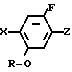
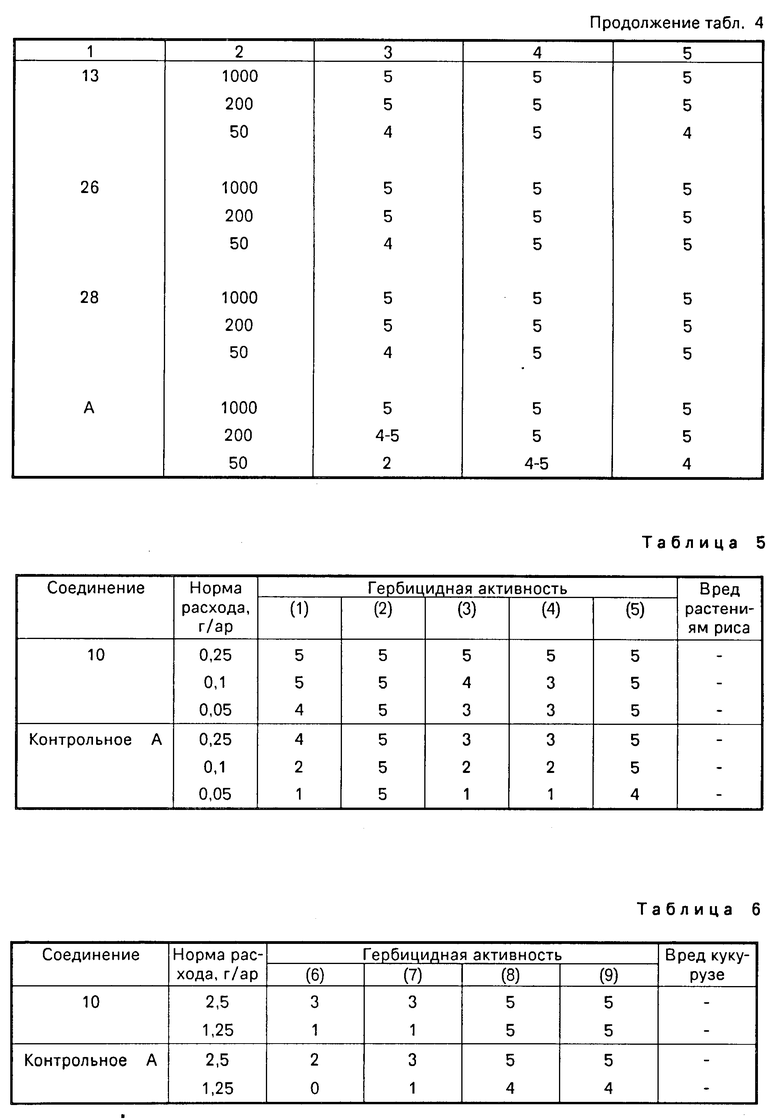
Изобретение относится к производным бензола, замещенным гетероциклическим кольцом, представленным общей формулой
X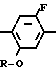 Z (1) где R циклоалкильная группа, имеющая 3-8 атомов углерода;
Z (1) где R циклоалкильная группа, имеющая 3-8 атомов углерода;
Х атом галогена;
Z группа формулы
-N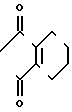 или -N
или -N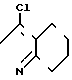 в которой циклоалкильная группа может быть замещена алкильной группой, имеющей 1-6 атомов углерода, к процессу их получения и к гербициду, содержащему их в качестве активного ингредиента. Более конкретно, изобретение относится к N-замещенным 3,4,5,6-тетрагидрофталимидным производным и N-замещенным фенил-4,5,6,7-тетрагидро-2Н-индазоль- ным производным, характеризующимся наличием циклоалкилоксигруппы в 5-положении фенильного кольца у атома азота.
в которой циклоалкильная группа может быть замещена алкильной группой, имеющей 1-6 атомов углерода, к процессу их получения и к гербициду, содержащему их в качестве активного ингредиента. Более конкретно, изобретение относится к N-замещенным 3,4,5,6-тетрагидрофталимидным производным и N-замещенным фенил-4,5,6,7-тетрагидро-2Н-индазоль- ным производным, характеризующимся наличием циклоалкилоксигруппы в 5-положении фенильного кольца у атома азота.
До настоящего времени в качестве производных N-замещенных фенил-3,4,5,6-тетрагидрофталимида, имеющих гербицидную активность, известны были, например, N-(2-фтор-4-хлор-5-изопропоксифенил)-3,4,5,6-тетрагидрофталимид (патент Японии N 63-20428) или N-2-фтор-4-хлор-5-алкилоксифенил)-3,4,5,6-тетрагидрофталимид (патент Японии N 58-72563), но не были известны соединения, имеющие циклоалкилокси группу в 5-положении фенильного кольца у атома азота.
Японские патентные заявки (Кокаи) NN 63-68562, 63-68563 и 63-280060 раскрывают, например, соединения, которые отличаются от соединений настоящего изобретения тем, что они имеют метильную группу в тетрагидрофталимидо фрагменте, и в которых фенильное кольцо у атома азота замещено 2,4-дагалоид-5-циклоалкоксигруппой, но не известны подробные примеры получения и испытаний их активности.
Японская патентная заявка (Кокаи) N 55-139359 раскрывает соединения, которые отличаются от соединений настоящего изобретения тем, что заместителем во 2-положении фенильного кольца является атом хлора.
Известные в технике производные тетрагидрофталимида проявляют гербицидную активность, но эти соединения не считаются достаточными для использования в качестве активных ингредиентов, используемых на практике гербицидных средств.
Обращаясь к 2N-замещенным производным фенил-4,5,6,7-тетрагидро-2Н-индазола, например, известно, что 2N-(2-фтор-4-хлор-5-изопропоксифенил)-4,5,6,7-тетрагидро-2Н-индазол (японская патентная заявка Кокаи N 59-59666) или 2N-(2,4-дихлор-5-метоксифенил)-4,5,6,7-те- трагидро-2Н-индазол (японская патентная заявка Кокаи N 52-51365) обладают гербицидной активностью. Однако эти соединения не считаются достаточными для использования в качестве активного ингредиента практических гербицидных средств.
В процессе недавних поисков и исследований новых сельскохозяйстенных агентов разрабатывали сельскохозяйственые агенты нового типа, которыми можно заменить обычные сельскохозяйственные агенты, требующие при обработке применения высоких доз, чтобы избежать загрязнения окружающей среды и нарушения экологии. Для данной цели важно исследовать и изучить новые соединения, которые обнаруживают превосходные действия в низких дозах. Кроме того, важной проблемой является поиск новых соединений, которые проявляют селективную гербицидную активность только против сорняков и обнаруживают превосходные эксплуатационные характеристики со значительно уменьшенным вредным воздействием в отношении важных сельскохозяйственных культур.
Предлагаемые производные бензола, замещенные гетероциклическим кольцом и представленые формулой
X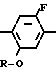 Z (1) где R циклоалкильная группа, имеющая 3-8 атомов углерода;
Z (1) где R циклоалкильная группа, имеющая 3-8 атомов углерода;
Х атом галогена;
Z группа
-N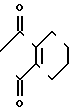 или -N
или -N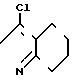 в которых циклоалкильная группа может быть замещена алкильной группой, имеющей 1-6 атомов углерода, обладают высоким гербицидным эффектом в отношении сорняков при обработке при низкой дозе и оказывают заметно уменьшенное вредное действие на основные культуры.
в которых циклоалкильная группа может быть замещена алкильной группой, имеющей 1-6 атомов углерода, обладают высоким гербицидным эффектом в отношении сорняков при обработке при низкой дозе и оказывают заметно уменьшенное вредное действие на основные культуры.
Соединения настоящего изобретения обладают превосходной гербицидной активностью при применении в низких дозах против разнообразных вредных сорняков, например широколистных сорняков, таких как обычная марь белая, слабостебельчатый амарант, лимнохарис, звездчатка средняя и др. и травянистых сорняков, таких как ежовник и щетинник зеленый и др. при применении на листве и на почве в полевых условиях и не обнаруживают при этом какого-либо гербицидного повреждения, которое вызывало бы проблемы, связанные с основными культурами, например широколистными культурами, такими как соя, хлопок, и др. и травянистыми культурами, такими как кукуруза и др.
Соединения настоящего изобретения проявляют также превосходную активность против различных пагубных сорняков на рисовых полях, например травянистых видов, таких как полевица ползучая и др. широколистных сорняков, таких как обыкновенный ложный очный цвет, ротала индийская, повойничек или сущеница топяная и др. осоковых сорняков, таких как японский камыш озерный, игольчатая болотница и др. и Sigitarria pygmaea Mig и др. при применении в низких дозах, при этом ущерб, причиняемый гербицидом пересаженному рису, является весьма незначительным. Высокая селективность соединений настоящего изобретения в отношении растений риса является совершенно непредсказуемой с учетом обычных тетрагидрофталимидных производных и производных тетрагидроиндазола. Данная характерная черта очевидно является следствием циклоалкокси группы, введенной в 5-положение фенильного кольца.
Ниже описываются способы получения соединений настоящего изобретения и промежуточных продуктов для них.
N-замещенные производные фенил-3,4,5,6-тетрагидрофталимида настоящего изобретения могут быть легко получены с помощью взаимодействия производного анилина, представленного формулой
X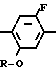 NH2 (2) где R циклоалкильная группа, имеющая 3-8 атомов углерода;
NH2 (2) где R циклоалкильная группа, имеющая 3-8 атомов углерода;
Х атом галогена, в которой циклоалкильная группа может замещаться алкильной группой, имеющей 1-6 атомов углерода, с 3,4,5,6-тетрагидрофталевым ангидридом в инертном растворителе. В качестве инертных растворителей могут использоваться такие растворители, как бензол, толуол, ксилол, хлорбензол, уксусная кислота и др. или смешанные растворители из указанных. Температура реакции выбирается из интервала между комнатной температурой и 150оС, реакция предпочтительно осуществляется при 50-120оС. После реакции требуемое соединение может легко выделяться с помощью приемов обработки в чистом виде и с помощью перекристаллизации из растворителя спиртового типа, такого как метанол и др.
Производное анилина, представленное общей формулой (2) являющееся промежуточным продуктом, может получаться, например, с помощью следующих альтернативных способов синтеза.
Способ синтеза I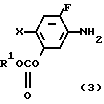
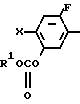 NH
NH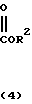 ______→
______→
X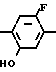 N
N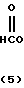 R2
R2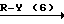
 OR2 ______→
OR2 ______→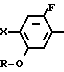 N
N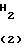 где R и Х указаны выше;
где R и Х указаны выше;
R1 алкильная группа, имеющая 1-6 атомов углерода;
R2- алкильная группа с 1-6 атомами углерода, алкильная группа с 3-4 атомами углерода или аралкильная группа, имеющая 7-8 атомов углерода;
Y атом хлора, брома или иода, метилсульфонилоксигруппа или n-толилсульфонилоксигруппа.
Способ синтеза II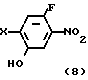
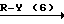
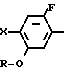 N
N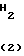 где R, X и Y значения, указанные выше.
где R, X и Y значения, указанные выше.
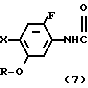
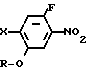
Более конкретно, согласно способу синтеза I производное анилина (3) подвергается реакции с хлорформатным эфиром в присутствии основания, такого как карбонат калия, карбонат натрия, окись магния и др. в растворителе, таком как ацетонитрил, ацетон, N,N-диметилформамид и др. с превращением его в карбаматное производное (4). Затем карбаматное производное (4) превращается в фенольное производное (5) с помощью селективного гидролиза карбонатной группы путем обработки в проточном растворителе в присутствии основания, такого как гидроокись натрия или гидроокись калия. Получающееся в результате фенольное производное (5) подвергается взаимодействию с соединением, представленным общей формулой R-Y (6) в присутствии основания, такого как карбонат калия, карбонат натрия, окись магния и др. посредством чего циклоалкилокси группа может бытиь введена в 5-положение фенильного кольца. Реакция предпочтительно проводится в соответствующем растворителе, кроме того, может использоваться такой растворитель, как ацетонитрил, ацетон, метанол, этанол, N, N-диметилформамид и др. Полученное таким образом карюаматное производное (7) может превращаться в производное анилина, представленное общей формулой (2) с помощью реакции в водном растворе гидроокиси натрия путем гидролиза карбаматного эфира или в случае когда R2 в общей формуле (7) представляет, например, бензильную группу с помощью гидрогенолиза с использованием каталитической реакции с палладием на угле.
Производные анилина, представленные общей формулой (3), являющиеся исходными материалами, описываются в японской патентной заявке (Кокаи) N 62-174065 и могут получаться с помощью процесса, показанного в примерах, описанных ниже. Соединения, представленные формулой (6), являются промышленно доступными или могут быть легко получены из промышленно доступных соединений.
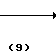
Согласно способу синтеза II нитрофенольное производное (8), описанное в японской патентной публикации N 1-61099, превращается в нитробензольное производное, представленное общей формулой (9) с помощью реакции его с соединением, представленныам формулой R-Y (6), в присутствии основания, такого как карбонат калия, карбонат натрия, окись магния, гидроокись калия, гидрид натрия, метилат натрия и др. а затем получающееся в результате нитробензольное производное может превращаться в производное анилина (2) с помощью процедуры, используемой обычно для восстановления ароматического нитросоединения в аминогруппу, например процесса, использующего сульфит натрия, восстанавливающее железо, порошок цинка, или процесса каталитического восстановления сиспользованием окиси платины или палладия на угле.
N-замещенное производное фенил-3,4,5,6-тетрагидрофталимида настоящего изобретения может получаться с помощью реакции фенольного производного, представленного общей формулой
X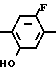 N
N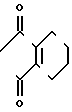 (10) где Х имеет те же значения, с соединением, представленным формулой (6). Реакция предпочтительно проводится в растворителе, таком как ацетонитрил, N,N-диметилформамид, ацетон, метанол и др. в присутствии основания, такого как карбонат калия, карбонат натрия, окись магния, метилат натрия и др.
(10) где Х имеет те же значения, с соединением, представленным формулой (6). Реакция предпочтительно проводится в растворителе, таком как ацетонитрил, N,N-диметилформамид, ацетон, метанол и др. в присутствии основания, такого как карбонат калия, карбонат натрия, окись магния, метилат натрия и др.
Фенольные производные общей формулы (10) как исходные материалы для указанной реакции описываются в японской патентной заявке (Кокаи) N 58-83672 и могут быть получены по способу синтеза III.
Cпособ синтеза III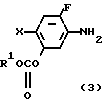 + O
+ O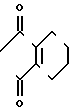 ______→
______→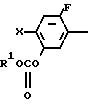 N
N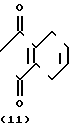 _____→ X
_____→ X N
N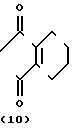 где Х и R1 имеют значения, определенные выше.
где Х и R1 имеют значения, определенные выше.
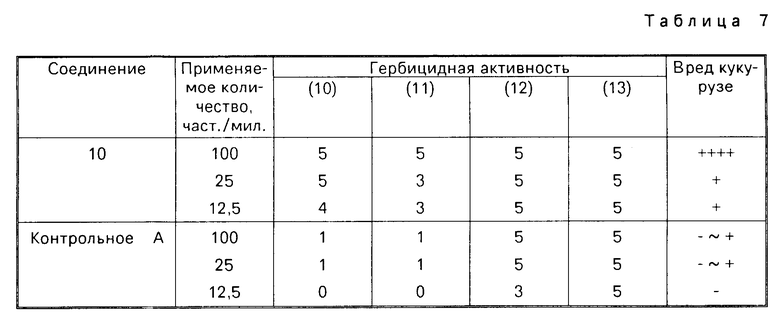
Более конкретно, производное анилина, представленное формулой (3), подвергается взаимодействию с 3,4,5,6-тетрагидрофта- левым ангидридом в инертном растворителе, таком как бензол, толуол, уксусная кислота и др. для превращения его в производное тетрагидрофталимида (11), а затем карбонатная группа в 5-положении фенильного кольца его селективно гидролизуется в присутствии основания, с помощью чего может получаться фенольное производное, представленное общей формулой (10). Примеры оснований, которые могут использоваться, включают карбонат калия, гидроокись калия, гидроокись натрия и др. и реакция предпочтительно проводится в протонном растворителе, таком как метанол, этанол, вода и др. при температуре от комнатной до примерно 100оС из соображений хороших показателей выхода продукта.
2N-замещенные производные фенил-4,5,6,7-тетрагидро-2Н-индазола настоящего изобретения могут получаться, например, согласно способу синтеза.
Способ синтеза IV
X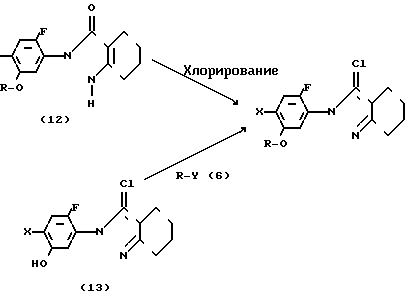 где R и Х имеют значения, указанные выше;
где R и Х имеют значения, указанные выше;
Y атом хлора, атом брома, иода, метилсульфонилоксигруппа или n-толуолсульфонилоксигруппа.
Более конкретно производное индазола настоящего изобретения может получаться с помощью хлорирования производного индазолинона, представленного общей формулой (12), с использованием хлорирующего агента, такого как хлорокись фосфора, пятихлористый фосфор и др. Реакция может осуществляться в органическом растворителе, но предпочтительно она проводится без растворителя для хороших выходов. Температура реакции выбирается в интервале 100-200оС. Кроме того, производное индазола настоящего изобретения может быть получено с помощью взаимодействия 3-хлор-2N-(2-фтор-4-галоид-5-гидроксифенил)-4,5,6,7-тетрагид- ро-2Н-индазола, представленного общей формулой (13), с соединением, представленным общей формулой (6), в соответствующем растворителе в присутствии основания при температуре от комнатной до температуры нагревания, предпочтительно при 30-100оС. Растворитель, который может использоваться, включает такой растворитель, как ацетонитрил, ацетон, тетрагидрофуран, диоксан, диметилсульфоксид, N,N-диметилформамид, метанол и др. и смешанные растворители из указанных. В качестве основания может использоваться карбонат калия, карбонат натрия, окись магния, метилат натрия, этилат натрия, гидрид натрия и др.
Производное индазола, представленое общей формулой (13), в качестве исходного материала известно из японской патентной заявки (Кокаи) N 59-170071 и японской заявкиN 62-30761.
Производное индазолона, представленное общей формулой (12), может быть получено согласно способу синтеза V.
Способ синтеза V
X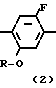 NH2
NH2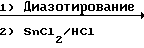 X
X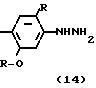
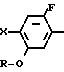 N
N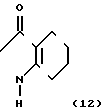 где Х, R и Y имеют те же значения, что определены выше;
где Х, R и Y имеют те же значения, что определены выше;
R3 алкильная группа, имеющая 1-6 атомов углерода.
Производное анилина (2), которое может быть получено с помощью процесса, показанного на схеме способа синтеза I или II, подвергается реакции с нитритом натрия в кислотных условиях с использованием соляной кислоты, серной или борфтористой кислоты для получения диазониевой соли, затем восстановлением диазониевой соли восстанавливающим агетом, таким как хлористое олово, для превращения его в производное гидразина (14). В описанной реакции без каких-либо проблем могут использоваться ацетон, ацетонитрил и др. которые используются при получении диазониевых солей.
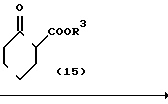
Получающееся в результате производное гидразина (14) может превращаться в 2N-замещенный фенил-1,2,4,5,6,7-гексагидро-3Н-индазол-3-он (12) с помощью подвержения его реакции циклизации-конденсации с 2-алкоксикарбонилциклогексаноном, представленным формулой (15). Реакция предпочтительно проводится в органическом растворителе, таком как бензол, толуол, ксилол, хлорбензол, уксусная кислота и др. Температура реакции предпочтительно выше, чем азеотропная температура используемого растворителя и воды ввиду хорошей эффективности реакции, и реакция предпочтительно проводится с использованием устройства для удаления воды, такого как аппарат Дина-Старка. Реакция протекает достаточно без использования катализатора, но предпочтительно она проводится в присутствии основного соединения, такого как триэтиламин, пиридин и др. поскольку при этом реакция может эффективно промотироваться.
Примеры соединений (1) настоящего изобретения включают соединения, показанные ниже.
 (1) Соединение Х R Соединения, где Z -N
(1) Соединение Х R Соединения, где Z -N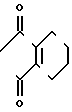
1 F Циклопропильная группа
2 F Циклопентильная группа
3 F 2-Метилциклопентильная
4 F 3-Метилциклопентильная
5 F Циклогексильная группа
6 F 2-Метилциклогексильная
7 F Циклогептильная группа
8 F Циклооктильная группа
9 Cl Циклопропильная группа
10 Cl Циклопентильная группа
11 Cl 2-Метилциклопентильная
12 Cl 3-Метилциклопентильная
13 Cl Циклогексильная группа
14 Cl 2-Метилциклогексильная
15 Cl Циклогептильная группа
16 Cl Циклооктильная группа
17 Br Циклопропильная группа
18 Br Циклопентильная группа
19 Br 2-Метилциклопентильная группа
20 Br 3-Метилциклопентильная группа
21 Br Циклогексильная группа
22 Br 2-Метилциклогексильная группа
23 Br Циклогептильная группа
24 Br Циклооктильная группа
Соединения, где Z -N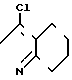
25 Сl Циклопропильная группа
26 Cl Циклопентильная группа
27 Cl 2-Метилциклопентильная группа
28 Cl 3-Метилциклопентильная группа
29 Cl Циклогексильная группа
30 Сl 2-Метилциклогексильная группа
31 Cl Циклогептильная группа
32 Cl Циклооктильная группа
33 Cl 2-Метилциклооктильная группа
34 F Циклопропильная группа
35 F Циклопентильная группа
36 F 2-Метилциклопентильная группа
37 F 3-Метилциклопентильная группа
38 F Циклогексильная группа
39 F 2-Метилциклогексильная группа
40 F Циклогептильная группа
41 F Циклооктильная группа
42 F 2-Метилциклооктильная группа
43 Br Циклопропильная группа
44 Br Циклопентильная группа
45 Br 2-Метилциклопентильная группа
46 Br 3-Метилциклопентильная группа
47 Br Циклогексильная группа
48 Br 2-Метилциклогексильная группа
49 Br Циклогептильная группа
50 Br Циклооктильная группа
51 Br 2-Метилциклооктильная группа
Соединения настоящего изобретения обладают отличительными эксплуатационными характеристиками в качестве гербицидного агента.
При использовании соединений настоящего изобретения в качестве гербицидного агента соединения могут быть применены сами по себе, но обычно они могут использоваться в качестве гербицидного агента в смеси с одним или более вспомогательными агентами. Обычно соединения могут предпочтительно использоваться в форме препаратов, например смачиваемых порошков, эмульгируемых агентов, порошков, гранул, текучих препаратов и др. известным способом с помощью преобразования соединений в готовые препаративные формы с различными носителями, разбавителями, растворителями, поверхностно-активными агентами, стабилизаторами и др. в качестве вспомогательных агентов.
Растворители в качестве одного из вспомогательных агентов в гербицидной композиции, включающей соединение настоящего изобретения в качестве активного ингредиента, включают, например, воду, спирты, кетоны, простые эфиры, алифатические и ароматические углеводороды, галоидированные углеводороды, амиды кислот, сложные эфиры, нитрилы. Растворитель может использоваться один или в смеси двух или более растворителей.
Разбавители, которые могут использоваться, включают минеральные порошки, например глины, такие как каолин и бентонит, тальк, такие как тальк или пирофиллит, окиси, такие как диатомовая земля и белая сажа, и растительные порошки, такие как соевая мука и СМС. Кроме того, поверхностно-активный агент может использоваться в качестве распределяющего агента, диспергирующего агента, эмульгирующего агента и агента, способствующего проникновению. Поверхностно-активные агенты включают, например, неионные поверхностно-активные агенты, катионные и амфотерные поверхностно-активные агенты. Эти поверхностно-активные агенты могут использоваться по одному или в виде смеси двух или более агентов в зависимости от полезности.
Предпочтительный способ применения гербицидного агента, содержащего соединение настоящего изобретения в качестве активного ингредиента, включает обработку почвы, водно-поверхностную обработку и применение на листве и проч. Особенно превосходный эффект может получиться с помощью применения перед прорастанием или на стадии прорастания сорняков, подлежащих уничтожению.
Гербицидный препарат, включающий соединение настоящего изобретения в качестве активного ингредиента, может использоваться в смеси или в сочетании с другими активными ингредиентами, которые не оказывают пагубного воздействия на гербицидную активность активных ингредиентов настоящего изобретения, например, другими гербицидными агентами, инсектицидами, противомикробными агентами, регуляторами роста растений и др.
Предлагаемое изобретение поясняется с помощью следующих примеров.
П р и м е р 1.
 C
C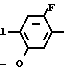 NH2 + O
NH2 + O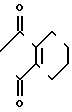
 C
C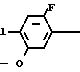 N
N
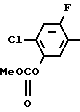
Раствор 2-фтор-4-хлор-5-циклопентилоксианилина (0,50 г, 2,18 ммоль) и 3,4,5,6-тетрагидрофталевого ангидрида (0,398 г, 2,61 ммоль) в уксусной кислоте (3,0 мл) перемешивался в течение 3 ч в условиях дефлегмации. Добавляли воду (20 мл) к получающейся реакционной смеси и смесь экстрагировали этилацетатом (20 мл х 3 раза). Органический слой сушили, а растворитель отгоняли при пониженном давлении. Получающееся в результате желтое маслянистое вещество очищалось с помощью хроматографии на силикагельной колонке (проявляющий растворитель: гексан/этил ацетат 8/1) с получением N-(2-фтор-4-хлор-5-циклопентилоксифенил)-3,4,5,6-тет- рагидрофталимида в виде бесцветного прозрачного маслянистого вещества (0,513 г, 1,41 ммоль, 65% выход). К нему добавлялся этанол (1,0 л) для перекристаллизации с получением продукта в виде белого твердого вещества. Т.пл. 69,0-75,2оС.
1Н-ЯМР спектр (СDCl3, ТМС, млн./дол.): δ 1,30-2,10 (12Н, м), 2,40 (4Н, м), 4,68 (1Н, м), 6,75 (1Н, д, IНF 7,0 Гц), 7,20 (1Н, д. IHF 9,0 Гц), ИК-спектр (КВr диск, см-1): 1725, 1505, 1430, 1385, 1200.
П р и м е р 2.
Cl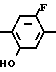 N
N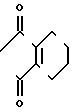 +
+  _ Br
_ Br  C
C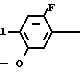 N
N
Циклопентилбромид (1,2 г, 8,1 ммоль) добавляли к раствору N-(2-фтор-4-хлор-5-гидроксифенил)-3,4,5,6-тетрагидрофталими- да (2,0 г, 6,76 ммоль) и карбоната калия (0,60 г, 4,34 ммоль) в ацетонитриле (50 мл) с последующим перемешиванием в течение 2 ч при температуре дефлегмации. После завершения реакции к полученной в результате смеси добавляли 1 н. соляную кислоту (20 мл) и смесь экстрагировали этилацетатом (20 мл х 3 раза). Органический слой промывали водой, сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. К полученному в результате реакции бледно-желтому маслянистому веществу добавляли этанол (5 мл). Выпавший в осадок N-(2-фтор-4-хлор-5-циклопентилоксифенил)-3,4,5,6-тетрагидрофталимид в виде белого твердого вещества (0,75 г, 2,06 ммоль, 30,5% выход) отделяли фильтрованием. Данные спектрального анализа и другие данные являются такими, как показаны в примере 1.
П р и м е р 3.
Cl N
N +
+  _ OTs
_ OTs  C
C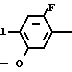 N
N
К раствору N-(2-фтор-4-хлор-5-гидроксифенил)-3,4,5,6-тетрагидрофталимида (2,0 г, 6,76 ммоль) и карбоната калия (0,60 г, 4,34 ммоль) в ацетонитриле (50 мл) добавляли циклопентил п-толуолсульфонат (1,90 г, 8,11 ммоль) с последующим перемешиванием в течение 2 ч при 80оС. По завершении реакции к полученной в результате смеси добавляли 1 н. соляную кислоту (20 мл) и смесь экстрагировали этилацетатом (20 мл х 3 раза). Органический слой промывали водой, сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. К полученному бледно-желтому маслянистому веществу добавляли этанол (5 мл) и выпавший в осадок N-(2-фтор-4-хлор-5-циклопентилоксифенил)-3,4,5,6-тетрагидрофталимид в виде белого твердого вещества (0,77 г, 2,12 ммоль, 31,4% выход) отделяли фильтрованием. Данные спектрального анализа и другие данные являются такими, как описаны в примере 1.
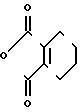
П р и м е р 4.
Me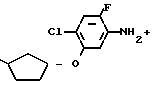
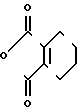
Раствор 2-фтор-4-хлор-5-(3-метилциклопентил)оксианилина (1,76 г, 7,22 ммоль) и 3,4,5,6-тетрагидрофталевого ангидрида (1,32 г, 8,68 ммоль) в уксусной кислоте (15 мл) перемешивался в течение 4 ч при пониженном давлении. Полученная в результате реакции смесь добавлялась к 1 н.соляной кислоте (50 мл) и экстрагировалась эфиром (3 порциями по 50 мл). Органический слой сушили, растворитель отгоняли и полученное в результате красно-коричневое маслянистое вещество очищали с помощью хроматографии на силикагельной колонке (проявляющий растворитель: гексан/этилацетат8/1). Полученный в результате N-(2-фтор-4-хлор-5)3-метилциклопентил (оксифенил)-3,4,5,6-тетрагидрофталимид в виде бесцветного прозрачного маслянистого вещества перекристаллизовывался из метанола, давая белое твердое вещество (0,93 г, 2,38 ммоль, 33,0% выход).
Т.пл. 68,0-70,0оС.
Спектр 1Н-ЯМР (СDCl3, ТМС, млн./дол.): δ 1,01 и 1,08 (всего 3Н, каждый д. I 6,0 Гц), 1,25-2,20 (11Н, м), 2,49 (4Н, м.), 4,70 (1Н, м.), 6,72 (1Н, д. IHF 6,0 Гц), 7,20 (1Н, д. IHF 9,0 Гц).
ИК-спектр (КВr диск, см-1): 1720, 1500, 1430, 1375, 1195.
П р и м е р 5.
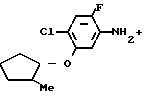

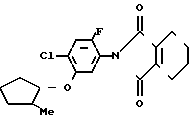
2-Фтор-4-хлор-5-(2-метилциклопентил) оксианилин (660 мг, 2,71 ммоль), 3,4,5,6-тетрагидрофталевый ангидрид (503 мг, 3,31 ммоль) и уксусная кислота (10 мл) загружали в круглодонную колбу (50 мл), нагревали в течение 5 ч при температуре дефлегмации. По завершении реакции реакционный раствор охлаждали до комнатной температуры и выливали в ледяную воду (100 мл). Смесь экстрагировали этилацетатом (30 мл х 3), органические слои объединяли, промывали водой и насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом магния.
Осушенный агент отделяли фильтрованием, растворитель отгоняли при пониженном давлении, полученное в результате бледно-коричневое маслянистое вещество очищали хроматографией на силикагельной колонке (проявляющий растворитель: гексан/этилацетат 9/1), в результате получали N-(2-фтор-4-хлор-5-)2-метилциклопентил (оксифенил)-3,4,5,6-тетрагидрофталимид (1,00 г, 2,65 ммоль, 98% выход).
Бесцветное прозрачное маслянистое вещество.
Cпектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): 1,13 (3Н, д. I 7,0 Гц), 1,40-2,15 (10Н, м.), 2,25-2,50 (4Н, м.), 4,25 (1Н, м.), 4,52 (1Н, м.), 6,72 (1Н, д. IHF 7,3 Гц), 7,30 (1Н, д. IHF 10,2 Гц).
ИК-спектр (чистый, см-1): 2970, 1725, 1500, 1425, 1375, 1195.
П р и м е р 6.
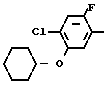 NH2+ O
NH2+ O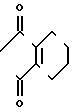
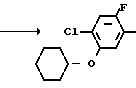 N
N
2-Фтор-4-хлор-5-циклогексилоксиани- лин (213 мг, 0,874 ммоль), 3,4,5,6-тетрагидрофталевый ангидрид (134 мл, 0,874 ммоль) и уксусная кислота (10 мл) загружали в круглодонную колбу (50 мл) и нагревали при температуре дефлегмации в течение 15 ч. После завершения реакции реакционный раствор охлаждался до комнатной температуры, к нему добавляли воду (50 мл) и смесь экстрагировали этилацетатом (20 мл х 3). Органические слои объединяли, промывали водой и насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом магния. Осушающий агент отделяли фильтрованием и растворитель отгоняли при пониженном давлении с получением неочищенного продукта (332 мг). Продукт очищали хроматографией на силикагельной колонке (проявляющий растворитель: гексан/этилацетат 19/1), в результате N-(2-фтор- 4-хлор-5-циклогексилоксифенил)-3,4,5,6-тетра- гидрофталимид в виде белого твердого вещества (230 мг, 0,609 ммоль, 70% выход). Путем перекристаллизации из смеси гексан/хлороформ продукт выделяли в виде бесцветных игольчатых кристаллов. Т.пл. 102-103оС.
Спектр 1Н-ЯМР (СDCl3, ТМС, част./млн.): δ 1,20-2,50 (4Н, м.), 4,18 (1Н, м.), 6,79 (1Н, д. IHF 7,3 Гц), 7,23 (1Н, д. IHF 10,2 Гц).
ИК-спектр (КВr диск, см-1): 2950, 1715, 1495, 1425, 1375, 1190.
П р и м е р 7.
 C
C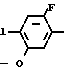 N
N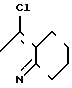
2-Фтор-4-хлор-5-циклопентилоксифе-нилгидразин (24,5 г, 0,10 моль), 2-этоксикарбонилциклогексанон (17,0 г, 0,10 моль) растворяли в уксусной кислоте (200 мл) и перемешивали в течение 2 ч при температуре дефлегмации. По завершении реакции растворитель отгоняли, при этом получали неочищенный продукт (37 г). Продукт промывали смешанным растворителем бензол/гексан, при этом получали 2N-(2-фтор-4-хлор-5-циклопентилоксифе- нил)-1,2,4,5,6,7-гексагидро-3Н-индазол-3-он в виде белого твердого вещества (25,3 г, 72,3% выход). Т.пл. 151-152оС.
Спектр 1Н-ЯМР (CDCl3 CF3CO2H, ТМС, млн./дол.): δ 1,40-2,03 (12Н, м.), 2,37 (2Н, м.), 2,55 (2Н, м.), 4,65 (1Н, м.), 6,97 (1Н, д. IHF= 9,0 Гц).
ИК-спектр (КВr, диск, см-1): 2950, 2400, 1770, 1600, 1505, 880.
МС-спектр (м/e): 352 (М+, 6%), 350 (М+, 18%), 284 (100%), 81 (38%), 41 (66%).
Хлорокись фосфора (6,6 г, 43,0 моль) добавляли к полученному в результате 2N-(2-фтор-4-хлор-5-циклопентилоксифенил)-1,2,4,5, 6, 7-гексагидро-3Н-идазол-3-ону (15,0 г, 42,8 ммоль) с последующим перемешиванием в течение 30 мин при нагревании при 160оС. После охлаждения реакционной смеси охлажденный льдом разбавленный водный раствор гидроокиси натрия (50 мл) добавляли к полученной смеси и смесь экстрагировали метиленхлоридом (200 мл х 4). Экстракт промывали разбавленной водной гидроокисью натрия (400 мл) и насыщенным водным раствором хлористого натрия (400 мл), сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и остаток очищали хроматографией на силикагельной колонке (проявляющий растворитель: этилацетат/гексан 1/9 1/5), при этом получали 3-хлор-2N-(2-фтор-4-хлор-5-циклопентилоксифенил)-4,5,6,7-тетрагидро-2Н-индаз ол в виде белого твердого вещества (4,3 г, 26,5% выход). Т.пл. 88-91оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,05-2,10 (12Н, м.), 2,30-2,85(4Н, м.), 4,73 (1Н, м.), 6,95 (1Н, д. IHF 6,0 Гц), 7,23 (1Н, д. IHF 9,0 Гц).
ИК-спектр (КВr диск, cм-1): 2960, 1510, 1195.
МС-спектр (м/e): 372 (М+, 12,1% ), 370 (М+, 11,6%), 368 (М+, 17,0%), 300(100%), 265 (52,7%), 41 (64%).
Элементный анализ (вычисленные значения: C18H19N2OClF, ): С 58,39 (58,55); Н 5,04 (5,19); N 7,49 (7,59).
П р и м е р 8.
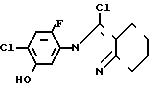
Раствор 3-хлор-2N-(2-фтор-4-хлор-5-гидроксифенил)-4,5,6,7-тетрагидро-2Н-ин- дазола (220 мг, 0,731 ммоль), 3-метилциклопентил n-толуолсульфоната (250 мг, 0,999 ммоль) и карбоната калия (120 мг, 0,870 ммоль) в ацетонитриле (25 мл) перемешивали в течение 4 ч при нагревании в условиях дефлегмации. После завершения реакции реакционный раствор выливали в 1 н. соляную кислоту (50 мл) и экстрагировали этилацетатом (50 мл х 3). Органический слой промывали насыщенным водным раствором хлористого натрия (100 мл) и сушили над сульфатом магния. Осушающий агент отделяли с помощью фильтрования и растворитель отгоняли из фильтрата с получением неочищенного продукта (0,29 г). Продукт очищали с помощью хроматографии на силикагельной колонке (проявляющий раствор: этилацетат/гексан 1/9) и получали 3-хлор-2N-(2-фтор-4-хлор-5-)3-метилцикло- пентил(оксифенил)-4,5,6,7-гексагидро-2Н-индазол в виде белого твердого вещества (242 мг, 86,4% выход).
1Н-ЯМР спектр (СDCl3, ТМС, млн.дол.):
δ 1,03 и 1,08 (общее 3Н, каждый д. I 6,0 Гц), 1,17-2,4 (11Н, м.), 2,4-2,83 (4Н, м.), 4,80 (1Н, м.), 6,98 (1Н, д. IHF 6,0 Гц), 7,38 (1Н, д. IHF 9,0 Гц).
ИК-спектр (чистый, см-1): 2970, 1505, 1200.
Ссылочный пример 1.
 NH2
NH2
 NH
NH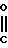 OEt
OEt
_____→ Cl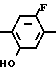 NH
NH OEt
OEt 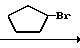
 C
C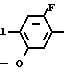 NH
NH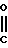 OEt
OEt C
C NH2
NH2
Этилхлорформат (16,3 г, 150 ммоль) добавляли к раствору 2-фтор-4-хлор-5-метоксикарбонилоксианилина (22,0 г, 100 ммоль) и карбоната калия (13,8 г, 100 ммоль) в ацетоне (300 мл) с последующим перемешиванием в течение 5 ч при 60оС. После завершения реакции растворитель отгоняли при пониженном давлении, остаток подкисляли с помощью добавления 1 н. соляной кислоты (100 мл) и экстрагировали этилацетатом (100 мл х 3). Органический слой промывали водой, сушили и растворитель отгоняли при пониженном давлении. Выпавшее в осадок твердое вещество отделяли фильтрованием.
Твердое вещество перекристаллизовывали из смеси хлороформа и гексана, получая этил N-(2-фтор-4-хлор-5-метоксикарбонилоксифенил)карбамат в виде белых кристаллов (23,3 г, 80,2% выход).
Т.пл. 143,8-147,2оС.
Спектр 1Н-ЯМР (СDCl3, ТМС, млн.дол.): δ 1,13(3Н, т. I 6,5 Гц), 3,92 (3Н, с), 4,23 (2Н, кв. I 6,5 Гц), 6,80 (1Н, шир.с.), 7,15(1Н, д. IHF 10,5 Гц), 8,12 (1Н, д. IHF 8,0 Гц).
ИК-спектр (КВr диск, см-1): 1770, 1730, 1545, 1290, 1235, 1215.
Полученный в результате этил N-(2-фтор-4-хлор-5-метоксикарбонилоксифенил) карбамат (45,2 г, 155 ммоль) подвергали реакции с карбонатом калия (21,4 г, 155 ммоль) и водой (100 мл) в течение 2 ч при нагревании в течение 2 ч при температуре дефлегмации. После завершения реакции реакционную смесь охлаждали до комнатной температуры, растворитель отгоняли при пониженном давлении. Остаток подкисляли путем добавления 1 н. соляной кислоты (300 мл) и экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали водой, сушили, растворитель отгоняли при пониженном давлении и осажденное твердое вещество отделяли фильтрованием. Твердое вещество перекристаллизовывали из смеси хлороформ-гексан с получением этил N-(2-фтор-4-хлор-5-гидроксифенил)-карбамата в виде белых кристаллов (35,2 г, 97% выход). Т.пл. 151,5-154,2оС.
Спектр 1Н-ЯМР (СDCl3, ТМС, част./млн.): δ 1,32 (3Н, т. I 7,2 Гц), 4,23 (2Н, кв. I 7,2 Гц), 5,84 (1Н, с.), 6,80 (1Н, шир. с.), 7,04 (1Н, IHF 10,5 Гц), 7,85 (1Н, д. IHF7,5 Гц).
ИК-спектр (КВr, см-1): 3440, 1710, 1560, 1430, 1250.
Затем раствор полученного в результате этил N-(2-фтор-4-хлор-5-гидроксифенил)карбамата (10 г, 42,8 ммоль) и карбоната калия (8,87 г, 64,2 ммоль) в ацетонитриле (150 мл) перемешивали в течение часа при 80оС. К полученной смеси добавляли по каплям циклопентилбромид (9,57 г, 84,2 ммоль) с последующим взаимодействием в течение 7 ч. После завершения реакции растворитель отгоняли при пониженном давлении, остаток подкисляли добавлением 1 н. соляной кислоты (100 мл) и экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали водой, сушили и растворитель отгоняли при пониженном давлении с получением N-(2-фтор-4-хлор-5-циклопентилоксифенил)-карбамата (12,7 г, 98% выход). Т.пл. 92,8-97,8оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, част. /млн. ): δ 1,33 (3Н, т. I 7,0 Гц), 1,40-2,10 (8Н, м. ), 4,32 (2Н, кв, I 7,0 Гц), 4,88 (1Н, м.), 4,32 (2Н, кв. шир.с.), 7,15 (1Н, д. IHF 10,5 Гц), 7,92 (1Н, д. IHF 7,0 Гц).
ИК-спектр (КВr, см-1): 1710, 1535, 1495, 1415, 1255.
При использовании n-толилсульфоната вместо циклопентилбромида в указанной реакции можно получить N-(2-фтор-4-хлор-5-циклопентилоксифенил)карбамат с выходом 95%
К полученному таким образом этил N-(2-фтор-4-хлор-5-циклопентилоксифенил)кар- бамату добавляли этиловый спирт (50 мл) и 2 н. водный раствор гидроокиси натрия (100 мл) с последующим перемешиванием в течение 4 ч при нагревании на масляной бане при 110оС. После завершения реакции растворитель отгоняли и остаток экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали насыщенным водным хлористым натрием, сушили и растворитель отгоняли при пониженном давлении с получением 2-фтор-4-хлор-5-циклопентилоксианилина (9,36 г, 40,8 ммоль, 97% выход).
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,40-2,07 (8Н, м.), 3,72 (2Н, шир.с.), 4,57 (1Н, м.), 6,35(1Н, д. IHF 8,0 Гц), 6,98 (1Н, д. IHF 10,5 Гц).
ИК-спектр (KBr, диск, см-1): 2980, 1635, 1510, 1423, 1250, 1190.
Ссылочный пример 2.
Этил N-(2-фтор-4-хлор-5-метоксикарбонилоксифенил)карбамат (1,45 г, 4,97 ммоль), полученный по ссылочному примеру 1, и раствор карбоната калия (1,03 г, 7,46 ммоль) в этаноле (5,0 мл) перемешивали в течение 1 ч при температуре дефлегмации и затем к смеси добавляли циклопентилбромид (1,11г, 7,46 ммоль) с последующим перемешиванием в течение 2 ч. После завершения реакции смесь выливали в 1 н. соляную кислоту (50 мл) и экстрагировали этилацетатом (50 мл х 3 раза). Органический слой сушили, концентрировали при пониженном давлении с получением этил N-(2-фтор-4-хлор-5-циклопентилоксифенил)карбамата в виде беловато-серых кристаллов (1,41 г, 4,69 ммоль, 94,4% выход). Данные спектрального анализа и другие данные являются такими же, как показаны в ссылочном примере 1.
Ссылочный пример 3.
Cl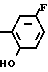
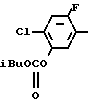 NO2
NO2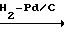
 NH2
NH2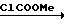
 NH
NH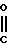 OMe
OMe Cl
Cl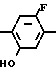 NH
NH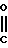 OMe
OMe 
 C
C NH
NH OMe
OMe C
C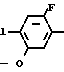 NH2
NH2
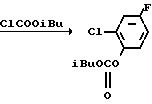
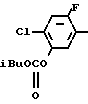
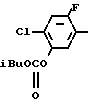
2-Хлор-4-фторфенол (29,3 г, 0,20 ммоль) загружали в круглодонную колбу (300 ммл), снабженную капельной воронкой, и к нему добавляли водный раствор 2 н. NaOH (100 мл) при охлаждении льдом с последующим перемешиванием в течение 30 мин. Затем к смеси добавляли по каплям изобутилхлорформат (30 мл, d 1,053, 31,6 г, 0,23 моль), смесь перемешивали в течение 2 ч при постепенном повышении температуры до комнатной. После завершения реакции смесь экстрагировали метиленхлоридом (100 мл х 3 раза) и сушили над безводным сульфатом магния. После удаления осушающего агента растворитель отгоняли при пониженном давлении с получением 2-хлор-4-фторфенил (изобутил)карбоната в виде бесцветного прозрачного масла (45,8 г, 0,186 ммоль, 93,0% выход).
1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,00 (6Н, д. I 6,9 Гц), 2,05 (1Н, т. и сеп. I 6,3 и 6,9 Гц), 4,05 (2H, д. I 6,3 Гц), 6,8-7,3 (3Н, м.).
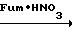
Затем в круглодонную колбу (200 мл) выливали дымящуюся азотную кислоту (100 мл, 98% d 1,52) и к ней медленно добавляли 2-хлор-4-фторфенил (изобутил)карбонат (10 г, 40,5 ммоль) при охлаждении льдом. После перемешивания смеси в течение 30 мин реакционную смесь выливали на лед. Выпавший в осадок в виде бледно-желтого твердого вещества 2-фтор-4-хлор-5-изобутилоксикарбонилоксинитробензол отфильтровывали и промывали водой. С помощью тщательной сушки получали желаемое соединение в виде белых кристаллов (10,8 г, 36,9 ммоль, 91,0% выход). Т.пл. 38,0-49,0оС.
1Н-ЯМР (CDCl3, ТМС,част./млн.): δ 1,00 (6Н, д. I 6,9 Гц), 2,07 (1Н, т. и сеп. I 6,3 и 6,9 Гц), 4,07 (2Н, I 6,3 Гц), 7,42 (1Н, д. IHF10,2 Гц), 8,02 (1Н, д. IHF 6,9 Гц).
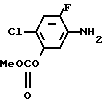
Полученный таким образом 2-фтор-4-хлор-5-изобутилоксикарбонилоксинитробе- нзол (10 г, 34,3 ммоль), толуол (100 мл) в качестве растворителя и 10% -ный палладий на угле (1,5 г) загружали в выдерживающий давление стеклянный автоклав (300 мл). После тщательного вытеснения внутреннего содержимого газообразным водородом начинали перемешивание при давлении водорода 4 атм. По мере протекания реакции выделялось тепло (приблизительно до 50оС), при этом продолжали перемешивание. Далее перемешивали при добавлении время от времени водорода до тех пор, пока не прекращалось поглощение водорода. После завершения реакции палладий на угле отделяли фильтрованием и освободившуюся воду удаляли с помощью осушающего агента. Растворитель отгоняли при пониженном давлении с получением количественно в основном чистого 2-хлор-4-фтор-5- изобутилоксикарбонилоксианилина (9,42 г) в виде желтого масла.
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,00 (6Н, д. I 6,9 Гц), 2,04 (1Н, т. и сеп. I6,3 и 6,9 Гц), 4,04 (2Н, д. I 6,3 Гц), 6,97 (1Н, д. IHF 6,9 Гц), 7,24 (1Н, д. IHF 9,0 Гц).
Метилхлорформат (23,4 г, 0,248 моль) добавляли к раствору 2-фтор-4-хлор-5-изобутилоксикарбонилоксианилина (65,0 г, 0,248 моль) и карбоната калия (32 мг, 0,232 ммоль) в ацетоне (300 мл), далее смесь перемешивали в течение 5 ч при температуре дефлегмации. После завершения реакции растворитель отгоняли при пониженном давлении, к остатку добавляли 1 н. соляную кислоту (300 мл) и выпавшее в осадок твердое вещество отфильтровывали. Твердое вещество тщательно промывали водой и сушили, получали метил N-(2-фтор-4-хлор-5-изобутилоксикарбонилоксифенил)карбамат в виде белых криталлов (56,6 г, 71,4% выход). Т.пл. 72,2-78,8оСМ.
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,00 (6Н, д. I 6,5 Гц), 2,05 (1Н, т. и септ. I 6,5 Гц), 3,78 (3Н, с.), 4,03 (2Н, д. I 6,5 Гц), 6,85 (1Н, шир. с.), 7,08 (1Н, д. IHF 10,2 Гц), 8,10 (1Н, д. IHF 7,5 Гц).
ИК-спектр (КВr, диск, см-1): 1773, 1733, 1545, 1285, 1235, 1180.
Метил N-(2-фтор-4-хлор-5-изобутилоксикарбонилоксифенил)-карбамат, полученный, как описано выше, растворяли в метаноле (100 мл), и затем к смеси добавляли карбонат натрия (7,26 г, 52,3 ммоль) с последующей реакцией при 50оС в течение 3 ч. По завершении реакции растворитель отгоняли при пониженном давлении, полученное твердое вещество растворяли в уксусной кислоте (20 мл). Раствор выливали в ледяную воду и выделившееся твердое вещество отделяли фильтрацией. Твердое вещество тщательно промывали водой и сушили, при этом получали метил N-(2-фтор-4-хлор-5-гидроксифенил)карбамат в виде белых кристаллов (9,60 г, 43,7 ммоль, 99,8%). Т.пл. 140,0-141,0оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,57 (3Н, с.), 3,78 (3Н, с.), 5,53 (1Н, с.), 6,75 (1Н, шир.с.), 7,05 (1Н, д. IHF10,5 Гц), 7,82 (1Н, д. IHF 7,5 Гц).
ИК-спектр (КВr, диск, см-1): 3440, 1717, 1560, 1430, 1250.
Раствор метил N-(2-фтор-4-хлор-5-гидроксифенил)карбамата (5,0 г, 22,8 ммоль), полученного, как описано выше, и карбоната калия (3,89 г, 28,1 ммоль), в ацетонитриле (50 мл) перемешивали в течение 1 ч при температуре дефлегмации. Затем бромциклопентан (4,07 г, 27,3 ммоль) добавляли по каплям к указанной смеси, которую после этого подвергали реакции в течение 3 ч. После завершения реакции растворитель отгоняли, остаток подкисляли добавлением 1 н. соляной кислоты (50 мл)и экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали водой и растворитель отгоняли при пониженном давлении с получением метил N-(2-фтор-4-хлор-5-циклопентилоксифенил)карбамата (5,56 г, 19,3 ммоль, 84,7% выход). Т.пл. 120,0-123,0оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,40-2,10 (8Н, м.), 3,77(3Н, с. ), 4,77 (1Н, м.), 6,82 (1Н, шир.с.), 7,07 (1Н, д.IHF 10,5 Гц), 7,83 (1Н, д.IHF 7,5 Гц).
ИК-спектр (КВr, диск, см-1): 1714, 1535, 1500, 1415, 1255, 1190.
К полученному метил N-(2-фтор-4-хлор-5-циклопентилоксифенил)карбамату добавляли этиловый спирт (30 мл) и 2 н. водный раствор гидроокиси натрия (50 мл) и смесь перемешивали в течение 4 ч при нагревании на масляной бане при 110оС. После завершения реакции растворитель отгоняли и остаток экстрагировали этилацетатом (100 мл х х 3 раза). Органический слой промывали насыщенным водным раствором хлорида натрия, сушили и растворитель отгоняли при пониженном давлении, при этом получали маслянистый 2-фтор-4-хлор-5-циклопентилоксианилин (4.36 г, 19,0 моль, 95,0% выход). Данные спектрального анализа и остальные данные являются такими. как показаны в ссылочном примере 17.
Ссылочный пример 4.
Cl NO2 +
NO2 +  - OTs
- OTs  C
C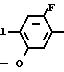 NO2
NO2 C
C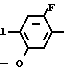 NH2
NH2
Раствор 2-фтор-4-хлор-5-гидроксинитробензола (7,1 г, 37,1 ммоль) и карбоната калия (5,1 г, 37,1 ммоль) в ацетонитриле (300 мл) перемешивали в течение 2 ч при температуре дефлегмации. К нему добавлялся циклопентил п-толуолсульфонат (10,3 г, 40,8 ммоль) с перемешиванием в течение 2 ч при температуре дефлегмации. После завершения реакции растворитель отгоняли из реакционной смеси при пониженном давлении. К нему добавляли 1 н. соляную кислоту (300 мл) и смесь экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали водным раствором бикарбоната натрия и водой, сушили, растворитель удаляли при пониженном давлении, при этом получали 2-фтор-4-хлор-5-циклопентилоксинитро- бензол в виде желтого твердого вещества (8,93 г, 34,3 ммоль, 92,6 выход). Т.пл. 58,0-62,6оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,41-2,26 (8Н, м.), 4,87(1Н, м.), 7,34 (2Н, д. IHF 10,5 Гц), 7,62 (2Н, д. IHF 7,5 Гц).
ИК-спектр (КВr, диск, см-1): 1535, 1490, 1350, 1205.
Полученный в результате 2-фтор-4-хлор-5-циклопентилоксинитробензол (13 г, 50,1 ммоль) растворяли в толуоле (100 мл), к смеси добавляли каталитическое количество 10% палладия на угле (0,5 г) и смесь перемешивали при температуре от комнатной до 70оС в условиях давления водорода 3-5 атм в стеклянном автоклаве. После того, как адсорбция газообразного водорода прекращалась, катализатор удаляли с помощью фильтрации, растворитель отгоняли при пониженном давлении из фильтрата с получением маслянистого 2-фтор-4-хлор-5-циклопентилоксианилина. Данные спектрального анализа и др. данные являются такими, как описаны в ссылочном примере 1.
Ссылочный пример 5.
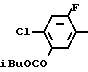 NH
NH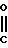 OMe +
OMe + 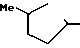 OTs
OTs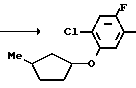 NH
NH OMe ____→
OMe ____→ 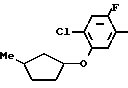 NH2
NH2
Раствор метил N-(2-фтор-4-хлор-5-изобутилоксикарбонилоксифенил) карбамата (5,37 г, 16,8 ммоль), приготовленого по способу, показанному в ссылочном примере 3, 3-метилциклопентил п-толуолсульфоната (5 г, 20,2 ммоль) и карбоната калия (2,32 г, 16,8 ммоль) в метаноле (50 мл) перемешивали в течение 5 ч при температуре дефлегмации. После завершения реакции реакционную смесь выливали в 1 н. соляную кислоту (100 мл) и смесь экстрагировали этилацетатом (50 мл х 3 раза). Органический слой сушили, растворитель удаляли при пониженном давлении, при этом получали N-[2-фтор-4-хлор-5-(3-метилциклопентил)оксифенил] карбамат в виде серо-белых кристаллов (3,81 г, 12,6 ммоль, 75,2% выход).
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,02 и 1,08 (общие 3Н, д. I6,0 Гц), 1,25-2,40 (7Н, м. ), 3,77 (3Н, с.), 4,75(1Н, м.), 6,68 (1Н, шир.с.), 7,05 (1Н, д. IHF= 10,5 Гц), 7,75(1Н, д. IHF 7,5 Гц).
Затем этиловый спирт (20 мл) и 2 н. водный раствор гидроокиси натрия (30 мл) добавляли к полученному в результате метил N-[2-фтор-4-хлор-5-(3-метилциклопентил)оксифенил] карбамату (3,45 г, 11,4 ммоль), после чего смесь перемешивали в течение 3 ч при температуре дефлегмации. После завершения реакции реакционную смесь экстрагировали этилацетатом (50 мл х 3 раза). Органический слой промывали насыщенным водным раствором хлористого натрия, сушили и растворитель отгоняли при пониженном давлении, в результате чего получали 2-фтор-4-хлор-5-(3-метилциклопентил)-оксианилин (1,77 г, 7,26 ммоль, 63,6% выход).
Спектр 1Н-ЯМР (CDCl3, ТМС, част./млн.): δ 1,02 и 1,10 (общее 3Н, д. I6,0 Гц), 1,22-2,58 (7Н, м.), 3,75(2Н, шир.с.), 4,65 (1Н, м.), 6,33 (1Н, д. IHF 8,0 Гц), 6,98 (1Н, д. IHF 10,0 Гц).
Ссылочный пример 6.
 +
+ ____→
____→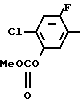 N
N ____→ Cl
____→ Cl N
N
Раствор 2-фтор-4-хлор-5-метоксикарбонилоксианилина (20 г, 91,1 ммоль) и 3,4,5,6-тетрагидрофталевого ангидрида (14 г, 92,0 ммоль) в уксусной кислоте (200 мл) подвергали реакции в течение 5 ч при нагревании при температуре дефлегмации. После завершения реакции смесь охлаждали до комнатной температуры, к ней добавляли воду (200 мл) и полученную смесь экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали водным раствором карбоната натрия и воды, сушили и растворитель отгоняли при пониженном давлении. Полученное в результате маслянистое вещество очищали хроматографией на силикагельной колонке (проявляющий растворитель: этилацетат/гексан 1/5), при этом получали N-(2-фтор-4-хлор-5-метоксикарбонилоксифе- нил)-3,4,5,6-тетрагидрофталимид в виде белого твердого вещества (26,2 г, 72,1 ммоль, 79,2% выход). Т.пл. 138,5-146,2оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, млн./дол.): δ 1,82 (4Н, м.), 2,42 (4Н, м.), 3,93 (3Н, с.), 7,21 (1Н, д. IHF 6,5 Гц), 7,33 (1Н, д. IHF 9,0 Гц).
ИК-спектр (КВr, диск, см-1): 1765, 1725, 1508, 1500, 1440, 1430, 1260, 1195.
Карбонат калия (4,6 г, 33,3 ммоль) добавляли к раствору N-(2-фтор-4-хлор-5-метоксикарбонилоксифенил)-3,4,5,6-тетрагидрофта- лимида (11,8 г, 33,4 ммоль), полученного, как описано выше, в метаноле (100 мл) с последующим перемешиванием в течение 5 ч при температуре дефлегмации. После завершения реакции смесь выливали в 1 н. соляную кислоту (200 мл) и экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали водой, сушили и растворитель отгоняли при пониженном давлении с получением неочищенного продукта (9,4 г). Продукт перекристаллизовывали из смеси простой эфир/гексан, при этом получали N-(2-фтор-4-хлор-5-гидроксифенил)-3,4,5,6-те- трагидрофталимид в виде белого твердого вещества (6,7 г, 22,7 ммоль, 67,8% выход). Т.пл. 145,5-156,4оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 1,80 (4Н, м.), 2,40 (4Н, м.), 6,00 (1Н, шир.с.), 6,85 (1Н, д. IHF 6,5 Гц), 7,17(1Н, д. IHF9,0 Гц).
ИК-спектр (КВr, см-1): 3440, 1785, 1720, 1530, 1430, 1395, 1185.
Ссылочный пример 7.
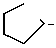 OH+Me
OH+Me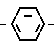
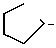
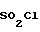 ___→
___→
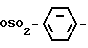 Me
Me
Циклопентанол (50 г, 0,58 ммоль) и n-толуолсульфонилхлорид (120г, 0,629 моль) растворяли в пиридине (200 мл), растор выливали в смесь льда и воды (примерно 1 л) с последующим тщательным перемешиванием. Выпавшее в осадок твердое вещество отфильтро- вывали, сушили и получали циклопентил n-толуолсульфонат в виде белого твердого вещества (94,9 г, 0,390 моль, 68,1% выход).
Т.пл. ниже 30оС.
Спектр 1Н-ЯМР (СDCl3, ТМС, млн.дол.): δ 1,23-2,07 (8Н, м.), 2,45 (3Н, с. ), 4,98 (1Н, м.), 7,38 (2Н, д. I 9,0 Гц), 7,85 (2Н, д. I 9,0 Гц).
Ссылочный пример 8.
 - OH + Me -
- OH + Me - - SO2Cl ___→
- SO2Cl ___→  - OSO2-
- OSO2- - Me
- Me
Циклопентанол (10 г, 0,116 моль), n-толуолсульфонилхлорид (24,3 г, 0,128 моль) и простой эфир (100 мл) загружали в круглодонную колбу (200 см3) и растворяли. Затем медлено к раствору при охлаждении ниже 10оС на водяной бане добавляли гидроокись калия (32,5 г, 0,58 моль) в виде порошка. После добавления смесь перемешивали в течение дополнительных 2 ч. После завершения реакции смесь выливали в ледяную воду (20 мл), органический слой и водный слой разделяли. Органический слой сушили и концентрировали при пониженном давлении, получая циклопентил n-толуолсульфонат в виде бледно-желтой вязкой жидкости (22,0 г, выход 81,8%).
Ссылочный пример 9.
 OH + Me -
OH + Me - - SO2Cl ___→
- SO2Cl ___→  OSO2-
OSO2- - Me
- Me
3-Метилциклопентанол (5,0 г, 49,9 ммоль) подвергали взаимодействию с n-толуолсульфонилхлоридом в пиридине (50 мл) таким же образом, как в ссылочном примере 7, с получением 3-метилциклопентил n-толуолсульфоната (11,7 г, 46,2 ммоль, выход 92,5%).
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 0,93 и 1,00 (общий 3Н, каждый д. I 6,0 Гц), 1,20-2,30 (7Н, м.), 2,48 (3Н, с.), 4,97 (1Н, м.), 7,38 (1Н, д. I 8,0 Гц), 7,85 (1Н, д. I 8,0 Гц).
Ссылочный пример 10
Cl  - NHCOOMe +
- NHCOOMe +  OTs
OTs  - NH2
- NH2
Раствор метил N-(2-фтор-4-хлор-5-гидроксифенил)карбамата (1,53 г, 6,98 ммоль), синтезированного с помощью процесса, описанного в ссылочном примере 3, 2-метилциклопентил n-толуолсульфоната (1.78 г, 6,99 ммоль) и N,N-диметилформамида (15мл) загружали в круглодонную колбу, (50 см3), затем к нему добавляли гидроокись калия (400 мг, 7,15 ммоль) в виде порошка с последующим перемешиванием в течение 7 ч при нагревании на масляной бане при 80-100оС. После завершения реакции реакционный раствор охлаждали до комнатной температуры, к нему добавляли 2 н. соляную кислоту (50 мл) и смесь экстрагировали этилацетатом (20 мл х 3). Органические слои объединяли, промывали насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли с помощью фильтрования, растворитель отгоняли при пониженном давлении, давая неочищенный продукт (1,51 г). Продукт очищали с помощью хроматографии на силикагельной колонке (проявляющий растворитель: гексан/этилацетат 17/3), получая 2-фтор-4-хлор-5-(2-метилциклопентил)оксианилин в виде бесцветного маслянистого вещества (666 мг, 2,73 ммоль, выход 40%).
Бесцветное прозрачное маслянистое вещество.
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 1,04 и 1,15 (общий 3Н, каждый д. I 7,0 Гц и 7,0 Гц), 1,40-2,40 (7Н, м.), 3,66(2Н, шир.с.), 4,15 и 4,42 (общий 1Н, каждый м.), 6,35 (1Н, д. IHF 9,2 Гц), 6,98 (1Н, д. IHF 11,9 Гц).
ИК-спектр (диск, см-1): 3425, 2980, 2900, 1630, 1510, 1245, 1190.
Ссылочный пример 11.
Cl 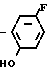 - NHCOOMe +
- NHCOOMe +  OTs ____→
OTs ____→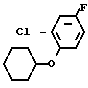 - NHCOOMe +
- NHCOOMe + 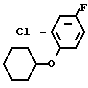 - NH2
- NH2
Метил N-(2-фтор-4-хлор-5-гидроксифенил)карбамат (1 г, 4,56 ммоль), синтезированный с помощью процесса, описанного в ссылочном примере 3, циклогексил n-толуолсульфоната (1,20 г, 4,73 ммоль), карбонат калия (635 мг, 4,95 ммоль), каталитическое количество йодистого калия и N,N-диметилформамид (20 мл) в качестве растворителя загружали в круглодонную колбу (100 см3) и перемешивали на масляной бане при 80оС. После завершения реакции реакционный раствор охлаждали до комнатной температуры, к нему добавляли 1 н. соляную кислоту (100 мл), и смесь экстрагировали этилацетатом (20 мл х 3). Органические слои объединяли, промывали водой и насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли с помощью фильтрования, растворитель отгоняли при пониженном давлении, давая неочищенный продукт (875 мг). Продукт отделяли и очищали с помощью хроматографии на силикагельной колонке (проявляющий растворитель: гексан/этилацетат 9/1), давая метил N-(2-фтор-4-хлор-5-циклогексилоксифенил)карбамат (12 мг, 0,46 ммоль, выход 8,1%), 2-фтор-4-хлор-5-циклогексилоксианилин (213 мг, 0,87 ммоль, выход 19%) и непрореагировавший исходный материал, метил N-(2-фтор-4-хлор-5-гидроксифенил)карбамат (540 мг, 2,46 ммоль, степень выделения 54%).
Метил N-(2-фтор-4-хлор-5-циклогексилоксифенил)карбамат.
Белые игольчатые кристаллы.
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 1,03-2,08 (10Н, м.), 3,80 (3Н, с.), 4,22 (1Н, м.), 7,10 (1Н, д. IHF 10,5 Гц), 7,88 (1Н, д. IHF= 7,3 Гц).
2-Фтор-4-хлор-5-циклогексилоксианилин.
Бесцветное прозрачное маслянистое вещество.
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 1,15-2,06 (10Н, м.), 3,46 (2Н, шир.с.), 4,10 (1Н, м.), 6,39 (1Н, д. IHF 9,0 Гц), 6,97 (1Н, д. IHF 11,5 Гц).
ИК-спектр (чистый, см-1): 3500, 3400, 2940, 2860, 1630, 1505, 1240, 1190.
Ссылочный пример 12.
Cl  - NHCOOMe +
- NHCOOMe + 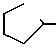 OTs
OTs 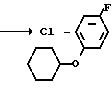 - NH2
- NH2
Метил N-(2-фтор-4-хлор-5-гидроксифенил)карбамат (2,03 г, 9,23 ммоль), циклогексил n-толуолсульфоната (2,51 г, 9,85 ммоль) и N,N-диметилформамид (30 мл) загружали в круглодонную колбу (100 см3). Затем в нее добавляли гидроокись калия (1г, 17,8 ммоль) в виде порошка. Смесь перемешивали в течение 4 ч на масляной бане при 80оС. После завершения реакции реакционный раствор охлаждали до комнатной температуры, к нему добавляли 1н. соляную кислоту (100 мл) и смесь экстрагировали этилацетатом (20 мл х х 3). Органические слои объединяли, промывали водой и насыщенным раствором хлористого натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрацией, растворитель отгоняли при пониженном давлении, при этом получали неочищенный продукт. Продукт отделяли, очищали с помощью хроматографии на колонке (проявляющий растворитель:гексан/этилацетат 9/1), получали 2-фтор-4-хлор-5-циклогексилоксианилин (574 мг, 2,36 ммоль, выход 26%) и непрореагировавший исходный материал, метил N-(2-фтор-4-хлор-5-гидроксифенил)карбамат (1,22 г, 5,54 ммоль, соотношение при выделении 60%). Данные спектров и др. показатели являются такими же, как показаны в ссылочном примере 11.
Ссылочный пример 13.
 NH2
NH2
 NH
NH OCH2Ph
OCH2Ph
____→ Cl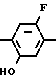 NH
NH OCH2Ph
OCH2Ph  l
l -
- - NH
- NH OCH2Ph
OCH2Ph l
l -
-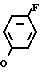 - NH2
- NH2
2-Фтор-4-хлор-5-метоксикарбонилокси- анилин (2 г, 6,86 ммоль), карбонат калия (1,42 г, 10,3 ммоль), бензилхлорформат (1,17 г, 6,86 ммоль) и ацетон (20 мл) в качестве растворителя загружали в двухгорлую колбу (100 см3) и нагревали в течение 2 ч в условиях дефлегмации. После завершения реакции смесь добавлялась к 1н. соляной кислоте (50 мл) и экстрагировали этилацетатом (50 мл х 3).После сушки органический слой концентрировали и получали чистый бензил N-(2-фтор-4-хлор-5-метоксикарбонилоксифенил)карбамат в виде твердого вещества (2,21 г, 91,1 выход).
Т.пл. 70-72оС.
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 3,62 (3Н, с.), 3,96 (2Н, с.), 6,59 (1Н, д. I 7,5 Гц), 7,01 (1Н, д. I 10,0 Гц), 7,20 (1Н, шир.с.), 7,10-7,50 (5Н, с.).
Бензил N-(2-фтор-4-хлор-5-метоксикарбонилоксифенил)карбамат (1,77 г, 5 ммоль), полученный, как описано выше, и карбонат калия (0,69 г, 5 ммоль) загружали в колбу (50 см3) и к ним добавляли метанол (30мл) в качестве растворителя с последующим перемешиванием при 50-60оС в течение 2 ч. После завершения реакции смесь охлаждали до комнатной температуры и выливали в охлажденую 2 н. соляную кислоту (60 мл). Смесь тщательно перемешивали, выпавший в осадок бензил N-(2-фтор-4-хлор-5-гидроксифенил)карбамат в виде белого твердого вещества (1,36 г, 4,60 ммоль, выход 92%) выделяли с помощью фильтрования и хорошо сушили.
Т.пл. 70-72оС.
Спектр 1Н-ЯМР (СDCl3 + DMCO-d6, ТМС, млн.дол.):
δ 5,25 (2Н), 7,10 (1Н. д. I 11,5 Гц), 7,49 (5Н, с.), 7,85 (1Н, д. I 8,6 Гц), 9,47 (1Н, шир.с.).
Затем бензил N-(2-фтор-4-хлор-5-гидроксифенил)карбамат (12,2 г, 41,3 ммоль), карбонат калия (5,97 г, 432,1 ммоль), циклопентил n-толуолсульфонат (11,2 г, 46,4 ммоль) и ацетон в качестве растворителя (150 мл) загружали в круглодонную колбу (500 см3) и нагревали в течение 9 ч в условиях дефлегмации. После завершения реакции реакционную смесь выливали в 1н. соляную кислоту. После тщательного перемешивания выпавший в осадок бензил N-(2-фтор-4-хлор-5-циклопентилоксифенил)карбамат в виде бледно-желтого твердого вещества (14,9 г, 40,9 ммоль, выход 99%) отделяли с помощью фильтрования и хорошо сушили.
Спектр 1Н-ЯМР (СDCl3, ТМС, млн.дол.): δ 1,48-2,07 (8Н, м.), 4,83 (1Н, м. ), 5,26( (2Н, с.), 6,92 (1Н, шир.с.), 7,14 (1Н, д. IHF 11,5 Гц), 7,47 (5Н, с.), 7,93 (1Н, д. IHF 8,3 Гц).
Бензил N-(2-фтор-4-хлор-5-циклопентилоксифенил)карбамат (2 г, 5,51 ммоль), синтезированный, как описано выше, 5% Рд/C (100 мл) в качестве катализатора и толуол (15 мл) в качестве растворителя загружали в круглодонную колбу (50 см3), при этом внутренняя часть колбы замещалась в достаточной степени водородным газом. Затем в атмосфере водородного газа смесь тщательно перемешивали в течение 3 ч при 50оС. После завершения реакции катализатор отделяли с помощью фильтрования, получающийся в результате фильтрат сушили с помощью безводного сульфата магния. После отделения осушающего агента с помощью фильтрования растворитель отгоняли при пониженном давлении из фильтрата и получали 2-фтор-4-хлор-5-циклопентилоксианилин (1,26 г) в виде бледно-коричневого маслянистого вещества. Спектральные данные и другие характеристики являются такими, как показаны в ссылочном примере 1.
Ссылочный пример 14.
 OH + Me -
OH + Me - - SO2Cl ___→
- SO2Cl ___→  SO2-
SO2- - Me
- Me
2-Метилциклопентанол (21,06 г, 0,210 моль) и n-толуолсульфонилхлорид (48,3 г, 0,252 моль) загружали в трехгорлую колбу, (500 см3), снабженную мешалкой, а затем к смеси при охлаждении льдом добавляли по каплям пиридин (170 мл). Смесь перемешивали в течение 10 ч при постепенном повышении температуры до комнатной. После завершения реакции к реакционной смеси добавляли холодную воду (500 мл) и смесь экстрагировали эфиром (200 мл х x 3). Органические слои объединяли, промывали последовательно 2н. соляной кислотой, водой и насыщенным водным раствором хлористого натрия и сушили безводным сульфатом магния. Осушающий агент отделяли с помощью фильтрования, растворитель отгоняли при пониженном давлении и получали по существу чистый 2-метилциклопентил n-толуолсульфонат в виде бесцветного прозрачного маслянистого вещества (49,7 г, 0,195 моль, выход 93%).
Бесцветное прозрачное маслянистое вещество.
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 0,85 (3Н, д. I 7,5 Гц), 1,41-2,10 (7Н, м.), 2,43 (3Н, с.), 4,42 и 4,80 (всего 1Н, каждый м.), 7,33 (2Н, д. IHF 9,0 Гц), 7,80 (2Н, д. IHF9,0 Гц).
Ccылочный пример 15.
 OH + Me -
OH + Me - - SO2Cl ___→
- SO2Cl ___→  OSO2-
OSO2- - Me
- Me
Циклогексанол (5,01 г, 50 ммоль) и n-толуолсульфонилхлорид (10,6 г, 55,6 ммоль) загружали в круглодонную колбу (100см3) и затем к смеси при охлаждении льдом по каплям добавляли пиридин (20 мл). Смесь перемешивали в течение 8 ч при постепенном повышении температуры до комнатной. После завершения реакции к реакционной смеси добавляли холодную воду (500 мл) и смесь экстрагировалась эфиром (100 мл х 3). Органические слои объединяли, промывали последовательно 2н. соляной кислотой, водой и насыщенным водным раствором хлористого натрия и сушили безводным сульфатом магния. Осушающий агент отделяли с помощью фильтрования и растворитель отгоняли при пониженном давлении, при этом получали по существу чистый циклогексил n-толуолсульфонат в виде белого твердого вещества (12,7 г, 49,9 ммоль, выход 99%).
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 1,00-1,93 (10Н, м.), 2,43 (3Н, с.), 4,30-4,64 (1Н, м.), 7,30 (2Н, д. IHF 9,0 Гц), 7,78 (2Н, д. IHF 9,0 Гц).
Ссылочный пример 16.
 l
l -
- - NH2
- NH2 l
l -
-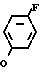 - N
- N l
l -
-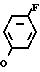 - NHNH2
- NHNH2
2-Фтор-4-хлор-5-циклопентилоксиани- лин (23 г, 0,1 моль), синтезированный с помощью процесса, описанного в ссылочном примере 1, 3, 4 или 13, растворяли в ацетонитриле (50 мл) и к нему добавлялии 42%-ную борфторную кислоту (150 мл). После перемешивания в течение 30 мин при комнатной температуре медленно при 0-5оС добавляли водный раствор (150 мл) сульфита натрия (20 г, 0,280 моль). После перемешивания в течение 2 ч при данной температуре образовавшееся твердое вещество отделяли с помощью фильтрования и промывали смесью льда и воды, а затем смесью этилацетат/гексан (1/6). С помощью тщательной сушки получали 2-фтор-4-хлор-5-циклопентилоксифенилдиазоний- фторборат в виде серо-белого твердого вещества (29,5 г, выход 89,4%).
Т.пл. 147-150оС (разложение).
Спектр 1Н-ЯМР (CDCl3, ТМС, млн.дол.): δ 1,38-2,17 (8Н, м.), 4,88 (1Н, м. ), 7,57 (1Н, д. IHF 9,0 Гц), 8,10 (1Н, д. IHF 5,0Гц).
ИК-спектр (чистый, см-1): 3130, 2975, 2280, 1490, 525.
К раствору моногидрата хлористого олова (II) (200 г, 0,886 моль), растворенного в концентрированной соляной кислоте (200 мл) и ТГФ (200 мл), добавляли ТГФ (200 мл), раствор 2-фтор-4-хлор-5-циклопентилоксифенил-диазонийфторбората (31,3 г, 0,094 моль) при охлаждении при 0-5оС. После перемешивания в течение 8 ч при данной температуре к нему добавляли водный раствор (1000 мл) гидроокиси калия (160 г). Твердое вещество, которое образовывалось, отделяли с помощью фильтрования и фильтрат экстрагировали этилацетатом (1000 мл х 3). Экстракт промывали насыщенным водным раствором хлористого натрия (1000 мл) и сушили над безводным сульфатом магния. Осушающий агент отделяли с помощью фильтрования и растворитель отгоняли при пониженном давлении, давая 2-фтор-4-хлор-5-циклопентилоксифенилгидразин в виде коричневатых кристаллов (20,9 г, 85,6% выход).
Т.пл. 77-79оС.
Спектр 1Н-ЯМP (СDCl3, ТМС, млн.дол.): δ 1,35-2,1 (8Н, м.), 3,85 (1Н, шир. с. ), 4,75 (1Н, м.), 6,60 (1Н, д. IHF 8,0 Гц), 6,95 (1Н, д. IHF 14,0 Гц).
ИК-спектр (КВr, диск, см-1): 3350, 2980, 1610, 1510, 1175, 855.
МС-спектр (м, е): 246 (М+, 5%), 244 (М+, 17%), 176 (100%), 41 (61%).
Элементный анализ. Вычисленные значения C11H14N2OClF, C 53,80 (53,99); H 5,74 (5,77); N 11,35 (11,45).
Далее изобретение иллюстрируется с помощью примеров препаратов гербицидного средства, включающего соединение настоящего изобретения в качестве активного ингредиента, и примеров, описывающих гербицидное действие, оказываемое гербицидным средством. Части даны по массе.
Пример препарата 1 (эмульгируемый препарат).
Соединение 10 настоящего изобретения (20 ч.) ксилол(35ч.), циклогексан (40 ч.), и Сорбол 900А (5 ч.) [Тохо Кемикал Ко. Лтд. равномерно смешивались с получением эмульгируемого препарата. Другие соединения настоящего изобрете- ния также обрабатывались, как описано выше, с получением эмульгируемых препаратов.
Пример препарата 2 (смачиваемый порошок).
Смесь 50 ч. соединения 10 настоящего изобретения, 25ч. диатомовой земли, 22 ч. глины и 3 ч. Лунокс Р100С (Тохо Кемикал Ко. Лтд.) равномерно смешивались и измельчались с получением смачиваемого порошка. Другие соединения настоящего изобретения также обрабатывались, как описано выше, с получением смачиваемых порошков.
Пример препарата 3 (гранулы).
Смесь 5 ч. соединения 10 настоящего изобретения, 35 ч. бентонита, 55 ч. талька и 5 ч. лигнин сульфоната натрия равномерно смешивались и измельчались, а затем перемешивалась с добавлением воды. Смесь гранулировалась экструдированием из гранулятора, сушилась и при регулировании размера зерен получались гранулы.
Пример получения 4 (порошкообразный агент). Смесь из 3,11 г соединения 10 данного изобретения, 5 г пропиленгликоля, 5 г Ньюкалгена FS-3 (Такемото Юси), 10 г Ньюкалгена D-3020 (Такемото Юси), 0,5 г Кунипиа G4 (Кунимайн Индастри), 1 г Ньюкалгена ЕХ-70 (Такемото Юси), 0,1 г Проналя ЕХ-300 (Тохо Кемикал) и 75,29 г ксантановой смолы измельчали в течение 1,5 ч в небольшой водяной мельнице с 100 мл стеклянных шариков (диаметр 1-2 мм).
Полученный порошок просевали с получением 86 г (текучего) порошкообразного агента. Содержание активного ингредиента, определенное с помощью жидкостной хроматографии, составляло 3,21%
Пример получения 5 (0,5%-й гранулированный агент). К 285,65 г глины УА (Канто Бентонайт Индастри) добавляли раствор, приготовленный путем растворения 1,6 г соединения 10 данного изобретения в 20 мл ацетона. Смесь хорошо смешивали и сушили при комнатной температуре. К высушенной смеси добавляли поверхностно-активные вещества, состоящие из 15 г Ньюкалгена G-285 (Такемото Юси) и 7,5 г Ньюкалгена DR-705 (Такемото Юси). Полученную смесь смешивали, гранулировали с помощью цилиндрического экструдера небольшого диаметра и сушили при 50оС. Полученные зерна обрабатывали для получения 212 г гранулированного агента с размером частиц 16-48 меш. Содержание активного ингредиента, определенное с помощью жидкостной хроматографии, составляло 0,503%
Пример получения 6 (эмульгируемый агент). К 56,55 г соединения 10 данного изобретения добавляли 66 г Ньюкалгена ST-30, 5,5 мл 1,3-диметил-2-имидазолидинона, 55 мл диметилсульфоксида и 390 мл Сольвессо 150 (ароматическая нафта, имеющая высокую температуру кипения). Смесь полностью перемешивали для получения эмульгирующего агента. Содержание активного ингредиента, определенное посредством жидкостной хроматографии, составляло 10,3%
Для определения гербицидной активности агентов полученные препараты испытывались следующим образом.
Пример испытания 1 (действие на сорняки на рисовом поле).
Горшки Вагнера 1/5000 ар заполнялись почвой рисовых полей и засевались семенами ранней горчицы полевой, Монохории и камыша японского и в них пересаживались растения риса (вид: Нихонбаре) на стадии 2-3 листа. Горшки выдерживались при поливе водой. Через 5 дн. поверхность воды обрабатывалась разбавленным раствором соединения настоящего изобретения, преобразованного в форму смачиваемого порошка или эмульгируемого препарата согласно примерам препаратов в заданных количествах 5, 2,5 и 0,5 г/ар. На 20 день после обработки исследовалось гербицидное действие на испытуемые сорняки и ущерб, причиняемый растениям риса, в соответствии со следующими стандартными критериями. Полученные результаты показаны в табл. 1 и 2.
В качестве контрольного соединения использовалось промышленно доступное соединение (А), при использовании тех же препаратов и методов обработки с помощью указанных стандартных критериев оценки исследовались активность по уничтожению сорняков и ущерб, причиняемый культуре. Результаты его также показаны.
Контроль, соединение А (Ронстар).
Пример испытания 2 (действие при обработке полевой почвы).
Емкости, имеющие площадь поверхности 16 х 10 см2 и глубину 7 см, заполняли полевой почвой и засевали семенами южно-африканского проса, обыкновенной мари белой, ежовника, сои и кукурузы. Емкости затем покрывали слоем почвы толщиной 1 см. На следующий день разбавленный раствор соединения настоящего изобретения, преобразованного в препаративную форму смачиваемого порошка или эмульгируемого препарата в соответствии с примерами препаратов, добавляли каплями равномерно на покрывающий слой почвы в заданных количествах 20, 10 и 5 г/ар. На 20-й день после применения так же как в примере испытания 1, исследовались гербицидная активность в отношении испытуемых сорняков и повреждения сои и кукурузы. Результаты испытаний показаны в табл. 3.
Пример испытания 3 (действие при применении на листве).
Емкости (лотки), имеющие площадь поверхности 16 х 11 см2 и глубину 7 см, заполняли полевой почвой и засевали семенами южно-африканского проса, обыкновенной мари белой, ежовника и сои. Спустя 15 дн. разбавленный раствор соединения настоящего изобретения, преобразованного в препаративную форму смачиваемого порошка или эмульгируемого препарата, наносили с помощью опрыскивания или обработки спреем в заданных концентрациях в воде в количестве 10 л/ар. На 20-й день после обработки исследовали эффект уничтожения сорняков на испытуемых сорных растениях и повреждения сои, таким же образом, как в примере испытания 1. Результаты показаны в табл. 4.
Пример испытания 4 (действие на сорняки на рисовом поле).
Горшки площадью 1/5000 ар заполняли почвой с рисового поля и затем засевали семенами Echinochlod oryzicola (I), Monochoria Vaginalis (2), Eleocharis acicularis (3), Scirpus jincoides (4) и других однолетних широколиственных сорняков и в них пересаживали растения риса (вид: Косихикари) на стадии 2-3 листа. Горшки содержались в условиях увлажнения водой. На следующий день производили обработку разбавленным раствором соединения данного изобретения, приготовленным в виде смачиваемого порошка или эмульгируемого агента, согласно примерам их получения в заданном количестве на ар.
На 15-й день после добавления раствора исследовали гербицидное действие на опытные сорняки и вред, наносимый растениям риса, ипользуя при этом указанные критерии оценки.
Аналогичное испытание проводили с использованием контрольной композиции А.
Полученные результаты показаны в табл. 5.
Пример испытания 5 (действие при обработке полевой почвы).
Емкости, имеющие площадь поверхности 10 х 10 см2 и глубину 7 см, заполняли почвой с поля и засевали семенами Еchinochlod crus-galli (6), Digitalia cibiaris (7), Amaranthus viridis (8), Chenopodium albuni (9) и кукурузы. Емкости затем покрывали слоем почвы толщиной 1 см. На следующий день на такую покрытую почву равномерно добавляли по каплям в заданном количестве на ар разбавленный раствор соединения данного изобретения, приготовленного в виде смачиваемого порошка или эмульгируемого агента, согласно примерам их получения.
На 15-й день после внесения раствора исследовали гербицидную активность по отношению к опытным сорнякам и вред, наносимый кукурузе. Полученные результаты показаны в табл. 6.
Пример испытания 6 (действие при применении к листве).
Емкости, имеющие площадь поверхности 16 х 11 см2 и глубину7 см, заполняли почвой с поля и засевали семенами Echinochloa erus-galli (10), Digitalia ciliaris (11), Amaranthus viridis (12), Chenopodium album (13) и кукурузы.
Через 15 дней листья выросших растений опрыскивали разбавленным раствором соединения данного изобретения, приготовленного в виде смачиваемого порошка или эмульгируемого агента, в заданных концентрациях в 20 л воды на ар.
На 10-й день после обработки исследовали гербицидное действие на опытные сорняки и вред, наносимый кукурузе.
Полученные результаты показаны в табл. 7.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЭПОКСИЦИКЛОГЕКСАНА И РЕГУЛЯТОРЫ РОСТА РАСТЕНИЙ | 1995 |
|
RU2126396C1 |
| ПРОИЗВОДНЫЕ 4,5,6,7-ТЕТРАГИДРОБЕНЗИМИДАЗОЛА | 1991 |
|
RU2033414C1 |
| АМИДЫ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2208608C2 |
| ПРОИЗВОДНЫЕ 2-ЗАМЕЩЕННОГО ФЕНИЛ-2-ОКСАЗАЛИНА ИЛИ 2-ЗАМЕЩЕННОГО ФЕНИЛ-2-ТИАЗОЛИНА И ИНСЕКТИЦИДНАЯ И/ИЛИ АКАРИЦИДНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1990 |
|
RU2029766C1 |
| ПРОИЗВОДНОЕ УРАЦИЛА | 1992 |
|
RU2040523C1 |
| ОПТИЧЕСКИ АКТИВНЫЕ ПРОИЗВОДНЫЕ ПИРИДОБЕНЗОКСАЗИНА ИЛИ ИХ СОЛИ | 1991 |
|
RU2029771C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТИАЗОЛА ИЛИ ИХ СОЛЕЙ С ГАЛОИДВОДОРОДНОЙ КИСЛОТОЙ | 1990 |
|
RU2010026C1 |
| ПРОИЗВОДНЫЕ АЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2161612C2 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИРРОЛИДИНА, ИМЕЮЩИЕ АРОМАТИЧЕСКИЕ ЗАМЕСТИТЕЛИ | 2000 |
|
RU2255938C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2043365C1 |
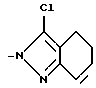
Использование: в сельском хозяйстве, т.к. обладают гербицидной активностью. Сущность изобретения: продукт - производные бензола, содержащие гетероцикл, ф-лы 1, где R-циклоалкил C3-C8 который может быть замещен алкилом C1-15 ; Z-группа ф-лы 2 или 3. Получают взаимодействием производного анилина с 3, 4, 5, 6-тетрагидрофталевым ангидридом или производного фенола с соединением R-Y. Гербицидное средство, в котором в качестве активного ингредиента используют соединение ф-лы 1 в количестве 0,5 - 50 мас.%, остальное - целевые добавки. Структура соединений ф-л 1, 2, 3: 
 6 с. и 6 з.п. ф-лы, 7 табл.
6 с. и 6 з.п. ф-лы, 7 табл.
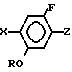
где R C3 C8-циклоалкильная группа, которая может быть замещена C1 C6-алкильной группой:
X галоген;
Z группа формулы
2. Соединение по п.1, отличающееся тем, что R циклопентильная группа, замещенная C1 C6-алкильной группой.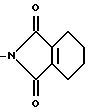
4. Соединение по п.1 или 2, отличающееся тем, что Z группа формулы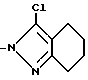
5. Способ получения N-замещенного производного фенил-3,4,5,6- тетрагидрофталимида общей формулы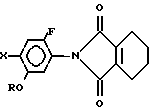
где R C3 C8-циклоалкильная группа, которая может быть замещена C1 C6-алкильной группой;
X галоген,
отличающийся тем, что производное анилина общей формулы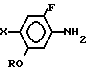
где R и X имеют указанные значения,
подвергают взаимодействию с 3, 4, 5, 6-тетрагидрофталевым ангидридом.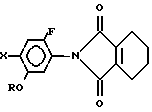
где R C3 C8-циклоалкильная группа, которая может быть замещена C1 C6-алкильной группой;
Х галоген,
отличающийся тем, что производное фенола общей формулы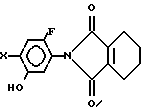
где Х имеет указанные значения,
подвергают реакции с соединением общей формулы
R Y,
где R имеет указанные значения;
Y хлор, бром, иод, метилсульфонилоксигруппа или п-толуолсульфонилоксигруппа,
в присутствии основания.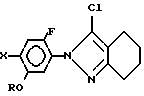
где R C3 C8-циклоалкильная группа, которая может быть замещена C1 C6-алкильной группой;
Х галоген,
отличающийся тем, что производное гидразина общей формулы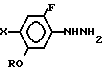
где R и X имеют указанные значения,
подвергают реакции с эфиром циклогексанон-2-карбоновой кислоты общей формулы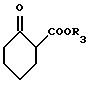
где R3 C1 C6-алкильная группа,
с получением производного индазолона общей формулы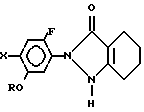
где R и X имеют указанные значения,
и затем хлорируют названное производное индазолона.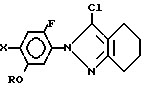
где R C3 C8 циклоалкильная группа, которая может быть замещена С1 C6 алкильной группой;
X галоген,
отличающийся тем, что осуществляют реакцию фенольного производного общей формулы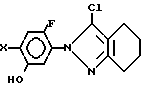
где X имеет указанные значения,
с соединением общей формулы
RY,
где R имеет указанные значения;
Y хлор, бром, иод, метилсульфонилоксигруппа или п-толилсульфонилоксигруппа,
в присутствии основания.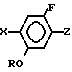
где R C3 C8-циклоалкильная группа, возможно C1 - C6-замещенная алкильной группой;
Х галоген;
Z группа формулы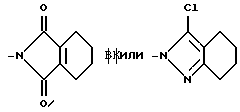
при следующем содержании ингредиентов, мас.
Активный ингредиент 0,5 50
Целевые добавки Остальное
10. Гербицидное средство по п.9, отличающееся тем, что R - циклопентильная группа, замещенная C1 C6-алкильной группой.
| Способ получения молочной кислоты | 1922 |
|
SU60A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Справочник по пестицидам | |||
| М.: Химия, 1985, с.82. | |||
