Предлагаемое изобретение относится к способам получения карбоната бария, применяемого в производстве электровакуумного стекла, и может быть использовано в химической промышленности.
Выпускаемый в настоящее время отечественный карбонат бария, согласно требованиям государственного стандарта, должен иметь массовую долю хлора (Cl) не более 0,05% По существующей технологии, где карбонат бария получают взаимодействием растворов хлорида бария и карбоната натрия, достигнуть этого уровня показателя по хлоридам не представляется возможным [1]
Достигнутый мировой уровень по данному показателю характеризуется еще более низким содержанием хлоридов. Гарантией достижения требуемой величины данного показателя может быть использование сырьевых компонентов, не содержащих хлоридов. Такая технология используется ведущими зарубежными фирмами производителями карбоната бария.
Известен способ получения карбоната бария, который включает в себя восстановление баритового концентрата углем и осаждение карбоната бария из раствора сульфида бария диоксидом углерода (см. Проспект фирмы "Kali Cemie A. G. ФРГ KC-Barium Carbonat, 1988). Качественный состав получаемого карбоната бария по данному способу следующий, мас. сульфиды в пересчете на SO3 0,3 0,4 или на SO4 0,36 0,48, нерастворимый в HCl остаток 0,8 1,2.
Основным недостатком этого способа является получение конечного продукта, загрязненного примесями серы.
Известен способ получения карбоната бария из нитратного щелока [2] Для производства карбоната бария используют раствор, содержащий 140 160 г/л Ba(NO3)2 осветленный нитратный щелок из производства азотнокислого бария. К щелоку после контрольной фильтрации на вакуум-фильтре добавляют раствор NaOH с концентрацией 500 г/л до свободной щелочности 1 2 г/л (в пересчете на NaOH). Подщелачивание ведут при 60 70oC в резервуаре с змеевиками для подогрева паром и барбатером для перемешивания воздухом. Затем из нитратного раствора удаляют сернистые соединения медленным введением раствора гипохлорита натрия (120 г/л активного хлора) до появления свободного активного хлора.
При этом идут следующие реакции:
BaS + 4NaClO BaSO4 + 4NaCl
S + 3NaClO + H2O H2SO4 + 3NaCl
H2SO4 + Ba(NO3)2 BaSO4 + 2HNO3
Осветленный раствор нитрата бария после отстаивания его от BaSO4 и других примесей приливают к перемешиваемому, я подогретому до 80 90oC раствору соды (200 г/л Na2CO3) для осаждения BaCO3.
Этот способ является наиболее близким к заявляемому и выбран в качестве прототипа.
Недостатком известного способа является введение гипохлорита натрия с целью очистки раствора нитрата бария от сернистых соединений, что сказывается на загрязнении целевого продукта ионами хлоридов (см. реакции 1, 2), а также громоздкость технологической схемы очистки от примесей с использованием дефицитного реагента гипохлорита натрия.
Целью изобретения является повышение качества целевого продукта за счет повышения массовой доли основного вещества и снижения примесей хлоридов с одновременным упрощением технологической схемы.
Поставленная цель достигается в описываемом способе получения карбоната бария, включающем получение барийсодержащего раствора обработкой водной суспензии плава сернистого бария азотной кислотой, разбавленной до массовой доли HNO3 20 23% при температуре 60 70oC с последующей обработкой полученной суспензии паром до температуры 85 95oC, очистку его от примесей, обработку очищенного раствора карбонатом натрия, отделение осадка карбоната бария, его промывку и сушку.
Отличительными от прототипа признаками являются:
получение раствора нитрата бария обработкой водной суспензии плава сульфида бария азотной кислотой, разбавленной до массовой доли HNO3 20 23% при температуре 60 70oC;
очистка полученного раствора нитрата бария от сернистых соединений и серы обработкой паром до температуры 85 95oC.
Применение разбавленной до 20 23% азотной кислоты и пониженной температуры 60 70oC для разложения водной суспензии плава сульфида бария позволяет направить процесс в сторону протекания основной реакции:
BaS + 2HNO3 _→ Ba(NO3)2 + H2S (4)
и значительно снизить возможность протекания окислительных реакций:
Подача пара в полученную суспензию позволяет отдуть сероводород и скоагулировать элементарную серу, которая после нейтрализации суспензии отфильтровывается вместе с основной массой шлама, что способствует снижению серосодержащих примесей и исключает возможность появления хлора в целевом продукте.
Из известного уровня техники предлагаемое решение является новым и обладает по сравнению с выбранным прототипом существенными отличиями.
Способ получения карбоната бария осуществляют следующим образом.
Плав сульфида бария непрерывно подают на выщелачивание в шаровую мельницу, куда одновременно подают горячую воду. Выходящую из шаровой мельницы суспензию сульфида бария подают в реактора кислотной обработки, куда одновременно вводят разбавленную до 20 23% азотную кислоту. Обработку суспензии проводят до PH 2 3 при температуре 60 70oC. Полученную суспензию после кислотной обработки направляют в реактор-коагулятор, куда одновременно вводят пар отдувки остаточного сероводорода и коагуляции серы. Температура в коагуляторе 85 95oC. Далее суспензию нейтрализуют в реакторах-нейтрализаторах 40%-ым раствором гидроксида натрия до pH 8,5 + 5 и фильтруют. Отфильтрованный раствор нитрата бария подвергают взаимодействию с раствором карбоната натрия, осаждая карбонат бария. Образовавшуюся суспензию карбоната бария сгущают, фильтруют, отделяя пасту карбоната бария. Отфильтрованную пасту карбоната бария промывают, сушат и гранулируют. Гранулированный продукт подвергают рассеву и дроблению. Полученный продукт карбонат бария анализируют на содержание основного вещества, хлоридов в пересчете на хлор-ион и общей серы в пересчете на SO4.
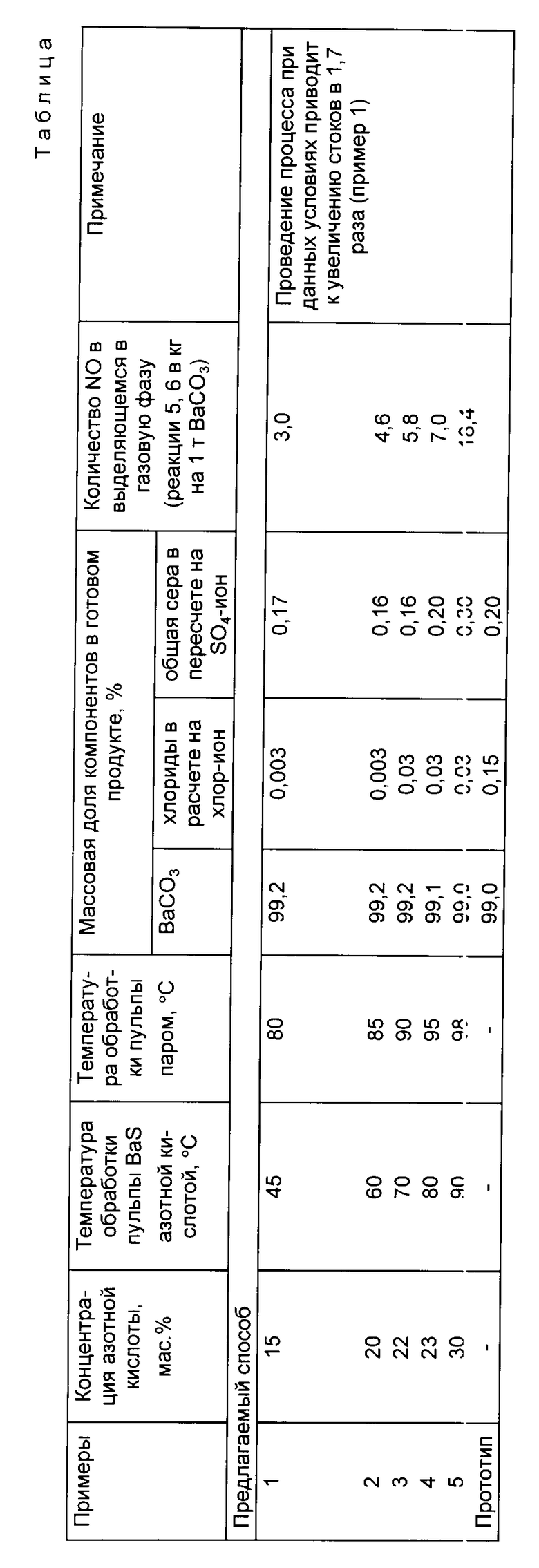
Пример 1. 850 кг/час плава BaS и 3400 кг/ч горячей воды непрерывно поступают в шаровую мельницу на выщелачивание. Полученную суспензию плава сульфида при температуре 60 70oC обрабатывают в реакторе с мешалкой азотной кислотой с массовой долей HNO3 20% в количестве 2550 кг/ч, доводят pH суспензии до 2 3. Суспензию нитрата бария подают в реактор-коагулятор, куда при перемешивании вводят пар в течение 10 минут с доведением температуры до 85 95oC. Далее суспензию нейтрализуют 40%-ым раствором едкого натра в количестве 40 кг/ч, доводя pH суспензии до 8,5, отделяют твердый остаток с влажностью 0,38% в количестве 0,264 кг/ч и выводят из процесса. К полученному раствору нитрата бария, массовая доля Ba(NO3)2 в котором 13,5% добавляют 40% -ый раствор едкого натрия до избыточной щелочности в растворе 10 г/л для очистки от примесей кальция. Осадок гидроксида кальция сгущают, затем фильтруют раствор нитрата бария на листовом гравитационном фильтре, а сгущенный шлам на барабанном вакуум-фильтре.
Полученный раствор нитрата бария в количестве 8375,2 кг/ч обрабатывают раствором карбоната натрия с массовой долей Na2CO3 18% в количестве 2500 кг/ч. Обработку проводят при температуре 80oC в реакторе, снабженном мешалкой и паровым обогревом. Полученную суспензию направляют на барабанный вакуум-фильтр, где отделяют пасту карбоната бария в количестве 1050 кг/ч от маточного раствора.
Далее пасту карбоната бария промывают, сушат в барабанной вращающейся сушилке и гранулируют во вращающейся барабанной печи при температуре 600 - 700oC. После рассева и дробления получают 790 кг/ч гранулированного продукта, в котором:
массовая доля BaCO3 99,2%
массовая доля хлоридов в пересчете на хлор-ион 0,003%
массовая доля общей серы в пересчете на SO4 -ион 0,15%
Сравнительные данные по предлагаемому способу и прототипу приведены в таблице.
Таким образом, по сравнению со способом-прототипом предложенный способ обладает рядом существенных преимуществ: повышается массовая доля основного вещества в продукте, снижается массовая доля хлоридов. Последнее является очень существенным для производителей электровакуумного стекла.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ β ФЕНИЛЭТИЛОВОГО СПИРТА | 1995 |
|
RU2086528C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТА БАРИЯ | 2001 |
|
RU2195428C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ АМИНОБЕНЗОЙНЫХ КИСЛОТ | 1995 |
|
RU2096403C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУСПЕНДИРОВАННОГО УДОБРЕНИЯ | 1992 |
|
RU2046115C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОБЕНЗОЙНЫХ КИСЛОТ | 1995 |
|
RU2110511C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТА БАРИЯ ВЫСОКОЙ ЧИСТОТЫ | 2009 |
|
RU2412906C1 |
| Способ получения водорастворимых солей бария из твердых отходов после выщелачивания плава сульфида бария | 1990 |
|
SU1824379A1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ ОКСИХЛОРИРОВАНИЯ ЭТИЛЕНА В 1,2-ДИХЛОРЭТАН | 1999 |
|
RU2148432C1 |
| НИКЕЛЕВЫЙ КАТАЛИЗАТОР ГИДРИРОВАНИЯ НА НОСИТЕЛЕ И СПОСОБ ПРИГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО НИКЕЛЕВОГО КАТАЛИЗАТОРА ГИДРИРОВАНИЯ НА НОСИТЕЛЕ | 1995 |
|
RU2095136C1 |
| СПОСОБ ПЕРЕРАБОТКИ ДИСТИЛЛЕРНОЙ СУСПЕНЗИИ АММИАЧНО-СОДОВОГО ПРОИЗВОДСТВА | 1993 |
|
RU2071940C1 |
Изобретение относится к получению карбоната бария и может быть использовано в химической промышленности. Сущность изобретения заключается в способе получения карбоната бария, включающем получение барийсодержащего раствора обработкой водной суспензии плава сернистого бария азотной кислотой, разбавленной до массовой доли HNO3 20 - 23% при температуре 60 - 70oC с последующей обработкой полученной суспензии паром до температуры 85 - 95oC, очистку его от примесей, обработку очищенного раствора карбонатом натрия, отделение осадка карбоната бария, его промывку и сушку. Использование изобретения позволит повысить качество целевого продукта за счет повышения массовой доли основного вещества и снижения примесей хлоридов с одновременным упрощением технологической схемы. 1 табл.
Способ получения карбоната бария, включающий получение барийсодержащего раствора, очистку его от примесей, обработку очищенного раствора карбонатом натрия, отделение осадка карбоната бария, его промывку и сушку, отличающийся тем, что барийсодержащий раствор получают обработкой водной суспензии плава сернистого бария азотной кислотой концентрации 20 23 мас. при 60 70oС с последующей обработкой полученной суспензии паром до 85 95oС.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Ахметов Т.Г | |||
| Химия и технология соединений бария | |||
| - М.: Химия, 1974, с.67 и 68 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Позин М.Е | |||
| Технология минеральных солей | |||
| - Л.: Химия, 1970, с.455. | |||

