Изобретение относится к усовершенствованному способу получения эфиров аминобензойных кислот, многие из которых являются биологически активными соединениями и находят широкое применение в медицине.
Известен способ получения эфиров п-аминобензойной кислоты химическим восстановителем соответствующих эфиров п-нитробензойной кислоты в присутствии активного металла, такого как железо или цинк в комбинации с минеральными или органическими кислотами [1-4] Процесс является длительным, трудоемким и отличается низкой селективностью: после окончания восстановления нитросоединения необходимо осадить растворенный металл (например, в виде карбонатов) и дополнительно осветлить реакционную массу для удаления из нее побочных продуктов. В продукте гидрирования, полученном таким способом, присутствуют продукты осмоления исходного нитроэфира, продукты неполного восстановления и другие органические примеси, наличие которых усложняет процесс получения чистого амина.
Для обеспечения более высокой селективности процесса восстановления нитросоединения до соответствующих аминов обычно используют палладиевые катализаторы, высокая активность которых делает процесс более эффективным.
Известен способ получения эфиров аминобензойных кислот гидрированием в ксилоле соответствующих эфиров нитробензойных кислот на палладиевых (1-25% благородного металла на носителе) катализаторах (0,5-3% от массы исходного нитроэфира) [5-7]
Активность используемых катализаторов в [5] составляет 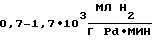 производительность соответственно
производительность соответственно 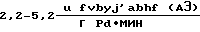 Удельный расход драгметалла при этом составляет 0,5-1,5•10-3 г на 1 г целевого АЭ.
Удельный расход драгметалла при этом составляет 0,5-1,5•10-3 г на 1 г целевого АЭ.
В [6] отсутствуют данные для расчета активности катализаторов и производительности, однако приведен выход АЭ 88% от теор. в виде гидрохлорида и 5,5% в виде основания дополнительным осаждением щелочью из маточника. При этом качество соли оценено только т.пл. (154,5-155,5oС), для АЭ-основания не приведены показатели качества.
В [7] активность используемых катализаторов составляет 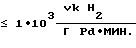 производительность
производительность 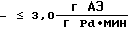 а удельный расход драгметалла - ≥0,8•10-3 г на 1 г АЭ.
а удельный расход драгметалла - ≥0,8•10-3 г на 1 г АЭ.
Основными недостатками известного способа [5-7] являются невысокие активность катализатора и производительность его по целевому АЭ, а также достаточно большой удельный расход драгметалла. Кроме того, при проведении процесса в ксилольном растворе выделение АЭ осуществляют многоступенчато: экстракцией водой продукта гидрирования из органического растворителя, обработкой водного экстракта соляной кислотой с целью выделения соли АЭ (при этом высоки потери продукта из-за высокой растворимости солей аминов в воде) и дополнительным осаждением щелочью из маточника раствора амина-основания.
В [7] предложено, кроме катализатора (5% Pd/C), в реакционную зону вводить осветляющий уголь (до 10-кратного избытка относительно катализатора), то есть одновременно с гидрированием ведут очистку. Катализатор, отделенный после гидрирования, разбавлен углем, что затрудняет его утилизацию.
Известен также способ гидрирования этилового эфира п-нитробензойной кислоты в органическом растворителе (например, толуоле, бензоле) в присутствии 5% Pd/C [8] Данные, позволяющие рассчитать активность катализатора и удельный расход драгметалла, отсутствуют, однако приведенная т.пл. полученного этилового эфира п-аминобензойной кислоты (86-87oC) свидетельствуют о его низком качестве.
Наиболее близким к предлагаемому является способ получения эфиров аминобензойных кислот гидрированием водных растворов соответствующих эфиров нитробензойных кислот, предварительно обработанных углем, в присутствии 1% Pd/C [9] Выход продукта 91% Активность катализатора в данном процессе составляет 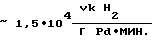 производительность соответственно
производительность соответственно 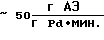 а удельный расход драгметалла -≈4•10-4 г Pd на 1 г АЭ.
а удельный расход драгметалла -≈4•10-4 г Pd на 1 г АЭ.
Недостатками данного способа являются относительно невысокие активность и производительность используемого в процессе катализатора, а также достаточно большой удельный расход драгметалла на единицу целевого продукта.
Цель изобретения снижение удельных затрат драгметалла, необходимых для получения эфиров аминобензойных кислот высокой степени чистоты с одновременным увеличением производительности процесса гидрирования соответствующих эфиров нитробензойных кислот.
Поставленная цель достигается использованием в процессе жидкофазного гидрирования эфиров нитробензойных кислот никелевого катализатора на носителе, модифицированного палладием и содержащего железо, или палладиевого катализатора на носителе, содержащего железо, причем никелевый катализатор содержит 0,07-0,25% никеля, 0,05-0,3% железа от массы носителя и соотношение никель палладий в катализаторе равно 1: 1-0,5, палладиевый катализатор содержит 0,07-0,6% палладия и 0,05-0,55% железа от массы носителя.
Активные металлы наносят на носитель адсорбцией соединений никеля, палладия, железа из их водных растворов, причем адсорбцию солей никеля и железа ведут в щелочной среде (pH 8-12), а палладий наносят на носитель адсорбцией из водного раствора его хлоргидроксокомплексов, полученных при массовом соотношении гидроксид щелочного металла:палладий, равном 0,25-0,65:1.
В качестве носителей для катализаторов используют известные с развитой удельной поверхностью (Sуд.) не менее 250 м2/г.
Исходный нитроэфир (НЭ) может вводиться на стадию гидрирования в виде раствора, эмульсии (суспензии) или плава.
Жидкофазный процесс гидрирования по предлагаемому способу осуществляют с использованием растворителя (разбавителя) или без него предпочтение тому или иному варианту отдается в зависимости от природы исходного НЭ, технологических и аппаратурных возможностей воплощения процесса. При проведении процесса в среде растворителя (разбавителя) предпочтительным является использование воды, что имеет очевидные преимущества по сравнению с органическими растворителями.
Температура и давление водорода не являются лимитирующими факторами в данном процессе (гидрирование НЭ по предлагаемому способу можно вести в широком интервале температуры и давления водорода), однако оптимальный режим гидрирования для каждого конкретного НЭ зависит от используемых катализатора и растворителя (разбавителя).
Выделение целевых АЭ осуществляют как в виде их солей, так и в "основной" форме в зависимости от потребности.
Катализаторы в предлагаемом способе обладают высокой активностью 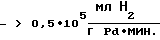 что позволяет снизить удельный расход драгметалла до <1•10-4 г Pd на 1 г АЭ и получить при этом целевые АЭ высокой степени чистоты с производительностью
что позволяет снизить удельный расход драгметалла до <1•10-4 г Pd на 1 г АЭ и получить при этом целевые АЭ высокой степени чистоты с производительностью 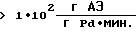
Следует отметить, что предлагаемые катализаторы проявляют высокую активность в различных растворителях, однако они особенно эффективны при проведении процесса в воде или водно-спиртовых средах, где имеют место конкурентные реакции превращения исходного НЭ гидрирование и гидролиз. Использование предлагаемых высокоактивных катализаторов позволяет вести гидрирование с высокой скоростью, при этом реакция гидролиза становится неконкурентноспособной, что ведет к увеличению выхода целевого АЭ и улучшению его качества.
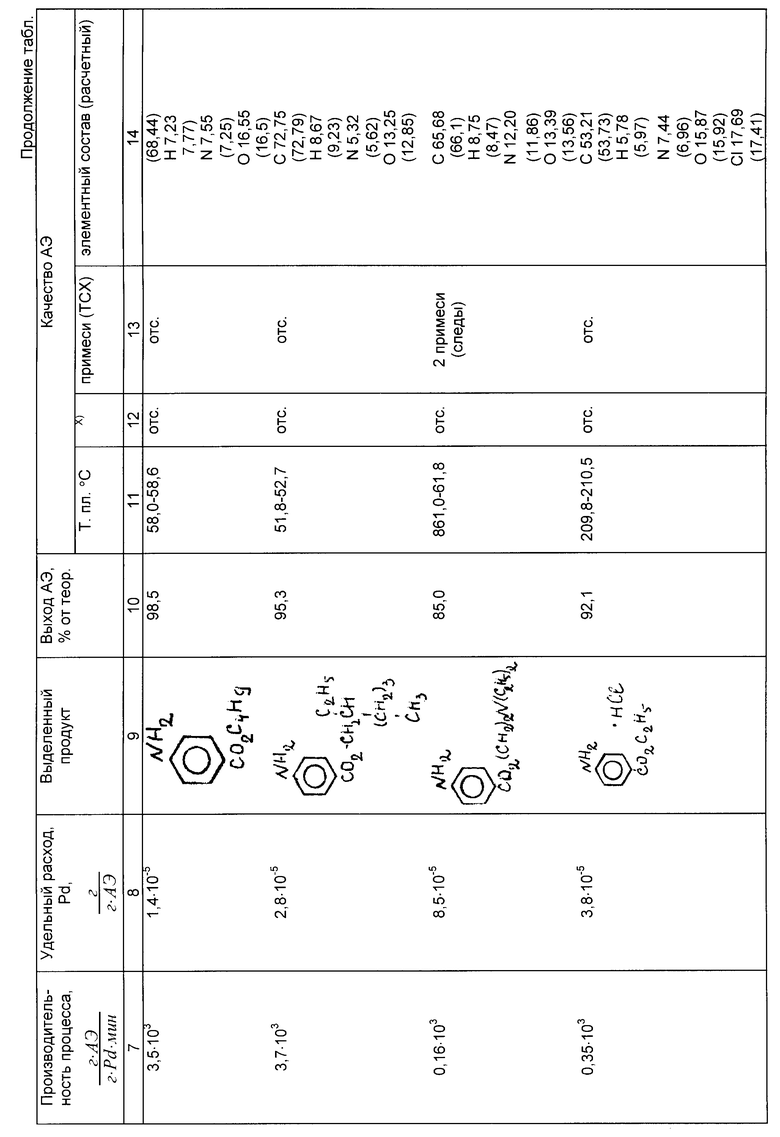
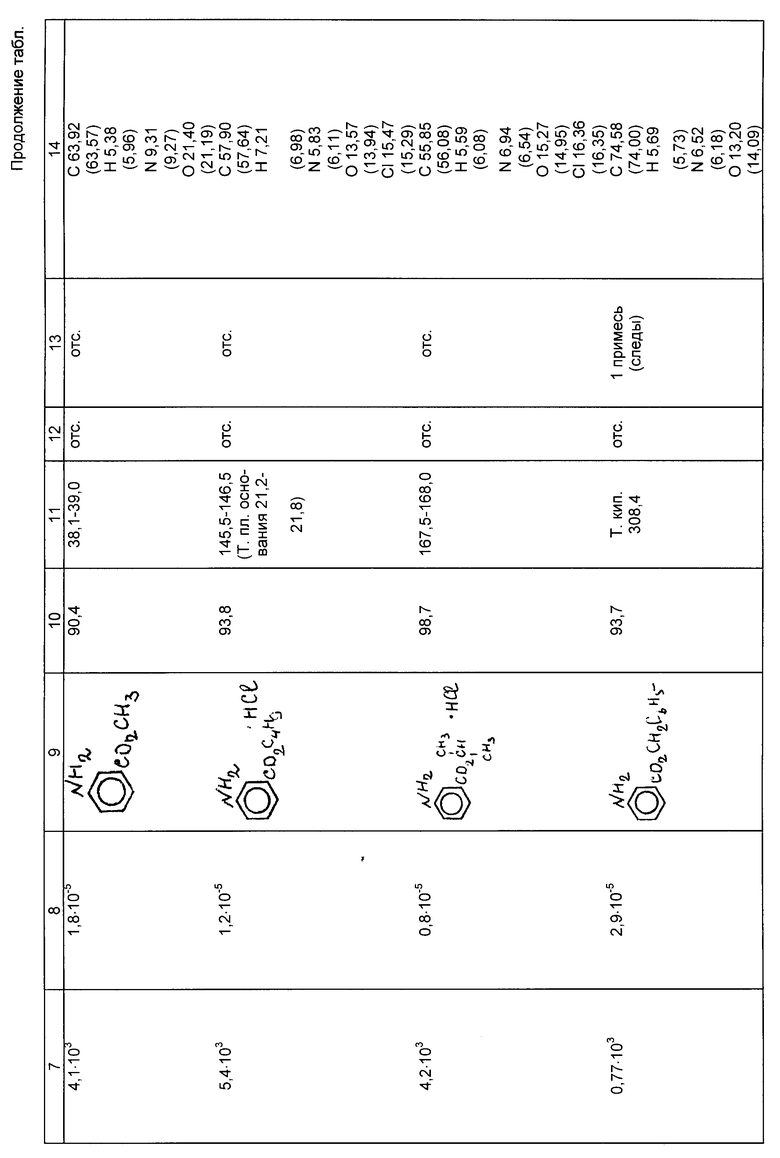
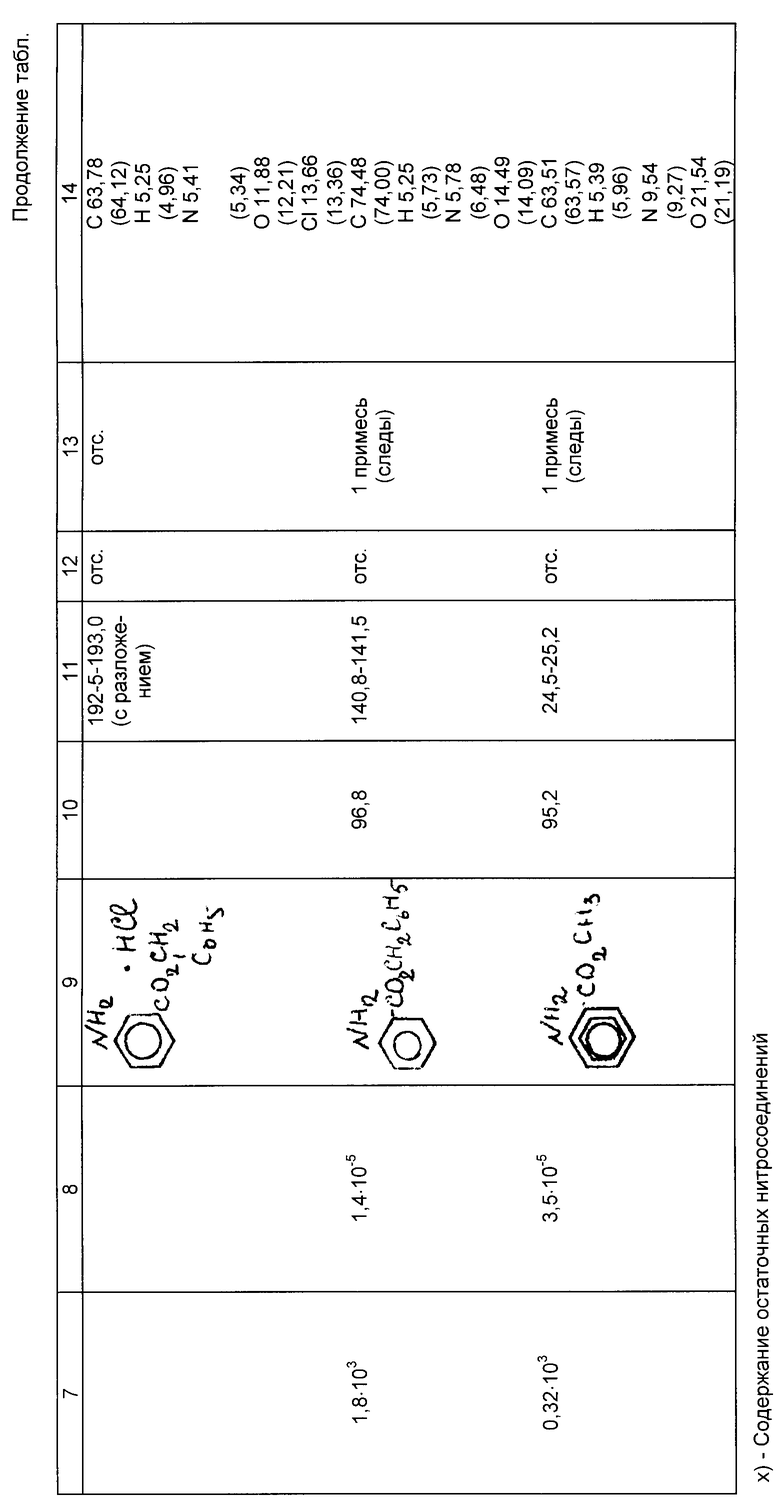
Эфиры аминобензойных кислот, полученные по предлагаемому способу, имеют температуры плавления (кипения), соответствующие литературным данным или превышающие их, по данным полярографического анализа они не содержат остаточных нитросоединений, по данным тонкослойной хроматографии (ТСХ) в них практически отсутствуют примеси.
Предлагаемый способ иллюстрируется следующими примерами.
А. Приготовление катализаторов.
Методика приготовления катализаторов.
В отдельных емкостях готовят растворы А, Б, В.
Раствор А раствор хлоргидроксокомплексов палладия (ХГКPd), полученный взаимодействием хлорида палладия и хлорида щелочного металла в кислой среде с последующим щелочным гидролизом, который ведут дозированным добавлением раствора щелочи до массового отношении гидроксида щелочного металла к палладию, равном 0,25-0,65.
Раствор Б водный раствор соли никеля с концентрацией 0,10-0,26 г•моль/л.
Раствор В водный раствор соли железа с концентрацией 0,01-0,27 г•моль/л.
Носитель суспендируют в нейтральном или щелочном водном растворе, концентрация носителя в суспензии варьируется в пределах 6,5-20 мас. щелочная среда достигается добавлением раствора щелочи (щелочная обработка носителя проводится при синтезе никелевого катализатора, модифицированного палладием).
Носитель, суспендированный в водной среде, обрабатывают в определенной последовательности растворами А, Б, В, варьируя их концентрации и объемы в зависимости от желаемого содержания металлов в катализаторе и условий его синтеза (например, емкости реакторов).
Пример 1а. Катализатор готовят на лабораторной установке, состоящей из круглодонной колбы емкостью 500 мл, снабженной мешалкой, термометром, капельной воронкой, холодильником и обогреваемой с помощью колбонагревателя, а также колбы Бунзена с воронкой для фильтрования под вакуумом.
Предварительно готовят раствор ХГКРd (раствор А). С этой целью в стаканчик емкостью 10 мл загружают 1 мл дистиллированной воды, 0,0014 мл концентрированной соляной кислоты, 0,017 г хлорида палладия (59,5% Pd) и 0,014 г хлорида калия. Смесь нагревают до 65-70oC и перемешивают на магнитной мешалке до полного растворения соли палладия. Полученный раствор охлаждают до 40-50oC и гидролизуют, дозируя медленно, по каплям, 1н. раствор гидроокиси калия до массового отношения KOH:Pd=0,65 с последующей выдержкой в течение 2 ч.
Также предварительно готовят раствор Б, растворяя 0,081 г хлорида никеля (NiCl2•6H2O) в 1,6 мл дистиллированной воды при комнатной температуре и раствор В раствор 0,048 г хлорида железа (FeCl3•6H2O) в 13,5 мл воды.
В колбу загружают при перемешивании 10 г (в расчете на сухой) активного угля АЦБ-0 (Sуд.=630 м2/г) и 100 мл дистиллированной воды. Уголь суспендируют в воде при комнатной температуре, после чего суспензию угля нагревают до 65-70oС и подщелачивают 1н. раствором гидрокиси калия до pH≥10, затем дозируют раствор Б и производят 30-минутную выдержку. Суспензию охлаждают до 30oС, дозируют раствор щелочи для поддержания pH на уровне 9,5, после чего дозируют раствор В с последующей 15-минутной выдержкой. Затем к суспензии дозирую раствор А и выдерживают ее в течение 30 мин.
Подученную таким образом суспензию катализатора фильтруют под вакуумом на воронке Бюхнера, пасту отмывают дистиллированной водой до отсутствия ионов хлора в промывной воде и сушат до остаточной влажности не более 30мас.
Готовый катализатор содержит по данным атомноабсорбционного анализа 0,2мас. никеля, 0,1мас. палладия и 0,18 мас. железа.
Пример 2а. Катализатор готовят на установке, состоящей из эмалированного реактора синтеза емкостью 160 л, снабженного рубашкой для обогрева, мешалкой и холодильником, нутч-фильтра и емкости-дозатора.
В реактор загружают при перемешивании 50-70 л дистиллированной воды и 5 кг (в пересчете на сухой продукт) активного угля ОУА. К суспензии дозируют из емкости-дозатора в течение ≈ 10 мин раствор 94,0 г нитрата железа Fe(NO3)3•6H2O в 1 л воды. Затем дозируют 10% раствор карбоната натрия до pH суспензии 8,5-9,0 и нагревают содержимое реактора до 65-70oC. Затем суспензию подщелачивают 1н. раствором NaOH до pH≥10, после чего дозируют раствор 62 г нитрата никеля Ni(NO3)2•6H2O в 1000 мл воды и выдерживают суспензию с течение 30 мин при температуре 65-70oС. Затем суспензию охлаждают до 50oС и дозируют раствор ХГКPd в течение 10 мин.
Раствор ХГКPd готовят предварительно в емкости с мешалкой растворением при нагревании до 60-70oС 8,5 г хлорида палладия, 5,6 г хлорида натрия в 250 мл дистиллированной воды, подкисленной 0,6 мл концентрированной соляной кислоты, после чего раствор охлаждают до 40-50oС и гидролизуют раствором NaOH до массового отношения NaOH:Pd=0,45 с последующей выдержкой в течение 3 ч.
После дозировки раствора ХГКPd суспензию выдерживают при перемешивании 30-40 мин, затем фильтруют на нутч-фильтре под вакуумом, отмывают пасту катализатора водой и отжимают вакуумом от избыточной влаги.
Готовый катализатор представляет собой пасту (40-50 мас. воды), содержащую по данным атомноабсорбционного анализа (в расчете на массу сухого продукта) 0,25% никеля, 0,12% палладия и 0,3% железа.
Пример 3а. Катализатор готовят на установке, описанной в примере 1а.
Предварительно готовят раствор ХГКPd: 0,0168 г хлорида палладия и 0,010 г хлорида натрия растворяют при 65-70oС (перемешивая на магнитной мешалке) в 1 мл воды, подкисленной 0,0015 мл концентрированной соляной кислоты и после охлаждения раствора до 40-50oС его гидролизуют 1н. раствором NaOH до массового отношения NaOH:Pd=0,55 и выдерживают полученный раствор ХГКPd 5 ч.
В колбе при включенной мешалке суспендируют 10 г мелкодисперсного силигагеля марки "КСК" (Sуд.=325 м2/г) в 100 мл дистиллированной воды и в полученную суспензию при комнатной температуре и нейтральном pH дозируют предварительно приготовленный раствор ХГКPd.
После 30-минутной выдержки в суспензию дозируют раствор 0,045 г хлорида железа (2+) в 10 мл воды, затем 15 мл 0,1% раствора карбоната натрия и 54 мл 10% раствора бикарбоната натрия. Суспензию выдерживают в течение 30 мин (pH ≈ 9,0), затем фильтруют вакуумом и отмывают пасту катализатора на фильтре до отсутствия ионов хлора в промывной воде, после чего подсушивают до остаточной влажности 20-30 мас.
По данным атомноабсорбционного анализа сухой катализатор содержит 0,12 мас. палладия и 0,17 мас. железа.
Пример 4а. Катализатор готовят на установке, описанной в примере 1а.
Предварительно готовят растворы А, Б и В.
Для получения раствора А 0,034 г хлорида палладия и 0,022 г хлорида натрия растворяют в 1 мл дистиллированной воды, подкисленной 0,0025 мл концентрированной соляной кислоты, как описано в примере 1а. После охлаждения раствора до 40-50oС его гидролизуют, приливая медленно 0,5н. раствор NaOH до массового отношения NaOH: Pd=0,25 и выдерживают полученный раствор ХГКPd в течение суток.
20 г (в расчете на сухой) порошкообразного углеродного носителя "Сибунит" (Sуд.=300 м2/г) суспендируют в 200 мл дистиллированной воды. Суспензию носителя нагревают до 65-70oС и с помощью 1н. раствора NaOH доводят ее pH до значения >10. После этого дозируют заранее приготовленный раствор 0,096 г сульфата никеля в 3,0 мл воды и выдерживают горячую суспензию в течение 30 мин. Затем суспензию охлаждают до 50oC и дозируют в нее раствор ХГКPd с последующей выдержкой в течение 25-30 мин, после чего к суспензии дозируют раствор сульфата железа FeSO4•7H2O (0,05 г в 1 мл воды) с последующей 30-минутной выдержкой.
Катализатор отфильтровывают под вакуумом, сушат в вакуумном эксикаторе.
Приготовленный таким образом катализатор содержит 0,07 мас. никеля, 0,07мас. палладия и 0,05мас. железа.
Пример 5а. Катализатор готовят на установке, описанной в примере 1а.
В колбу загружают при перемешивании 10 г (в расчете на сухой) активного угля ОУБ и 100 мл дистиллированной воды. Уголь суспендируют.
Предварительно готовят раствор ХГКPd, растворяя в 3 мл дистиллированной воды, подкисленной 0,006 мл концентрированной соляной кислоты, 0,085 г хлорида палладия и 0,28 г хлорида натрия. Смесь нагревают до полного растворения соли палладия. Гидролиз осуществляют 0,25н. раствором гидроокиси натрия до отношения NaOH:Pd=0,5. После 30-минутной выдержки раствор ХГКPd дозируют при комнатной температуре к суспензии угля и выдерживают ≈ 1 ч.
Суспензию катализатора фильтруют под вакуумом, отмывают пасту на фильтре до отсутствия ионов хлора в промывной воде и подсушивают в эксикаторе до остаточной влажности 20-30%
Полученный катализатор содержит 0,45мас. палладия и 0,25мас. железа (из исходного угля).
Пример 6а. Катализатор готовят на установке, описанной в примере 2а.
В качестве носителя используют активный уголь ОУБ с содержанием железа 0,23мас.
Суспендируют 5 кг (в расчете на сухой) угля ОУБ в 70 л воды. Суспензию подщелачивают при постоянном перемешивании и температуре 50oС до pH≥10, после чего дозируют раствор 62,0 г нитрата никеля Ni(NO3)2•6H2O в 1000 мл воды. После получасовой выдержки дозируют в течение ≈ 10 мин раствор ХГКPd (16,8 г хлорида палладия, 11,0 г хлорида натрия, 1,2 мл концентрированной соляной кислоты в 500 мл воды, гидролиз раствором NaOH до массового отношения NaOH:Pd=0,5).
Готовый катализатор содержит (в расчете на массу сухого продукта) 0,25% никеля, 0,18% палладия и 0,23% железа.
Пример 7а. Катализатор готовят на установке, описанной в примере 1а.
В колбу, где в 100 мл воды, нагретой до 50-60oС, суспендируют 10 г хроматографической окиси алюминия, дозируют 90 мл раствора хлорида железа (c= 0,01 г-моль/л), затем 34 мл 1% раствора карбоната натрия и 11 мл 10% раствора бикарбоната натрия. После выдержки в течение 30-40 мин к суспензии дозируют раствор ХГКPd (3 мл воды, 0,1 г хлорида палладия, 0,066 г хлорида натрия, 0,0075 мл соляной кислоты, гидролиз до массового отношения NaOH:Pd=0,4, выдержка 4 ч).
По данным атомноабсорбционного анализа сухой катализатор содержит 0,6мас. палладия и 0,55мас. железа.
Пример 8а. Катализатор готовят на установке, описанной в примере 1а.
В суспензию 10 г "сибунита" в 100 мл воды при комнатной температуре дозируют (при постоянном перемешивании) раствор ХГКPd (1 мл воды, 0,012 г хлорида палладия, 0,005 г хлорида калия, 0,001 мл соляной кислоты, гидролиз 0,25н. раствором KOH до отношения KOH:Pd=0,6, выдержка 10 ч).
После дозировки раствора ХГКPd и получасовой выдержки дозируют раствор 0,024 г хлорида железа в 1 мл воды, затем 0,25н. раствор KOH до pH ≈ 9,0 с последующей выдержкой при перемешивании.
По данным атомноабсорбционного анализа сухой катализатор содержит 0,07мас. палладия и 0,05мас. железа.
Б. Получение эфиров аминобензойных кислот.
Для получения эфиров аминобензойных кислот используют три типа установок.
Тип I лабораторная кинетическая установка гидрирования при постоянных температуре и давлении водорода.
Реактор представляет собой стеклянную ампулу объемом 45-55 см3, герметизируемую в стальном термостатированном кожухе встряхивающего устройства, соединенном через систему вентилей с баллонами водорода и азота, буферной емкостью и измерительной системой. Обогрев системы производят с помощью термостата. Теплоносители силиконовое масло, глицерин или вода, температура регулируется контактным термопметром. Наличие буферной емкости в системе обеспечивает ведение процесса практически при постоянном давлении.
После окончания процесса водород замещают в системе азотом, катализатор отделяют от реакционной массы "горячей" вакуумной фильтрацией в приемник (охлаждаемый при необходимости).
Тип II опытная установка периодического действия, включающая реактор гидрирования, друк-фильтр для отделения катализатора, кристаллизатор (с рубашкой для охлаждения), в котором можно при необходимости проводить нейтрализацию солей АЭ с целью выделения АЭ в основной форме, нутч-фильтр для отделения пасты эфиров аминобензойных кислот от маточника.
Реактор аппарат объемом 25 л с турбинной мешалкой (п=2800 об/мин) снабженный рубашкой для обогрева, встроенным змеевиком для охлаждения, загрузочным люком и нижим штуцером для выпуска продукта.
Гидрирование ведут при постоянном давлении водорода и в политермическом режиме. После прекращения поглощения водорода и выдержки заменяют водород на азот и под давлением 3-4 атм реакционную массу переливают через нагретый друк-фильтр (объем 100 л, п=43 об/мин), где можно проводить и нейтрализацию. Кристаллизатор охлаждают до нужной температуры подачей в рубашку хладогента. Выпавший осадок отфильтровывают и промывают на нутч-фильтре.
Тип III опытно-промышленная установка периодического действия, состоящая из суспензатора, реактора гидрирования, мешочного фильтра, кристаллизатора, фильтр-пресса.
Суспензатор стальной аппарат с мешалкой (п=170 об/мин), емкостью 3,2 м3, снабженный люком для загрузки НЭ и катализатора. Приготовленный раствор (эмульсию, суспензию) центробежным насосом перекачивают в реактор гидрирования, представляющий собой вертикальный аппарат объемом 2,5 м3 с циркуляционной трубой, нижним герметичным электроприводом и мешалкой (п=2800 об/мин). Реактор снабжен рубашкой для обогрева паром и охлаждения оборотной водой.
Гидрирование проводят в политермическом режиме при постоянном давлении водорода. После окончания процесса и выдержки из реактора вытесняют водород путем многократного набора азота давлением 5 атм и сброса его до 3-4 атм, катализат передавливают через обогретый фильтр в кристаллизатор. Кристаллизатор стальной аппарат с мешалкой (п=110 об/мин) емкостью 10м3. Охлажденная рассолом суспензия поступает на автоматический фильтр-пресс типа ФПАКМ с механизированной выгрузкой осадка.
Пример 1б. Гидрирование метилового эфира п-НБК ведут на лабораторной установке типа I.
Загрузка: 12 мл водного метанола (H2O:CH3OH=95:5 об.), 3 г НЭ, 0,04 г катализатора, приготовленного по примеру 1а (Ni 0,2% Pd 0,1% Fe 0,18%).
Процесс поглощения водорода при постоянных температуре (100oC) и давлении (10 атм) завершается за 10 мин. После "горячей" фильтрации для отделения катализатора и охлаждения фильтрата в осадок выпадают бесцветные кристаллы метиллового эфира п-АБК с выходом 94,3% теор.
По данным полярографического анализа и ТСХ в продукте гидрирования отсутствуют исходное нитросоединение и примеси, Т.пл. 113,8-114,5oС. Элементный состав, C 64,03; H 5,81; N 9,35; O 20,81 (Рассчитано: C 63,57; H 5,96; N 9,27; O 21,19).
Активность катализатора в процессе 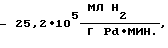 производительность процесса
производительность процесса  удельный расход палладия
удельный расход палладия 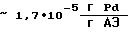
Пример 2б. Гидрирование гидрохлорида диэтиламиноэтилового эфира (ДЭАЭЭ) п-НБК ведут на установке типа II.
Загрузка: 15 л водного раствора (C ≈3,5мас.) NaOH, 4,1 кг гидрохлорида ДЭАЭЭ п-НБК, 36 г катализатора, содержащего Pd 0,45% и Fe - 0,25% (по примеру 5а).
При постоянном перемешивании под постоянным давлением водорода 20 атм содержимое автоклава нагревают подачей теплоносителя до 50oС, далее температура повышается до 85oС за счет тепла реакции. Через 20 мин поглощение водорода заканчивается. После выдержки и отделения катализатора фильтрацией катализат передавливают в кристаллизатор, где под азотной "подушкой" его охлаждают до 5oС. Затем проводят нейтрализацию раствора соли ДЭАЭЭ п-АБК 10% водным раствором NaOH до pH>8,0 (при постоянном охлаждении и перемешивании). После 30-минутной выдержки фильтрацией отделяют белый осадок АЭ. Выход 92,5% Т. пл. 61,8-62,6oС. По данным полярографического анализа в продукте гидрирования отсутствует исходный НЭ, по данным ТСХ отсутствуют примеси. Элементный состав, C 66,5; H 8,32; N 11,52; O 13,65 (Рассчитано: C 66,11; H 8,47; N 11,86; O 13,56). Полученный амин-основание можно перевести в соль любым известным способом, например при подкислении его раствора в изопропаноле концентрированной HCl до pH<5,0 получены белые кристаллы гидрохлорида ДЭАЭЭ п-АБК с Т.пл. 156,2-157oC (без очистки).
Активность катализатора в процессе 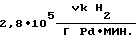 производительность процесса
производительность процесса 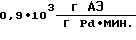 удельный расход палладия
удельный расход палладия 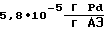
Пример 3б. Гидрирование ведут на опытно-промышленной установке типа III.
В реактор загружают 1600 л воды, 353 кг пасты этилового эфира п-НБК (с влагой 15мас.) и 18 кг пасты (влага 50мас.) катализатора, содержащего Ni - 0,25% Pd 0,18% и Fe 0,23% (по примеру 6а).
Гидрирование ведут при давлении водорода 15 атм. Начальная температура 60oС за счет тепла реакции к концу процесса поглощения водорода (15 мин) повышается до 90oС, после выдержки катализат, подогретый до 100oС, передавливается через фильтр для отделения катализатора в кристаллизатор с подогретой до 50oС сульфитной водой. Кристаллизатор постепенно (при постоянном перемешивании суспензии) охлаждают до 5-7oС, затем отфильтровывают белый кристаллический осадок этилового эфира п-АБК с выходом 98,4% от теор. В продукте гидрирования по данным полярографического анализа отсутствуют остаточные нитросоединения (100% конверсия исходного НЭ), по данным ТСХ ("силуфол", хлороформ: этанол= 100: 1) отсутствуют примеси, Т.пл. 91,6-92,2oС. Элементый состав, C 66,83; H 6,41; N 8,02; O 18,75 (Рассчитано: C 65,45; H 6,67; N 8,48; O 19,39).
Активность катализатора в процессе 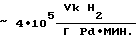 производительность процесса
производительность процесса  удельный расход палладия
удельный расход палладия 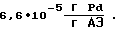
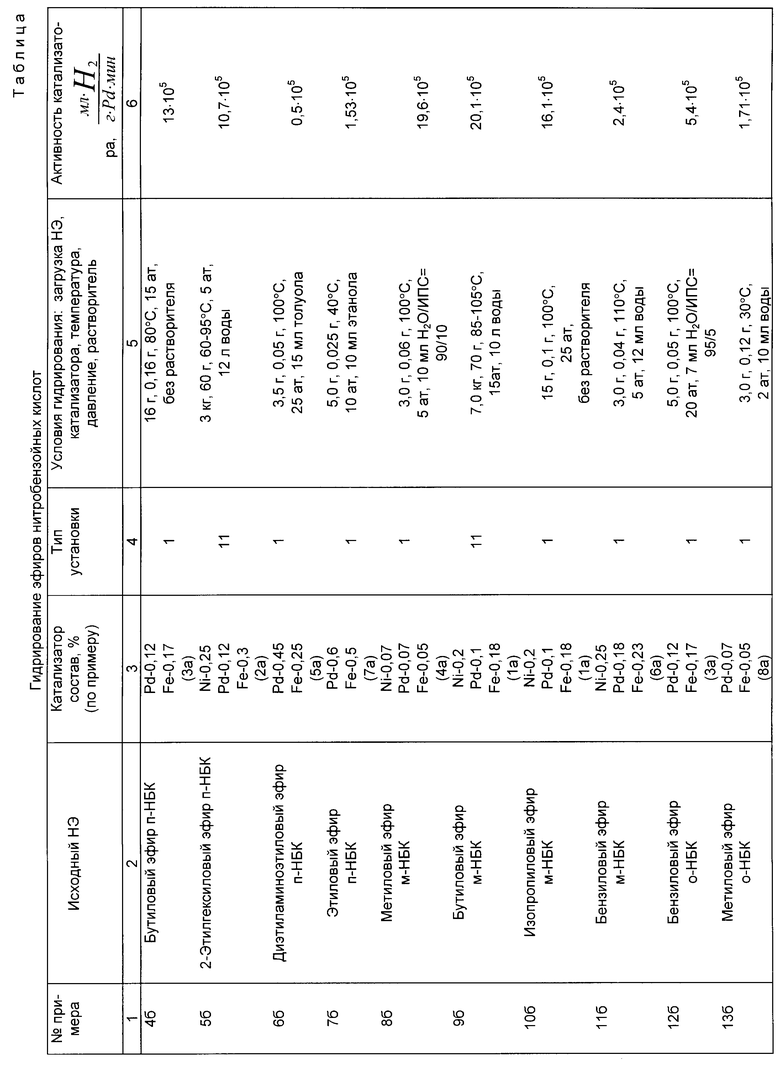
Примеры 4б-13б. Условия и результаты гидрирования эфиров нитробензойных кислот различного строения по предлагаемому способу представлены в таблице.
Предлагаемый способ получения эфиров аминобензойных кислот имеет следующие преимущества по сравнению с прототипом:
позволяет значительно сократить удельные затраты драгметалла;
позволяет получить целевые продукты высокой степени чистоты без дополнительной очистки;
увеличивает (на несколько порядков) производительность процесса гидрирования.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АМИНОБЕНЗОЙНЫХ КИСЛОТ | 1995 |
|
RU2110511C1 |
| НИКЕЛЕВЫЙ КАТАЛИЗАТОР ГИДРИРОВАНИЯ НА НОСИТЕЛЕ И СПОСОБ ПРИГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО НИКЕЛЕВОГО КАТАЛИЗАТОРА ГИДРИРОВАНИЯ НА НОСИТЕЛЕ | 1995 |
|
RU2095136C1 |
| СПОСОБ ПОЛУЧЕНИЯ β ФЕНИЛЭТИЛОВОГО СПИРТА | 1995 |
|
RU2086528C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЯНТАРНОЙ КИСЛОТЫ ИЛИ ЕЕ СОЛЕЙ | 1997 |
|
RU2129540C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2`, 4`, 4-ТРИАМИНОБЕНЗАНИЛИДА | 1992 |
|
RU2041200C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХЛОРИДА β -ДИЭТИЛАМИНОЭТИЛОВОГО ЭФИРА П-АМИНОБЕНЗОЙНОЙ КИСЛОТЫ | 1994 |
|
RU2083557C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОМЕРОВ ФТАЛЕВЫХ КИСЛОТ С ВЫСОКОЙ СТЕПЕНЬЮ ЧИСТОТЫ | 1993 |
|
RU2047595C1 |
| СПОСОБ ОЧИСТКИ СТОЧНЫХ ВОД ОТ РАСТВОРИМЫХ СОЕДИНЕНИЙ РТУТИ | 1997 |
|
RU2114065C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОМЕРОВ БЕНЗОЛДИКАРБОНОВЫХ КИСЛОТ С ВЫСОКОЙ СТЕПЕНЬЮ ОЧИСТКИ | 1993 |
|
RU2047594C1 |
| СПОСОБ РЕГЕНЕРАЦИИ КАТАЛИЗАТОРА ОКИСЛЕНИЯ АЛКИЛАРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 1998 |
|
RU2155098C2 |
Изобретение относится к усовершенствованному способу получения эфиров аминобензойных кислот, многие из которых являются биологически активными соединениями и находят широкое применение в медицине. Усовершенствование состоит в том, что процесс жидкофазного гидрирования эфиров нитробензойных кислот ведут в присутствии нанесенных на носитель палладийсодержащих катализаторов: никелевого катализатора, модифицированного палладием и содержащего железо, или палладиевого катализатора, содержащего железо. При этом никелевый катализатор содержит 0,07-0,25% никеля, 0,05-0,3% железа от массы носителя и соотношение никель - палладий в катализаторе равно 1:1-0,5, палладиевый катализатор содержит 0,07-0,6% палладия и 0,05-0,55% железа от массы носителя. Палладий в катализаторе нанесен на носитель адсорбцией из водного раствора его хлоргидроксокомплексов, полученных при массовом соотношении гидроксид щелочного металла - палладий, равном 0,25-0,65:1. Изобретение позволяет значительно сократить удельные затраты драгметалла, получить целевые продукты высокой степени чистоты без дополнительной очистки и увеличить (на несколько порядков) производительность процесса гидрирования эфиров нитробензойных кислот. 3 з.п. ф-лы, 1 табл.

