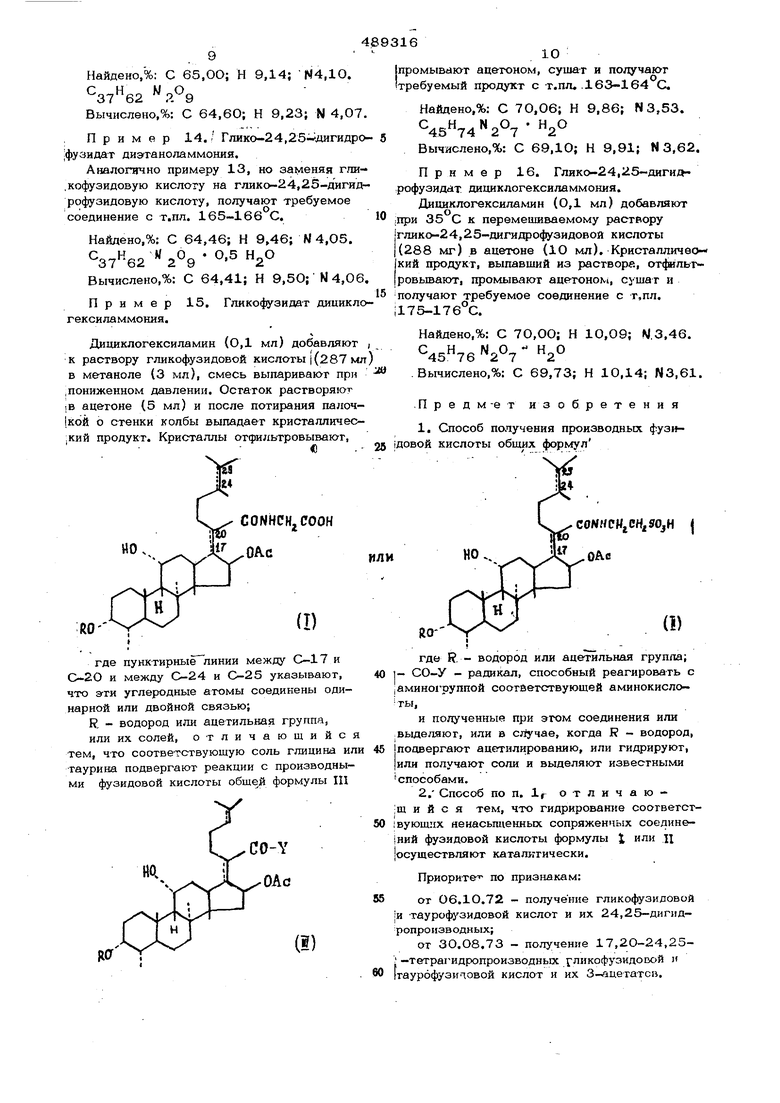
(54) СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФУЗИДСВОЙ КИСЛОТЫ R - водород или ацетильная группа, или их солей, заключающийся в том, что соответствующую соль глилина или таурина подвергают реакции с производным фузидо вой кислоты общей формулы где R - водород или ацетильная группа; -СО- Y - радикал, способный реагировать с аминогруппой соответствующей амино кислоты. Полученные при этом соединения выделяют или в случае, когда R - водород, подвергают ааетилированию, или гидрируют, или получают соли и выделяют известными способами. . Радикал СО-У моясет быть радикалом ангидрида, смешанного ангидрида, хлорангид рида кислоты или радикалом, полученным по реакшш свободной кислоты с карбодиимидом или N , N - карбонил Блимидазолом или подобным функциональным соединением. Реакцию проводят преимущественно в инертном растворителе, например диметилформамиде, те-трагидрофуране и т,п. Исходные соединения формулы Щ можно получать из известной фузидовой кислоты, ее 24,25-дигидро- и 17,20-24,25-тетрагидропроизводных или 3- ацетатов этих соединений .известным методом, и1гидро- и тетрагидрЬсоедиаения можно получать каталитическим гидрированием соответствующих ненасыщенных соединений формулы или 11 3- ацетаты - ацетили рованием соответствующих неацетилирован- ных соединений. П р и м е р 1, Таурофузидат натрия. К суспензии таурина (25 г) в диметилформамиде (40О мл) добавляют триэтиламин (28 мл) и после перемещивания в течение 30 мин при комнатной темпе ратуре добавляют фузидовый ангидрид (51,6 г). После перемещивания в течение 7О час при комнатной температуре смесь разбавляют этилацетатом (1,2 л), водой (4ОО мл) и рН среды водной фазы доводят до 2 добавлением 4 н. соляной кислоты при перемещивании. Еюдный слой отделяют, органическую фазу промывают водой (3 х X 200 мл), сушат и выпаривают в вакууме Получают полукрисгалличесхий остаток, ко- рый состоит главным образом из фузидоой кислоты, как это было показано тонколойной хроматографией (система раствориеля метиленхлорид: метанол 9:1; проявитель: ернаякислота 98%г-ная.К объединенным водым фазам и промывным водам (около 1,6 л) обавляют хлористый натрий (60 г) и -бутанол (400 мл ), и смесь сильно встряхивают. Органический слой отделяюти водную фазу снова экстрагируют JJ-бутанолом (2 х. X 200 мл). Объединенные Jt-бутанольные экстракты промывают насыщенным водным раствором хлористого натрия (3 х 10О мл), фильтруют и упаривают до объема около 200 мл при пониженном давлении. Образо- вавшийся кристаллический осадок хлористого натрия от4 1Льтровывают, фильтрат выпаривают досуха и в вакууме получают вязкое масло, которое растворяют в метаноле / ,, Р доводят до 10,8 дс бавлением 5 н. водного раствора едкого нат ра при перемешивании. Смесьвыпариваютдосуха в вакууме. Остаток снова растворяют в 99%-ном этаноле (15О мл) и фильтруют через декалит для удаления нерастворившегося хлористого натрия. Фильтрат снова выпаривают в вакууме и получают желтое масло. Этот остаток растворяют в метаноле (35 мл), добавляют воду (3,6 моля) и после потирания палочкой начинается кристаллизация бесцветного вещества. После выдержки в холодильнике в течение ночи смесь .разбавляют охлажденным на льду ацетоном (14О мл), кристаллы отфильтровывают, промывают ацетоном, сущат и получают таурофузидат натрия с т.пл. 220222 С. Выпаривание маточной жидкости при пониженно л давлении дает вторую порцию нужного продукта с т.пл. 216-218°С. Обе порции кристаллического таурофузидата натрия перекрис- таллизовывают из смеси метанол-ацетон и подучают чистое соединение с т.пл. 224 226°С;-,20. - 11°(,5; в метаноле). С 55,64; Н 8,35; N 2,03; Найдено, %: 8,56 54,52; N0 с 55,91; Н 8,39; N 1,98; Вычислено, 8,90 54,52: Пример 2. Тауро-24,25-дигидрофузидат натрия. К раствору таурофузидата натрия (3,54 г ;в (-ном этаноле (50 мл) добавляют 5% палладия на сульфате бария (катализатор, 0,7 г) и взбалтывают в атмосфере водорода в течение 3,5 час. Катализатор отфиль1 ровывают, промывают 96%-ным этанолом и фильтрат выпаривают досуха. К раствору остатка (4,6 мл) добавляюТ воду (0,4 мл) затем ацетон. (30 мл) и после потираний палочкой начинается кристаллизация. После перемешивания в течение часа при комнатной температуре смесь разбавляют ацетоном (25 мл) и держат в холодильнике в течение ночи. Кристаллы отфильтровывают, промывают ацетоном, сушат и получают тауро-24,25 -дигидрофузидат натрия с т.пл. 198-202 С; - 14 i(,5; в метаноле). Найдено,%: С 56,70; Н 8,43; N1,99; 54,63; 7,92. Вычислено,%: С 56,47; Н 8,62-;М2,00; S4,57: . 7,92 После выпаривания маточного раствора при пониженном давлении получают вторую порцию требуемого соединения с т.пл. 194- 200°С.С Пример 3. Гликофузидовая кислота К суспензии глицина (15 г) в диметилформамиде (4ОО мл) добавляют триэтиламин (28 мл) и после перемешивания в течение 30 мин при комнатной температуре добавляют фузидовый ангидрид (51,6 г). После перемешивания в течение 70 час при комна тной температуре смесь разбавляют этилацетатом (1,2 л) и добавляют воду (400 мл). рН водной фазы доводят до 2 добавлением 4 Н. соляной кислоты при перемешивании. С ганическую фазу отделяют, промывают водой (З X 2ОО мл) и упаривают при понижен ном давлении до объема около 25О мл. К концетрату добавляют воду (200 мл) и рН водной фазы доводят до 7,5 добавлением при перемешивании насыщенного водного раствора бикарбоната натрия. Водную фазу отделяют и органическую фазу промывают водой (2 X 1ОО мл), сушат и выпаривают в вакууме. Полученный полукристаллический продукт, согласно тонкослойной хроматографии, состоит из смеси фузидовых кислоты и ангидрида. Объединенные водные фазы и промывные воды смешивают с этилацетатом (400 мл) и рН водной фазы доводят до 2 добавлением к, соляной кислоты при перемешивании Органическую фазу отделяют и водную фазу скова экстрагируют этилацетагом (2ОО мл). Объединенные органические экстракты промывают водой (2 X 1ОО мл), сушат и упаривают при пониженном давлении до объема около 100 мл. Выпавшее кристаллическое вещество отфильтровьшают, промывают эти- шацетатом, сушат и получают гликофузидовую кислоту с т.пл. 234-236 С, Упаривание маточной жидкости дае-т вторую порцию тре буемого продукта с т.пл. 228-2ЗО С.Лерек- ристаллизация из метанола-зтилацетата дает аналитическую пробу с т.пл. 236-238 С; 20 21 ;(,5; в метаноле). Найдено,%: С 68,91; Н 9,04; N2,51. .7 Вычислено,%: С 69,08; Н 8,96;N2,44. Пример 4. Глико-.24,25-дигидро|фузиловая кислота. I К раствору гликофузидовой кислоты (33,44 г) в 96%-ном этаноле (70 мл) добавляют 5% палладия на сульфате бария .(катализатор, О,7 г) и смесь взбалтывают в атмосфере водорода в течение 2,5 час. Ка тализа тор от(|щльтровьтают, промывают :96%-ным этанолом, и фильтрат выпарива- :ют досуха. кристаллизуют из эти- лацетата и получают требуемое соединение 1с т.пл. 215-217 С. Перекристаллизация из метанола-этилацетата повысила т.пл. до 219-220% C-flJ ,27 ,5; в ме1таноле). Найдено,%: С 68,57; Н 9,23; N2,39. 7 Вычислено,%: С 68,84; Н 9,28; N2,43. Пример 5. Глико-17,20-24,25гeтpaгидpoфyзидoвaя кислота. К раствору гликофузидовой кислоты (2,87 г) в 96%-ном этаноле (50 мл) до|бавляют окись платины (О,3 г) и смесь взбалтывают в атмосфере водорода в течение часа. Катализа тор отфильтровывают, |Промывают 96%-ным этанолом, вываривают досуха в вакууме. Аморфный оста- гток растворяют в метаноле (15 мл), добав- |ляют этилацетат (©О мл) и смесь выпарива- ЮТ приблизительно до 15 мл при понижен|ном давлении. После потирвния палочкой (требуемое соединение кристаллизуется. Крио таллический осадок отфильтровьюают, промы- ;вают этилаце татом и сушат, получают гликоi-17, 2О-24,25-тетрагидрофузидовую кислоту. JC т.пл. 205-209 С. Две перекрксталлгзации |ИЗ метанола-этилацетата повышают т.пл. до 213-215°С; Мд (,5; в метаноле). Найдено,%: С 68,37; Н 9,58; N 2,43. Вычислено,%: С 68,60; Н 9,59; N2,42. Пример 6. Тауро 17,20-24,25;-тетрагигпофузидат натрия. Раствор таурофузидага натрия (7,0 г) в 96 ь-ном эганоле (100 мл) взбалтыраюг прн комнатной температуре при 1 атм водорода в присутствии окиси платины (0,7 г), Чероз 90мин псглощается 500 мл водорсьда и потребление водорода прекращается. Катализатор удаляют фильтрованием, фильтрат вьптаривают досуха в вакууме. Кристаллизация остатка из метанола-ацетона дает требуемое соединение. Пример 7„ З-Ацетил-глико-24,25 дигидрофузидовая кислота. , Глико-24,25-дигидрофузидовую кислоту (2,88 г) растворяют в смеси уксусного ангидрида (З мл) и пиридина (3 мл). Посла выстаивания в течение 1,5 час при когинат- ной темпера туре добавляют воду (6О мл) и смесь экстрагирук/т этнлацетатом (3x30 м Объединенные экстракты промывают разбавленной водной соляной кислотой для удаления пиридина, затем РОДОЙ до ней ральной реакции. Органический слой сушат и выпаривают досуха в вакууме, из остатка требуемое сбединение можно получи ть очист К.ОЙ на хроматографической колонке на сили- кагеле. Пример 8. Сопряженные соединения таурина и 24,25-дигидро- и 17,20-24,25-тетрагидрофузидовой кислотыи IX натриевые соли. Аналогично примеру 1, но заминяя фузидовый ангидрид аа 24,25-дигидрофузидовый или 17,20-24,25-тетрагидрофузидовый ангидрид, получают требуемое соединение с такой же точкой плавления и физическими свойствами, как в примерах 2 и 6. Пример 9. Сопряженные соединения глицина и 24,25-дигидро- и 17,2О-.24,25-тетрагидрофузидовой кислоты Аналогично примеру 3, но заменяя фузидовый ангидрид на 24,25-дигидрофуэидовый или 17,2О-24,25-тетрагидрофузидовый- ангидрид, получают требуеглое соединение с такой жэ точкой плавления и физическими свойствами, как в примерах 4 и 5. Пример Ю. 3-Аиетилтаурофу аидат натрия. 3-Лцвтилфузидовый ангидрид (11 г) доубавляют к перемешиваемой суспензии -гаурина (2,5 г) и т)эиэтиламина (2,8 мл) в смеси диме тилформамида (40 мл) и гексаметилфосфорного триамида (4О мл). После перемешива)1ия в течение 72 час при ком- натной Температуре добавляют этиладетат (240 мл) и воду (8О мл). рН смеси дозбдя т до 2 добавлением 4 н. соляной кислоть при перемешивании. Водную фазу отделяют, органическую фазу промыва1от воде и t3 х 4О -мл), сушат и выпаривают в вакууме получаюТ полукристаллический остаток, оторый, по данным тонкослойной хроматогафии, состоит главным образом из 3-аце- тилфузидовой кислоты. Добавляют хлористый натрий (20 г) и М-бутанол (150 мл) к водной фазе, и смесь сильно встряхивают. Органический слой отделяют и водвдю фазу снова экстрагируют Н-бутанолом (2x75 мл). Объединенные И-бутанольные экстракты тшательно промывают водой, фильтруют, выпаривают досуха в вакууме. Маслянистый остаток растворяют в метаноле (40 мп), йерастворимую соль отфильтровывают и рН фильтрата доводят до 10,8 добавлением 5 н. водного раствора едкого натра при перемешивании. Смесь выпаривают досуха при пониженном давлении и получают желтоватое масло. Остаток растворяют в метаноле (5 мл), добавляют ацетон (75 мл) и из полученного таким образом раствора после потирания палочки о стенки колбы выпадают бесцветные кристаллы требуемого соединения, их отфильтровываю т, промывают ацетоном и сушат. Пример 11. Гликофузидат натрия. Раствор гликофузидовой кислоты (574 г) в метаноле (5 мл) осторожно нейтрализуют 2 н, раствором едкого натра (индикатор фенол4)талеин) и смесь выпаривают в ме. Полученный аморфный растворяют в метаноле (О,5 мл), добавляют цетон (5 мл) и вызывают кристаллизацию поти1 анием о стенки. После стояния в течение часа при комнатной температуре добавляют еще ацетон (5 мл), кристаллы отфильтровывают, промывают ацетоном, сушат и получают требуемое соединение. Пример 12. Глико-24,25-дигидрофузида т натрия. Аналогично примеру 11, но заменяя гликофузидовую кислоту на глико-24,25-длгид- рофузидовую кислоту, получают требуемое соединение. Пример JL3. Гликофузидат диэта- ноламмония. К. перемешиваемому раствору гликофузи- довой кислоты. (287 мг) в метаноле (3 мл) добавляют 0,5 М раствор диэтаноламлна в ацетоне (1 мл), смесь выпаривают в вакууме. Мас; 1янистый остаток растворяют в ацетоне (5 мл), и после потираний палочкой о стенки колбы выпадает кристаллический продукт. Кристаллы отфильтровывают, промывают ацетоном, сушат и получаю требуемое соединение с т.пл. 179-1-8О°С.