Изобретение относится к аналитической и неорганической химии, в частности спосбб применим для определения процентного содержания бора в неорганических соединениях, например в нитриде бора.
Известен способ определения бора в борорганических соединениях с помощью реагента азометина Н. Однако органические соединения бора легко переводятся в водорастворимое состояние в отличие от борсодержащих неорганических соединений, каким является нитрид бора.
Наиболее близким к изобретению является Д-маннитный способ определения бора, по которому навеску нитрида бора (100-200 мг) выдерживают в платиновом тигле при 800°С со смесью карбонатов щелочных металлов в течение 2 ч В тигель насыпают смесь карбонатов натрия и калия, затем помещают навеску нитрида бора и засыпают ее снова такой же смесью, чтобы общее количество смеси составляло (5±0,5) г Затем добавляют азотнокислый
калий в количестве 0,05-0,1 г Сплавление проводят в печи. После охлаждения плав растворяют в стакане в 10 мл воды, туда же добавляют2 капли индикатора-метилового красного и 20 мл раствора соляной кислоты. Затем раствор количественно переносят в мерную колбу и доводят до метки дистиллированной водой. Из мерной колбы отбирают 2 пробы по 50 мл в конические колбы. Отобранный раствор нитрида бора кипятят в течение 10 мин для удаления углекислого газа, охлаждают и нейтрализуют насыщенным раствором гидроокиси бария Количество бора в нитриде бора определяют методом титрования образовавшейся борной кислоты гидроокисью бария определенной концентрации. Для установления титра раствора гидроокиси бария выполняют пять параллельных определений. Конец титрования определяют при помощи добавленного реагента Д-маннита
В таких же условиях проводят титрование контрольной пробы
сл
С
-ч
00 СО СО Ю СО
Количество бора (% В) в нитриде бора вычисляют по формуле
K(Vi-V2V0.001082-4-100 Л вm
где Vi.Va - объем 0,05 моль/л раствора гидроокиси бария, пошедшего на титрование исследуемой и контрольной пробы соответ- CTBeYmo, мл;
К - поправочный коэффициент к титру раствора гидроокиси бария;
fh - масса навески нитрида бора;
4 - коэффициент пересчета при отборе проб (50 мл от 200 мл);
0,001082 - количество бора, соответствующее 1 мл 0,05 моль/л раствора гидроокиси бария, г.
За результат анализа принимают среднее арифметическое двух параллельных определений. Абсолютное значение суммарной погрешности результата анализа составляет 0,6 %.
К недостаткам этого способа относятся: необходимость брать для анализа больше навески нитрида бора (100-200 мг); использование для сплавления трех реагентов. При этом время разложения нитрида бора составляет 2 ч; в ходе анализа приходится готовить растворы строго определенной концентрации с дальнейшей проверкой их титра; в процессе анализа исследуемый раствор нитрида бора подвергают кипячению. Способ отличается трудоемкостью, большим количеством последовательно выполняемых операций;сам процесс титрования визуальный.
Целью изобретения является повышение точности определения количества бора в нитриде бора и упрощение процесса анализа. Указанная цель достигается тем, что после сплавления навески нитрида бора с реагентом, охлаждения, количественного переноса плава, отбора пробы для анализа, образующуюся после разложения нитрида бора борную кислоту количественно связывают в устойчивый окрашенный комплекс желтого цвета цветореатентом азоме- тином Н, измеряют оптическую плотность при длине волны 415 нм и определяют содержание бора расчетным путем. Цвето- реагент азометин Н представляет со- брй продукт конденсации салицилового альдегида и аш-кислоты. Его химическое название: 4-(2-гидроксибензилиденамйно)-5- гидроксинафталин-2 6-лисульфокислота.
S03H.
-ОН
S03H
Спектр поглощения раствора комплекса азометина Н с борной кислотой имеет четко выраженный максимум при длине волны 415 нм. Способ осуществляют следующим образом.
Навеску нитрида бора (3-8 мг) сплавляют в герметически закрытом никелевом реакторе с едким кали (8-10 гранул) при температуре 850°С в течение 30 мин. Плав
затем количественно переносят в мерную колбу. Для анализа из мерной колбы отбирают 2 пробы исследуемого раствора нитрида бора. В процессе проведения анализа используют следующие растворы: буферный раствор (рН 5,0-5,3); раствор цветоре- агента азометина стандартный раствор борной кислоты с содержанием бора 100 мкгВ/мл.
К отобранной пробе исследуемого раствора нитрида бора добавляют раствор азометина Н, буферный раствор и доводят до метки дистиллированной водой. Аналогично готовят для анализа стандартный раствор: в мерную колбу отбирают пробу
раствора борной кислоты с известным содержанием бора, добавляют буферный раствор и раствор азометина Н. Приготовленные для анализа растворы помещают на 2 ч в темное место, после чего растворы
спектрофотометрируют при длине волны 415 нм. Оптическую плотность исследуемого и стандартного растворов измеряют относительно холостого опыта.
Процентное содержание бора в нитриде бора вычисляют по формуле
o/oB DncaLVcI V.j(1)
DCT Уиссл ГЛ
Оиссл, DCT - оптическая плотность исследуемого и стандартного растворов соответст- венно;
УИССЛ , VCT -аликвотные объемы исследуемого и стандартного растворов соответственно, мл;
V- разведение исходного раствора нит- рида бора;
m - навеска образца нитрида бора, мг. Стандартный и исследуемый растворы готовят таким образом, чтобы концентрация бора в фотометрируемом растворе не превышала 1 мкгВ/мл, так как область концентрации бора, где наблюдается ли- нейная зависимость оптической плотности раствора от концентрации, составляет 0,1- 1,0 мкг В/мл.
5 Добавление специального буферного раствора обусловлено тем, что образование комплекса борной кислоты с азометином Н и развитие устойчивой окраски комплекса происходит в строго определенных условиях, а именно если рН раствора составляет 5,0-5,3.
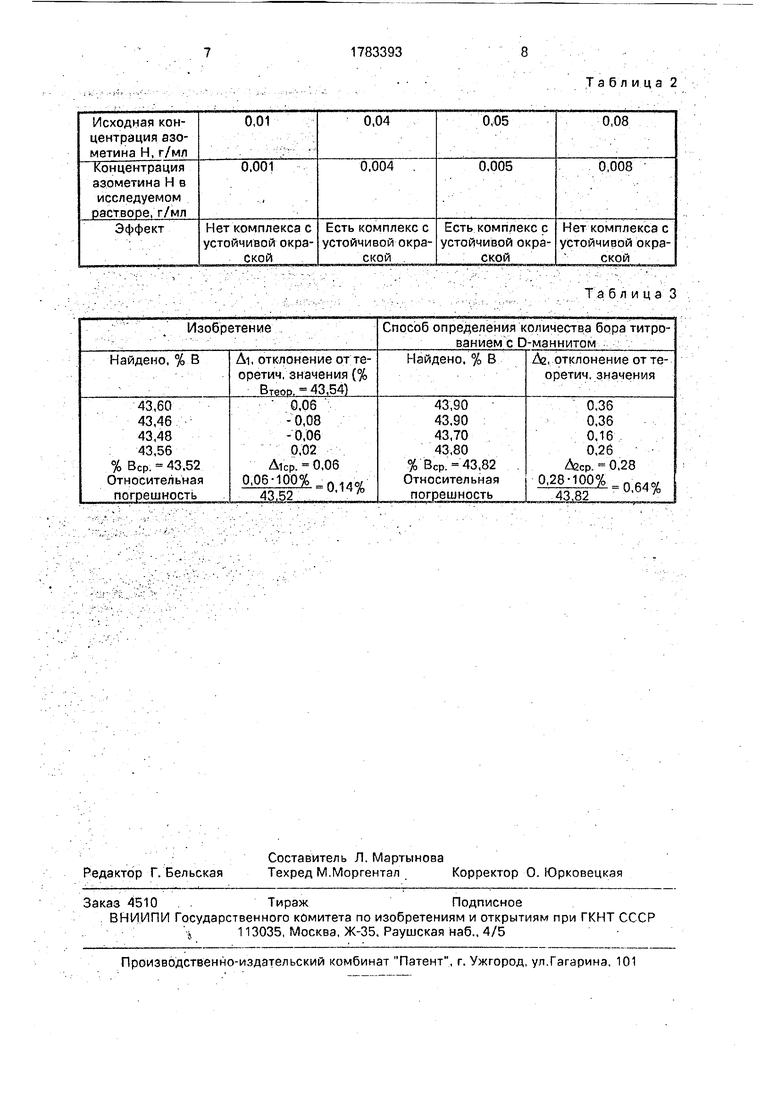
Реакция образования комплекса борат- иона с реагентом азометином Н обратима, поэтому необходимо к анализируемому раствору нитрида бора прибавлять точно измеренное количество цветореагента азо- метина Н строго определенной концентрации, В частности, для анализа нитрида бора подобранная область концентраций азоме- тина Н составляет 0,02-0,06 г/мл. Кроме того, интенсивность окраски комплекса зависит от рН анализируемого раствора нитрида бора и является устойчивой, если рН раствора составляет 5,0-5,3; такое значение рН поддерживают буферным раствором специально подобранного состава. Результаты анализа приведены в табл.1,2.
Для анализа использованы образцы пи- ролитического нитрида бора, полученного методом химического осаждения из газовой фазы на графитовую подложку
П р и м е р 1. Навеску нитрида бора 3,810 мг сплавляют с едким кали (0,5 г) в течение 30 мин при 850°С в герметически закрытом никелевом реакторе. Плав охлаждают и количественно переносят в коническую колбу промыванием реактора водой. Из колбы отбирают 10 мл и переносят в мерную колбу на 100 мл. туда же добавляют 10 мл раствора азометина Н и 10 мл буферного раствора; доводят до метки дистиллированной водой. Таким образом приготавливают два исследуемых раствора.
Приготовление стандартного раствора. В мерную колбу на 100 мл отбирают 10 мл раствора борной кислоты с известным содержанием количества бора (10 мкг В/мл), добавляют 10 мл раствора азометина Н и буферный раствор: доводят до метки дистиллированной водой
Приготовление холостого раствора. В мерную колбу на 100 мл отбирают 10 мл раствора азометина Н, добавляют буферный раствор и доводят до метки дистиллированной водой.
Все приготовленные растворы помещают на 2 ч в темное место, после чего растворы спектрофотометрируют. Оптическая
плотность растворов измерена на спектрофотометре марки Specord M 40 (производство ГДР).
Процентное содержание бора в исследованном образце нитрида бора, рассчитанного по формуле (1), составляет
% В
0,3648-10мл-168.43 0,3700-3,81-10мл
43,59%.
где Оиссл. 0,3648;
Ост 0,3700;
VCT УИССЛ. 10 мл;
т 3,81;
V 168,43.
П р и м е р 2. Аналогичные операции по анализу произведены с навеской нитрида бора, равной 4,299 мг:
% В
0.4210-10-193.09 0,4341-10-4,299
43,56%,
5
0
5
0
5
где Оиссл 0,4210;
Ост.-0,4341;
VCT УИССЛ 10 мл;
V 193,09;
m 4,299 мг.
В табл. 3 представлены результаты сравнительного анализа образцов нитрида бора по известному способу и изобретению.
Формула изобретения
Способ количественного определения бора в пиролитическом нитриде бора, включающий сплавление навески нмтрида бора с реагентом, охлаждение, количественный перенос плава, отбор пробы для анализа, получение исследуемого раствора, добавление цветореагента и определение содержания бора расчетным путем, отличающееся тем. что, с целью повышения точности определения количества бора и упрощения процесса анализа, в качестве реагента добавляют раствор цветореагента азометина Н концентрации 0,02-0,06 г/мл и буферный раствор рН 5,0-5,3 и измеряют оптическую плотность раствора при длине волны 415 нм.
50


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения бора | 1990 |
|
SU1797022A1 |
| Способ количественного определения сульфаниламидных лекарственных препаратов | 1983 |
|
SU1105791A1 |
| ОПРЕДЕЛЕНИЕ ДИМЕДРОЛА ИЛИ ПАПАВЕРИНА | 2002 |
|
RU2237237C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЭТИОНАМИДА | 1991 |
|
RU2027170C1 |
| Способ определения кислорода в графитоподобном нитриде бора | 1987 |
|
SU1465760A1 |
| Способ количественного определения аскорбиновой кислоты | 1990 |
|
SU1778645A1 |
| Способ количественного определения цистеина | 1984 |
|
SU1168834A1 |
| Способ количественного определения тиамина бромида | 1984 |
|
SU1163225A1 |
| Способ количественного определения тропацина | 1987 |
|
SU1456853A1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ АЛИФАТИЧЕСКИХ АМИНОКИСЛОТ | 1999 |
|
RU2167410C2 |
Сущность изобретения: образующаяся после разложения нитрида бора борная кислота количественно связывается цветореа- гентом - азометином Н - в устойчивый комплекс, имеющий максимум поглощения при длине волны 415 нм. 3 табл
Таблица 1
Таблица 2
Таблица 3
| Васильева М.Г | |||
| и др | |||
| Анализ бора и его неорганических соединений М | |||
| Приводный механизм в судовой турбинной установке с зубчатой передачей | 1925 |
|
SU1965A1 |
| Шанина Т.М., Гельман Н Э., Михайловская В,С | |||
| Количественный анализ элементо- органических соединений,- Журнал аналитической химии, 1967, т | |||
| ХХН, вып 5, с 782. | |||
