Изобретение относится к усовершенствованному процессу получения замещенных изофлавоновых производных высокой чистоты, которые пригодны для приготовления фармацевтических композиций, в частности для приготовления имприфлавона (ОsteоchinR), который является общим названием 7-изопропокси-изофлавона, пригодного для лечения остеопороза (НU-РS 162 377). При описании замещенные характеризуются следующим образом:
R обозначает водород или изопропил;
R2 и R3 обозначают водород или С1-2-алкокси.
В соответствии с изобретением чистые изофлавоновые производные общей формулы (I)
 (I) могут быть получены взаимодействием резорцинольных производных общей формулы (II)
(I) могут быть получены взаимодействием резорцинольных производных общей формулы (II)
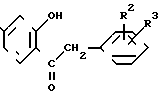 (II) и этил-орто-формиата
(II) и этил-орто-формиата
(С2Н5О)3СН (III) в присутствии основного соединения и по желанию алкилированием полученных продуктов, тогда как соединения общей формулы (II) и (III) подвергаются циклизации в присутствии органического растворителя, предпочтительно диметилформамида и/или изопропанола, и/или в объеме 0,3-2-кратного количества к рассчитанному объему по отношению к резорцинольным производным общей формулы (II), и/или в присутствии избытка эфира формулы (III) при температуре 70-100оС, после чего реакционная смесь становится пересыщенной (20-70% по массе) по отношению к продукту общей формулы (IV).

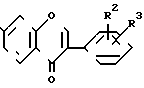 (IV) и таким образом продукт формулы (IV) непрерывно высаждается из реакционной смеси в процессе реакции. После охлаждения реакционной смеси продукт общей формулы (IV) фильтруется без добавления или с добавлением растворителя или добавляется почти эквивалентное количество безводного карбоната калия и выделяется кристаллизующаяся двойная соль общей формулы (V)
(IV) и таким образом продукт формулы (IV) непрерывно высаждается из реакционной смеси в процессе реакции. После охлаждения реакционной смеси продукт общей формулы (IV) фильтруется без добавления или с добавлением растворителя или добавляется почти эквивалентное количество безводного карбоната калия и выделяется кристаллизующаяся двойная соль общей формулы (V)
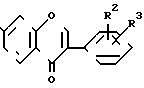
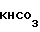 (V) в то время как загрязняющие примеси общей формулы (VI)
(V) в то время как загрязняющие примеси общей формулы (VI)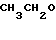
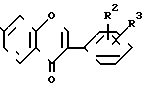 (VI) остаются в растворе, соответственно они селективно растворяются, после чего продукт общей формулы (V) или (I), где R Н, взаимодействует с изопропилгалоидом и по желанию выделяется чистый продукт, содержащий до 0,5 мас. загрязнения общей формулы (VI).
(VI) остаются в растворе, соответственно они селективно растворяются, после чего продукт общей формулы (V) или (I), где R Н, взаимодействует с изопропилгалоидом и по желанию выделяется чистый продукт, содержащий до 0,5 мас. загрязнения общей формулы (VI).
Как известно, соответствующие замещенные 7-гидрокси-изофлавоновые производные являются подходящими промежуточными соединениями при синтезе 7-алкоксиизофлавонов, которые являются эффективными лекарственными средствами в терапевтическом лечении и в ветеринарии. Таким образом, желательным является получение 7-гидроксиизофлавоновых производных общей формулы (I) такой степени химической чистоты, чтобы эти соединения были пригодны для приготовления 7-алкилированных конечных продуктов соответствующей чистоты, т.е. важным является требование подавления образования загрязняющих производных общей формулы (VI).
7-Гидроксипроизводные общей формулы (I) могут быть получены в промышленности циклизацией резорцинольного производного общей формулы (II) и эфира орто-муравьиной кислоты общей формулы (III).
Процессы циклизации выполняются при 110-150оС в присутствии смеси растворителя, кипящего при температуре выше 100оС (гомологи пиридина, диметилформамид и т.д.), и вторичного амина (пиперидин, морфолин, пирролидин) предпочтительно при температуре кипения смеси. В некоторых случаях образованный спирт отгоняется в процессе реакции вероятно для того, чтобы повысить конверсию, или для того, чтобы повысить температуру.
При воспроизведении названных процессов было установлено, что образуется значительное количество близкого к 7-гидроксиизофлавоновым производным общей формулы (VII) 7-этокси-изофлавонового производного (в некоторых случаях 2-10 мас. измеряемых методом ВЭЖХ) общей формулы (VI), близкого к другим побочным продуктам. При снижении молярного избытка этил-орто-формиата выход его значительно снижается, но образование этокси-изофлавоновой примеси не может быть полностью устранено. 7-Гидрокси-изофлавоновое производное общей формулы (VII), полученное этим способом, может быть очищено только с применением дорогостоящего метода при использовании нескольких обработок растворителем.
Было установлено, что в отличие от реакции, описанной в литературе, осуществляемая в умеренных условиях циклизация может быть проведена таким образом, что образованный 7-гидрокси-изофлавон общей формулы (I) начинает кристаллизоваться из реакционной смеси вскоре после начала реакции. Чистые 7-гидрокси-изофлавоновые производные общей формулы (I), (VII), выделенные из реакционной смеси, содержат такое небольшое количество примеси общей формулы (VI) (около 0,1-0,5 мас. найденных по методу ВЭЖХ) и других побочных продуктов, которое может быть устранено при желании осуществлением одностадийной очистки. Кроме того, загрязняющая примесь общей формулы (VI) может быть удалена из продукта выделением новой двойной калиевой соли. При проведении реакции циклизации предпочтительным является осуществление ее при 80-90оС и при применении теплового воздействия в течение 6-10 ч. Далее, предпочтительно оставлять этанол, образующийся в реакции в процессе циклизации. Более предпочтительно использовать основной катализатор для осуществления реакции циклизации такой как вторичный амин, преимущественно морфолин, пиперидин или пирролидин.
При осуществлении реакции циклизации установили, что наиболее предпочтительным является взаимодействие кетона общей формулы (I) с 20% молярного избытка эфира орто-муравьиной кислоты и с 0,3-2-кратным количеством растворителя и приблизительно с 20 мол. вторичного амина при 80-90оС. Спустя 30-60 мин из реакционной смеси начинает кристаллизоваться 7-гидрокси-изофлавон общей формулы (I). Реакция продолжается до полной конверсии исходного кетона общей формулы (II). Выход составляет свыше 90% и полученный продукт содержит загрязнений общей формулы (VI) менее, чем 0,1-0,5 мас. устанавливаемых по методу ВЭЖХ.
Двойная соль общей формулы (V) образуется из реакционной смеси при использовании неполярного растворителя, предпочтительно толуола безводным карбонатом калия при 40-80оС, предпочтительно при 60оС. Дальнейшая очистка и отделение проводится выделением двойной соли. Двойная соль может быть непосредственно алкилирована алкил-галоидом в отсутствии агента, связывающего кислоту, в подходящем растворителе, таком как диметилформамид или кетон. Таким образом могут быть получены 7-изопропоксиизофлавоновые производные в чистом состоянии.
Последним этапом процесса в соответствии с данным изобретением является (по желанию) алкилирование чистого 7-гидрокси-изофлавона. Предпочтительно алкилирование осуществляется алкил-бромидом в присутствии карбоната калия в качестве кислотосвязывающего агента в среде ацетона или диметилформамида. При соответствующих условиях продукт, полученный после циклизации, содержит менее, чем 0,1 мас. 7-этоксиизофлавона, и этот продукт может быть использован для приготовления фармацевтических композиций.
Другим преимуществом способа является то, что процесс обеспечивает выход, превышающий более, чем на 10% выход, полученный в известном способе, а также то, что могут быть получены особенно чистые химические соединения, которые по качеству являются пригодными для фармацевтического использования.
П р и м е р 1. 62,5 г (0,274 моль) 2,4-дигидроксифенилбензил кетона, 105 мл изопропанола, 5 мл морфолина и 49,7 г (0,33 моль) этил-орто-формиата перемешиваются в течение 7 ч при 80-90оС. В первые полчаса протекания реакции начинают выделяться кристаллы. В конце реакции закристаллизованная суспензия охлаждается до -5оС и фильтруется. После сушки получается 59,1 г 7-гидроксиизофлавона. Выход 90,6% Содержание активного ингредиента продукта, определенное спектроскопически, составляет 98% Загрязнение (примесь) 7-этокси-изофлавона, определяемое по методу ВЭЖХ 0,2-0,4 мас.
П р и м е р 2. Действовали как описано в примере 1. Когда реакция заканчивается, из кристаллической суспензии отгоняется 50 мл растворителя и при перемешивании добавляется 160 мл метанола. Смесь перемешивается в течение 20 мин при 58-60оС, после чего она кристаллизуется при -5оС. Осажденное вещество фильтруется и сушится. Получается 58,8 г 7-гидроксиизофлавона. Выход 90,1% Содержание активного ингредиента составляет свыше 98% (спектроскопически). Загрязнение 7-этоксиизофлавоном по методу ВЭЖХ 0,2-0,3 мас.
П р и м е р 3. Из реакционной смеси, полученной в примере 1, отгоняется 90 мл растворителя, после чего добавляется 37,8 г (0,274 моль) безводного карбоната калия и 200 мл толуола, и реакционная смесь перемешивается в течение 30 мин при 60-65оС, за которым следует перемешивание при 0 (-5оС) в течение 2 ч. Двойная соль, содержащая 7-гидрокси-изофлавон-калиевую соль и калийгидрокарбонат, отфильтровывается и высушивается. Получается 98,5 г двойной соли. Выход: 95%
Анализ: С15Н9О3К.КНСО3.
Молекулярная масса 376.
Вычислено, С 51,06; Н 2,66; К 20,7.
Найдено, С 51,9; Н 2,76; К 21,8.
ЯМР на спектрофотометре Bruker WР-80 в растворителе ДМСО-d6 при использовании в качестве внутреннего стандарта ТМС. 1Н "двойная соль" 7-гидрокси-изофлавон 5 С-Н 7,50 м.д. (д)3 J=9 Гц 8,00 м.д. (д)3 J=9 Гц 6 С-Н 6,13 м. д. (дд) 6,90 м.д. (дд) 8 С-Н 5,77 м.д. (д)4 J=2 Гц 6,87 м.д. (д)4 J=2 Гц 13С 7С 174,93 м.д. 156,82 м.д.
Полученная двойная соль растворяется в трехкратном количестве метанола и воды при 50-60оС. Раствор осветляется и фильтруется. Величина рН фильтрата доводится до 1 с использованием разбавленного 1:1 водного раствора соляной кислоты, осажденное вещество фильтруют, промывают до нейтральной реакции и сушат. Получают 58,7 г 7-гидрокси-изофлавона. Продукт содержит 98 мас. чистого продукта по спектроскопическому определению. Содержание 7-этоксиизофлавона, определяемого по методу ВЭЖХ, 0,1 мас. Выход 90%
П р и м е р 4. 50 г (0,219 моль) смеси 2,4-дигидроксифенилбензилкетона, 20 мл диметилформамида, 2,6 мл морфолина и 39,06 г (0,26 моль) этил-орто-формиата перемешиваются в течение 7 ч при 80-90оС. Через 25 мин наблюдается кристаллизация. К концу реакции закристаллизованная суспензия разбавляется 120 мл хлороформа и она кристаллизуется при 0оС в течение 2 ч. После фильтрации продукт дважды обрабатывается 45 мл хлороформа и высушивается. Получается 47,9 г 7-гидрокси-изофлавона. Выход 91,9% Загрязнение 7-этокси-изофлавона по методу ВЭЖХ 0,1-0,3 мас.
П р и м е р 5. 25 г (0,1096 моль) смеси 2,4-дигидроксифенилбензилкетона, 12,5 мл диметилформамида, 2 мл пиперидина и 19,7 г (0,33 моль) этил-орто-формиата перемешиваются в течение 16 ч при 80-90оС и разбавляются 65 мл хлороформа. Высадившееся вещество выделяется и кипятится со смесью хлороформа и метанола 8:1, фильтруется и высушивается. Получается 23,5 г 7-гидрокси-изофлавона. Выход 90% Содержание продукта, определяемое спектроскопически, 98,5 мас. Содержание 7-этокси-изолфлавона 0,2-0,4 мас. (ВЭЖХ).
П р и м е р 6. Смесь из 20 г (0,0877 моль) 2,4-дигидроксифенилбензилкетона, 20,7 г (0,14 моль) этил-орто-формиата и 1 мл морфолина перемешивается на горячей водяной бане. Кристаллизация начинается после нагревания в течение 25 мин. В процессе реакции внутренняя температура резко падает с 96 и до 87оС. После перемешивания в течение 5 ч реакционная смесь разбавляется 48 мл хлороформа и затем процесс продолжается так, как описано в примере 5. Получается 18,9 г 7-гидрокси-изофлавона. Выход 90,6% Содержание продукта, определяемое спектроскопически, 99 мас. содержание 7-этоксифлавона: 0,1-0,2 мас. (ВЭЖХ).
П р и м е р 7. Смесь, состоящая из 100 кг (438,5 моль) 2,4-дигидроксифенилбензилкетона, 38 кг диметилформамида, 5,2 кг морфолина и 75 кг (506 моль) этил-орто-формиата перемешивается при 80-90оС, при этом в течение часа начинается кристаллизация. После 7 ч выдерживания при 60оС к суспензии добавляется 360 кг хлороформа. После охлаждения кристаллическое вещество центрифугируется, промывается хлороформом, фильтруется и сушится. По- лучается 94,5 кг 7-гидроксиизофлавона, содержание 7-этокси-изофлавона 0,1 мас. выход 90,5%
П р и м е р 8. 98,5 г двойной соли суспензируется в 100 мл диметилформамида. Добавляется 44 г (0,36 моль) изопропилбромида и реакционная смесь перемешивается при 75-80оС в течение 2 ч, а затем выливается в 250 мл воды. Высадившееся вещество фильтруется, промывается водой до нейтральной реакции и сушится при 60оС. Получается 66 г 7-изо-пропокси-изофлавона, содержание активного ингредиента 99,5% потери при высушивании составляют 0,1% содержание 7-этоксиизофлавона 0,1% Выход 86,1% в расчете на 2,4-дигидроксифенилбензилкетон.
П р и м е р 9. 14,4 г (0,05 моль) 2,4-дигидроксифенил-(34 -диметоксибензил)кетона взаимодействуют с 10,5 г (0,07 моль) этил-орто-формиата в 10 мл диметилформамида в присутствии 1 мл морфолина. Реакционная смесь выдерживается при температуре 80-85оС и в течение второго часа высаждается твердое вещество. Через 6 ч к смеси добавляется 100 мл хлороформа, высажденное вещество фильтруется и сушится. Получается 7-гидрокси-3 4 -диметоксиизофлавон. Т. пл. 259-262оС.
После перекристаллизации из диметилформамида продукт плавится при 263-264оС.
Анализ: С17Н14О5 Молекулярная масса 298.
Вычислено, С 68,46; Н 4,69.
Найдено, С 68,30; Н 4,72.
Продукт идентифицирован в соответствии с ЯМР-исследованием.
Тонкослойная хроматография.
Проявляющая система: толуол: н-бутилацетат-уксусная кислота
а=8:2:1.
Адсорбент: Кизельгель 60 F254 (ф. merk).
Наносимое количество: 0,2 г (10 мл диметилформамида 100 мкг).
Фронт: 16 см.
Исследование: в УФ-свете при 254 нм
Rf=0,4.
П р и м е р 10. 47,4 г (0,15 моль) 2,4-дигидроксифенил-3,4-этоксибензилкетона взаимодействуют с 31,5 г (0,21 моль) этил-орто-формиата в 20 мл диметилформамида в присутствии 3 мл морфолина. Реакционная смесь выдерживается в течение 6 ч при 80-85оС. После охлаждения до 60оС добавляется 100 мл хлороформа. Высадившееся вещество фильтруется и сушится. Продукт: 7-гидрокси-34 -диэтоксиизофлавон. Т.пл. 189-191оС.
После перекристаллизации из диметилформамида т.пл. 192-193оС.
Анализ из формулы С19Н18О5.
Рассчитано, С 69,93; Н 5,52.
Найдено, С 69,31; Н 5,63.
Молекулярная масса 326.
Идентично в соответствии с ЯМР-анализом.
Исследование методом тонкослойной хроматографии. См. пример 8. Rf=0,5.
П р и м е р 11. Смесь, состоящая из 75 кг диметилформамида, 100 кг (420 моль) 7-гидроксиизофлавона и 76 кг (550,7 моль) безводного карбоната калия и 73 кг (598,3 моль) изопропилбромида, реагирует в течение 2 ч при 75-95оС, затем выдерживается в течение 10 мин при 100оС. После охлаждения в реакционную смесь добавляется 45 кг изопропанола и 350 кг воды. Кристаллическая взвесь фильтруется и промывается при 25оС до нейтральной реакции. Влажный продукт кристаллизуется в 4,4-кратном количестве безводного этанола в расчете на содержание сухого вещества. Продукт промывается этанолом и сушится при 60оС.
Получается 112,9 кг 7-изопропокси-изофлавона. Т.пл. 118-119оС. Содержание активного ингредиента: свыше 99,8% (ВЭЖХ), содержание 7-этокси-изофлавона менее, чем 0,1% он не содержит загрязнений. Выход 96%
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КЕТОНОВ ИЛИ ИХ СОЛЕЙ | 1990 |
|
RU2065848C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНКАРБОНОВЫХ КИСЛОТ | 1988 |
|
RU2014331C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНКАРБОНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1988 |
|
RU2049783C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПАРГИЛАММОНИЙХЛОРИДА | 1994 |
|
RU2130450C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ И СОЕДИНЕНИЕ | 1990 |
|
RU2044734C1 |
| АНТИМИКРОБНАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2030913C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АМИДОКСИМА О-(2-ГИДРОКСИ-3-ПИПЕРИДИНО-1-ПРОПИЛ)-НИКОТИНОВОЙ КИСЛОТЫ И ИХ СОЛЕЙ (ВАРИАНТЫ), ЧИСТОЕ КРИСТАЛЛИЧЕСКОЕ ОСНОВАНИЕ АМИДОКСИМА О-(2-ГИДРОКСИ-3-ПИПЕРИДИНО-1-ПРОПИЛ)-НИКОТИНОВОЙ КИСЛОТЫ | 1990 |
|
RU2074854C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАЛКИЛАМИНОВ ИЛИ СОЛЕЙ ЭТИХ СОЕДИНЕНИЙ | 1990 |
|
RU2015960C1 |
| КЛАТРАТ 7-ИЗОПРОПОКСИИЗОФЛАВОНА И ЦИКЛОДЕКСТРИНОВОГО ПОЛИМЕРА, ПРОЯВЛЯЮЩИЙ АКТИВНОСТЬ ПРОТИВ ОСТЕОПОРОЗА И ОСТЕОМАЛЯЦИИ | 1987 |
|
RU2022970C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАЛКИЛАМИНОВ ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1989 |
|
RU2007384C1 |
Использование: в медицине, в частности в способе получения имприфлавона, пригодного для лечения остеопороза. Сущность изобретения: способ предусматривает получение чистых изофлавоновых производных общей ф-лы I, приведенной в описании, где R - водород или изопропил; R2 и R3 - водород или C1-C2 -алкоксигруппа. Снтез ведут циклизацией резорцинольного производного ф-лы II, приведенной в описании с этил-орто-формиатом при 70 - 100°С (лучше 80 - 90°С) в присутствии органического растворителя (лучше диметилформамида и/или изопропанола) в 0,3 - 2-кратном количестве по отношению к рассчитанному объему резорциональных производных ф-лы III, приведенной в описании, и/или в присутствии избытка этил-орто-формиата для получения 20 - 70%-ного пересыщенного раствора продукта ф-лы III. Последний непрерывно выделяют из смеси с последующим охлаждением реакционной смеси и отделением фильтрованием соединения ф-лы II, где R1 - водород; R2 и R3 см. выше, и/или добавлением неполярного или полярного растворителя к реакционной смеси с селективным растворением образованного таким образом побочного продукта ф-лы, приведенной в описании, где R1 - водород; R2 и R3 см. выше, и/или добавлением к реакционной смеси почти эквивалентного количества безводного карбоната калия и выделением выкристаллизовавшейся двойной соли ф-лы IV, приведенной в описании, где R2 и R3 см. выше. После этого соединение ф-лы IV или 1, где R1 - водород, подвергают алкилированию галоидалкилом с выделением чистого целевого продукта ф-лы I, где R1 - изопропил, содержащего не больше 0,5% загрязняющей примеси соединения ф-лы V. Образование двойной соли происходит после добавления неполярного растворителя (лучше толуола) и безводного карбоната калия при 40 -80°С (лучше 60°С). Этанол, образованный в реакции циклизации оставляют в реакционной среде. Чистота полученного целевого продукта выше 99,8%. 3 з.п. ф-лы.
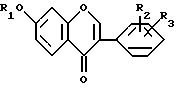
где R1 водород или изопропил;
R2 и R3 водород или С1-С2-алкоксигруппа,
циклизацией резорцинольного производного общей формулы II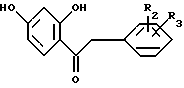
где R2 и R3 имеют указанные значения,
действием этилортоформиата формулы III
(C2H5O)3CH
в присутствии основного соединения и при необходимости алкилированием полученного соединения формулы I, где R1 водород, отличающийся тем, что циклизацию проводят при 70 100oС в среде органического растворителя, предпочтительно диметилформамида и/или изопропанола, взятого в 0,3 2-кратном расчетном объеме по отношению к объему резорцинольного производного формулы II и/или в среде избытка эфира формулы III и из полученного 20 70%-ного раствора (пересыщенного) соединения формулы IV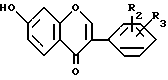
где R2 и R3 имеют указанные значения, непрерывно выделяется соединение формулы IV, где R2 и R3 имеют указанные значения, с последующим охлаждением реакционной смеси и отделением путем фильтрации соединения формулы I, где R1 водород, R2 и R3 имеют указанные значения, и/или добавлением полярного или неполярного растворителя в реакционную смесь для селективного растворения таким образом побочного продукта в выделенном фильтрованием соединения формулы I, где R1 - водород, R2 и R3 имеют указанные значения, и/или добавлением к реакционной смеси почти эквивалентного количества безводного карбоната калия и выделением кристаллической двойной соли общей формулы V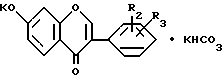
где R2 и R3 имеют указанные значения,
и при необходимости соединения формулы I, где R1 водород, или соединения формулы V, где R2 и R3 имеют указанные значения, подвергают алкилированию действием изопропилгалогенида и выделяют чистый целевой продукт формулы I, где R1 изопропил, R2 и R3 имеют указанные значения, содержащий не более 0,5 мас. загрязняющего соединения общей формулы VI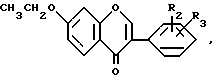
где R2 и R3 имеют указанные значения.
| Патент США N 3340276, кл | |||
| ЭЛЕКТРОМЕХАНИЧЕСКИЙ СНЕГООЧИСТИТЕЛЬ ДЛЯ ЖЕЛЕЗНЫХ ДОРОГ | 1922 |
|
SU549A1 |
