Изобретение относится к новым сериям тетрациклических соединений, обладающих двумя или тремя атомами азота, включенными в кольцо, которые обладают значительной антиаллергической и антиастматической активностями, содержит методики и композиции их использования, также как и технологии их получения.
Соединения данного изобретения могут быть в целом описаны как дибензопиразиноазепин или производственные бензопиридопиразиноазепина. Некоторые соединения этого типа известны [1-4] и имеют различную активность, включая антидипрессантную активность и антигистаминовую активность. Соединения, на которые ссылались ранее и которые имеют антиаллергическую активность, как было установлено, не достаточно удовлетворительны из-за того, что интенсивность активности меньше, чем было бы желательно для использования их в качестве коммерческого продукта, и часто проявляются побочные эффекты, такие как раздражение или угнетение центральной нервной системы. Поэтому было бы желательно разработать терапевтические агенты, которые наряду с проявлением отличных антигистаминовых, антиаллергических и антиастматических активностей, также не обладали бы побочными неблагоприятными реакциями, такими как угнетение или раздражение центральной нервной системы. Изобретение описывает новые серии тетрациклических соединений, не содержащих этих недостатков.
Соединения данного изобретения обладают отличной антигистаминовой, антиаллергической и антиастматической активностями без таких побочных реакций, как сонливость, и более того показывают ингибиторный эффект на выделение (медленно реагирующего вещества анафилаксии).
Соединения изобретения соответствуют формуле I
R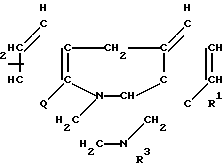 (I)
(I)
где Q представляет собой атом азота или группу формулы -СН-; R1 и R2являются одинаковыми или различными и каждый представляет собой атом водорода или атом галогена; R3 является замещенной алкильной группой, содержащей от 2 до 6 атомов углерода и имеющей, по крайней мере, один заместитель, выбранный из гидроксильных групп, атомов галогена и групп формулы -СОСR4 (где R4 представляет собой атом водорода, или алкильную группу, содержащую от 1 до 4 атомов углерода), групп формулы -NR5R6 (где R5 и R6 определены ниже) и групп формулы OCONR5R6 (где R5 и R6определены ниже), группой формулы [CH2CH2O]nCH2R8 в которой n 1-3, R8представляет гидроксиметил, группу формулы -СООR4 (где R4 определено выше) или группу формулы -CONR5R6 (где R5 и R6 определены ниже); группой формулы E-O-G-COOR4, в которой Е представляет собой алкиленовую группу, содержащую от 2 до 6 атомов углерода; G является группой, содержащей от 1 до 3 атомов углерода, а R4 является таким, как определен выше; R5 и R6 являются одинаковыми или различными и каждый представляет собой атом водорода, незамещенную алкильную группу, содержащую от 1 до 4 атомов углерода, циклоалкильную группу, имеющую 3-7 атомов углерода в кольце; фенильную группу; бензильную группу; 2-диметиламиноэтил или 2-[4-(n, n1-дифторбензгидрил)пиперазино] этил или R5 и R6 вместе с атомом азота, к которому они присоединены, представляют собой морфолино группу или 4-n- хлорбензгидрилпиперазин.
Соединения изобретения могут образовывать соли. Нет существенных ограничений по природе этих солей, однако терапевтического использования необходимо, чтобы они были фармацевтически приемлемыми. Когда они предназначаются для нетерапевтического использования, т. е. в качестве промежуточных соединений в технологии получения других и возможно более активных соединений, эти ограничения не существуют. Соединения изобретения включают некоторые основные атомы азота и могут, таким образом, образовывать кислые соли. Примеры таких дополнительных солей включают соли с минеральными кислотами, такими как плавиковая кислота, бромистоводородная кислота, иодистоводородная кислота, соляная кислота, азотная кислота, угольная кислота, серная кислота или фосфорная кислота; соли низших алкилсерных кислот, такие как метансульфокислота, трифторметансульфокислота, или этансульфокислота; если с арилсульфокислотами, такие как бензол- сульфокислота или р-толуолсульфокислота; соли с органическими карбоновыми кислотами, такие как муравьиная кислота, винная кислота, щавелевая кислота, малеиновая кислота, янтарная кислота или лимонная кислота; и соли с аминокислотами, такими как глютаминовая кислота или аспарагиновая кислота. Также соединения могут содержать свободные карбоксильные группы и в этом случае могут образовывать соли с основаниями. Примеры таких солей включают: соли со щелочными или щелочно-земельными металлами, такими как натрий, калий, литий, барий, кальций или магний, и соли органических оснований, такие как соль дициклогексиламина.
Соединения изобретения обязательно содержат несколько ассиметричных углеродных атомов в молекулах и таким образом могут образовывать оптические изомеры. Хотя все соединения представлены здесь одной молекулярной формулой, изобретение включает как индивидуальные изолированные изомеры, так и смеси, включая их рацематы. Индивидуальные изомеры могут быть получены направленно при использовании методик стереоспецифического синтеза или при использовании в качестве исходного материала оптически активных соединений, с другой стороны, если получена смесь изомеров, индивидуальные изомеры могут быть получены по общепринятым методикам.
Соединения изобретения могут быть получены по различным широко известным методикам получения соединений такого типа. В общем, один из методов включает в себя реакцию соединения, соответствующего формуле II
R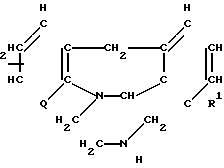 (II)
(II)
(в которой R1, R2 и Q представлены ранее), с соединением, соответствующим формуле III
R3 X (III)
(в которой R3 определено ранее и Х являются атомом галогена, преимущественно хлором, бромом или иодом), и затем, если требуется, превращение группы, представленной R3, в любую другую группу, соответствующую определенным для R3. Реакция идет нормально и эффективно в присутствии основания или инертного растворителя.
Нет существенных ограничений по природе оснований, используемых в данной реакции, при условии, что оно выполняет функции дезактивирующего агента и не имеет побочного влияния на другие части молекулы. Примеры предпочтительных оснований включают: органические амины, такие как триэтиламин,  -метилморфолин, пиридин, 4-(
-метилморфолин, пиридин, 4-( ,
,  -диметиламино) пиридин,
-диметиламино) пиридин,  ,
,  -диметиланилин и 1,8-диазобицикло (5.4.0)ундец-7-ене (DBu), и щелочи, включая карбонаты щелочных металлов, щелочно-земельных металлов, бикарбонаты и гидроксиды, такие как карбонат натрия, карбонат калия, бикарбонат калия, бикарбонат натрия, гидроокись натрия, гидроокись калия и гидроокись бария. Из них мы предпочитаем карбонаты щелочных металлов и гидроокиси щелочных металлов.
-диметиланилин и 1,8-диазобицикло (5.4.0)ундец-7-ене (DBu), и щелочи, включая карбонаты щелочных металлов, щелочно-земельных металлов, бикарбонаты и гидроксиды, такие как карбонат натрия, карбонат калия, бикарбонат калия, бикарбонат натрия, гидроокись натрия, гидроокись калия и гидроокись бария. Из них мы предпочитаем карбонаты щелочных металлов и гидроокиси щелочных металлов.
Нет существенных ограничений по природе используемого растворителя, при условии, что он не имеет побочного влияния на реакцию или на входящие реагенты. Примеры подходящих растворителей включают спирты, такие как метанол, этанол и пропанол, кетоны, такие как ацетон, 2-бутанол, 4-метил-2- пентанон, и амиды, особенно амиды жирных кислот, также как диметилформамид или диметилацетамид. Из которых кетоны наиболее предпочтительны. Реакция может проходить в широком интервале температур, и точная температура реакции не является критической в данном изобретении. В общем, установили, что реакция может проходить при температуре от 0 до 150оС (наиболее предпочтительно 60-140оС). Время реакции также может широко меняться в зависимости от многих факторов в особенности от температуры реакции и природы реагентов. Однако в случае, если реакция проходит в предпочтительных условиях, описанных выше, она обычно занимает период от 3 до 20 ч. Реакция может проходить в присутствии небольших количеств иодидов щелочных металлов, таких как иодид натрия или иодид калия, которые могут выполнять функцию катализатора.
Соединения, полученные по такой технологии, могут быть выделены из реакционной смеси общепринятыми методами. Для примера, соединения могут быть выделены путем отгонки растворителя из реакционной смеси или, если необходимо, после отгонки растворителя из реакционной смеси выливая остаток в воду и затем экстрагируя его с помощью не смешивающегося с водой органического растворителя и наконец отгонки растворитель из экстракта. Более того, при необходимости, результирующий остаток может быть далее очищен по различным хорошо известным методикам, таким как перекристаллизация, переосаждение, или различными хроматографическими методами, особенно колоночной хроматографией или лучше тонкослойной хроматографией.
Те соединения формулы (I), в которых R3 является алкильной группой, имеющей карбокси заместитель или группой с формулой E-O-G-COOH (в которой E и G описаны выше), т. е. карбоксильными кислотами, могут быть получены гидролизом соответствующих соединений, в которых R3 является алкильной группой, имеющей заместитель с формулой -СOOR4a или является группой с формулой -Е-О-G-COOR4a (в которой Е и G определены ранее и R4aявляется алкильной группой, имеющей 1-4 атомов углерода). Гидролиз может приводиться обычным способом, например, по реакции соответствующего эфира с основанием в инертном растворителе.
Не существует существенных требований по природе оснований, используемых в данной реакции гидролиза и любое общеиспользуемое основание в реакции такого типа может также использоваться при условии, что оно не оказывает побочного влияния на другие части молекулы. Примеры пригодных оснований включают: карбонаты щелочных металлов, такие как карбонат натрия или карбонат калия, гидроокиси щелочных или щелочно-земельных металлов, такие как гидроокись лития, гидроокись натрия, гидроокись калия или гидроокись бария. Из них гидроокиси щелочных металлов, такие как гидроокись натрия и гидроокись калия, наиболее предпочтительны. Нет существенных ограничений по природе используемого растворителя, при условии, что нет побочного влияния на реакцию или на другие используемые реагенты. Примеры пригодных растворителей включают: спирты, такие как метанол, этанол или пропанол; кетоны, такие как ацетон, 2-бутанон или 4-метил-2- пентанон, и эфиры, такие как диоксан или тетрагидрофуран. Из них наиболее предпочтительны спирты. Реакция может проходить в широком интервале температур и точная температура реакции является критической в изобретении. В общем, обнаружили, что реакция проходит в интервале температур от 0 до 120оС (более предпочтительно 0-80оС). Время реакции также может широко варьироваться, в зависимости от многих факторов, особенно от температуры реакции и природы реагентов. Однако при условии, что реакция проходит в предпочтительных условиях, оговоренных выше, время реакции 1-10 ч.
Соединения, полученные таким образом, могут быть выделены из реакционной смеси обычными методиками. Например, реакционная смесь может быть концентрирована отгонкой растворителя или, при необходимости, после отгонки растворителя из реакционной смеси остаток выливается в воду, и водная фаза подкисляется. Альтернативно, подкисленная водная фаза экстрагируется несмешивающимся с водой растворителем, после чего желаемое соединение может быть получено отгонкой растворителя из экстракта. Дополнительно, при желании, продукт может быть в дальнейшем очищен различными хорошо известными методами, такими как перекристаллизация, переосаждение или различными хроматографическими методами, особенно колоночной хроматографией или препаративной тонкослойной хроматографией.
Соединения общей формулы (II) хорошо известны или могут быть получены по методу, аналогичному известным методам, которые описаны, например, в Filer et al. J. Org. Chem, 46, 3344 (1981); C.A.A. van Boeekel et. al. Red Trav. Chim. Pays. Bas, 104, 259 (1985); u A. Org. Lee et. al. J. Heterocyclic Chem. 20, 1565 (1983).
Подобно этому исходные материалы формулы (III) известны или могут быть получены по методам, известным для получения известных аналогичных соединений. Однако соединение формулы (III), где R3 является группой формулы -E-O-G-COOR4, в которой Е и R4 описаны ранее и G является алкилоновой группой, имеющей 1-3 углеродных атома, т. е. соединение формулы (III), альтернативно может быть получено по следующей реакции: X E OH + X1 G1 COOR4 ____ X E O G1 COOR4
В описанных формулах R4, E, G1 и Х описаны ранее и Х1 является атомом галогена, предпочтительно бромом или йодом. Реакция нормально и предпочтительно проходит в присутствии основания или растворителя. Не существует существенных требований к природе используемого основания, при условии, что оно не оказывает побочного влияния на другие части молекулы или реагенты, и любое основание, обычно используемое для реакции такого типа, может также быть использовано здесь. Примеры подходящих оснований включают: гидриды щелочных металлов, такие как гидрид лития, гидрид натрия и гидрид калия, щелочные металлы, такие как натрий, калий, карбонаты щелочных или щелочно-земельных металлов, такие как карбонат лития, карбонат натрия, карбонат калия и карбонат бария, гидрированные карбонаты, такие как гидрированный карбонат натрия и гидрированный карбонат калия, алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия, и органические амины, такие как триэтиламин, пиридин, 4-диметиламинопиридин и DBu. Из них гидриды щелочных металлов и щелочные металлы предпочтительны.
Нет существенных ограничений по природе используемого растворителя, при условии, что он не оказывает побочного действия на реакцию или на реагенты. Примеры подходящих растворителей включают алифатические углеводороды, такие как пентан и гексан, алициклические углеводороды, такие как циклогексан, ароматические углеводороды, такие как бензол, толуол и ксилен, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан, амиды, особенно амиды жирных кислот, такие как диметилформамид и диметилацетат, и кетоны, такие как ацетон, металиэтилкетон и метилизобутил кетон. Один из этих растворителей или смесь одного или двух из них могут быть использованы. Из них предпочитают алифатические углеводороды, циклические углеводороды, ароматические углеводороды, простые эфиры и амиды. Реакция может проходить в широком интервале температур, и точная температура реакции не является критической в данном изобретении. В общем, обнаружили, что удобно проводить реакцию при температурах от -110оС до 130оС, более предпочтительно при 50оС до комнатных температур, и еще более предпочтительно при возрастании температуры с шагом от -50 до -20оС, от -10 до +10оС и при комнатной температуре. Однако предпочтительная температура реакции зависит от природы исходного материала. Время реакции также широко изменяется в зависимости от многих факторов, особенно от температуры реакции и природы реагентов. Однако при условии, что реакция проводится при предпочтительных условиях, оговоренных ранее, реакция проходит в течение 1-20 ч (более предпочтительно 1-6 ч). После завершения реакции продукт может быть выделен из реакционной смеси обычными методами, например выпариванием растворителя при пониженном давлении, при необходимости после удаления нерастворимой фазы фильтрацией или добавлением к осадку, экстракцией с помощью несмешивающегося с водой органического растворителя, и в конце отгонкой растворителя. После этого, при желании, продукт может быть очищен такими общепринятыми методами, как перекристаллизацией или различными хроматографическими методами, особенно препаративной тонкослойной хроматографией или колоночной хроматографией.
Соединение формулы (III), где R4 является атомом водорода может быть получено гидролизом сложных эфиров формулы (III), где R4 является R4a аналогичным описанному ранее.
Гетеротетрациклические соединения изобретения обладают, как это показано в данных по биологической активности, отличными антигистаминовой и антиаллергической активностями и отличной ингибиторной активностью против получения SRS-А. Соответственно соединения используются как терапевтические агенты для лечения или профилактики аллергических заболеваний или астмы.
Соединения изобретения таким образом могут быть использованы в лечении таких недугов, и для этих целей могут быть сформованы как удобные фармацевтические препараты.
Получение соединений изобретения будет в дальнейшем показано на примерах, получение некоторых соединений, используемых как исходный материал, описано в последующем получении. Биологическая активность некоторых соединений изобретения показана в последующих тест-примерах.
П р и м е р 1. Этиловый эфир 2-(1, 2, 3, 4, 10, 14в- гексагидродибензо (с, f)пиразино (1,2-а)азепин-2-ил) этоксиуксусной кислоты.
К 80 мл 4-метил-2-пентанона добавляют 2,5 г 1, 2, 3, 4, 10, 14в- гексагидродибензо (с, f(пиразино) 1,2-а)азепина, 2,4 г этилового эфира 2-хлорэтоксиуксусной кислоты (полученного как описано в методике 1 или 2), 3,82 г карбоната натрия и 0,14 г иодида натрия, и смесь кипятят в емкости с обратным холодильником в течение 18 ч. После этого смесь фильтруют и растворитель удаляют из фильтрата перегонкой под пониженным давлением. Остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, получая при этом из фракций, элюированных этилацетатом, 3,2 г (выход 84%) целевого соединения в виде масла светло-коричневого цвета. ИК-спектр (CHCl3), νmaxсм-1: 1495, 1750, 2830, 2950. К раствору целевого соединения в этаноле добавляют эквимолекулярное количество щавелевой кислоты, и смесь перемешивают при комнатной температуре в течение 30 мин. После этого растворитель удаляют перегонкой под пониженным давлением, и остаток перекристаллизовывают из этанола, получая оксалат целевого соединения с температурой плавления 141-144оС.
П р и м е р 2. 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)этоксиуксусная кислота.
К раствору 3,2 г этилового эфира 2-(1, 2, 3, 4, 10, 14в- гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты (полученного как описано в примере 1) в 20 мл этанола добавляют 7 мл 10 мас./об. водного раствора гидроокиси натрия и 10 мл воды. Смесь затем перемешивают при комнатной температуре в течение 1 ч, после чего смесь упаривают до половины ее первоначального объема перегонкой под пониженным давлением 10 мас./об. водного раствора хлористоводородной кислоты, и смесь экстрагируют хлороформом. Концентрирование экстракта упариванием под вакуумом дает 2,96 г (количественный выход) целевого соединения в виде вспененного вещества, которое перекристаллизовывают из воды, получая бесцветные иглы с температурой плавления 135оС (с разложением).
ИК-спектр (КВr) νmax см-1: 1426, 1450, 1491, 1602, 2820, 2940.
По методике, описанной в примере 1, затем получают следующие соли этого соединения:
натриевую соль с температурой плавления 140-145оС (с разложением);
фумарат с температурой плавления 187-188оС (с разложением);
оксалат с температурой плавления 184-186оС (с разложением).
П р и м е р 3. Метиловый эфир 2-(1, 2, 3, 4, 10, 14в- гексагидродибензо (с, f)пиразино (1,2-а)азепин-2-ил)этоксиуксусной кислоты.
3а. 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино (1,2-а)азепин-2-ил)этанол.
К 30 мл этанола добавляют 2 г 1, 2, 3, 4, 10, 14в- гексагидродибензо (с, f)пиразино(1,2-а)азепина, 0,84 г 2-хлороэтанола, 3,09 г карбоната калия и 0,13 г иодида натрия, и смесь кипятят в емкости с обратным холодильником в течение 16 ч. После этого смесь фильтруют, и растворитель удаляют из фильтрата перегонкой под пониженным давлением, получая 1,81 г (выход 77%) названного соединения с температурой плавления 123-125оС. ИК-спектр (KBr), νmax см-1: 1446, 1491, 2810, 3300, 3380.
Гидрохлорид этого соединения с температурой плавления 252-254оС (с разложением) получают по методике, аналогичной описанной в примере 17.
Это соединение может быть получено по методикам, представленным в следующих примерах 3в и 3с.
3в. Этиловый эфир (1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)уксусной кислоты.
К 20 мл 4-метил-2-пентанона добавляют 0,546 г 1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепина, 0,436 г этилового эфира бромоуксусной кислоты, 0,692 г карбоната натрия и 0,016 г иодида натрия, и смесь кипятят в емкости с обратным холодильником в течение 16 ч. После этого смесь разбавляют водой и затем экстрагируют этилацетатом. Растворитель затем удаляют упариванием под пониженным давлением. Получающийся в результате остаток подвергают затем разделению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента смесь гексана и этилацетата, взятых в объемном соотношении 4:1. При этом получают 0,602 г (выход 82%) названного соединения в виде масла бледно-желтого цвета. Гидрохлорид этого соединения с температурой плавления 187-190оС может быть получен по способу, аналогичному способам, описанным в примере 17, ИК-спектр (КВr) νmax, см-1: 1450, 1550, 1600, 1745, 2870, 2950.
3с. 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино (1,2-а)азепин-2-ил) этанол.
Суспензию 0,251 г литийалюминийгидрида в 20 мл тетрагидрофурана в течение 10 мин добавляют к охлаждаемому льдом раствору 2,22 г этилового эфира (1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f) пиразино(1,2-а)азепин-2-ил)уксусной кислоты полученного по методике, аналогичной описанной в примере 3в в 15 мл тетрагидрофурана в атмосфере азота. Смесь азота перемешивают при 0оС в течение 30 мин и в течение 2 ч при комнатной температуре, после чего добавляют 2 мл насыщенного водного раствора хлорида аммония. Смесь фильтруют, и фильтрат экстрагируют этилацетатом. Экстракт промывают водой и концентрируют упариванием под вакуумом, получая 1,65 г (выход 85%) названного соединения с температурой плавления 123-125оС.
3d. Метиловый эфир 2-(1, 2, 3, 4, 10, 14-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)этоксиуксусной кислоты.
0,478 г 55 мас./мас. дисперсии гидрида натрия в минеральном масле добавляют к раствору 1,5 г 2-(1, 2, 3, 4, 10, 14в- гексагидро- дибензо (с, f)пиразино(1,2-а)азепин-2-ил)этанола (полученного как описано на стадии а или с в 20 мл толуола в атмосфере азота). Затем смесь перемешивают при 40оС в течение 2 ч. после чего добавляют 0,924 г метилового эфира бромуксусной кислоты, охлаждая при этом смесь льдом, затем ее перемешивают при 40оС еще в течение 4 ч. После этого реакционную смесь фильтруют, фильтрат концентрируют упариванием под пониженным давлением. Остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента смесь гексана и этилацетата, взятых в объемном соотношении 1:1. При этом получают 0,58 г (выход 31%) названного соединения в виде масла. ИК-спектр (СНСl3), νmax, см-1 1450, 1495, 1600, 1655, 2820, 2950, 3000.
По методике, описанной в примере 1, из этого соединения может быть получен фумарат с температурой плавления 135оС (с разложением).
П р и м е р 4. Этиловый эфир 2-(3-хлоро-1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино (1,2-а)азепин-2-ил) этоксиуксусной кислоты.
Следуя методике, аналогичной описанной в примере 1, но используя 8-хлоро-1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино (1,2-а)азепин, названное соединение получают с выходом 52% ИК-спектр (СНСl3), νmax см-1: 1490, 1600, 1745, 2820, 2950.
Следуя методике, описанной в примере 1, получают оксалат названного соединения с температурой плавления 191-192оС (с разложением).
П р и м е р 5. Метиловый эфир 2-(3-бромо-1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты.
Следуя методике, аналогичной описанной в примере 1, но используя 8-бромо-1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин и метиловый эфир 2-хлороэтоксиуксусной кислоты, названное соединение получают с выходом 33%
ИК-спектр (СНСl3), νmax см-1: 1455, 1490, 1715, 1760, 2850, 2975.
Следуя методике, описанной в примере 1, также получают оксалат названного соединения с температурой плавления 183-185оС (с разложением).
П р и м е р 6. 2-(2-гидроксиэтокси)этил-1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин.
Следуя методике, описанной в примере 1, но используя 2-(2-гидроксиэтокси)этилхлорид, названное соединение получают с выходом 65%
ИК-спектр (СНСl3), νmax см-1: 1455, 1500, 1600, 2850, 2960.
Следуя методике, описанной в примере 1, также получают фумарат с температурой плавления 145-159оС (с разложением).
П р и м е р 7. 2-(2-/2-гидроксиэтокси)этокси)этил-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин.
Следуя методике, описанной в примере 1, но используя 2-(2-(2-гидроксиэтокси)этокси)этилхлорид, названное соединение получают с выходом 83%
Следуя методике, описанной в примере 1, также получают оксилат с температурой плавления 82-85оС (с разложением).
П р и м е р 8. Метиловый эфир 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с)пиразино(1,2а)пиридо(5,6-с) азепин-2-ил)этоксиуксусной кислоты.
Следуя методике, описанной в примере 1, но используя 1, 2, 3, 4, 10, 14в-гексагидробензо(с)пиразино(1,2-а)пиридо (1,2-а)пиридо(5,6-с)азепин и метиловый эфир 2-хлороэтоксиуксусной кислоты, названное соединение получают с выходом 67% ИК-спектр (СНСl3), νmax, см-1: 1455, 1595, 1755, 2970.
Следуя методике, описанной в примере 1, также получают оксалат с температурой плавления 130-132оС (с разложением).
П р и м е р 9-11. Используя методику, аналогичную описанной в примере 1, синтезируют следующие соединения, исходя из соответствующего соединения из примера 4, 5 или 6 соответственно.
П р и м е р 9. 2-(8-хлоро-1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)этоксиуксусную кислоту с выходом 94%
ИК-спектр (КВr) νmax, см-1: 1488, 1586, 2360, 2480, 2900, 2950.
Также получают оксалат названного соединения с температурой плавления 167-169оС (с разложением).
П р и м е р 10. 2-(8-бромо-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусную кислоту с количественным выходом.
ИК-спектр (КВr), νmax, см-1: 1426, 1450, 1490, 1600, 2830, 2940.
Также получают оксалат названного соединения с температурой плавления 177-178оС (с разложением).
П р и м е р 11. 2-(1, 2, 3, 4, 10, 14в-гексагидробензо(с)пиразино(1,2-а)пиридо(5,5с) азепин-2-ил)этоксиуксусную кислоту с выходом 72%
ИК-спектр (КВr) νmax, см-1: 1445, 1500, 1600, 2460, 2970.
Также получают оксалат названного соединения с температурой плавления 197-198оС (с разложением).
П р и м е р ы 12 и 13. Используя методику, описанную в примере 3d, синтезируют следующие соединения, исходя из соответствующих соединений из примеров 6 и 7 соответственно.
П р и м е р 12. Этиловый эфир 2-(7-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси) этоксиуксусной кислоты с выходом 35%
ИК-спектр (СНСl3), νmax см-1: 1495, 1600, 1750, 2825, 2950, 3010.
Масс-спектр (m/Z):424/56, M+/, 263/100/.
П р и м е р 13. Этиловый эфир 2-(2-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо)(с, f)пиразино(1,2-а) азепин-2-ил)этокси)этоксиуксусной кислоты с выходом 24%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1490, 1600, 1750, 2820.
Масс-спектр /m/Z/:468/29,M+/, 263/100/.
П р и м е р ы 14 и 15. Используя методику, описанную в примере 2, синтезируют следующие соединения, исходя из соответствующего соединения из примера 12 или 13 соответственно.
П р и м е р 14. 2-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)-азепин-2-ил)этокси)этоксиуксусную кислоту с количественным выходом.
ИК-спектр (СНСl3), νmax, см-1: 1455, 1500, 1600, 1740, 2560, 2980.
Также получают фумарат названного соединения с температурой плавления 169-172оС (с разложением).
П р и м е р 15. 2-(2-(2-(1, 2, 3, 4, 10, 14а-гексагидродибензо) (с, f)пиразино(1,2-а)азепин-2-ил)этокси)этоксиуксусную кислоту с выходом 97%
ИК-спектр (СНСl3), νmax, см-1: 1455, 1495, 1600, 2810, 2960, 3010.
Также получают гидрохлорид названного соединения с температурой плавления 130-133оС (с разложением).
П р и м е р 16. 3-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)пропанол.
К 30 мл этанола добавляют 1,5 г 1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино)1,2-а)азепина, 0,74 г 3-хлорпропанола, 2,32 г карбоната калия и 0,10 г иодида натрия, смесь кипятят в емкости с обратным холодильником в течение 20 ч. После этого реакционную смесь фильтруют, растворитель затем удаляют из фильтрата перегонкой при пониженном давлении. Остаток затем подвергают разделению на хроматографической колонке, наполненной силикагелем, и целевое соединение получают в кристаллическом виде из фракций, элюированных хлороформом, содержащим 5 об. этанола. Вещество затем перекристаллизовывают из этилацетата, получая 1,28 г (выход 69%) названного соединения с температурой плавления 127-128оС. ИК-спектр (КВr), νmax, см-1: 1448, 1492, 2821, 2895, 2956, 3194.
П р и м е р ы 17 и 18. Следуя методике, описанной в примере 16, но используя 4-хлорбутанол или 6-хлоргексанол, синтезируют следующие соединения.
П р и м е р 17. 4-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)бутанол с выходом 51%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1600, 2830, 2950.
Названное соединение затем растворяют в этилацетате, к полученному в результате раствору добавляют раствор хлористого водорода в этилацетате. Растворитель затем удаляют перегонкой в вакууме, получая гидрохлорид названного соединения с температурой плавления 233-235оС (с разложением).
П р и м е р 18. 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)гексанол с выходом 34%
ИК-спектр (КВr), νmax, см-1: 1440, 1494, 1590, 2813, 2944, 3204.
Также получают гидрохлорид названного соединения с температурой плавления 192-193оС (с разложением).
П р и м е р ы 19-21. Следуя методике, аналогичной описанной в примере 3в, но используя соответствующие бромосодержащие сложные эфиры, синтезируют следующие соединения, и затем получают их соли.
П р и м е р 19. Этиловый эфир 4-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f) пиразино(1,2-а)азепин-2-ил) бутановой кислоты с выходом 94%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1500, 1600, 1735, 2840, 2960.
П р и м е р 20. Этиловый эфир 5-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил) пентановой кислоты с выходом 87%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1600, 1730, 2820, 2950.
П р и м е р 21. Этиловый эфир 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)гексановой кислоты с выходом 99%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1600, 1730, 2820, 2950.
Следуя методике, описанной в примере 1 или примере 17, но используя соответствующие кислоты, получают следующие соли.
Фумарат этилового эфира 4-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)бутановой кислоты с температурой плавления 139-140оС.
Гидрохлорид этилового эфира 5-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)пентановой кислоты с температурой плавления 167-169оС.
Гидрохлорид этилового эфира 6-(1, 2, 3, 4, 10, 14в гексагидродибензо (c, f)пиразино(1, 2, -a)азепин-2-ил)гексановой кислоты с температурой плавления 150-152оС.
П р и м е р ы 22-24. Следуя методике, описанной в примере 2, но используя соответствующий сложный эфир из примера 19, 20 или 21 соответственно, синтезируют следующие карбоновые кислоты.
П р и м е р 22. 4-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)бутановая кислота с выходом 63%
П р и м е р 23. 5-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)пентановую кислоту с температурой плавления 240-243оС (с разложением) с выходом 54%
ИК-спектр (КВr), νmax, см-1: 1445, 1492, 1744, 2596, 2683, 2939, 3020.
П р и м е р 24. 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)гексановую кислоту с количественным выходом. ИК-спектр (СНСl3), νmax, см-1: 1450, 1500, 1600, 1720, 2450, 2970.
Следуя методике, описанной в примере 17, получают следующие соли.
Гидрохлорид 4-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)бутановой кислоты с температурой плавления 188-189оС.
ИК-спектр (КВr), νmax см-1: 1447, 1492, 1595, 1729, 2362, 2949, 2981.
Гидрохлорид 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)гексановой кислоты с температурой плавления 193-195оС (с разложением).
П р и м е р 25. Этиловый эфир 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) гексилоксиуксусной кислоты.
0,060 г дисперсии гидрида натрия в минеральном масле с концентрацией 55 мас. /мас. добавляют к раствору 0,40 г 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а) азепин-2-ил)гексанола (полученного как описано в примере 18) в 10 мл толуола в атмосфере азота. Смесь перемешивают при 40оС в течение 2 ч и затем охлаждают с помощью льда, после чего добавляют 0,229 г метилового эфира бромоуксусной кислоты, и смесь перемешивают при 40оС в течение еще 4 ч. После этого реакционную смесь фильтруют, и растворитель удаляют из фильтра упариванием под пониженным давлением. Остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента смесь этилацетата и гексана, взятых в объемном соотношении 1:1. При этом выделяют 90 мг (выход 18%) названного соединения в виде масла. Спектр ЯМР (CDCl3), δ, миллионные доли: 1,26 (3Н, триплет), 1,53-1,72 (3Н, мультиплет), 2,31-2,45 (4Н, мультиплет), 2,94 (2Н, дублет из дублетов), 3,26-3,37-(3Н, мультиплет), 3,56-3,63 (2Н, мультиплет), 4,06-4,25 (4Н, мультиплет), 4,82 (IH, дублет), 6,86 (IH, триплет) 7,01-7,20 (3Н, мультиплет).
ИК-спектр (СНСl3), νmax, см-1: 1445, 1490, 1595, 1745, 2800, 2925.
П р и м е р 26. 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этоксиацетамид.
К 15 мл тетрагидрофурана добавляют 0,358 г 2-(1, 2, 3, 4, 10, 14в-геноагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты (полученной как описано в примере 2) и 0,11 г триэтиламина. Затем к смеси по каплям при охлаждении льдом добавляют раствор 0,11 г этилхлороформиата в 2 мл тетрагидрофурана, и смесь перемешивают при комнатной температуре в течение 30 минут. Затем реакционную смесь смешивают с водой и экстрагируют хлористым метиленом. Экстракт концентрируют упариванием под пониженным давлением, и остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента хлористый метилен, содержащий 5 об. метанола. При этом выделяют 0,29 г (выход 82%) названного соединения с температурой плавления 66-68оС.
ИК-спектр (КВr), νmax cм-1: 1450, 1492, 1684, 2814, 2942, 3100, 3276.
П р и м е р ы 27-29. Следуя методике, описанной в примере 26, но используя соответствующий амин, синтезируют следующие соединения.
П р и м е р 27. α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси)-N,N-Диметилацетамид с выходом 79%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1600, 1645, 2820, 2950, 3005.
П р и м е р 28. α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси)-N-фенилацетамид с темпера- турой плавления 132-133оС с выходом 74%
ИК-спектр (СНСl3), νmax см-1: 1445, 1490, 1530, 1600, 1680, 2820, 2950, 3400.
П р и м е р 29. N-бензил-α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси)ацетамид с температурой плавления 92-93оС с выходом 74%
ИК-спектр (КВr), νmax, см-1: 1453, 1491, 1516, 1676, 2813, 2938.
Следуя методике, описанной в примере 17, получают гидрохлорид соединения из примера 27 с температурой плавления 80-82оС (с разложением).
П р и м е р 30. N-циклогексил-α- (2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси)ацетамид.
К 10 мл тетрагидроферана добавляют 0,358 г 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты (полученной как описано в примере 2), 0,1 г циклогексамина и 0,11 г триэтиламина. К полученной смеси при охлаждении ее льдом каплями добавляют 0,17 г диэтилцианофосфоната. Затем смесь перемешивают при комнатной температуре в течение 5 ч, после чего ее смешивают с водой и затем экстрагируют хлористым метиленом. Экстракт концентрируют упариванием под пониженным давлением, и остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента этилацетат, содержащий 10 об. этанола. При этом выделяют 0,4 г (выход 91%) названного соединения.
Следуя методике, описанной в примере 17, получают гидрохлорид названного соединения с температурой плавления 110-112оС (с разложением).
ИК-спектр (СНСl3), νmax, см-1: 1449, 1493, 1537, 1663, 2853, 2931, 3258.
П р и м е р ы 31-39. Следуя методике, описанной в примере 30, но используя соответствующий амин, синтезируют следующие соединения.
П р и м е р 31. α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси)-N,N-дипропилацетамид с выходом 61%
ИК-спектр (СHCl3), νmax, см-1: 1455, 1500, 1645, 2345, 2900, 2980, 3020.
П р и м е р 32. N-трет-бутил-α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этокси)ацетамид с температурой плавления 115-116оС с выходом 53%
ИК-спектр (KBr), νmax, см-1: 1449, 1491, 1523, 1673, 2004, 2951, 2969, 3401.
П р и м е р 33. N-циклопропил-α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этокси-ацетамид с выходом 83% ИК-спектр (СНСl3), νmax, см-1: 1450, 1600, 1670, 1720, 2830, 3000, 3440.
П р и м е р 34. N-циклобутил-α- (2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этокси)ацетамид с температурой плавления 113-115оС с выходом 88%
ИК-спектр (KBr), νmax, см-1: 1445, 1491, 1508, 1651, 2806, 2947, 3246.
П р и м е р 35. N-циклопентил-α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси) ацетамид с выходом 88%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1530, 1600, 1665, 2825, 2960, 3220.
П р и м е р 36. N-циклогептил-α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси) ацетамид с температурой плавления 97-99оС с количественным выходом.
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1530, 1600, 1665, 2860, 3330.
П р и м е р 37. 4-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f) пиразино(1,2-а)азепин-2-ил)этоксиацетил)морфолин с выходом 84%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1600, 1640, 1730, 2820, 2860.
П р и м е р 38. N-(2-диметиламиноэтил)-α-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а) азепин-2-ил)этокси)ацетамид с выходом 84%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1530, 1600, 1670, 2840, 2960, 3430.
П р и м е р 39. N-(2-(4-пара, пара-дифторбензгидрилпиперазин-2-ил)этил)-2-) (1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f) пиразино (1,2-а)азепин-2-ил-этокси) ацетамид с выходом 93%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1505, 1605, 1665, 2820, 2950, 3410.
Следуя методике, описанной в примере 17, получают следующие соли:
Гидрохлорид соединения из примера 31 с температурой плавления 82-84оС.
Гидрохлорид соединения из примера 35 с температурой плавления 124-127оС (с разложением).
Гидрохлорид соединения из примера 37 с температурой плавления 110-112оС (с разложением).
Дигидрохлорид соединения из примера 38 с температурой плавления 133-135оС (с разложением).
Тригидрохлорид соединения из примера 39 с температурой плавления 176-178оС (с разложением).
П р и м е р 40. 2-(2,3-дигидроксипропил)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиравино(1,2-а)азепин.
К охлажденному льдом раствору 1 г 1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепина в 2 мл этанола добавляют смесь 0,41 г глицидного спирта и 0,9 мл воды, и смесь перемешивают при комнатной температуре в течение 5 ч. Затем смесь концентрируют упариванием под пониженным давлением. Остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента смесь этилацетата и этанола, взятых в объемном соотношении 10:1. При этом выделяют 0,72 г (выход 56%) названного соединения в виде бесцветной пены. ИК-спектр (СНСl3), νmax, см-1: 1455, 1500, 1605, 1740, 2850, 2980, 3040.
Следуя методике, описанной в примере 17, получают гидрохлорид этого соединения с температурой плавления 205-207оС (с разложением).
П р и м е р 41. 2-(3-хлоро-2-гидроксипропил)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин.
К смеси 4,5 мл этанола и 1,5 г 1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепина при охлаждении льдом добавляют 0,832 г эпихлоргидрина, и смесь перемешивают при комнатной температуре в течение 5 ч. После этого смесь концентрируют упариванием под пониженным давлением. Остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента гексана смесь и этилацетата, взятых в объемном соотношении 1:1. При этом выделяют 1,56 г (выход 76%) названного соединения в виде бесцветной пены.
ИК-спектр (СНСl3), νmax, см-1; 1498, 1600, 3200.
Спектр ЯМР (СDCl3), δ, миллионные доли: 2,30-3,20 (5Н, мультиплет), 3,20-3,50 (3Н, мультиплет), 3,50-3,70 (2Н, дублет), 3,78-4,24 (2Н, мультиплет), 4,82 (IH, дублет), 6,70-7,40 (9Н, мультиплет).
П р и м е р 42. 2-(2-гидрокси-3-морфолинопропил) 1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин.
К 10 мл 4-метил-2-пентанола добавляют 0,5 г 2-(3-хлоро-2- гидроксипропил)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепина (полученного как описано в примере 41), 2,7 г карбоната натрия, 0,014 г иодида натрия и 0,254 г морфолина, и смесь кипятят в емкости с обратным холодильником в течение 2 ч. После этого реакционную смесь фильтруют, а фильтр затем концентрируют упариванием под пониженным давлением. Остаток подвергают разведению на хроматографической колонке, наполненной силикагелем, используя в качестве элюента этилацетат. При этом выделяют 0,50 г (выход 87%) названного соединения в виде бесцветной пены.
ИК-спектр (нуйол), νmax, см-1: 1454, 1491, 2853, 2951, 3400.
Следуя методике, описанной в примере 17, получают гидрохлорид названного соединения с температурой плавления 223-226оС (с разложением).
П р и м е р ы 43-46. Следуя методике, описанной в примере 42, но используя соответствующий амин, синтезируют следующие соединения.
П р и м е р 43. 2-(3-(4-пара-хлорбензгидрилпиперазин-1-ил) -2-гидроксипропил)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин с выходом 99%
ИК-спектр (СНСl3), νmax, см-1: 1455, 1495, 1600, 2830, 2950, 3400.
П р и м е р 44. 2-(2-гидрокси-3-циклогексиламинопропил)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин с выходом 87%
ИК-спектр (СНСl3), νmax, см-1: 1450, 1495, 1600, 2850, 2940, 3400.
П р и м е р 46. 2-(2-гидрокси-3-фениламинопропил)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин с выходом 69%
Следуя методике, описанной в примере 1 или примере 17, но используя соответствующую кислоту, получают следующие соли. Гидрохлорид соединения из примера 43 с температурой плавления 213-215оС (с разложением). Малеат соединения из примера 44 с температурой плавления 195-197оС (с разложением). Гидрохлорид соединения из примера 46 с температурой плавления 223-226оС.
ИК-спектр (КВr), νmax, см-1: 1445, 1493, 1603, 2608, 2712, 2969, 3319.
П р и м е р 47. Этиловый эфир 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а) азепин-2-ил)этоксиуксусной кислоты.
К охлаждаемому льдом раствору 0,5 г 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты (полученной, как описано в примере 2) в 10 мл хлороформа, содержащего 2 об. этанола, добавляют 0,11 г хлороэтилкарбоната, и смесь перемешивают при комнатной температуре в течение 1 ч. После этого смесь промывают водой, и растворитель удаляют перегонкой под пониженным давлением. Получающийся в результате остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, элюируя этилацетатом. При этом выделяют 0,25 г названного соединения в виде бесцветного масла. ИК-спектр этого соединения идентичен спектру соединения, полученного как описано в примере 1.
Способом, аналогичным описанному в примере 17, это соединение переводят в его гидрохлорид с температурой плавления 170-171оС.
П р и м е р ы 48 и 49. Следуя методике, аналогичной описанной в примере 8, но используя соответствующее хлороэтокси-соединение, синтезируют следующие соединения.
П р и м е р 48. 2-(2-гидроксиэтокси)этил-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)пиридо(5,6-с)азепин с выходом 84%
ИК-спектр (СНСl3), νmax, см-1: 1440, 1590, 1730, 2820, 2870, 2950, 3300.
Дихлоргидрат, плавящийся при 132-135оС (с разложением) получался с помощью приемов, аналогичных описанным в примере 17.
П р и м е р 49. 2-(2-(2-(2-гидроксиэтокси)этокси)этил)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с)пиразино(1,2-а)пиридо(5,6-с) азепиндигидрохлорид с температурой плавления 102-104оС (с разложением) с количественным выходом.
ИК-спектр (СНСl3), νmax, см-1: 1440, 1590, 1705, 2870, 2950, 3450.
П р и м е р 50. 3-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)пропилкарбамат.
К охлаждаемому льдом раствору 4 мг 3-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)пропанола (полученного как описано в примере 16) в 20 мл метиленхлорида добавляют 0,2 г трихлороацетилизоцианата, и смесь перемешивают при комнатной температуре в течение 2 ч. После этого растворитель удаляют перегонкой под пониженным давлением. Получающийся в результате остаток растворяют в 22 мл метанола, к раствору добавляют 9,0 г силикагеля, и смесь перемешивают при комнатной температуре в течение 15 ч. После этого смесь фильтруют, используя ускоритель фильтрования из "Целита" (торговая марка), и фильтрат концентрируют упариванием под пониженным давлением. Остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, элюируя этилацетатом, получая, после перекристаллизации из этилацетата, 260 мг (выход 59%) названного соединения в виде кристаллов с температурой плавления 160-161оС.
ИК-спектр (КВr), νmax см-1: 1638, 1722, 3119, 3313.
П р и м е р 51. 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепино-2-ил)этилкарбамат.
Следуя методике, описанной в примере 50, но используя 2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепино-2-ил) этанол, получают названное соединение с температурой плавления 182-184оС (с разложением) с выходом 62%
ИК-спектр (КВr), νmax, см-1: 1611, 1695, 1723, 3278, 3396.
П р и м е р ы 52-57. Следуя методике, аналогичной описанной в примере 1, исходя из соответствующих хлоросодержащих сложных эфиров, синтезируют следующие соединения.
П р и м е р 52. Метиловый эфир 3-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) пропоксиуксусной кислоты в виде масла с выходом 48%
ИК-спектр (СНСl3), νmax, см-1: 1495, 1600, 1758, 2830, 2970.
П р и м е р 53. Этиловый эфир 4-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) бутоксиуксусной кислоты в виде масла с выходом 50%
ИК-спектр (СНСl3), νmax, см-1: 1500, 1600, 1750, 2840, 2960.
П р и м е р 54. Метиловый эфир 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) гексилоксиуксусной кислоты в виде масла с выходом 72%
ИК-спектр (СНСl3), νmax, см-1: 1435, 1600, 1750, 2830, 2950.
П р и м е р 55. Этиловый эфир 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) гексилоксиуксусной кислоты в виде масла с выходом 75% ИК-спектр этого соединения идентичен спектру соединения, полученного как описано в примере 25.
П р и м е р 56. Этиловый эфир 2-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этокси) пропионовой кислоты в виде масла с выходом 77%
ИК-спектр (СНСl3), νmax, см-1: 1495, 1600, 1740, 2840, 2960.
П р и м е р 57. Этиловый эфир 2-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этокси)бутоновой кислоты в виде масла с выходом 68%
ИК-спектр (СНСl3), νmax, см-1: 1495, 1750, 2830, 2950.
Следуя методике, описанной в примере 1, получают следующие соли:
Оксалат этилового эфира 4-(1, 2, 3, 4, 10, 14-вгексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) бутоксиуксусной кислоты с температурой плавления 157-160оС.
Оксалат метилового эфира 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) гексилоксиуксусной кислоты с температурой плавления 140-141оС.
Оксалат этилового эфира 6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) гексилоксиуксусной кислоты с температурой плавления 115-120оС.
Оксалат этилового эфира 2-(2-(1, 3, 4, 10, 14в-гексагидродибензо (с, f)пиразино(1,2-а)азепин-2-ил)этокси)пропионовой кислоты с температурой плавления 160-162оС (с разложением).
Оксалат этилового эфира 2-(2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этокси)бутановой кислоты с температурой плавления 160-162оС (с разложением).
Оксалат метилового эфира 3-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) пропоксиуксусной кислоты с температурой плавления 102-104оС.
П р и м е р 58. Этиловый эфир (S) -2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты.
К 340 мл 4-метил-2-пентанона добавляют 32,48 г (S)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин (полученного как описано в методике 3), 22,7 этилового эфира 2-хлорэтоксиуксусной кислоты, 48,9 г карбоната натрия и 0,79 г иодида натрия, смесь кипятят в емкости с обратным холодильником в течение 16 ч. После этого смесь охлаждают до комнатной температуры и фильтруют, используя целитовый ускоритель фильтрования, фильтрат концентрируют упариванием под пониженным давлением. Получающийся в результате остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, элюируя смесью этилацетата и гексана, взятых в объемном соотношении 1:1. При этом получают 44,85 г (выход 91%) названного соединения в виде масла слабожелтого цвета.
ИК-спектр (СНСl3), νmax, см-1: 1495, 1600, 1750, 2820, 2950, [α]D25= +275o (c 1,0; метанол).
П р и м е р 59. (S)-2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)этоксиуксусная кислота.
К раствору 44,85 г этилового эфира (S)-2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты (полученного как описано в примере 58) и 450 мл этанола добавляют 65 мл 10 мас. /об. водного раствора гидроокиси натрия и 200 мл воды, и смесь перемешивают при комнатной температуре в течение 2 ч. После этого рН смеси доводят до 4 добавлением концентрированной соляной кислоты, смесь концентрируют упариванием под пониженным давлением. Выпадающие кристаллы собирают фильтрованием и перекристаллизовывают из воды, получая 39,20 г (выход 90%) названного соединения в виде моногидрата с температурой плавления 105-108оС.
ИК-спектр (КВr), νmax, см-1: 1446, 1492, 1598, 1633, 3404, 3449 [α]D25 +325о (с 1,0, диметилформамид).
К раствору названного соединения в этаноле добавляют 10 об./об. раствор соляной кислоты в этаноле, и смесь перемешивают при комнатной температуре в течение 30 мин. После этого растворитель удаляют упариванием в вакууме, и остаток перекристаллизовывают из воды, получая гидрохлорид названного соединения с температурой плавления 210-212оС. [α]D25 230о (с 1,0, диметилформамид). К раствору названного соединения в этаноле добавляют эквимолекулярное количество фумаровой кислоты, и смесь перемешивают при комнатной температуре в течение 30 мин. После этого растворитель удаляют упариванием в вакууме, и к остатку добавляют этилацетат, получая гемифумарат названного соединения с температурой плавления 161-163оС. [α]D25 +265о (с 1,0, диметилформамид).
П р и м е р ы 60 и 61. Следуя методике, описанной в примере 58, но используя соответствующий бромосодержащий сложный эфир, синтезируют следующие соединения.
П р и м е р 60. Этиловый эфир (S)-4-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(2,2-а)азепин-2-ил)бутановой кислоты в виде масла с выходом 97%
ИК-спектр (СНСl3), νmax, см-1: 1495, 1680, 1730, 2830, 2960. [α]D25= +301o (c 1,0:метанол).
Следуя методике, аналогичной описанной в примере 1, получают фумарат названного соединения с температурой плавления 139-141оС. [α]D25= +215o (c 1,0, метанол).
П р и м е р 61. Этиловый эфир (S)-6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) гексановой кислоты в виде масла с выходом 98%
ИК-спектр (СНСl3), νmax, см-1: 1495, 1600, 1730, 2840, 2955.
[α]D25 +284o (c 1,0, метанол).
П р и м е р 62. Следуя методике, аналогичной описанной в примере 58, но используя (R)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин (полученный как описано в методике 4) и соответствующий хлорид, получают этиловый эфир (R)-2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты в виде масла с выходом 91%
ИК-спектр (СНСl3), νmax, см-1, 1495, 1600, 1750, 2820, 2950 [α]D25= -303o (c 1,0, метанол).
Следуя методике, описанной в примере 1, получают фумарат названного соединения с температурой плавления 128-130оС. [α]D25 241o (c 1,0, диметилформамид).
П р и м е р ы 63 и 64. Следуя методике, аналогичной описанной в примере 58, но используя (R)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин (полученный как описано в методике 4) и соответствующий бромосодержащий сложный эфир, синтезируют следующие соединения.
П р и м е р 63. Этиловый эфир (R)-4-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) бутановой кислоты в виде масла с выходом 99%
ИК-спектр (СНСl3), νmax, см-1: 1495, 1600, 1730, 2830, 2960.
[α]D25 -296o (c 1,0, метанол).
Следуя методике, описанной в примере 1, получают фумарат названного соединения с температурой плавления 139-141оС.
[α]D25 -216о (с 1,0, метанол).
П р и м е р 64. Этиловый эфир (R)-5-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)гексановой кислоты в виде масла с выходом 98%
ИК-спектр (СНСl3), νmax, см-1: 1495, 1600, 1730, 2840, 2955.
[α]D25 -283о (с 1,0, метанол).
П р и м е р 65. Следуя методике, аналогичной описанной в примере 59, получают моногидрат (R)-2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты с температурой плавления 104-107оС и выходом 88% исходя из этилового эфира (R)-2-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил) этоксиуксусной кислоты (полученного, как описано в примере 62).
[α]D25 -322 (с 1,0, диметилформамид).
Следуя методике, описанной в примере 1 или примере 17, получают гидрохлорид и фумарат названного соединения с температурой плавления 209-211оС и 154-156оС, соответственно. [α]D25 -225о (с 1,0, диметилформамид) для гидрохлорида.
[α]D25 -207о (с 1,0, диметилформамид) для фумарата.
П р и м е р ы 66 и 67. Следуя методике, аналогичной описанной в примере 59, а затем методике, аналогичной описанной в примере 17, синтезируют следующие гидрохлориды, исходя из соответствующих сложных эфиров, полученных как описано в примерах 61 и 64.
П р и м е р 66. (R)-6-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)гексановой кислоты гидрохлорид с выходом 85% плавящийся при температуре 255-259оС (с разложением).
[α]D25 +256о (с 1,0, диметилформамид).
П р и м е р 67. (R)-8-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)гексановой кислоты гидрохлорид с выходом 92% плавящийся при температуре 243-250оС (с разложением).
[α]D25 -254o (c 1,0, диметилформамид).
Методика 1. Этиловый эфир 2-хлорэтоксиуксусной кислоты.
Суспензию 25,0 г гидрида натрия (в виде 60 мас./мас. суспензии в минеральном масле) в 100 мл диметилформамида по каплям добавляют к раствору 50 г 2-хлорэтанола и 104 г этилбромацетата в 350 мл диметилформамида при температуре, лежащей между -45 и -40оС, и смесь перемешивают при этой же температуре в течение 1 ч, при температуре от -30 до -25оС в течение 2 ч, при температуре от -5 до 5оС в течение 1 ч и при комнатной температуре в течение 2 ч. После этого смесь концентрируют упариванием в вакууме, к остатку добавляют 100 мл толуола. Затем раствор промывают водой, растворитель удаляют упариванием в вакууме. Перегонка дает 77,6 г (выход 75%) названного соединения, кипящего при температуре 57-58оС при 2,5 мм рт. ст. (33 Па).
Спектр ЯМР (CDCl3), δ, миллионные доли: 1,30 (3Н, триплет, J 7,0 Гц), 3,70 (2Н, триплет, J 4,5 Гц), 3,84 (2Н, триплет, J 4,5 Гц), 4,15 (2Н, синглет), 4,22 (2Н, квартет, J 7,0 Гц).
Методика 2. Этиловый эфир 2-хлороэтоксиуксусной кислоты.
Раствор 30 г 2-(2-хлороэтокси)этанола в 300 мл ацетона по каплям добавляют к смеси 240 мл реагента Джонса, приготовленного растворением 133,5 г триоксида хрома путем добавления 115 мл концентрированной серной кислоты и воды с последующим добавлением дополнительного количества воды до суммарного объема в 500 мл и 1050 мл ацетона при температуре, лежащей в интервале от -5 до 0оС, и смесь перемешивают при этой же температуре в течение 30 мин. После этого к смеси добавляют 150 мл изопропанола, смесь затем перемешивают при комнатной температуре в течение 1 ч. После этого смесь фильтруют, фильтрат концентрируют упариванием при пониженном давлении, рН доводят до 3 добавлением водного раствора бикарбоната натрия. Смесь экстрагируют этилацетатом, и растворитель упаривают из экстракта, получая 24,9 г (выход 75%) 2-хлорэтоксиуксусной кислоты в виде бледно-зеленого масла. Через раствор 24,9 г этой кислоты в 250 мл этанола пропускают газообразный хлористый водород, смесь кипятят в емкости с обратным холодильником в течение 3 ч. После этого смесь концентрируют упариванием при пониженном давлении. Перегонка дает 29,2 г (выход 95%) названного соединения в виде бесцветного масла, кипящего при температуре 96оС при 14 мм рт. ст. (1866 Па).
Методика 3. (S)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин).
3а. Этиловый эфир (S)-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин)карбоновой кислоты.
Раствор 5,32 г (S)-1, 2, 3, 4, 10, 14-гексагидро-2- метилдибензо(с, f)пиразино(1,2-а)азепина (полученного как описано в методике 6) в 30 мл толуола по каплям добавляют к раствору 6,8 г этилхлороформиата в 50 мл толуола при 80оС в течение 10 мин, смесь кипятят в емкости с обратным холодильником в течение 3 ч. После этого выпадающие кристаллы удаляют фильтрованием, а растворитель упаривают из фильтрата при пониженном давлении. При этом получают 5,57 г (выход 98%) вызванного соединения в виде масла бледно-желтого цвета.
ИК-спектр (СНСl3), νmax, см-1: 1240, 1435, 1495, 1600, 1690, 3010, [α] D25 +286о (с 1,0, метанол).
3в. (S)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f),пиразино)1,2-а)азепин.
Раствор 10,2 г гидроокиси калия в 34 мл воды добавляют к раствору 6,54 г этилового эфира (S)-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f) пиразино(1,2-а)азепин-2-ил) карбоновой кислоты (полученного как описано в методике 3а) в 85 мл этиленгликоля, смесь кипятят в емкости с обратным холодильником в течение 16 ч. После этого смесь выливают в ледяную воду и экстрагируют этилацетатом. Упаривание растворителя из экстракта дает 4,71 г (выход 93%) названного соединения в виде бесцветных кристаллов, плавящихся при температуре 122-124оС.
ИК-спектр (КВr), νmax, см-1: 1489, 2791, 2899, 3191, [α]D25 +488о(с 1,0, метанол).
Методика 4. (R) 1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин,
4а. Этиловый эфир (R)-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)карбоновой кислоты.
Следуя методике, описанной в методике 3а, но используя (R)-1:2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепин, получают названное соединение с выходом 98%
ИК-спектр (СНСl3), νmax, см-1: 1240, 1435, 1495, 1600, 1690, 0,3010.
[α]D25 -255о (с 1,0, метанол).
4в. (R)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин.
Следуя методике, описанной в методике 3в, но используя этиловый эфир (R)-(1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин-2-ил)карбоновой кислоты, получают названное соединение с выходом 95% плавящееся при температуре 122-124оС. [α]D25 -486о (с 1,0, метанол).
Методика 5. (S)-1, 2, 3, 4, 10, 14в-гексагидродибензо(c, f)пиразино(1,2-а)азепин и (R)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепин.
12,01 г хлорангидрида -α-метокси-α-(трифторометил) фенилуксусной кислоты по каплям добавляют к раствору 10,81 г рацемата 1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепина и 8,77 г триэтиламина в 90 мл хлороформа в течение 15 мин, причем раствор перемешивают и охлаждают льдом, затем смесь перемешивают при температуре 0-5оС в течение 1 ч. После этого в смесь добавляют воду, смесь экстрагируют хлороформом. Упаривание растворителя из экстракта дает остаток. Этот остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, элюируя смесь этилацетата и гексана, взятых в объемном соотношении 1:19. При этом получают 8,39 г (выход 42% ) менее полярного вещества в виде бесцветных игл, плавящихся при температуре 159-161оС, и 9,06 г (выход 45%) более полярного вещества в виде бесцветных призм, плавящихся при температуре 224-227оС [α]D25 +171о (с 1,0, диметилформамид) для менее полярного вещества. [α]D25 -211о (с 1,0, диметилформамид) для более полярного вещества.
23,7 мл 1,5 М раствора диизобутилалюминийгидрида в толуоле по каплям добавляют к раствору 8,30 г менее полярного соединения, полученного как описано выше, в 140 мл толуола при -160оС в течение 40 мин, смесь перемешивают при этой же температуре в течение 1,5 ч. После этого к смеси добавляют насыщенный водный раствор хлористого аммония, температуру поднимают до комнатной. Затем смесь фильтруют, фильтрат экстрагируют толуолом. Растворитель удаляют из экстракта упариванием под пониженным давлением, остаток подвергают разделению на хроматографической колонке, наполненной силикагелем, элюируя смесью метанола и метиленхлорида, взятых в объемном соотношении 1:19. При этом получают 2,30 г (выход 52%) (S)-1, 2, 3, 4, 10, 14в-гексагидробензо (с, f)пиразино(1,2-а)азепина, плавящегося при температуре 128-130оС. [α]D25= +504о (с 1,0, диметилформамид).
Следуя описанной выше методике, но используя 9,00 г более полярного соединения, получают 3,12 г (выход 65%) (R)-1, 2, 3, 4, 10, 14в-гексагидродибензо(с, f)пиразино(1,2-а)азепина, плавящегося при температуре 129-131оС.
ИК-спектр (КВr), νmax, см-1: 1489, 2791, 2898, 3189, [α]D25 -483o(c 1,0, диметилформамид).
Методика 6. (R)-1, 2, 3, 4, 10, 14в-гексагидро-2- метилдибензо(с, f)пиразино(1,2-а)азепин и (S)-1, 2, 3, 4, 10, 14в-гексагидро- 2-метилдибензо(с, f)пиразино(1,2-а)азепин.
Раствор 10,8 г дибензоил L-винной кислоты в 20 мл метанола добавляют к раствору 15 г рацемата 1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина в 180 мл метанола. Выпадающие кристаллы отфильтровывают и сушат, получая соль, плавящуюся при температуре 186оС (с разложением), [αD25 -266o (c 1,0, диметилформамид).
Всю эту соль суспендируют в 10 мас./об. водном растворе карбоната калия и экстрагируют этилацетатом. Испарение растворителя из экстракта дает 4,62 г (выход 31%) (R)-1, 2, 3, 4, 10, 14в-гексагидро-2-метилбензо(с, f)пиразино(1,2-а)азепина.
[α]D25 -457o (c 1,0, метанол). Раствор 10,8 г дибензоил-L- винной кислоты в 30 мл метанола добавляют к раствору 15 г рацемата 1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина в 270 мл метанола. Выпадающие кристаллы отфильтровывают и сушат, получая соль, плавящуюся при температуре 186оС (с разложением).
[α] D25 +243o (c 1,0, диметилформамид). Всю эту соль суспендируют в 10 мас. /об. водном растворе карбоната калия и экстрагируют этилацетатом. Испарение растворителя из экстракта дает 4,79 г (выход 32%) (S)-1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина.
[α]D25 +469о (с 1,0, метанол).
Методика 7. (R)-1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепин и (S)-1, 2, 3, 4, 10, 14в-гексагидро-2- метилдибензо(с, f)пиразино(1,2-а)азепин.
Раствор 2,66 г диацетил-L-винной кислоты в 20 мл этанола добавляют к раствору 5 г рацемата 1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина в 180 мл этанола. Выпадающие кристаллы отфильтровывают и сушат, получая соль, плавящуюся при температуре 188-189оС (с разложением). [α]D25 -274o (c 1,0, диметилформамид). Всю эту соль перекристаллизовывают из метанола, суспендируют в 10 мас./об. водного раствора карбоната калия и экстрагируют этилацетатом. Испарение растворителя из экстракта дает 1,5 г (выход 30% ) (R)-1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина, [α]D25= 474o (c 1,0, метанол).
Маточные растворы, получающиеся в результате перекристаллизации названных солей, упаривают, и остатки обрабатывают карбонатом калия, как указано выше, получая при этом 2,73 г (R) и (S) смеси 1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина. Смесь и 2,27 г рацемата 1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина объединяют и растворяют в 180 мл этанола. К раствору добавляют раствор 2,66 г диацетилвинной кислоты в 20 мл этанола. Выпадающие кристаллы отфильтровывают и сушат, получая соль, плавящуюся при 189-190оС (с разложением). [α]D25 +250o (c 1,0, диметилформамид). Всю эту соль перекристаллизовывают из метанола и обрабатывают водным карбонатом калия способом, аналогичным описанному выше, получая 1,65 г (выход 33%) (S)-1, 2, 3, 4, 10, 14в-гексагидро-2-метилдибензо(с, f)пиразино(1,2-а)азепина. [α]D25 +467o (c 10,0, метанол).
Контрольный пример 1. Ингибирование пассивной кожной анафилаксии (РСА) у крыс.
В соответствии с методом Моты (И.Мота, Immunology 681-699, 1964 готовят антисыворотку (РСА-титр, умноженный на 256) крыс против яичного альбумина и разбавляют ее в четыре раза физиологическим солевым раствором. В качестве подопытных животных в группах, каждая из которых включает 4 животных, используют самцов крыс вида SD в возрасте 5 недель). Крыс сенсибилизируют внутрикожной инъекцией 0,05 мл разбавленного раствора антисыворотки в положении лежа на спине. Спустя 48 ч после инъекции крысам, голодавшим в течение 1 дня, вводят через рот суспензию испытываемого соединения в 0,5 мас./об. водном растворе трагаканта и 60 мин спустя крысам в каудальную вену из расчета 5 мл/кг массы тела вводят физиологический солевой раствор, содержащий 0,4 мас./об. личного альбумина и 0,1 мас./об. голубого Эванса. Спустя 30 мин после этой последней инъекции крыс умерщвляют с помощью двуокиси углерода и в соответствии с методом Харады (Харада и др. J. Pharm. Pharmach. 23, 218-219, 1971 определяют проступивший голубой Эванса в дорсальном внутрикожном участке.
Результаты, полученные на испытываемых группах, которых были обработаны испытываемыми соединениями, анализируют с целью определения степени ингибирования путем сравнения со средним количеством красителя, проступившего в контрольной группе, которую не обрабатывали испытываемым соединением.
Степень ингибирования рассчитывают по следующему уравнению. Степень ингибирования (% )- /I-B/A/ x 100, где А количество проступившего красителя в контрольной группе; В количество проступившего красителя в испытываемой группе. (Натриевая соль (1, 2, 3, 4, 10, 14-тетрагидро-2Н, 10Н-пиразино(1,2-а)пирроло (2,1-с)(1,4)бензодиа- зепин-2-ил)уксусной кислоты.
Известные соединения и определены ранее при обсуждении современного состояния проблемы.
Контрольный пример 2. Влияние на антигениндуцированный бронхостеноз у сенсибилизированных морских свинок.
В качестве подопытных животных используют самцов морских свинок линии Hartley массой около 400-500 г. Этих животных сенсибилизируют по методу Морриса (Г.Р. Моррис, Br. Pharmac. 67, 179-184, 1979). Морским свинкам подкожно и внутрибрюшинно дважды вводят (каждый раз по 25 мг) яичного альбумина (сорт 5 производства фирмы "Сигма") через недельные интервалы. Семь дней спустя после второй из этих еженедельных инъекций животных не кормят в течение одного дня и затем подвергают воздействию аэрозоля яичного альбумина (10 мг/мл). Все обработанные таким образом животные реагируют на это конвульсиями, что указывает на расстройство дыхания, обусловленное сужением дыхательных путей, в продолжении 6 мин. За 60 мин до обработки яичным альбумином каждому из животных перорально вводят одно из испытываемых соединений, представленных в табл. 1. Соединение считается эффективным, если животное не реагирует конвульсиями в течение 6 мин ингаляции. Результаты показаны в табл. 1-2.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЙ ПИВАЛОИЛОКСИМЕТИЛ (IR, 5S, 6S)-[(4R)-2-ОКСО-4-ПИРРОЛИДИНИЛТИО]-6-[(IR)-1-ГИДРОКСИЭТИЛ]-1-МЕТИЛ-1-КАРБАПЕН-2-ЕМ-3-КАРБОКСИЛАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2090567C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| ЧЕТЫРЕХКООРДИНАЦИОННЫЕ КОМПЛЕКСЫ ДВУХВАЛЕНТНОЙ ПЛАТИНЫ | 1992 |
|
RU2039064C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОТИВООПУХОЛЕВОГО КОМПЛЕКСА ПЛАТИНЫ | 1988 |
|
RU2007413C1 |
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-АКРИЛОИЛПИПЕРАЗИНА | 1990 |
|
RU2024513C1 |
| ПРОИЗВОДНЫЕ АМИДОВ ТИАЗОЛИДИНКАРБОНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВОАЛЛЕРГИЧЕСКОЙ И ПРОТИВОАСТМАТИЧЕСКОЙ АКТИВНОСТЬЮ И СПОСОБНОСТЬЮ СНИЖАТЬ ФАКТОР АКТИВАЦИИ ТРОМБОЦИТОВ | 1991 |
|
RU2030409C1 |
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2072997C1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА АМИДНОГО ТИПА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 2001 |
|
RU2232761C2 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047602C1 |
Использование: в качестве антиаллергических и антиастматических препаратов. Сущность изобретения: продукт-азетидиновое производное формулы I с определенными значениями радикалов. Реагент 1: соединение формулы I, где R3 H. Реагент 2: соединение R3 X, где R3 указано, X атом галогена. Условия реакции: с последующим, если необходимо, преобразованием группы R3 в любое другое значение группы R3 2 табл. Структура соединения ф-лы I 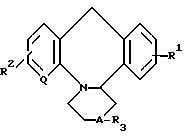
Способ получения азетидинового производного общей формулы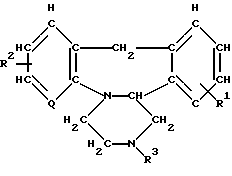
где Q азот или группа формулыCH-;
R1, R2 одинаковые или различные и каждый водород или галоген;
R3 замещенная алькильная группа, содержащая 2 6 атомов углерода и имеющая по крайней мере один заместитель, выбранный из гидроксильных групп, атомов галогена и групп формулы -COOR4 (где R4 водород или алкильная группа, содержащая 1 4 атома углерода), групп формул -NR5R6 -OCONR5R6 (где R5 и R6 имеют указанные значения), группа формулы [СН2СН2О]nСН2R8, где n 1 3;
R8 гидроксиметил, группа формулы -COOR4(где R4 имеет указанное значение) или группа формулы -CONR5R6 (где R5 и R6 имеют указанные значения), группа формулы -E-O-G-COOR4, где Е - алкиленовая группа, содержащая 2 6 атомов углерода; G алкиленовая группа, содержащая 1 3 атома углерода;
R4 имеет указанное значение;
R5 и R6 одинаковые или различные и каждый водород, назамещенная алкильная группа, содержащая 1 4 атома углерода, циклоалкильная группа, имеющая 3 7 атома углерода в кольце; фенильная группа; бензильная группа; 2-диметиламиноэтил или 2-[4-(п, п′ -дифторбензгидрил)пиперазино]этил
или R5 и R6 вместе с атомом азота, к которому они присоединены, морфолиногруппа или 4-п-хлорбензгидрилпиперазин,
или его фармацевтически приемлемой соли, отличающийся тем, что осуществляют взаимодействие соединения общей формулы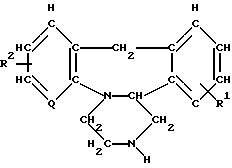
где R1, R2 и Q имеют указанные значения,
с соединением общей формулы
R3 X,
где R3 имеет указанные значения;
X галоген,
и затем, если требуется, преобразуют группу, представленную R3, в любую другую группу, включенную в определение R3-групп.
Приоритет по признакам:
05.10.89 при R3 группа общей формулы [СН2СН2О]СН2R8, где n 1 3; R8 - гидроксиметил, группа общей формулы -COOR4, где R4 водород или С1 С4-алкильная группа.
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Шланговое соединение | 0 |
|
SU88A1 |
