(5) ГЕРБИЦИДНЫЙ СОСТАВ














| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения диариловых эфиров -фосфонометилглицинонитрила или их солей | 1978 |
|
SU704457A3 |
| Гербицидный состав | 1977 |
|
SU730273A3 |
| Способ получения сложных триэфиров -фОСфОНОМЕТилглициНА | 1977 |
|
SU843755A3 |
| ТРИАЗИНСОДЕРЖАЩИЕ АНИОННЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2170731C2 |
| СМЕСЬ БЛОК-ОЛИГОМЕРОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ И КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭТУ СМЕСЬ | 1996 |
|
RU2175660C2 |
| СПОСОБ ПОЛУЧЕНИЯ 0,0-ДИАЛКИЛ-0- | 1973 |
|
SU367105A1 |
| Способ получения производных тетрагидро-1,3,5-тиадиазин-4-она или их солей | 1978 |
|
SU876057A3 |
| Способ получения замещенных 4,6-диарил-3-оксо-3,4-дигидро-1-фосфа-2,4,5-триазинов | 1983 |
|
SU1081170A1 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА | 1992 |
|
RU2062777C1 |
| КОМПОЗИЦИЯ ДЛЯ ПОКРЫТИЯ | 1992 |
|
RU2107704C1 |
Изобретение относится к химическим средствам борьбы с нежелательно растительностью, а именно к гербицид ному составу на основе производных N-фосфонометилглицинонитрила. Известен гербицидный состав, срде жащий N-фосфонометилглицинонитрил и обычные добавки l. Однако азктивность его недостаточна. Цель изобретения - изыскание ново го дербицидного состава на основе произ водных N-фосфонометилглицинонитрила, обладающего усиленным гербицидным действием. Указанная цель достигается исполь зованием в качестве производного N-фосфонометилглнцинонитрила соединения общей формулы г я ,ч II (арш1--Ла -o)j.i-P-CHa-l f-CHi-CH- Л Ц где арил - фенил, р-нафтил, бифенил X - хлор, фтор, метил, метокси, метилтио, метилендиок си, циан, трифторметил, нитро; Z - кислород или сера; а - целое число от О до 3; b - целое число О или 1; R - хлористоводородная, бромистоводородная, йодистоводородная, хлорная, фторборная, азотная, серная, фосфорная, щавелевая, п-толуолсульфоновая, пхлорбензосульфоновая, трихлорметилфосфоновая, трихлоруксусная, трифторуксусная кислоты или трифторметансульфокислота, метансульфокислота; X - О или 1 при условии, что X равен О, если Ь равно 1, в количестве 5-95 вес.ч. ормы применения препаратов обычоединения формулы (I) могут быть чены взаимодействием диарилфосс 1,3,5-трицнанометилгексагид,3,5-триазином. -фосфонометилглицинонитрилы ктурной формулы (1), где х и Ь-О, чают путем образования смеси, оящей, в основном, из сложного эфира фосфорной кислоты структурной формулы (врил-ХВ-.0) (П) где X, Z и а имеют вышепрьведенные значения, и 1,3,5-трицинометилгексагидро-1,3,5-триаэина структурной формулы у . (П1) . нс-снг- Nftj -CHjCK ,с нагревом этой смеси до температуры достаточной для инициирования реакции взаимодействия и поддержания ее для протекания взаимодействия сложного эфира фосфорной кислоты с триаЭином для получения N-фосфонометилглицинонитрила структурной форму-; лы (I). . В некоторых случаях рекомендуется работать с растворителем для прос тоты и удобства протекания реакции. Температура реакции может колебаться от 25 до . Соотношение сложного эфира фосфорной кислоты и триаэина в целях достижения оптимальных результатов должно быть 3:1. Для получения соединений структурной формулы (1), где b 1, х « образуют просто раствор соединения структурной формулы (я/)ал-ха-о)г-р-cHj-i-CHi-cH k где X, Z и а имеют вьшеприведенные значения, в растворителе, содержащем по меньшей мере, 1 моль/экв; воды, поддерживая раствор при комнатной температуре, при которой одна из (арил-Хс(-О)-групп гидролизуется. Пре почтительным растворителем для этого является ацетон. Желательный продукт выделяют обычными приемами - фракционированная кристаллизация или вакуумная выпарка растворителя и других летучих продуктов гидролиза; при этом желательный продукт криста лизуют из соответствующего раствори теля . Для получения соединений структу ной формулы (1), где Ь - О, X 1, т.е. соединений структурной формулы г к, V (V) (o/Jun-Xa-o)j--P-CKj-V-CHj-CK -R соединение структурной формулы (IV) растворяют в безводном растворителе как хлороформ, добавляя в этот раст вор сильную кислоту, либо в раство рителе, в некоторых случаях, в чистом виде с размешиванием при комнат of: температуре в течение времени, достаточного для взаимодействия соединения структурной формулы {IV) с кислотой. Желательный продукт формулы (v) осаждается в кристаллической форме. В некоторых случаях добавляют смесь хлороформа с диэтиловым эфиром (50/50% по объему), чтобы вызвать кристаллизацию продукта или его выделение из реакционной смеси в виде масла. При получении солей сильных кислот структурной формулы (I) применяют сложные диэфиры фосфоновой кислоты и сильную кислоту в эквимолярном соотношении относительно каждой вьоделяемой соли сильной кислоты. Сложные арильные диэфиры структурной формулы (IV), предпочтительно получают согласно одному из двух нижеописываемых способов. А. Ацетонитрильный раствор (50 мл) 1,3,5-трицианометилгексагидро-1,3,5-триазина 3,4 г (0,0167 моль) и диарилфосфита (0,050 моль) смешивают в реакционном.сосуде, нагревая 1-90 ч при 45-85 С до расхода всего фосфита или триазина, что проверяют по ЯМР-анализу. Если по ЯМР-анализу больше не содержится примесей, продукт выделяют концентрированием в ваку-уме. Если же примеси еще есть, продукт вьщеляют и очищают кристаллизацией или хроматографией, в некоторых случаях сложный диэфир.выделить трудно в высокочистом виде, из-за происходящего во время очистки гидролиза. Б: Смесь диарилфосфита (0,05 моль) и 1,3,5-трицианометил-гексагидро-триазина 3,4 г (0,0167 моль) загружают в реактор и нагревают при 60lOO C 20-60 мин до расхода всего -фосфита или триазина, что определяют по ЯМР-анализу. Продук-ты очищают кристаллизацией или хроматографией. Моноарильные сложные эфиры N-фосфонометилглицинонитрила получают путем растворения диарильного сложного эфира в ацетоне, содержащем небольшое количество воды (обычно около 2% по весу воды) с перемешиванием реакционной смеси 18-72 ч. Сложные моноэфиры обычно кристалличны по форме; их собирают фильтрацией, промывают ацетоном и сушат на воздухе. Сильные кислотные соли диарильных сложных зфиров получают по следующему общему методу. Раствор сильной кислоты (или кислоту в чистом виде) (0,01 моль) по каплям добавляют в хлороформный раствор сложного диэфира, выдерживая при комнатной температуре. В случае образования кристаллов их собирают фильтрацией, промывают 50% по объему смесью .хлороформа и эфира. В другом случае 50% по--объему смесь добавляют в целях достижения кристаллизации соли или ее вьаделения из раствора в виде масла. Пример 1. Ди(пара-хлорфени фосфит 23,32 г, 78% чистоты 0,Обмол смешивают в реакторе при комнатной температуре, нагревая до 20 м Получают 27 г О,0-ди-(пара-хлорфенил )-М-фосфонометилглицинонитрил а. 1,5747. Выход 100%. Пример 2. Ацетонитрильный раствор (10 мл) ДИ(3,4-диметилфенил)-фосфита 8,7 г (0,03 моль) добавляют в ацетонитрильный раствор (50 мл) 1,3,5-трицианометил-гекса.гидро-1,3,5-триазина 2, 04 г (О,01моль нагревая смесь при 90 ч. Фильт рацией образовавшегося твердого про дукта и выпаркой растворителя получают масло цвета красного бургундск го вина, которое согласно ЯМР-анали зу содержит желательный продукт и аминаль зтого продукта. Хроматографией масла (8,0 г), на силикагеле (450 г)/с циклогекса- ном/этилацетатом (50/50%, 60 мл фракции) получают 0,0-ди(3,4-диметилфенил)-N-фосфонометилглицинонитрил во фракциях 30-41, т.пл. 61-64 по удалении растворителя. Твердый продукт перекристаллизовывают из тетрахлорметана/изооктана, получая продукт с т.пл. бЗ-бб С. Выход 40%. Пример 3. Перемешиваемую смесь 0,02 моль ди(пара-метилтиофенил)-фосфита и 0,0067 моль 1,3,5трицианометил-гексагидро-1,3,5-три азина 1 ч нагревают до 80с, получая темное красно-коричневое масло. Половину пробы помещают в холодильник на 8 дней, получая полутвердую масс Пробу перекристаллизовнвают из 70 мл тетрахлорметана, получая тверд1лй продукт розового цвета. Его растворяют в 100 мл горячего Тетрахлормета на и фильтруют через целит, покрытый 5,0 г силикагеля. Фильтрат концентрируют до 50 мл и помещают на ночь в холодильник. Суспензию фильтруют, получая 1,8 г белого твердого продук та 0,0-ди(пара-метилтиофенил)-М-фосфонометилглицинонитрил, т.пл. 64 65°С. Выход 45%. Найдено, %: С 51,7; Н 4,9; N 7,1 Вычислено, %: С51,8; Н 4,9; N 7,1. Пример 4. Раствор ди(орто-метоксифенил)фосфита 8,5 г, 91% чистоты (0,025 моль) и 1,3,5-трицигт анометил-гексагидро-1,3,5-триазин 1.7г (0,0083 моль) нагревают до 55°С 73 ч, затем фильтруют. Фильтрат концентрируют до получения темнокоричневого масла (9,6 г). Масло 5.8г за счет адгезии наносят на 8 г силикагеля, зкстрагируют 80 мл этилацетата. Этилацетатный раствор концентрируют с нанесением полученного маслаза счет адгезии на 4,0 г силикагеяя. Силикагель экстрагируют 70 мл этилацетата, раствор концентрируют в вакууме, получая бледножелтый раствор, Пр 1,5542. Желтое масло является 0,0-ди(орто-метоксифенил)-N-фосфонометилглицинонитрилом, содержащим небольшое количество орто-метоксифенола. Пример 5. Раствор 1,3,5трицианометил-гексагидро-1,3,5триазина 13,6 г (0,06б моль) и дифенилфосфита 46,8 г (0,2 моль) в ацетонитрипе (100 мл) нагревают при 55°С 48 ч ЯМР-спектр показывает полное превращение в О,О-дифенил-N-фосфонометилглицинонитрил. Ацето.нитрил удаляют в вакууме, получая 57 г (94,4%) вязкого черного масла, которое растворяют в хлороформе, добавляя 114 г силикагеля; смесь выпаривают досуха в вакууме. Пропитанный продуктом силикагель помещают в колонну, содержащую шлам хлороформа и силикагеля (200 г), и элюируют до тех пор, пока по ЯМР-спектру в элюате более не окажется продукта. Хлороформные элюен ты концентрируют, растворяют в хлористом метилене и дважды промывают холодной 5%-ной КОН (100 мл), затем водой. Метиленхлоридный слой высушивают сульфатом магния, фильтруют и выпаривают с образованием 57,9 г светло-желтого масла, которое постепенно затвердевает. Твердый продукт является согласно определению О,О-дифенил-N-фосфонометилглицинонитрилом, т.пл. 64-67,. Выход 75%. Пример 6. Ацетонитрильный раствор (100 мл) ди(мета-толил)-фосфита 10,7 г (0,04 моль) и 1,3,5-трицианометил-гексагидро-1,3,5-триазина- 2,72 г (0,0133 моль) нагревают до 3 дня. Раствор принимает цвет красного бургундского вина, растворитель выпаривают с получением 12,4 г красного масла (92; 4%-ная регенерация). Масло (9,0 г) хроматографируют на силикагеле, элюируя циклогексаном/этилацетатом (60/40%) с получением фракций по 60 мл. Фракции 45-63 являются чистым О,0-ди(мета- толил)-N-фосфонометилглицинонитрилом, п25 1,54567. Выход 1,25 г. (14%). ° Найдено, %: С 61,75; Н 5,81; N 8,41. Вычислено, %: С 61,81; Н 5,80; N 8,48. Пример 7. Раствор ди(метанитрофенил)фосфита 15,2 г,83% чисоты (0,0392 моль) и 1,3,5-трицианоетил-гексагидро-1,3,5-триазина 2,66 г (0,013 моль) в ацетонитриле агревают 20 ч до 50°С.-.ЯМР-анализ: ревращение полное. Раствор фильтуют, растворитель удаляют в вакууме. олучают 13 г масла янтарного цвета ,О-ди(мета-нитрофенил)-N-фосфономеилглицинонитрила.
С 45,80; Н 3,39; N
Найдено, %: 4,27. С 45,93; Н 3,34;
Вычислено, %: 14,28.
ил)фосфат 15,63 г, 94% чистоты (0,05 моль) и 1,3,5-трицианометилгексагидро-1,3,5-триазин 3,4 г (0,0167 моль) растворяют в ацетонитиле, раствор нагревают 1 ч с обратым холодильником. Растворитель выаривают в вакууме, получая темно-роовое масло (19,0 г). 5 г масла подвергают жидкостной хроматографии под высоким давлением с применением смеси циклогексана/этилацетата (40/60%по объему), получая 4,1 г О,0-ди(пара-метоксифенил)-N-фосфо- . нометилглицинонитрила в виде масла, 1,5541. Выход 82%.
Пример 9. Смесь.. ,3,5-трицианометил-гексагидро-1,3,5-триазина 2,04 г (0,01 моль) и ди(пара-фторфенил)фосфита 8,8 г, 91,6%-чистоты (0,03 моль) в ацетонитриле нагревают 70 ч до 55°С. Реакционную смесь фильтруют, растворитель удаляют в вакууме, получая коричневое масло, которое является чистым 0,0-ди(парафторфенил)-N-фосфонометилглицинонитрилом, п25 1,5270. Степень чистоты 92%.
Пример 10. Ди(метйл-хлорфенил).фосфит 9,93 г, 91,5% чистоты (0,03 моль), растворенного в 20 мл ацетонитрила добавляют к 1,3,5-трицианометил-гексагидро-1уЗ,5-триазину 2,04 г (0,01 моль), растворенного в 50 мл ацетонитрила. Смесь нагревают 70 ч до 55с. Ацетонитрил удаляют в вакууме с получением светло-розового масла, которое является О,0-ди(мета-хлорфенил)-N-фосфонометилглицинонитрилом, 1,5656. Степень чистоты 92%.
Вышеописанными способами могут быть получены:
О,0-ди(пара-цианофенил)-N-фосфог нометилглицинонитрил;
О,0-ди(пара-бифенил)-N-фосфонометилглицинонитрил.
Пример 11. Сложный диэфир 4,0 г (0,099 моль), полученный по примеру 10, растворяют в 100 мл 2%-ного водного ацетона, перемешивают раствор 6 дней при комнатной температуре. Образуется твердый продукт. Его собирают, промывают ацетоном и сушат, получая 1,55 г О-мета-хлорфенил-М-фосфонометилглицинонитрила в виде твердого вещества, т.пл. 181-182°С. Выход 60%.
С 41,5; Н 3,9; N 10,8.
Найдено, . : С 41,5; Н 3,-9;N
Вычислено, 10,3.
Пример 12. Полученный по примеру 9 сложный диэфир 2,38 г (0,069 моль) растворяют в 2%-ном водном ацетоне (100 мл) и перемешивают 3 дня при комнатной температуре. Полученный шлам фильтруют, твердый продукт промывают ацетоном, получая 0,87 г рыжеватого твердого продукта, т.пл. 258-262°С. Маточный раствор выдерживают 6 недель, полученный твердый продукт собирают и промывают водным ацетоном, получая дополнительные 0,8 г материала, который является 0-пара-фторфенил-N-фосфонометилглицинонитрил с той же T.njf. Выход 98%.
Найдено, %: С 44,3; Н 4,2; N 11,5
Вычислено, %: с 44,3; Н 4,1; N 11,5.
Пример 13. 0,0-Дифeнил-N-фосфонометилглицинонитрил 1,51 г (0,005 моль) перемешивают в 50 мл 2 Н.соляной кислоте с нагревом до полного растворения всего продукта (2 ч). На дне колбы образуется масло янтарного цвета, являющееся фенолом. Колбу охлаждают до комнатной температуры, соляно-кислый раствор дважды промывают хлористым метиленом (25 мл в целях удаления любого исходного продукта и фенола, полученного во время реакции. Соляно-кислый раствор охлаждают на ледяной -бане; во время этого начинают образовываться кри таллы. Кристаллы собирают, промывают холодной водой и сушат на воздухе, они являются 0-фeнил-N-фocфoнoметилглицинонитрил без определенной т.пл.
4,93; N
С 47,52; Н
Найдено, %: 12,12.
Вычислено, %: С 47/79; N 4,90; N 12,39.
Пример 14. О,0-Ди(м-толил)-М-фосфонометилглицинонитрил 4,0 г (0,012 моль) растворяют в ацетоне (50 мл), содержащем воду (1 мл), перемешивают 60 дней при комнатной температуре. Получает три порции кристаллов. Первые две с примесями, т.пл. 161-166 с. Третья порция является аналитически чистым 0-м-толил-N-фосфонометилглицинонитрилом, ;т.пл. 179-179,5°С. Выход 53%.
Найдено, %: С 40,0; Н 3,4; N 15,5
Вычислено, %: С 40,0; Н 3,4; N 15,6.
Пример 15. О,0-Ди(м-нитpoфeнил)-N-фocфoнoмeтилглицинoнитpил 3,15 г (0,008 моль) растворяют в 50 мл ацетона и 1 мл воды, перемешивая при комнатной температуре 16 ч Получают твердый продукт, который собирают и промывают ацетоном с получением 1,1 г О-м-нитрофенил-Н-фосфонометилглицинонитрил, т.пл. 17417бс (с разложением). Выход 51%.
Найдено, %: С 50,0; Н 5,5; N 11,7.
С 50,0; Н 5,5; N
Вычислено, % 11,7.
Пример 16. Ацетонитрильный раствор (100 мл) ди(.м-три(|)тортолил)-фосфита 1Г,64 г (0,0314. моль) и 1,3,5-трицианоМетил-гексагидро-1,3,5-триазина 2,15 г (0,0105 моль нагревают в течение ночи при . Ацетонитрил выпаривают в вакууме, начинает образовываться твердый продукт. Остаточный материал растворяют в 50 мл ацетона и 1 мл воды, перемешивая затем в течение ночи при комнатной температуре. Образуется твердый продукт. Его собирают и про мывают ацетоном, получая 3,5 г белого твердого продукта, т.пл. 195ige C, определяемого как О-м-трифторметил-М-фосфонометилглицинонитрил. Выход 39,5%.
Найдено, %:C41,0;H3,5;N9,7.
Вычислено, %: С 40,8; Н 3,4; N 9,5.
Пример 17. О,0-Ди(п-xлopфeнил ) -N-фосфонометилглицинонитрил 9,0 г (0,024 моль) растворяют в 50 м ацетона и 1 мл воды, перемешивая при комнатной температуре два дня. Образуется 2,35 г твердого продукта 0-п-хлорфенил-N-фосфонометилглицинонитрил, т.пл. 170с (с разложением). Маточный раствор выдерживают несколько недель, затем собирают еще 0,85 г Общий выход 3,2 г (51%).
П р и м ер 18. 21 г раствора, содержащего 83,8 вес.% ди(3-метил-4-нитрофенил)фосфита (0,05 моль) и 1,3,5-трицианометил-гексагидро-1,3,5-триазина 3,4 г (0,0167 моль), растворяют в 100 мл ацетонитрила и нагревают 1 ч до 70°С. Затем ацетонитрильный растворитель удаляют в вакууме, остаток растворяют в 50 мл ацетона, содержащего 1 мл воды, и перемешивают при комнатной температуре. Кристаллы (4,3 г) определяют как О-(3-метил-4-нитрофенил)-Н-фосфонометилглицинонитрил, т.пл. 181182°С. Выход 30%.
Найдено, %: С 42,2;Н 4,3;N 14,7.
Вычислено, %: С 42,1; Н 4,2; N 14,7.
Пример 19. 0,0-Ди(п-метоксифенил)-N-фосфонометилглицинонитрил 3,0 г (0,0082 моль) растворяют в 50 мл ацетона и 1 мл воды, перемешивая три месяца при комнатной температуре. Образуется твердый продукт, который удаляют фильтрацией, промывают ацетоном и сушат. Твердый продукт определяют как 0-п-метоксйфеНИЛ-N-фосфонометилглицинонитрил, т.пл. 185-195с (с разложением).
Найдено, %: С 47,1; Н 5,2; N 10,8.
Вычислено, %: С 46,9; Н 5,1;N 11,0.
Пример 20. Ди(о-хлорфенил)фосфит 19,5 г, 80% по весу (0,05 моль) добавляют в ацетонитрильный раствор (50 мл)1,3,5-трицианометилгексагидро-1, 3, 5-триазина 3, 4 г (0,01640 моль) , и нагревают до 70°С 2 ч. Концентрируют раствор до 15 мл, растворяют
в 50 мл ацетона и 1 мл воды и в течение ночи перемешивают. ООразуется твердый продукт. Его собирают, промывают ацетоном и сушат, получая 3,2 г продукта, определяемого как О-(о-хлорфенил)-N-фосфонометиЛглицинонитрил, т.пл. 170-171 С. Выход 82%.
Найдено, %: С 41,4; Н 3,9; N 10,
Вычислено, %:C41,5;H3,9;N 10,8.
П р и ме р 21. 0-0-Ди(пара-фторфенил)-N-фосфонометиглицинонитрил 2,38 г (0,069 моль) перемешивают в 50%-ной по объему смеси тетрахлорметана и хлористого метилена, фильтруют с добавлением метансульфо кислоты 0,67 г (0,069 моль). Раство вьадерживают в течение ночи, образовавшиеся кристаллы собирают фильтрацией и промывают тетрахлорметаном, получая 2,68 г белого кристаллического продукта, определяемого как соль метансульфокислоты 0,0-ди(пфторфенил1-М-фосфонометилглицинонитрила, т.пл. 132-132,5°С.
Найдено, %: С 44,0; Н 4,0; N 6,6 S 7,5.
Вычислено, %: С 44,2; Н 4,0; N 6,5; S 7,4.
Пример 22. п-Толуолсульфокислоту 1,9 г (0,01 моль) нагревают с обратным холодильником в 100 мл бензола, удаляя присутствующую воду азеотропным путем с применением бензола. Бензольный раствор добавляют в бензол-метиленхлоридный раствор (50/50% по объему, 100 мл) 0,0-дифенил-М-фосфонометилглицинонитрила 3,02 г (0,1 моль). Смесь перемешивают 1 мин при комнатной температуре; за это время происходит кристализация. Полученный шлам перемешивают в течение ночи при комнатной температуре, затем фильтруют, получая 4,38 г белого твердого продукта, определяемого как соль п-толуолсульфокислоты О,О дифенил-М-фосфонометилглицинонитрила, т.пл. 152-153°С. Выход 92,4%.
Найдено, %: С 55,4; Н 4,9; N 5,7.
Вычислено,%: С 55,7;Н 4,9; N 5,9.
Пример 23. Хлороформный раствор п-хлорбензосульфокислоты 1,92 г (0,01 моль) добавляют в хлороформный раствор О,О-дифенил-N-фосфонометилглицинонитрила 3,0 г (0,01 моль). Смесь перемешивают, через 10 мин начинается кристаллизация. Шлам перемешивают в течение ночи, фильтруют, твердый продукт промывают хлороформом с получением 4,0 белого твердого продукта, оп1 еделяемого как соль п-хлорбензосульфокислоты О, О-дифенил.-Ы-фосфонометилглицинонитрила, т.пл. 149-151°С. Выход 81i.
Найдено, %: С 50,7; Н 4,1; N 5,7.
Вычислено, %: С 51,0; Н 4,1;N 5,7 Пример 24 . Хлорофо1змный рас твор (20 мл) трихлоруксусной кислоты 1,63 г (0,01 моль) добавляют в хлоро формный раствор (100- мл) 0,0-дифенил -N-фосфонометилглицинонитрила 3,0 г (0,01 моль) и в течение ночи перемешивают при комнатной температуре. Кристаллизация не начинается, растворители удаляют в вакууме с получением 3,75 г светло-желтого масла . 1,5410, определяемого как соль трихлоруксусной кислоты 0,0-дифенил-N-фосфонометилглицинонитрила. Вы ход 80%. Найдено, %: С 43,9; Н 3,5; N 5,9. Вычислено, %: С 43,9; Н 3,5;N 6, П р и м .е р 25. Ацетонитрильный раствор (25 мл) дигидрата щавелевой кислоты 1,2G г (10 моль) добавляют в ацетоновый раствор 0., О-дифенил-N-фосфонометилглицинонитрила 3,02 г (10- моль). Через 10 мин соль начинает выкристаллизовываться из раствора Раствор перемешивают в течение ночи охлаждают, твердый продукт собирают (-1 , 9 г) и промывают ацетоном Вто рую порцию получают путем кондентрир вания маточного раствора (0,8 г). Кристаллы определяют как соль щавеле вой кислоты О,О-дифенил-Н-фосфонометилглицинонитрила, т.пл. (с разложением). Выход 2,7 г (69%). Найдено, %: С 52,1; Н 4,4; N 7,1 Вычислено., %: С 52,1; Н 4,4; N 7,1. Пример 26. Эфирный раствор хлорной кислоты добавляют в хлороформно-эфирный раствор 0,0-дифенил-N-фосфонометилглицинонитрила 3,0 г (10 моль). Перхлорат медленно выкристаллизовывается в виде белых призм. Твердый продукт, определяемый как соль хлорной кислоты 0,0-дифенил-N-фосфонометилглицинонитрила, собирают и промывают эфиром-хлороформом получая 0,73 г, т.пл. 166-168С. Выход 18%. Найдено, %: С 44,8; Н 4,0; N 7,0 Вычислено,%: С 44,7; Н 4,0; N 7,0 Пример 27. Хлороформно-мета нольный раствор трихлорметанфосфоновой кислоты 1,99 г (0,01 моль) добавляют в хлороформный раствор 0,0-дифенил-М-фосфонометилглицинонитрила 3,0 г (10 моль). Через 10 мин добавляют эфир, кристаллов не образуется. Затем добавляют петролейный эфир до достижения точки помутнения Через 10 мин начинают образовыватьс кристаллы,раствор вьщерживают еще 10 мин. Кристаллы собирают в виде двух порций (2,9 г). Кристаллы опре деляют как соль трихлорметанфосфоно вой кислоты О,О-дифенйл-М-фосфонометилглицинонитрила, т.пл. 145146 С. Выход- 58%. Найдено, %: С 38,3; Н 3,5; N 5,6 Вычислено, %: С 38,3; Н 3,4; . N 5,6. Пример 28. Эфирный раствор фторборной кислоты добавляют в хлороформно-эфирный раствор 0,0-дифенил-N-фосфонометилглицина 3,0 г (10 моль). Раствор перемешивают в течение ночи, твердый продукт фильтруют, промывают эфиром/хлороформом (50/50%) с получением белых кристаллов (1,1 г), определяемых как соль фторборной кислоты О,О-дифенил-N-фосфонометилглицинонитрила, т.пл. 156-158с. Выход 28%. Найдено, %: С 46,0; Н 4,2; N 7,2.. Вычислено, %: С 46,2; Н 4,1; N 7,2.. . Пример 29. -Газообразный броистый водород вводят в хлороформный раствор О,О-дифенил-Н-фосфонометилглицинонитрила 3,0 г (10 моль). Раствор вьщерживают в течение ночи после того, как гидробромид.кристаллизуется, кристаллы собирают и промывают эфиром с получением 3,0 г соли бромистого фодорода О,О-дифенил-М-фосфонометилглицинонитрила. Выход 78%. Найдено, %: С 47,1; Н 4,3; N 7,4. Вычислено, %: С 47,0;Н 4,2; N 7,3. Пример 30, 57%-ный раствор йодистоводородной кислоты (2 мл) добавляют в хлороформный раствор О,0-дифенил-N-фосфонометилглицинонитрила 3,О- г (10 моль). Раствор мутнеет и становится золотым. Через 2 ч трведый продукт не образуется, так что добавляют эфир до достижения точки помутнения и начала кристаллизации. Раствор перемешива}от еще 1 ч, твердый продукт, определяемый как соль йодистоводородной кислоты О,0-дифенил-N-фосфонометилглицинонитрила, собирают в виде светло-желтых плиток, т.пл. 153-164°С (2,4 г. Выход 56%. Найдено, %: С 41,8; Н 3,8; I 29,3. Вычислено, %: С 41,9; Н 3,8; I 29,5. Пример 31. Трифторуксусную кислоту 1,13 г (10 моль) добавляют в хлороформный раствор 0,0-дифенил-N-фосфонометилглицинонитрила 3,0 г (10 моль). Раствор перемешивают в течение ночи, растворитель выпаривают в вакууме с получением 4,0 г светло-желтого масла, п 1,5172, определяемого как соль трифторуксусной кислоты О, О-дифенил-Ы-фосфонометил- .. глицинонитрила. Выход 96%. П. р и м е р 32. Трифторметансульфокислоту (дымящуюся) 1,50 г (Юмоль) добавляют в хлороформный раствор О,0-дифенил-М-фосфонометилглицинонитрила 3,О г (10 моль). Реакционную смесь перемешивают 2 ч при ком.натной температуре, затем добавляют эфир до достижения точки помутнения. Продукт кристаллизуется. После выдерживания 1 ч твердый продукт собирают и промывают хлороформом-эфиром (50%), получая 3,8 г соли трифтормеггансульфокислоты о,О-дифенил-М-фосфон метилглицинонитрила, т.пл. 119-120 Выход 84%. Найдено, %: С 42,7; Н 3,6; N 6, Вычислено, %: С 42,5; Н 3,6; N 6,2. Пример 33. В хлороформный раствор О,О-дифенил-М-фосфонометил глицинонитрила 15,1 г (0,05 моль) добавляют метансульфокислоту 5,0 г (0,051 моль). Раствор перемешивают 2 ч при комнатной температуре. Оседает твердый продукт, который соби (рают, промывают эфиром и сушат. Тве дый продукт весит 15,90 г и являетс солью метансульфокислоты 0,0-дифенил-Ы-фосфонометилглицинонитрила, т.пл. 147-150с. Выход 82,1%. Найдено, %: С 44,0; Н 4,0; N 6, S 7,5. Вычислено, %: С 44,2;Н 4,0; N 6,5; S 7,4. Пример 34. Эфирный раствор (10 мл) азотной кислоты 0,9 г 70% по весу (0,01 моль) добавляют в хл роформный раствор (100 мл) добавляют в хлороформный раствор (100 мл) содержащий О,О-дифенил-М-фосфономет глицинонитрила 3,0 г (0,01 моль). Помутнения не наблюдается. Добавляют диэтилового эфира, затем изооктан (20 мл); после этого твердый продукт начинает выкристаллизовываться. Смесь перемешивают 30 мин при комнатной температуре; кристалл собирают, промывают хлороформом и суш на воздухе. Кристаллы весят 2,66 г и представляют собой соль азотной кислоты О,О-дифенил-М-фосфонометилг глицинонитрила, т.пл. 116-116,. Выход 72%. Найдено, %: С 49,2; Н 4,42; N 11,6. Вычислено, %: С 49,32; Н 4,42; N 11,5. Пример 35. В раствор 0,0-дифенил-М-фосфонометилглицинонитрила 3,0 г (0,01 моль) в хлороформе (100 мл) добавляют эфирный раствор 98%-ной серной кислоты 1,01 г (0,01 моль). Затем добавляют дополнительное количество хлороформа, смесь перемешивают 2 ч. Твердый про дукт удаляют фильтрацией и промывают хлороформом, эфиром, потом сушат получая 3,9 г продукта, определяемо го как соль серной кислоты О,0-дифе нил-ы-фосфонометилглицинонитрила, т.пл. 151-151,5°С. Выход 100%. Найдено, %: С 44,90; Н 4,27; S 8,05. Вычислено, %: С 45,0; Н 4,28; S 8,01. Пример 36. Эфирный раствор фосфорной кислоты (0,01 моль) добавляют в хлороформный раствор 0,0-дифенил-Н-фосфонометилглицинонитрила 3,0 г (0,01 моль) при комнатной температуре. Раствор моментально мутнеет. На дне колбы образуется масло. По охлаждении растворитель сливают, выпаривают досуха и сушат безводным сульфатом магния. Твердый материал определяют как соль фосфорной кислоты.О,О-дифенил-Н-фосфонометилглицинонитрила, т.пл. 74,578,5°С.. Выход 25%. Найдено, %: С 44,8; Н 4,6; N 7,1. Вычислено, %: С 45,0; Н 4,5; N 7,0. П р и м-е р 37. Гетерогенный раствор О,О-дифенил-Мтфосфонометилглицинонитрила 60,4 г (0,2 моль) в этаноле (500 мл) охлаждают на ледяной бане с пропусканием через него сухого газообразного хлористого водорода. Раствору дают стоять, добавляют этиловый эфир, после чего отсосной фильтрацией собирают белый твердый продукт. При пропускании через .:. этанол-эфирный маточный раствор приблизительно при 0°С сухого хлористоводородного газа образуется дополнительное количество белого твердого продукта, который собирают и промывают эфиром, он является гидрохлоридной солью О,О-дифенил-Ы-фосфонометилглицинонитрила, т.пл. 112.-123°С. Выход62,7 г (93%). Найдено, %: С 53,51; Н 4,78; N 8,30. Вычислено, %: С 53,19; Н 4,79; N 8,27 Пример 38. Ди(2,4,6-триметилфенил)фосфит 17,8 г (0,05 моль) добавляют в ацетонитрильный раствор (50 мл) 1,3,5-тpициaнoмeтил-гeкcaгидpo-l, 3,5-триазин 3,4 г (0,0164 моль) с нагревом смеси 18 ч при . Образовавшийся черный раствор фильтруют и концентрируют с получением масла. 7 г этого масла хроматографируют на силикагеле (4.50 г) с применением циклогексана/этилацетата (70/30%, фракции по 60 мл), получая 1,0 г (14%) 0,0-ди(2,4,6-триметилфенил)-Мфосфонометилглицинонитрила, т.пл. 118-120°С в фракциях 28-40, которые со времененм кристаллизуются. Найдено, %: С 65,38; Н 7,07; N 7,18. Вычислено, %: С 65,27; Н 7,04; N 7,25. Пример 39. Раствор сложного иэфира по примеру 3 (0,025 моль) о влажном ацетоне (50 мл) нагревают обратным холодильником 2 ч, затем ыдерживают при комнатной температуе 5 дней. Взвесь фильтруют с получеием розоватого продукта с примесями (0/9 г). Фильтрат помещают в закрыую колбу и вьщ-зрживают При комнатой температуре еще 30 дней. Полученую взвесь фильтруют, твердый продукт ромывают 50 мл ацетона. Получают ,5 г О-(пapa-мeтилтиo)-N-фocфoнoетилглицинонитрила в виде белого
твердого продукта, т.пл. 250-253С (с разложением). Выход 66%.
Найдено, %: С 44,26; Н 4,86; N1.0,22.
Вычислено, %: С 44,12; Н 4,81; N 10,29.
П р И м е р .40. Дифенилтиофосфит .8,2 г (0,0246 моль) и 1,3,5-трицианометилгексагидро-1,3,5-триазин 1,68 г (0,00823 моль) растворяют в 50 мл ацетонитрила с нагревом при 60-65°С 2 дня. Полученное масло хроматографируют на силикагеле(450 г), элюиру с применением циклогексана/этилацетата (60/40%, фракции по 60 мл),получая 1 , 6 г (20%) О,О-дифенил-Ы-тиолг
фосфонометилглицинонитрила, 1,5847 во фракции 40.
Найдено, %: С 56,40; Н 4,80; N 8,73; S 10,26.
Вычислено, %: С 56,60; Н 4,75; N 8,80; S 10,07.
Пример 41. Ацетонитрильный раствор (100 мл) ди((}-нафтил)фосфита 33,5 г (0,1 моль) и 1,3,5-трицианометил-гексагидро-1,3,5-триазина 20,4 г (0,1 моль) нагревают с обратным холодильником 1 ч, концентрируя затем с получением красно-коричневого масла. 10 г масла очищают жидкостной хроматографией под высоким давлением, элюируя с применением циклогексана/этилацетата (60/40%, фракции по 20 мл). Фракции 45-64 соединяют и концентрируют, полученное масло кристаллизуют из тетрахлорметана, получая 1,1 г О , 0-ди(|3-нaфтил)-N-фocфoнoмeтилглицинoнитpилa в виде темно-желтого твердого продукта, т.пл. 104-105°С.Найдено, %: С 68,58; Н 4,79; N 6,92.
Вычислено, %: С 68,65; Н 4,76; N 6,96.
Пример 42. Перемешиваемый раствор ди(3,4-метилендиоксйфенил)фосфита (0,05 моль) и 1,3,5-трицианометил-гексагидро-1,3,5-триазин (0,0167 моль) в 75 мл ацетонитрила нагревают 3 ч до 75С, вьщерживая затем в течение ночи при комнатной температуре. Полученный раствор концентрируют с получением масла янтарного цвета. В хлороформный раствор (100 мл) этого масла 7,6.г (0,02 моль) по каплям добавляют метансульфокислоту 1,92 г (0,02моль После перемешивания 1-5 мин добавля. ют эфир (200 мл); оседает твердый белый продукт Его перекристаллизоввают два раза из ацетона, получая 4,6 г соли метансульфокислоты 0,0ди(3,4-метилендиоксифенил)-Н-фосфонометилглицина, т.пл.. 135-136,. Выход 47%.
Найдено, %:С 44,26; Н 3,94; N 5,71.
Вычислено, %: С 44,45; Н 3,94; . N 5,76.
Пример 43. Раствор 0,01 мол масла янтарного цвета по примеру 42 во влажном ацето {е (70 мл) 4 дня обрабатывают с обратным холодильником, выдерживают 1 день при комнатной температуре. Полученную взвесь фильтруют с получением 1,7 г 0-(3,4-метилендиоксифенил)N-фосфонометилглицинонитрила в виде твердого продукта белого цвета, т.пл. 160-161°С.
Пример 44. Перемешиваемый раствор ди-(3,4-дихлорфенил)фосфита (о,04 моль) и 1,3,5-трицианометил-гексагидро-1,3,5-триазина (О,013моль В ацетонитриле (40 мл) нагревают до 80°Сf выдерживая затем 18 ч. Полученный раствор концентрируют с получением масла, добавляя влажный ацетон (80 мл), после чего смесь нагревают с обратным холодильником 80 ч. Полученную взвесь фильтруют, получая тведый белый продукт, который промывают 50 мл ацетона, поопучая 6,3 г 0-(3,4-дихлорфенил)-М-фосфонометилглицинонитрила, т.пл. 169-170°С. Выход 53%.
Пример 45. Получают 0,0-ди(4-хлор-м-толил)-N-фосфонометилглицинонитрил в виде розового масла, п 1,5588.
Пример 46. Получают соль метансульфокислоты О,0-ди(4-xлop-м-тoлил)-N-фocфoнoмeтилглицинoнитpил, т.пл. 143-147°С.
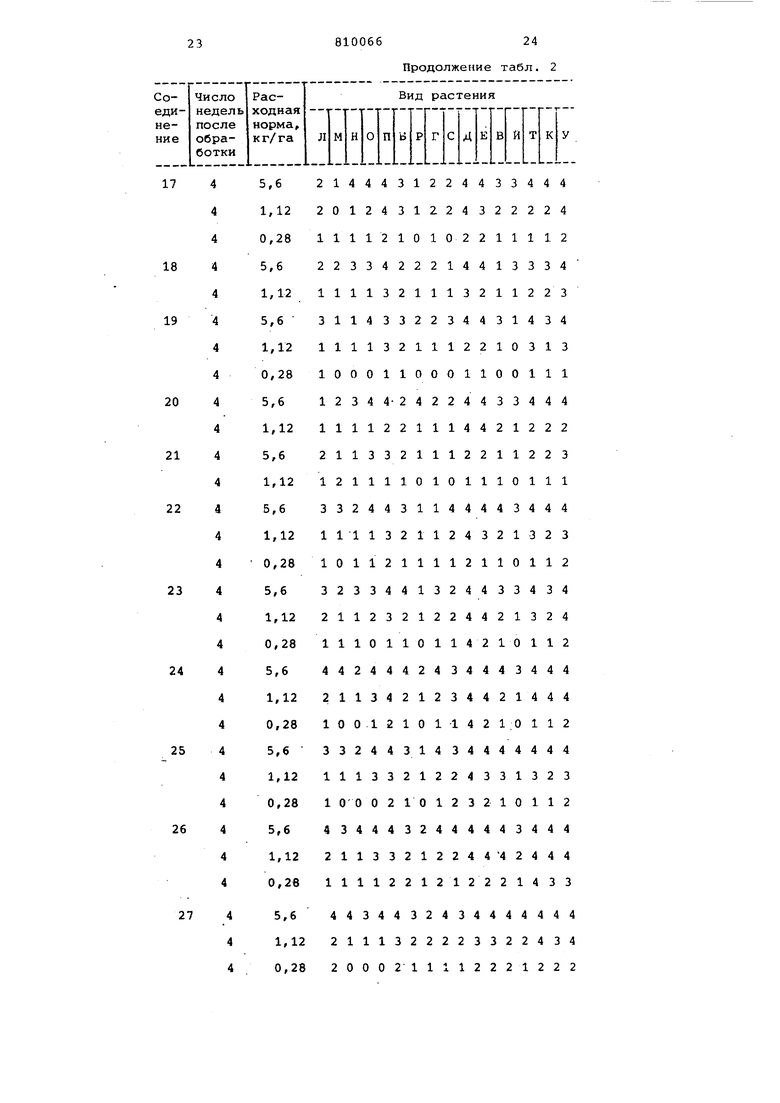
Послевсходовое гербицидное действие различных соединений согласно изобретению демонстрируют следующим образом.
Активные ингредиенты наносят в виде раствора для опрыскивания на различные виды растений в стадии развития 14-21 дней. Раствор для опрыскивания - водный или раствор органического растворителя в воде, содержащий активный ингредиент и ПАВ (35 ч. бутиламиновой соли додецилбензолсульфокислоты и 65 ч. таллового масла, конденсированного с окисью этилена в соотношении 11 моль окиси этилена и 1 моль таллового масла) наносят на растения в различных сериях вегетационных сосудов плоской формы с применением различной расходной нормы (кг/га) активного ингредиента. Обработанные растения помещают в парниковые условия, действие регисрируют по истечении примерно двух или четырех недель.
Послевсходовое действие дов представлено в табл. 1 менением следующего кода:
Повреходение растеНИИ, %
0-24
25-49
50-74
75-99 Все растения
уничтожены
Во время испытания не присутствовавшие растения+ Виды испытуемых растений в табл.1,
2и 3 обозначены буквами: А - бадяк полевой; Б - дурнишник; В - лимнохарис; Г - ипомея; Д - марь белая; Егорец перечный; Ж - Cyperus rotundus;
3- пырей; И - гумай; И - В готus tec to г urn; К - ежовник; Л.- соя; М свекла сахарная; Н - пшеница; О рис; П - сорго; Р - гречиха полевая; С - Hemp sesbania; Т - просо; У росичка кровяная. 1411,234414432333 45,63342442112.3 2411,2 44324332234 45,633324421143 3411,2 33334423323 45,634124422212 4411,2443444 22243 45,643323421134 5411,2 24344322334 44,48 34334421213 6411,23443441 2223 43,6 241344 11313 7411,2 23214321422 . 4 . 5,6 32213311413 8411,2 24214423413 45,6333244 2. 1413 ..9 4 11,244434413234. 45,644424413234 10 411,2443344233 4 4 45,6 333244-12134
Продолжение табл. 1.
10 11,2 44444433234 5,6 44424423134 11,2 44324423424 5,6 44324433314 11,2 2+444333434 4,48 3+33423 3333 11,2 44434442234 5,6 33324411234 11,2 44424443344 5,6 .34013422433 11,2443 144 34424 5,6 44214 423403 11,2 44434433434 5,6 34434422134 11,2 33324422323 5,6 4 3 2 2 4 4 2 1 3 1 3 11,244422231302 5,6 24112231102 11,23422342 3323 5,6 33124421323 11,2 44324423433 5,6 23324422233 1,2 44434444444 5,6334 2 4433324 1,2 44444444244 1,2 24344412223 5,6 23223310412
Продолжение табл. 1
Продолжение табл. 1
21
81006622
Продолжение табл. 2
81006624
23
РасЧислоходная недельнорма, после к г/га
Л
м обработки
45,62144431224433444 41,122012431224322224 40,281111210102211112
45,62233422214413334 41,121111321113211223
45,63114332234431434 41,121111321112210313 40,281000110001100111
45,612344-24224433444 41,121111221114421222
45,62113321112211223 41,121211110101110111
45,63324431144443444 41,121111321124321323 40,281011211112110112
45,63233441324433434 41,122112321224421324 40,281110110114210112
45,64424442434443444 41,122113421234421444 40,281001210114210112
45,63324431434444444 41,121113321224331323 40,281 00 0210123210112
45,64344432444443444 41,1221133212244 4 2444 40,261111221212221433
4 5,64434432434444444
4 1,122111322223322434
4 0,282000211112221222
Продолжение табл. 2 Вид растения
и
к
Е
В
н
25
28
29
30
31
32
33
34
35
36
37
81006626
Продолжение табл. 2
3433432434444444
2112321223322433
1011221122221222
4443341434444444
2212321224422334
1011211122210222
4424444444444444
1111211224441224
1111211122220222
3322331334444444
2311442224443333
1001211113211112
4434442344444444
2111231143231233
1101110011110122
2324431214434443
0211421102111112
1100210101100002
33-34442224213444
2110431114212222
1 + 00211111100111
2214432224422434
1211221113211222
1000100000000100
3233433223433334
1111 2 21212111122
1001110111001001
3113431214321334
Ч
1111321103211233 1011321102101122
27
ТаблицаЗ
81006628
Продолжение табл. 2.
Продолжение табл. 3 29810066 Продолжение табл. 3Вид растания Расходнаянорма, кг/га
11,220000000000
11,220010001100
11,220000000000
11,230000000000
11,230000000100
11,230010000100
11,230000022300
11,230000011000
11,230001001000
11,200000000000
11,230000000000 в. Предвсходовое гербицидное действие различных соединений демонстрируется следующим образом. Соответствующее количество пахотной почвы помещают в алюминиевые ве- гетационные сосуды плоской формы и утрамбовывают с достижением уровня 3/8 - 1/2 дюйма от верхней части каждого сосуда. Заранее определенное число семян или сеянцев каждого вида растений помещаю т на поверхность почвы в каждом сосуде, вдавливая затем в почву. Верхний слой почвы обрабатывают гербицидными составами, описанными в примере А, на основе активных ингредиентов согласно изобретению . Согласно предлагаемому способу почву, необходимую для покрытия семян и сеянцев, взвешивают и смешивают с гербнцидным составом, содержащим известное количество активного ингредиента. После этого вегетационные сосуды наполняют смесью, выравнивая поверхность почвы. Полив осуществляют с помощью отверстий на дне сосудов, через которые почва может впитывать влагу. Сосуды с семенами и сеянцами помещают на влажный песок и выдерживают примерно две недели в нормальных условиях при солнечном свете с поливом. К концу этого периода сравнивают число, взошедших растений с необработанным контролем. Данныо приведены в табл. 3 с примененном следующего кода: 45
11,200000001100
40 41
11,200000000000
10
11,230000000000 . 42 43
11,230003001000
15 44 45 46
11,230000000300
11,230001000200
11,230000100000
20
о ;: p-CHiUHCHiCM
CtHsO. CjHjOСиз25вестно) 5,6
00000000000 ге ше дей изв рил пы тел что дей изв ла (Ч где Продолжение табл. 3 Вид растания асходная. Уничтожение растений,% 0-25 26-50 51-75 76-100 таким образом, предлагаемый бицидный состав обладает повыной гербицидной активностью. Формула изобретения Гербицидный состав, содержащий ствующее начало на основе проодных М-фосфонометилглицинонита и добавку, выбранную из групноситель, разбавитель и наполниь, отли чающийся тем, , с целью усиления гербицидного ствия, он содержит в качестве проодного N-фосфонометилглициноиитрисоединения общей формулы IV о л-Ха-0),.-Р-CHj-Н-ОЦ-СИ Их арил - фенил, jj-нафтил, Сифенил; X - хлор, фтор, метил, метокси, метилтио, метилендиокси, циан, трифторметил, нитро; Z - кислород или сера; а - целое число от О до 3; b - целое число О или 1; 31. 81006 R - хлористоводородная,Сромистоводородная,йодистоводородная,хлорная,фтррборная, азотная,серная,фосфорная, щавелевая,п-толуолсульфоновая,п-хлОрбензолсульфоновая, УКсусГа ° т;руксусная кислоты или трифторметансульфокислота, метансульфокислота; 632 X - о или 1 при условии что х равен О, если % равно 1, в количестве 5-95 вес.ч. Источники информации, принятие во внимание при экспертизе 1. Патент США 3923877, кл. 260-502.5, опублик. 02.12.75.
