Изобретение относится к синтезу лекарственных препаратов, конкретно к способу получения азотнокислой соли 1-[2-(2,4-дихлорфенил)-2-(2,4-дихлорфенил-метокси)] -эти- лимидазола,(1-(2,4-дихлорфенил)-1-(2,4-дихлорбензилокси)-2-(имидазол-1-ил)эта- на (миконазола нитрата))}-эффективного противогрибкового препарата широкого спектра действия:
Cl CH
CH N
N N·HNO3
N·HNO3
Миконазол и его соли (нитрат, сульфосалицилат) широко используются в медицине в составе различных лекарственных форм. Методы получения миконазола описаны в ряде источников. Согласно указанным источникам получение миконазолнитрата осуществляется в несколько стадий
алкилирование имидазола α-бром-(α -хлор-)- 2,4-дихлорацетофеноном (I)
восстановление полученного фенацилимидазола (II) боргидридом натрия до спирта (III)
алкилирование полученного спирта (III) 2,4-дихлорбензилбромидом, 2,4-дихлорбензилхлоридом или 2,4-дихлорбензиларилсульфонатом (получение миконазола) (IV)
обработка миконазола концентрированной азотной кислотой (получение миконазол нитрата (V) согласно схеме I (см.далее).
Возможны варианты осуществления этой схемы. Например, в известных работах порядок стадий (1) и (2) обратный, т.е. вначале проводится восстановление галогенацетофенона I, а затем алкилирование образовавшимся спиртом имидазола (схема 2).
Ключевым моментом обоих вариантов схемы является получение 1-(2,4-дихлорфенил)-2-(имидазол-1-ил)этанола (соединение III), которое проводится в 2 стадии (1 и 2). Этим определяются и недостатки указанной схемы:
1) использование высокотоксичных 2,4-дихлор-α -бром (или α-хлор) ацетофенона (соед. I). В существующих условиях это соединение труднодоступно, поскольку для его синтеза используется м-дихлорбензол, который не производится в стране, и большое количество безводного хлористого алюминия, что создает экологические проблемы.
2) При восстановлении фенацилимидазола (II) до спирта (III) используется дефицитный, дорогостоящий боргидрид натрия, причем технологические процессы с его использованием пожароопасны.
Прототипом изобретения является способ получения миконазола нитрата, согласно которому получение миконазола ведут в несколько стадий. Исходным соединением является 2,4-дихлорацетофенон. Его подвергают бромированию в кипящем метаноле. Полученный 2-бром-2,4-дихлорацетофенон (I) вводят в реакцию алкилирования с имидазолом в среде метанола. После проведения реакции алкилирования 2,4-дихлорфенацилимидазол (II) выделяют экстракцией хлористым метиленом, с последующей отгонкой растворителя и обработкой остатка азотной кислотой (соединение II) выделяется в виде нитрата). Затем 2,4-дихлор-фенацилимидазол II подвергают восстановлению боргидридом натрия в среде абсолютного метанола или этанола. Полученный спирт (III) алкилируют 2,4-дихлорбензилхлоридом или дихлорбензилбромидом в среде диметилформамида или тетрагидрофурана в присутствии оснований (гидрид натрия, едкий натр), образующийся миконазол IV выделяют при выливании реакционной смеси в воду, экстракцией бензолом или ксилолом и упариванием растворителя. Конечный продукт миконазола нитрат (V) получают при обработке соединения IV азотной кислотой.
Недостатки способа-прототипа:
малый суммарный выход целевого продукта (11%) на исходный 2,4-дихлорацетофенон;
использование в качестве одного из исходных продуктов-α-бром(или α-хлор)-2,4-дихлорацетофенона, являющегося сильным отравляющим веществом кожно-нарывного и раздражающего действия;
многостадийность процесса, использование широкого набора исходных продуктов, в том числе малодоступных или требующих отдельных операций по их получению (ацилирование 1,3-дихлорбензола хлористым ацетилом в присутствии AlCl3, бромирование 2,4-дихлорацетофенона, восстановление 2,4-дихлорфенацилимидазола боргидридом натрия).
Сущность предлагаемого способа заключается в том, что в способе получения миконазол нитрата используется новый алкилирующий агент 2,4-дихлорфенилоксиран (VI), алкилирование ведут в среде толуола и/или диметилформамида (диметилацетамида) с последующим алкилированием полученного спирта (III) 2,4-дихлорбензилхлоридом (2,4-дихлорбензилбромидом) в присутствии оснований и переводом образовавшегося миконазола (IV) в азотнокислую соль (V) (схема 3).
Предлагаемый способ позволяет организовать процесс получения миконазол-нитрата в нашей стране за счет использования доступных исходных продуктов, упрощения схемы получения, улучшения экологической обстановки и условий труда.
Кроме того, предлагаемый способ более технологичен (ключевой полупродукт III получается в одну стадию, а не в две, как в прототипе) и более эффективен с экономической точки зрения, так как позволяет получить высокий выход целевого продукта и снизить расходные коэффициенты по сырью.
Схема 1.
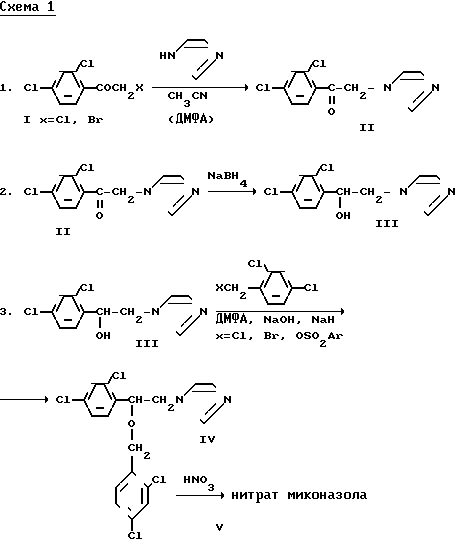
Схема 2.
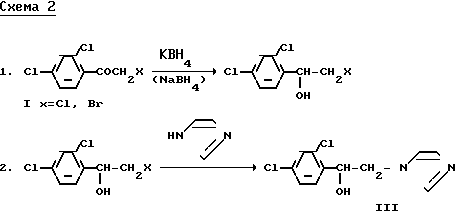
Схема 3.
Выходы промежуточных и конечных продуктов по стадиям в предлагаемом методе и по методике прототипа* представлены в таблице.
Сравнительный анализ доступности исходных продуктов 2,4-дихлор-2-бромацетофенона (прототип) и 2,4-дихлорфенилоксиpана (предлагаемый способ) для отечественной промышленности показывает очевидные преимущества 2,4-дихлорфенилоксирана.
Метод получения соединения I состоит из двух стадий:
а) ацилирование метадихлорбензола ацетилхлоридом в присутствии хлористого алюминия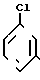

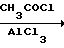 Cl
Cl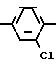 COCH3
COCH3
Данный процесс приводит к образованию большого количества нерегенерируемых отходов хлористого алюминия. Кроме того, необходимо учитывать, что сырье для данного процесса метадихлорбензол в стране не производится.
б) бромирование 2,4-дихлорацетофенона:
Cl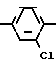 COCH3
COCH3 Cl
Cl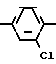 COCH2Br
COCH2Br
Бром является весьма дефицитным видом сырья, а продукт реакции сильным лакриматором и отравляющим веществом кожно-нарывного действия.
Синтез 2,4-дихлорфенилоксирана осуществляется также в две стадии исходя из 2,4-дихлортолуола, многотоннажное производство которого имеется в стране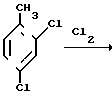
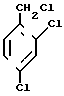
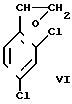
Хлорирование толуола в боковую цепь хорошо отработанный в промышленности процесс. На второй стадии в качестве реагента SR2 диалкилсульфида может быть использовано любое из производящихся промышленностью соединений этого класса.
В результате реакции 2,4-дихлорфенилоксиран получается в виде толуольного экстракта и может быть выделен дистилляцией.
Отличительными признаками предлагаемого способа являются: использование в качестве алкилирующего агента 2,4-дихлорфенилоксирана и среда алкилирования.
Использование совокупности признаков позволяет получить промежуточный продукт III не в две, а в одну стадию.
Известны реакции имидазола и других азолов с оксиранами. В большинстве случаев данный процесс производится в сильно полярных протонных или апротонных растворителях (спирты, ДМФА) в присутствии основных катализаторов. Например, алкилирование производных 1,2,4-триазола оксидами алкенов осуществлялось в этаноле в присутствии щелочей, а алкилирование имидазола оксидами циклоалкенов в этаноле путем кипячения в течение 70 ч, выход 75%
Взаимодействие имидазола с незамещенным по бензольному ядру фенилоксираном требует 50-часового нагревания до 90оС в среде ДМФА (выход 74%), либо нагревания до 75оС в абсолютном этаноле в присутствии пиридина в качестве катализатора (выход 39% ); либо использования в качестве катализатора металлического калия в среде ДМФА (выход 70%).
Необходимо отметить, что раскрытие оксиранового цикла при взаимодействии с гетероциклами может протекать в двух направлениях: по правилу Красуского (размыкание связи О-СН2)
R-C CH2 + Ht ___→ R-
CH2 + Ht ___→ R-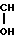 -CH2-Ht или с размыканием связи О-СН
-CH2-Ht или с размыканием связи О-СН
R-C CH2 + Ht ___→ R-
CH2 + Ht ___→ R-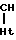 -CH2OH Кроме того, проведение процесса в условиях основного катализа может приводить к протеканию полимеризации оксиранов, изомеризации их в альдегиды и кетоны, а также к образованию стирилпроизводных. Возможность протекания названных побочных реакций в значительной мере зависит от природы субстрата и реагента и условий проведения реакции, причем для фенилоксиранов значительное влияние имеют также заместители в бензольном ядре.
-CH2OH Кроме того, проведение процесса в условиях основного катализа может приводить к протеканию полимеризации оксиранов, изомеризации их в альдегиды и кетоны, а также к образованию стирилпроизводных. Возможность протекания названных побочных реакций в значительной мере зависит от природы субстрата и реагента и условий проведения реакции, причем для фенилоксиранов значительное влияние имеют также заместители в бензольном ядре.
Примеры проведения алкилирования гетероциклов оксиранами, в частности фенилоксиранами в неполярных апротонных растворителях, в литературе отсутствуют.
Таким образом, успешное проведение реакции согласно предлагаемому методу в среде апротонного малополярного растворителя толуола или в смеси толуол-ДМФА является неочевидным.
Синтез 1-(2,4-дихлорфенил)-2-(имидазол-1-ил)-этанола
I. К раствору 8,2 г имидазола (0,12 моль) в 30 мл толуола при 85оС в течение 1 ч дозировали раствор 18,9 г (0,1 моль) 2,4- дихлорфенилоксирана в 40 мл толуола. После завершения дозировки выдерживали раствор при 95оС в течение 2 ч. Затем реакционную массу охлаждали и выливали в 100 мл воды, тщательно перемешивали и фильтровали. Осадок промывали диэтиловым эфиром и сушили. Получено 20,0 (78%) порошка белого цвета. Т.пл. 132-133оС.
ПМР, δ м. д. 4,15 (2Н,СН2); 5,2 т (1Н, СН); 6,9 с (1Н, СН); 7,1 с (1Н, СН); 7,55 м (3Н, СН); 7,7 с (1Н, СН).
Строение продукта доказано пробой смешения с образцом, полученным встречным синтезом.
II. К раствору 8,2 г имидазола (0,12 моль) в 15 мл ДМФА при 75оС в течение 1 ч дозировали раствор 18,9 г (0,1 моль) 2,4-дихлорфенилоксиран в 40 мл толуола. После завершения дозировки раствор выдерживали при 95оС в течение 1 ч, охлаждали, выливали в 100 мл воды, тщательно перемешивали и фильтровали. Осадок промывали диэтиловым эфиром и сушили. Получено 21,1 г (81.8%) белого порошка с т.пл. 131-132оС. Физико-химические характеристики аналогичны предыдущему.
III. К раствору 8,2 г (0,12 моль) имидазола в 15 мл ДМФА при 75оС в течение 1 ч дозировали раствор 18,9 г (0,1 моль) 2,4-дихлорфенилоксирана в 40 мл ДМФА. После завершения дозировки раствор выдерживали 1 ч при 95оС, охлаждали и выливали в смесь 100 мл воды и 50 мл толуола. Суспензию тщательно перемешивали и фильтровали. Осадок промывали эфиром и сушили. Получено 21,2 г (82%) белого порошка. Т.пл.129-131оС. Физико-химические характеристики аналогичны приведенным выше.
IV. Синтез 1-(2,4-дихлорфенил)-2-(имидазол-1-ил)-этанола в среде диметилацетамида аналогичен предыдущему.
V. Синтез миконазола нитрата.
10 г (0,078 моль) 1-(2,4-дихлорфенил)-2-(имидазол-1-ил)-этанола растворяли в 80 мл ДМФА, затем при Т=10-15оС в реактор добавляли 7,8 г (0,195 моль) едкого натра и перемешивали до растворения. В полученный раствор в течение 1,5 ч дозировали раствор 16,2 г (0,830 моль) 2,4-дихлорбензилхлорида в 80 мл ДМФА. По окончании дозировки выдержка 0,5 ч при 25оС.
Затем реакционную массу выливали в 400 мл воды и экстрагировали бензолом 4 х 100 мл. Бензол отгоняли, остаток растворяли в 300 мл этилацетата и при перемешивании добавляли 5 мл НNO3(конц.). Выпавший осадок отфильтровывали, промывали эфиром и сушили.
Получено: 34 г (91%) миконазола нитрата. Т.пл. 183-184оС. ПМР, δ м.д. 4,5 с (2Н, СН2); 4,65 с (2Н, СН2); 5,3 т (1Н, СН); 7,6 м (5Н, СН); 7,8 с (2Н, СН); 9,3 с (1Н, СН).
Полученная субстанция по всем тестам соответствовала требованиям проекта ВФС.
Элементный анализ.
Вычислено, C 45,09; H 3,13; N 8,77; Cl 29,65
Найдено, C 44,91; H 3,05; N 8,72; Cl 29,53.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИН-2,4-ДИОНОВ | 2013 |
|
RU2650678C2 |
| R-(-)-1-[2-(7-ХЛОРБЕНЗО[b]ТИОФЕН-3-ИЛМЕТОКСИ)-2-(2,4-ДИХЛОРФЕНИЛ)ЭТИЛ]-1H-ИМИДАЗОЛ И ЕГО СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОГРИБКОВЫМ ДЕЙСТВИЕМ | 2003 |
|
RU2276152C1 |
| Способ получения производных имидазола или их фармацевтически приемлемых солей присоединения кислот | 1987 |
|
SU1572415A3 |
| Аллиламиноэтилазолы, а также их фармакологически переносимые или переносимые растениями кислотно-аддитивные соли, обладающие фунгицидной и противогрибковой активностью | 1991 |
|
SU1834661A3 |
| Способ получения 3-азолилпропанолов | 2022 |
|
RU2786670C1 |
| Способ получения производных 1-(2-арил-1,3-диоксолан-2-илметил)- @ -имидазолов и @ -1,2,4-триазолов,их кислых аддитивных солей,комплексных солей металлов или стереомеров | 1982 |
|
SU1192625A3 |
| (-)-ЭНАНТИОМЕР 2-[2-(1-ХЛОРЦИКЛОПРОПИЛ)-3-(2-ХЛОРФЕНИЛ)-2-ГИДРОКСИПРОПИЛ]-2,4-ДИГИДРО-[1, 2,4]-ТРИАЗОЛ-3-ТИОНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2000 |
|
RU2238270C2 |
| Способ получения 4,5-диарил-2-нитроимидазолов | 1979 |
|
SU940647A3 |
| Способ получения имидазола | 1990 |
|
SU1747442A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(АЗОЛ-1-ИЛ)ЭТАНАМИНОВ | 2006 |
|
RU2317984C2 |

Использование: в химии гетероциклических активных веществ, в частности в способе получения нитрата 1-[2-(2,4-дихлорфени)-2-(2,4-дихлорфенилметокси)] этилимидазола с противогрибковыми широкого спектра действия свойствами. Сущность изобретения: способ получения нитрата 1-[2-(2,4-дихорфенил)-2-(2,4-дихлорфенилметокси)] этилимидазола ведут алкилированием имидазола 2,4-дихлорфенилоксираном в среде тролуола и/или диполярных апротонных растворителях с последующим алкилирование полученного продукта 1-(2,4-дихлорфенил)-2-(имидазол-1-ил)этанола.
СПОСОБ ПОЛУЧЕНИЯ НИТРАТА 1-[2-(2,4-ДИХЛОРФЕНИЛ)-2-(2,4-ДИХЛОРФЕНИЛМЕТОКСИ)] -ЭТИЛИМИДАЗОЛА, включающий алкилирование имидазола, алкилирование полученного 1-(2,4-дихлорфенил)-2-(имидазол-1-ил)этанола и выделение целевого продукта в виде соли, отличающийся тем, что алкилирование имидазола проводят 2,4-дихлорфенилоксираном в среде толуола и/или диполярных апротонных растворителей.
| G | |||
| Cooper, W | |||
| I | |||
| Yzvin, I Chem | |||
| Soe | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
