫ И СПОСОБ ЕЕ ОЧИСТКИ | 2010 |
|
RU2464225C2 |
Сущность изобретения: продукт - хлормолочная кислота БФ СзН СЮз. Чистота 96,0%. Реагент 1: эпихлоргидрин. Реагент 2: азотная кислота. Условия реакции: проведение в две стадии, при смешении на первой стадии эпихлоргидринз и концентрированной азотной кислоты с массовой долей 60-80% в их мольном соотношении 1:(2,0-2,8) при 8-20°С и проведение на второй стадии при температуре 60-80°С и давлении 0,3-0,6 МПа с введением разбавленной 10-40%-ной азотной кислоты, предварительно обработанной отходяи.имк газами при 20-30°С, а также подачей воздуха на заключительную стадию процесса окисления. При этом частицы упаривания проводят при углублении вакуума, а повторное упаривание проводят при вакууме от 100 мм рт.сг. до 40 мл, рт.ст.
Даухстедийный синтез р -хлормолочной кислоты под давлением е рециклом р««б« лвнной мотной кислоты и с отделением побочного продукта - ««««левой кислоты нижотемпературной кристаллизацией
t.12
| СПОСОБ ПОЛУЧЕНИЯ р ХЛОРМОЛОЧНОЙ КИСЛОТЫ | 1972 |
|
SU426999A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ получения на волокне оливково-зеленой окраски путем образования никелевого лака азокрасителя | 1920 |
|
SU57A1 |
| кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
