Изобретение относится к оксазолидиноновым производным, имеющим замещенную диазиновую часть, связанную с N-арильным кольцом. Эти соединения обладают противомикробной активностью и являются эффективными против ряда патогенов человека и животных, включая полирезистентные стафилококки и стрептококки, а также анаэробные организмы, такие как бактероиды и клостридии, и кислотоустойчивые организмы, такие как Mycobacterium tuberculosis и Mycobacterium avium. Эти соединения являются особенно эффективными против последних из указанных микроорганизмов, которые, как известно, ответственны за инфицирование индивидуумов СПИДом.
В заявке РСТ/US 89/03548 раскрываются 5'индолинил-5β-амидометилоксазолидиноны; 3-(замещенный у конденсированного ядра) фенил-5β-амидометилоксазолидиноны; и 3-(азотзамещенный)фенил-5β-амидометилоксазолидиноны, которые могут быть использованы в качестве антибактериальных агентов.
Различные оксазолидиноны описаны также и в других работах, включая, патент США N 5801600, патент США N 4921869, Gregoty W.A. И др. J. Med. Chem., 32, 1673-81 (1989); Wang C., и др. Tetrahedron, 45, 1323-26 (1989); и Brittelli и др. J. Med. Chem., 35 (1992).
В публикации Европатента N 352781 раскрываются фенил- и пиридил-замещенные фенилоксазолидиноны.
В публикации Европатента N 316594 раскрываются 3-замещенные стирилоксазолидиноны.
В публикации Европатента N312000 раскрываются фенилметил- и пиридинилметил-замещенные фенилоксазолидиноны.
В одном из своих вариантов настоящее изобретение относится к соединению структурной Формулы I: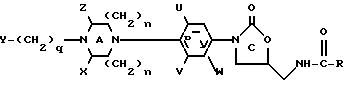
или к их фармацевтически приемлемым солям,
где Y представляет собой:
a) водород;
b) -C1-6-алкил или -арил;
c) -OH, -O-C1-6-алкил, -O-винил, -O-фенил, -O-C(O)-C1-6-алкил, -O-C(O)-фенил (фенил может быть замещен 1-3 группами, выбранными из F, Cl, -OCH3, -OH, NH2 или C1-4-алкила); или -O-C(O)-O-CH3;
d) -S-C1-6-алкил;
e) -SO2-C1-6-алкил, -SO2-N(R3)2 (где R3 независимо представляет собой водород, C1-4-алкил, или фенил, который может быть замещен 1-3 группами, выбранными из F, Cl, OCH3, OH, NH2, или -C1-4-алкила);
f) -C(O)-C1-6-алкил, -C(O)-O-C1-6-алкил, -C(O)-N(R3)2, -C(O)-CH(R4)N(R3)2, или -C(O)-CH(R4)NH-C(NH)-NH2 (где R4 представляет собой боковую цепь аминокислоты);
g) -N(R3)2, -N(CH2)m (где m = 2-6 и образует циклическую структуру вместе с атомом азота и один или несколько атомов углерода могут быть замещены S, O, или NR3), или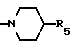 ,
,
(где R5 представляет собой OH, OCH3, CH2OH, CH2OCH3, CO2CH3 или CO2C2H5);
h) -C(CH3)=N-OR;
i) 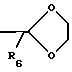 (где R6 является CH3 или водородом);
(где R6 является CH3 или водородом);
j) 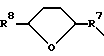 (где R7 является CH2 или C(O), а R8 является -H или = O);
(где R7 является CH2 или C(O), а R8 является -H или = O);
k) 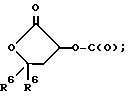 ;
;
l) 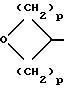 (где p=1 или 2)
(где p=1 или 2)
m) 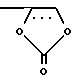 ;
;
n) 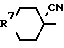 (где R7 представляет собой O, S, S(O), SO2, CH2, NH, NCH3, NC2H5, NCHO, NCOCH3 или NCO2CH3);
(где R7 представляет собой O, S, S(O), SO2, CH2, NH, NCH3, NC2H5, NCHO, NCOCH3 или NCO2CH3);
причем, в каждом случае указанные C1-6-алкил может быть замещен одним или несколькими заместителями, выбранными из F, Cl, Br, I, OR1, CO2R1, CN, SR1 или R1 (где R1 представляет собой водород или C1-4-алкил);
X и Z независимо представляют собой C1-6-алкил, C3-12-циклоалкил или водород; либо X и Z, взятые вместе образуют мостиковую C0-3-группу; однако предпочтительно, если X и Z являются водородом;
U, V и W независимо представляют собой C1-6-алкил, F, Cl, Br, водород или C1-6-алкил, замещенный одним или несколькими атомами F, Cl, Br или I, а предпочтительно, если U и V являются фтором, а W является водородом;
R представляет собой водород; C1-12-алкил; C3-12-циклоалкил; C1-6-алкокси; C1-6-алкил, замещенный одним или несколькими заместителями, выбранными из F, Cl, Br, I или OH;
q равно 0-4 включительно.
В формуле I предпочтительно, если U и V являются фтором, а W является водородом; либо U является фтором, а V и W - водородом. Y предпочтительно выбирают из группы, включающей в себя: H, метил, этил, изопропил, трет-бутил, бензил, фенил, пиридил, ацетил, дифтороацетил, гидроксиацетил, бензоил, метоксикарбонил, этоксикарбонил, 2-хлороэтоксикарбонил, 2-хлороэтоксикарбонил, 2-гидроксиэтоксикарбонил, 2-бензолоксиэтоксикарбонил, 2-метоксиэтоксикарбонил, 2,2,2-трифтороэтоксикарбонил, цианометил, 2-цианоэтил, карбометоксиметил, 2-карбометоксиэтил, 2-фтороэтоксикарбонил, бензилоксикарбонил, трет-бутоксикарбонил, метилсульфонил, фенилсульфонил или паратолуолсульфонил; а более предпочтительно, если Y является метоксикарбонилом или цианометилом. Предпочтительно также, если R является метилом, H, метокси или CHCl2, а n=1. Кроме того, предпочтительно, чтобы соединения формулы I представляли собой оптически чистые энантиомеры, имеющие S-конфигурацию у C5 оксазолидинонового кольца.
Предпочтительными соединениями настоящего изобретения являются следующие:
(a) Метиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты;
(b) Этиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2- фторофенил)-1-пиперазинкарбоновой кислоты;
(c) Метиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)фенил)-1-пиперазинкарбоновой кислоты;
(d) N-((2-оксо-3-(4-(4-(фенилкарбонил)-1-пиперазинил)фенил)-5-оксазолидинил)метил) ацетамид;
(e) N-((3-(4-(3-фторо-4-(4-(2-цианоэтил)-1-пиперазинил))-фенил)-2-оксо-5-оксазолидинил) метил)-ацетамид;
(f) N-((3-(4-(3-фторо-4-(4-(2-гидроксиэтил)карбонил-1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)-ацетамид;
(g) N-((3-(4-(3-фторо-4-((фенилкарбонил)-1-пиперазинил))-фенил)2-оксо-5-оксазолидинил)метил)-ацетамид;
(h) 2-метоксиэтиловый сложный эфир 4-[4-[5-[(ацетиламино)метил]-2-оксо-3-оксазолинидинил]-2-фторофенил]-1-пиперазинкарбоновой кислоты;
(i) 4-[4-[5(ацетиламино)метил] -2-оксо-3-оксазолидинил]-2-фторофенил]-1-пиперазинацетонитрил;
(j) (+/-)-N-[[3-[4-[4-] 4-(1,4-Дизоксопентил)-1-пиперазинил]-3-фторофенил]-2-оксо-5-оксазолидинил]метил)-ацетамид;
(k) (S)-N-[[3-[3-фторо-4-[4-(2-метоксиэтил)-1-пиперазинил]-фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(l) (S)-N-[[3-[3,5-дифторо-4-[4-(2-метоксиэтил)-1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Более предпочтительными являются соединения:
(a) 4-[4-[5-(ацетиламино)метил]-2-оксо-3-оксазолидинил]-2-фторофенил]-1-пиперазининкарбоновой кислоты метиловый сложный эфир;
(i) 4-[4-[5-(ацетиламино)метил]-2-оксо-3-оксазолидинил]-2-фторофенил]-1-пиперазинацетонирил.
В другом своем варианте настоящее изобретение относится к способу лечения микробных инфекций у теплокровных животных путем введения теплокровному животному, нуждающемуся в таком лечении, эффективного количества соединения формулы I, описанного выше. Предпочтительно, если указанное соединение вводят в количестве от около 0,1 до около 100 мг/кг веса тела в день, а более предпочтительно от около 3,0 до около 50 мг/кг веса тела в день.
Изобретение относится к диазинилоксазолидинонам структурной формулы I, представленной выше. Указанные соединения обладают противомикробной активностью и являются эффективными против ряда патогенов человека и животных, включая полирезистентные стафилококки и стрептококки, а также анаэробные организмы, такие как бактероиды и клостридии, и кислотоустойчивые организмы, такие как Micobacterium tuberculosis и Micobacterium avium.
Используемое в настоящем описании определение "C1-6 или C1-12-алкил" означает метил, этил, пропил, бутил, пентил, гексил и т.п., а также из изомерные формы.
"Циклоалкил" означает C3-12-циклопропил, -циклобутил, -циклопентил, -циклогексил, и т.п., а также их изомерные формы.
"Алкокси" означает такие группы, как метокси, этилокси, бутилокси, и т. п. , в которых 1-6 атомов углерода связаны с атомом кислорода, а также их изомерные формы. Кроме того, в некоторых случаях группы, определенные в настоящем описании как "алкоксикарбонил", в номенклатуре химических соединений обозначаются сложными алкилэфирами (например метоксикарбонил и метиловый сложный эфир).
"Арил" означает фенильную, пиридильную или нафтильную часть, необязательно замещенную одним или несколькими заместителями, выбранными из F, Cl, Br, I, OR1, CO2R1, CN, SR1, или R1 (где R1 является водородом или C1-4 алкилом).
Термин "фармацевтически приемлемые соли" означает соли, которые могут быть использованы для введения соединений настоящего изобретения и которые представляют собой гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат, ацетат, пропионат, лактат, мезилат, малеат, малат, сукцинат, тартрат, цитрат, 2-гидроксиэтилсульфонат, фумарат, и т.п. Эти соли могут присутствовать в виде гидратов.
Кольцо A может иметь 6-8 атомов, а более крупные кольца могут содержать два или три атома углерода между атомами азота, например: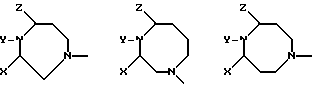
В случае кольца более крупных размеров это кольцо может иметь внутренний мостик и образовывать бициклическую систему, как, например, показано ниже: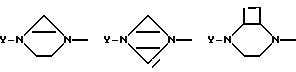
Если размер кольца составляет 6 атомов, то это кольцо может быть необязательно замещенным в положениях X и Z алкильными группами, циклоалкильными группами, фторо-группами или мостиковыми алкильными группами, как, например, показано ниже: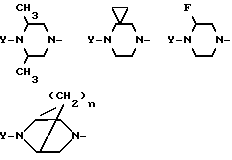
В дополнение к вышеуказанным примерам ниже приводится пример альтернативной бициклической системы: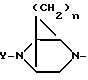
Кольцо B может быть незамещенным или замещенным одним или несколькими атомами галогена, например фтором, хлором, или бромом. Так, например, группы U, V, и W на кольце B могут в различных моделях замещения независимо представлять собой либо атомы водорода, либо атомы галогена.
Группа Y у атома азота кольца A может быть введена стандартными методами (описанными ниже) с использованием коммерчески доступных реагентов. Предпочтительно, если Y выбирают из группы, включающей в себя H, метил, этил, изопропил, т-бутил, бензил, фенил, пиридил, ацетил, дифтороацетил, гидроксиацетил, бензоил, метоксикарбонил, этоксикарбонил, 2-хлороэтоксикарбонил, 2-гидроксиэтоксикарбонил, 2-бензолоксиэтоксикарбонил, 2-метоксиэтоксикарбонил, 2,2,2-трифтороэтоксикарбонил, цианометил, 2-цианоэтил, карбометоксиметил, 2-карбометоксиэтил, 2-фтороэтоксикарбонил, бензилоксикарбонил, третбутоксикарбонил, метилсульфонил, фенилсульфонил или паратолуолсульфонил; а более предпочтительно, если Y представляет собой метоксикарбонил или цианометил.
R-заместитель является предпочтительно метилом, но он может также представлять собой H, метокси или CHCl2.
Наиболее предпочтительные соединения могут быть получены в виде оптически чистых энантиомеров, имеющих (S)-конфигурацию у C5 оксазолидинового кольца.
Оптически чистый материал может быть получен одним из методов асимметрического синтеза либо альтернативным методом разделения рацемической смеси путем селективной кристаллизации солевой формы соединения; например, оптически чистый амин может быть получен исходя из промежуточного амина 12 (как описано в примере 1 и показано на схеме 1) с использованием соответствующей оптически активной кислоты, такой как дибензоилтартрат или 10-камфосульфоновая кислота, с последующей обработкой основанием.
Оптически чистый материал может быть получен и другим способом (например как описано в схемах). Путем обработки коммерчески доступного 3-фторофенилизоцианата коммерчески доступным (R)-глицидилбутиратом в условиях Herweh и Kauffmann (Tetrahedron Letters, 1871, 809) может быть получен соответствующий оптически чистый оксазолидинон с требуемой (S)-конфигурацией в 5-положении оксазолидинонового кольца. После удаления бутиратной группы путем обработки карбонатом калия в метаноле или метоксидом натрия в метаноле получают соответствующий спирт, который может быть затем дериватизирован стандартными методами, например, путем мезилирования с последующим замещением с использованием азида натрия, в результате получают азидометилоксазолидинон. После восстановления азида путем гидрогенизации с последующим ацилированием полученного амина посредством обработки уксусным ангидридом и пиридином получают целевой оптически активный ацетиламинометилоксазолидинон. Что касается указанного ацетиламинометилоксазолидинона, то в данном случае необходимо образование пиперазиновой части. Нитрование фторооксазолидинонового производного дает преимущественно нитро-группу в пара-положении у атома азота оксазолидинонового кольца и в орто-положении кольцевого атома фтора. В результате восстановления нитро-группы путем гидрогенизации получают соответствующее анилиновое производное и после его обработки гидрохлоридом бис-(2-хлороэтил)амина в присутствии карбоната калия в кипящем диглиме получают оптически активное пиперазиновое производное N-[3-[4-[3-фторо-4-(1-пиперазинил)]фенил-2-оксо-5-оксазолидинил]метил]-ацетамид (22), которое может быть затем использовано для получения некоторых соединений, примеры которого раскрываются в настоящем описании.
Эти соединения могут быть использованы для лечения микробных инфекций у человека и других теплокровных животных путем парентерального и перорального введения указанных соединений человеку или животным. Из соединений формулы I наиболее активными, а поэтому наиболее активными и предпочтительными являются метиловый сложный эфир 4-[4-[5-(ацетиламино)метил-2-оксо-3-оксазолидинил] -2-фторофенил] -1-пиперазинкарбоновой кислоты (23) и 4-[4-[5-(ацетиламино)метил]-2-оксо-3-оксазолидинил]-2-фторофенил]-1- пиперазинацетонитрил. В указанных соединениях общей формулы I кольцо A является пиперазиновой частью.
Фармацевтические композиции настоящего изобретения могут быть получены путем смешивания соединений aормулы I настоящего изобретения с твердым или жидким фармацевтически приемлемым носителем и необязательно с фармацевтически приемлемыми адъювантами и наполнителями при помощи обычно используемых стандартных методик. примерами твердых композиций являются порошки, таблетки, диспергируемые гранулы, капсулы, облатки и суппозитории. Твердый носитель представляет собой по крайней мере одно вещество, которое может также служить в качестве разбавителя, ароматизирующего агента, растворителя, смазывающего агента, суспендирующего агента, связующего вещества, дезинтегрирующего агента и инкапсулирующего агента. примерами инертных твердых носителей являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, целлюлозные материалы, низкоплавкий воск, масло-какао и т. п. Примерами жидких композиций являются растворы, суспензии, и эмульсии. Эти композиции могут быть, например, получены в виде растворов соединений настоящего изобретения в воде или в системах растворителей, таких как вода-пропиленгликоль и вода-полиэтиленгликоль, необязательно содержащих обычно используемые окрашивающие агенты, ароматизирующие агенты, стабилизаторы и загустители.
Фармацевтические композиции изготавливаются предпочтительно с использованием стандартных методик в виде единичной дозированной формы, содержащей эффективное или подходящие количества активного ингредиента, т. е. соединения настоящего изобретения формулы I.
Количество активного ингредиента, т. е. соединения настоящего изобретения формулы I, в фармацевтической композиции или ее унифицированной форме может варьироваться в широком диапазоне в зависимости от конкретного применения данного соединения, его эффективности и нужной концентрации. В основном количество активного ингредиента составляет от 0,5 до 90% по весу композиции.
При терапевтическом использовании в целях лечения или борьбы против бактериальных инфекций у теплокровных животных соединения настоящего изобретения или их фармацевтические композиции могут быть введены перорально и/или парентерально в дозе, обеспечивающей получение и поддержание соответствующих концентраций (т.е., количества) или уровней активного соединения в крови теплокровного животного, подвергаемого лечению, которые являются эффективными против бактериальной инфекции. В основном такие антибактериально эффективные дозы активного ингредиента составляют от около 0,1 до около 100, а более предпочтительно от около 3,0 до 50 мг на 1 кг веса тела в день. При этом следует отметить, что указанные дозы могут варьироваться в зависимости от потребностей пациента, тяжести бактериальной инфекции и конкретно используемого соединения. Следует также отметить, что для более быстрого достижения желаемого уровня активного ингредиента в крови пациента первоначально вводимая доза может превышать верхний предел вышеуказанного диапазона; либо эта первоначально вводимая доза может быть меньше оптимального значения, но при этом суточная доза может постепенно возрастать в течение курса лечения в зависимости от конкретной ситуации. Если требуется, суточная доза может быть разделена на несколько доз, например, для введения 2-4 раза в день.
Соединения настоящего изобретения формулы I могут быть введены парентерально, т.е., путем инъекций, например путем внутривенных инъекций, или каким-либо другим способом парентерального введения. Фармацевтические композиции для парентерального введения содержат в основном фармацевтически приемлемое количество соединения формулы I в виде растворимой соли (кислотноаддитивной соли или основной соли), растворенной в фармацевтически приемлемом жидком носителе, например, таком, как вода для инъекций, и буфер для получения соответствующим образом забуференного изотонического раствора, имеющего, например, pH около 3,5-6. Подходящими забуферивающими агентами являются, например, тринатрийортофосфат, бикарбонат натрия, цитрат натрия, N-метилглюкамин, L(+)-лизин и L(+)-аргинин и некоторые другие известные забуферивающие агенты. Обычно соединения формулы I растворяют в соответствующем носителе в количестве, достаточном для обеспечения фармацевтически приемлемой для инъекции концентрации в диапазоне от около 1 мг/мл до около 400 мг/мл раствора. Полученную жидкую фармацевтическую композицию вводят в таком количестве, чтобы в результате получить нужную антибактериально эффективную дозу. Соединения настоящего изобретения формулы I вводят преимущественно перорально в виде твердой или жидкой дозированной формы.
Противомикробную активность соединений настоящего изобретения испытывали in vivo с использованием мышиной модели. Группе мышей-самок (шесть мышей по 18-20 г каждая) вводили внутрибрюшинную инъекцию бактерий, которые перед их использованием размораживали в экстракте "мозгового" сердца, содержащем 4% пивных дрожжей (Staphylococcus aureus) или в экстракте "мозгового" сердца (стрептококки). Через 1 ч и через 5 ч после инъекции бактерий (т.е. после инфицирования) проводили обработку антибиотиками в шести различных дозах либо путем пероральной интубации, либо путем подкожного введения. Ежедневно в течение шести дней регистрировали число выживших животных. Затем, исходя из относительного количества погибших животных и с использованием пробит-анализа, вычисляли значение ED50. В качестве контроля и для проведения сравнения с соединениями настоящего изобретения использовали хорошо известные противомикробные средства. Результаты представлены в таблице.
В таблице каждый из примеров относится к следующим соединениям:
Пример 1. 9-(4-(5-(Ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1- пиперазинкарбоновой кислоты метиловый сложный эфир (23);
Пример 3. 4-(4-(5-Ацетиламино)метил)-2-оксо-3-оксазолидинил)-1-пиперазинкарбоновой кислоты метиловый сложный эфир;
Пример 5. N-((3-(4-(3-фторо-4-(4-(2-цианоэтил)-1-пиперазинил))фенил-2-оксо-5- оксазолидинил)метил)-ацетамид;
Пример 6. 4-(4-(5((Ацетиламино)метил-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты 2-гидроксиэтиловый сложный эфир;
Пример 7. N-((3-(4-(3-фторо-4-((фенилкарбонил)-1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)-ацетамид;
Пример 10. (+/-)-N-[[3-[4-[4-(1,4-диоксопентил)-1-пиперазинил]-3-фторофенил]-2-оксо-5-оксазолидинил]метил)-ацетамид;
Пример 36. (S)-N-[[3-[3-фторо-4-[4-(2-метоксиэтил)-1-пиперазинил]фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
Пример 37. (S)-N-[[3-[3,5-дифторо-4-[4-(2-метоксиэтил)-1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Общий метод синтеза метилового сложного эфира 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1- пиперазинкарбоновой кислоты, метилового эфира (23) и этилового сложного эфира 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты (24) описан в примерах 1 и 2, а также на структурных схемах 1 и 2 (для простоты используемые в этих схемах соединения имеют цифровые обозначения, соответствующие конкретным химическим названиям). В соответствии с этими схемами, коммерчески доступный дифторонитробензол (2) обрабатывают избыточным количеством пиперазина, в результате чего получают продукт замещения 3. После реакции защиты с образованием трет-бутоксикарбонильного (ВОС) производного 4 проводят реакцию восстановления нитрогруппы с использованием системы реагентов, содержащей формат аммония и Pd/C, в результате чего получают анилиновое производное 5. После реакции защиты полученного соединения 5 получают бензилоксикарбонильное (CBZ) производное 6, которое затем подвергают аллилированию с образованием соединения 7. В результате осмилирования соединения 7 по методу Kelly и Van Rheenen (Tetrahedron Letters, 1973 (1976) получают диол 8, который после обработки карбонатом калия в кипящем ацетонитриле циклизуют с образованием оксазолидинона 9. После мезилирования оксазолидинона 9 в классических условиях получают мезилат 10, который затем подвергают мягкой реакции замещения с использованием азида натрия, в результате чего получают азид 11. Этот азид 11 подвергают реакции восстановления путем гидрогенизации в присутствии Pd/C и получают амин 12, который затем подвергают ацилированию in situ с использованием уксусного ангидрида и пиридина, в результате чего получают ВОС-защищенное оксазолидиноновое промежуточное соединение, а именно 1,1-диметилэтиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты.
После реакции снятия защиты с использованием трифтороуксусной кислоты получают ключевое промежуточное соединение N-((3-(4-(3-фторо-4-(1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил) -ацетамид (22), который затем используют для получения аналогов. После обработки соединения 22 либо метилхлороформатом, либо этилхлороформатом, предпочтительно в условиях Schotten-Baumann (NaHCO3/ацетон-вода), получают метиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты 23 и этиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты (24) соответственно.
Хотя описанный выше способ, который раскрывается в примере 1, может быть использован для получения всех целевых соединений, однако существует и другой способ (хотя и менее эффективный), который заключается в получении промежуточных соединений для получения целевых соединений таких, как N-((2-оксо-3-(4-(4-((фенилкарбонил)-1-пиперазинил)фенил)-5-оксазолидинил)метил)ацетамид (20) и метиловый сложный эфир 4-(4-(5((ацетиламино)метил-2-оксо-3-оксазолидинил)фенил)-1-пиперазинкарбоновой кислоты (19). Например, диол 13, полученный исходя из пиперазина и п-фторонитробензола способом, аналогичным способу, описанному для получения диола 8 (см.схему 1), обрабатывают одним эквивалентом мезилхлорида или тозилхлорида, в результате чего получают моно-дериватизированное соединение 14 вместе с неизменным исходным материалом и бисдериватизированным материалом. После хроматографического выделения мезилата 14a или тозилата 14b и обработки любого из этих материалов азидом натрия получают азидоспирт 15. После обработки этого азидоспирта 15 основанием (для осуществления циклизации) получали оксазолидинон 16, который может быть затем превращен в ацетамидное производное 17 в соответствии с методикой восстановления-ацилирования (проводимой в одном резервуаре), описанной в примере 1. Как показано на схеме, после сольволитического снятия защиты у соединения 17 получают N-((2-оксо-3-(4-(1-пиперазинил)-фенил)-5-оксазолидинил)-метил)-ацетамид (18), который затем подвергают ацилированию с получением двух нефторированных аналогов, а именно: метиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-4-оксазолидинил)фенил-1-пиперазинкарбоновой кислоты (19) и N-((2-оксо-3-(4-(фенилкарбонил)-1-пиперазинил)фенил)-5-оксазолидинил)метил)- ацетамид (20).
Получение аналогов метилового сложного эфира 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2- фторофенил)-1-пиперазинкарбоновой кислоты (23) и этилового сложного эфира 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторо-фенил)-1- пиперазинкарбоновой кислоты (24) может быть легко осуществлено путем простой замены пиперазина другими циклическими аминами, и соединения 2 другими нитробензоловыми производными; либо путем обработки N-((3-(4-(3-фторо-4-(1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)-ацетамида (22) (или их аналогов) другими ацилирующими или алкилирующими агентами.
Пример 1. 4-(4-(5-(Ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1- пиперазинкарбоновой кислоты метиловый сложный эфир (23).
(a) Получение 1-(2-фторо-4-нитрофенил)пиперазина (3):
Раствор 12,0 г (75,42 мМ) 3,4-дифторонитробензола (2) в 150 мл ацетонитрила обрабатывали 16,24 г (188,6 мМ) пиперазина, а затем нагревали с обратным холодильником в течение 3 ч. Полученный раствор охлаждали до комнатной температуры и концентрировали в вакууме. Остаток разводили 200 мл воды и экстрагировали этилацетатом (3х250 мл). После этого объединенные органические слои экстрагировали водой (200 мл) и насыщенным раствором хлорида натрия (200 мл), а затем осушали сульфатом натрия. Полученный раствор концентрировали в вакууме, в результате чего образовывалось оранжевое маслообразное вещество, которое хроматографировали на 450 г силикагеля (230-400 меш), элюируя сначала дихлорметаном до тех пор, пока не были проэлюированы наименее полярные фракции. Затем продолжали элюирование 2%-ным (об. /об.) метанолом/хлороформом и 10%-ным (об./об.) метанолом/хлороформом. В результате этих стадий получали 13,83 г (81%) нужного пиперазинового производного (3), т.пл. 68,5-71oC.
(b) Получение 1-(трет-бутоксикарбонил)-4-(2-фторо-4-нитрофенил)пиперазина (4):
Раствор 12,0 г (53,29 мМ) нитропроизводного (3) в 110 мл тетрагидрофурана по капле обрабатывали раствором 14,53 г (66,61 мМ) ди-третбутилдикарбоната в 110 мл тетрагидрофурана. После добавления раствор перемешивали при комнатной температуре в течение 24 ч. После этого раствор концентрировали в вакууме, и остаток хроматографировали на 450 г силикагеля (230-400 меш), элюируя 20%-ным (об./об.) этилацетатом в гексане, 30%-ным (об./об.) этилацетатом в гексане, а затем 50%-ным (об./об.) этилацетатом в гексане. В результате получали 16,6 г (96%) ВОС-производного (4) в виде желтого твердого вещества, т.пл. 151-153,5oC.
(c) Получение 1-(трет-бутоксикарбонил)-4-(2-фторо-4-аминофенил) пиперазина (5):
Раствор 1,73 г (5,32 мМ) нитросоединения (4) в 30 мл метанола и 20 мл тетрагидрофурана и 10 мл ацетата обрабатывали 1,68 г (26,59 мМ) формата аммония и 200 мг 10%-ного палладированного угля. Сразу после обработки наблюдалось выделение газа, которое приблизительно через 30 мин прекращалось. После этого смесь перемешивали в течение ночи, а затем фильтровали через целит, и образовавшийся осадок на фильтре промывали метанолом. Фильтрат концентрировали в вакууме, растворяли в 50 мл этилацетата и экстрагировали водой (2х30 мл) и насыщенным раствором хлорида натрия (30 мл). После осушки сульфатом натрия и концентрирования в вакууме получали 1,6 г (прибл. 100%) амина (5) в виде коричневого твердого вещества, которое очищали для использования в последующей стадии.
(d) Получение 1-(трет-бутоксикарбонил)-4-(2-фторо-4-бензилоксикарбониламино)пиперазина (6):
Раствор 1,57 г (5,32 мМ) амина 5 и 806 мг (0,84 мл, 6,65 мМ) диметиланилина в 25 мл тетрагидрофурана при -20oC по капле обрабатывали бензилхлороформатом (1,0 г, 0,84 мл, 5,85 мМ). После этого раствор перемешивали при -20oC в течение 30 мин, а затем нагревали до комнатной температуры. Образовавшуюся смесь разводили 125 мл этилацетата и экстрагировали водой (2х50 мл) и насыщенным раствором хлорида натрия (50 мл). После осушки сульфатом натрия и концентрирования в вакууме образовывался негомогенный материал. Этот материал адсорбировали на слое силикагеля и хроматографировали на 115 г силикагеля (230-400 меш), элюируя 18%-ным (об./об.) этилацетатом в гексане, затем 25%-ным (об. /об. ) этилацетатом в гексане и наконец 30%-ным (об./об.) этилацетатом в гексане. В результате получали 1,15 г (50%) CBZ-производного (6) в виде белого твердого вещества, т.пл. 150-153oC.
(e) Получение 1-(трет-бутоксикарбонил)-4-(2-фторо-4-бензилоксикарбонилаллиламино)пиперазина (7):
Раствор 1,15 г (2,68 мМ) CBZ-производного 6 в 10,2 мл диметилформамида порциями обрабатывали 77 мг (129 мг 60%-ного раствора в масле, 3,21 мМ) гидрида натрия, а затем перемешивали при комнатной температуре в течение 20 мин. После этого раствор обрабатывали 356 мг (0,26 мл, 2,95 мМ) аллилбромида, а затем перемешивали при комнатной температуре в течение 18 ч. Полученный раствор осторожно обрабатывали 75 мл воды и экстрагировали диэтиловым эфиром (3х100 мл). Объединенные органические слои экстрагировали насыщенным раствором хлорида натрия (100 мл) и осушали сульфатом натрия. После концентрирования в вакууме получали негомогенный материал, который растворяли в дихлорметане и осушали сульфатом натрия. Остаток концентрировали в вакууме и получали маслообразное вещество янтарного цвета, которое хроматографировали на 60 г силикагеля (230-400 меш), элюируя 25%-ным (об./об.) этилацетатом в гексане. В результате получали 1,12 г (90%) аллилового производного (7) в виде маслообразного вещества.
(f) Получение 1-(трет-бутоксикарбонил)-4-[2-фторо-4-бензилоксикарбонил(2,3-дигидроксипроп-1-ил)аминофенил]пиперазина (8):
Раствор 2,18 г (4,64 мМ) аллилового соединения 7 и 3,26 г (27,86 мМ) N-оксида N-метилморфолина в 21 мл ацетона и 6,4 мл воды обрабатывали 5 мл 2,5%-ного (по массе) раствора тетроксида осмия в трет-бутиловом спирте. Полученный раствор перемешивали при комнатной температуре в течение 24 ч. После этого раствор охлаждали до 0oC и добавляли 25 мл насыщенного раствора NaHSO3, после чего перемешивали при 0oC в течение 15 мин и нагревали до комнатной температуры в течение 2 ч. Полученную смесь разводили 50 мл воды и 50 мл насыщенного раствора хлорида натрия, а затем экстрагировали этилацетатом (5х100 мл). Объединенные органические слои осушали сульфатом натрия и концентрировали в вакууме с получением коричневого маслообразного вещества. Это вещество хроматографировали на 150 г силикагеля (230-400 меш), элюируя 10%-ным (об./об.) метанолом в хлороформе. В результате получали 2,0 г (86%) диола (8) в виде беловатой гигроскопичной жесткой пены.
(g) Получение 3-[3-фторо-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил] -5-гидроксиметил-2-оксазолидинона (9):
Раствор 2,0 г (4,01 мМ) диола 8 в 20 мл ацетонитрила обрабатывали карбонатом калия (1,1 г, 8,02 мМ), а затем нагревали с обратным холодильником в течение 3 ч. После этого раствор охлаждали и концентрировали в вакууме. Остаток растворяли в 100 мл этилацетата и полученный раствор экстрагировали водой (2х50 мл), а затем насыщенным раствором хлорида натрия (50 мл). После осушки сульфатом натрия и концентрирования в вакууме образовывалось маслообразное вещество, которое хроматографировали на 80 г силикагеля (230-400 меш), элюируя 20%-ным (об./об.) ацетоном в дихлорметане. В результате получали 1,6 г (100%) оксазолидинона 9 в виде белого твердого вещества, т.пл. 144-146,5oC.
(h) Получение 3-[3-фторо-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил] -5-метансульфонилоксиметил-2-оксазолидинона (10):
В раствор 375 мг (0,95 мМ) оксазолидинона 9 и 144 мг (0,20 мл, 1,42 мМ) триэтиламина в 3,8 мл дихлорметана при 0oC по капле добавляли 130 мг (0,09 мл, 1,14 мМ) метансульфонихлорида, а затем перемешивали при 0oC в течение часа. Полученный раствор разводили 30 мл дихлорметана, после чего экстрагировали водой (2х25 мл) и насыщенным гидрокарбонатом натрия (25 мл). Образовавшийся остаток осушали сульфатом натрия и концентрировали в вакууме, в результате чего получали 440 мг (98%) мезилата 10 в виде белого твердого вещества, которое было достаточно чистым, чтобы можно было использовать это соединение в следующей стадии.
(i) Получение 3-[3-фторо-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил] -5-азидометил-2-оксазолидинона (11):
Раствор 440 мг (0,93 мМ) мезилата 10 в 22 мл ацетона обрабатывали раствором азида натрия (604 мг, 9,29 мМ) в 6,4 мл воды. Полученную смесь нагревали с обратным холодильником в течение 18 ч. Затем смесь охлаждали, добавляли раствор 600 мг азида натрия в 6 мл воды, а затем нагревали с обратным холодильником еще 18 ч. После этого смесь снова охлаждали, добавляли раствор 1,2 г азида натрия в 12 мл воды, а затем нагревали с обратным холодильником в течение 24 ч. После охлаждения смесь разводили 60 мл воды и экстрагировали этилацетатом (3х75 мМ). Объединенные органические слои экстрагировали 100 мл насыщенного раствора хлорида натрия, после чего осушали сульфатом натрия. После концентрирования в вакууме получали 358 мг (92%) азида 11 в виде белого твердого вещества (т.пл. 130,5-132,5oC), которое было достаточно чистым для использования его в последующей стадии.
(j) Получение 3-[3-фторо-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил] -5-аминометил-2-оксазолидинона (12) и 3-[3-фторо-4-трет-бутоксикарбонилпиперазин-1-ил)фенил]-5- ацетиламино-метил-2-оксазолидинона (21).
Раствор 1,42 г (3,38 мМ) азида 11 в 200 мл этилацетата обрабатывали 400 мг 10%-ного палладированного угля, а затем гидрогенизировали при атмосферном давлении в течение 48 ч. Полученный этилацетатный раствор (12) обрабатывали 1,34 г (1,37 мл, 16,9 мМ) пиридина и 870 мг (0,80 мл, 8,5 мМ) уксусного ангидрида, а затем перемешивали при комнатной температуре в течение 48 ч. После этого раствор обрабатывали 1,37 мл пиридина и 0,8 мл уксусного ангидрида, а затем перемешивали при комнатной температуре еще 48 ч. Этот раствор фильтровали через целит, а образовавшийся осадок на фильтре промывали этилацетатом. Фильтрат промывали водой (4х50 мл), 1,0М раствором сульфата меди (50 мл) и снова водой (50 мл). После осушки сульфатом натрия и концентрирования в вакууме получали пенообразное вещество, которое растворяли в дихлорметане и перемешивали в течение часа с насыщенным раствором гидрокарбоната натрия.
Полученную смесь экстрагировали дихлорметаном, а объединенные органические слои осушали сульфатом натрия и концентрировали в вакууме, в результате чего образовывалось маслообразное вещество янтарного цвета. Это вещество хроматографировали на 74 г силикагеля (230-400 меш), элюируя 2%-ным (об./об. )метанолом в дихлорметане, а затем 5%-ным (об./об.) метанолом в дихлорметане. В результате получали 1,19 г (81%) 3-(3-фторо-4-трет-бутоксикарбонилпиперазин-1-ил)фенил)-5-ацетиламинометил-2-оксазолидинона (21) в виде беловатого жесткого пенопласта (т.пл. 162-164oC).
(k) Получение N-((3-(4-(3-фторо-4-(1-пиперазин))фенил)-2-оксо-5-оксазолидинон)метил)ацетамида (22).
Раствор 1,19 г (2,73 мМ) ВОС-производного [3-(3-фторо-4-трет-бутоксикарбонилпиперазин-1-ил)фенил)-5-ацетиламинометил-2-оксазолидинона (21)] в 40 мл дихлорметана при 0oC обрабатывали 15 мл трифтороуксусной кислоты. Этот раствор перемешивали 30 мин при 0oC, а затем нагревали до комнатной температуры, после чего реакция была полностью завершена. Полученный раствор концентрировали в вакууме, а остаток разводили этилацетатом и насыщенным раствором гидрокарбоната натрия. Водный слой экстрагировали этилацетатом, после чего было видно, что большая часть продукта оставалась в водном слое. После этого водный слой доводили до pH=14 путем добавления 50%-ного раствора NaOH. После экстрагирования этилацетатом, осушки сульфатом натрия и концентрирования в вакууме получали 179 мг маслообразного вещества янтарного цвета. Это вещество подвергали круговой хроматографии на 2 мм-пластинах, элюируя 10%-ным (об./об.) метанолом в хлороформе, а затем 15%-ным (об./об.) метанолом в хлороформе. В результате получали 125 мг (79%) N-((3-(4-(3-фторо-4-(1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)- ацетамида (22) в виде беловатого жесткого пенопласта.
(I) Получение 4-(4-(5-((Ацетиламино)метил)-2-оксо-3-оксазолидинил)-2- фторофенил)-1-пиперазинкарбоновой кислоты метилового сложного эфира (23).
Раствор 120 мг (0,36 мМ) N-((3-(4-(3-фторо-4-(1-пиперазинил))фенил)-2- оксо-5-оксазолидинил)метил)-ацетамида и 60 мг (0,71 мМ) твердого гидрокарбоната натрия в 1,5 мл ацетона и 0,7 мл воды при 0oC обрабатывали 37 мг (30 мкл, 0,39 мМ) метилхлорформата. После этого раствор перемешивали в течение 1 ч при 0oC, а затем разводили 20 мл воды. Полученную смесь экстрагировали этилацетатом (30 мл), а органический слой экстрагировали водой (2х10 мл) и насыщенным раствором гидрокарбоната натрия (10 мл). Затем раствор осушали сульфатом натрия и концентрировали в вакууме с получением 95 мг неочищенного продукта. Этот неочищенный продукт подвергали радиальной хроматографии на 2 мм-пластинах, элюируя 33%-ным (об./об.) ацетоном в дихлорметане, а затем 50% (об./об.) ацетоном в дихлорметане. В результате получали 81 мг (57%) метилового сложного эфира 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинила)-2-фторофенил)-1-пиперазинкарбоновой кислоты в виде белого твердого вещества, т.пл.177-179oC.
Пример 2. Этиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты (24).
Были использованы те же самые методики, описанные в примере 1 (стадии a-k). Раствор 100 мг (0,30 мМ) N-(33-(4-(3-фторо-4-(1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)-метил)ацетамида (23) (продукта из стадии (k), описанной выше) и 50 мг (0,59 мМ) твердого гидрокарбоната натрия в 2 мл ацетона и 1 мл воды при 0oC обрабатывали 35 мг (31 мкл, 0,33 мМ) этилхлороформата. Полученный раствор перемешивали при 0oC в течение 2 ч, а затем нагревали до комнатной температуры в течение 18 ч. Этот раствор разводили 30 мл воды и экстрагировали этилацетатом (40 мл). Органический слой промывали 30 мл воды и 30 мл насыщенного раствора гидрокарбоната натрия. После осушки сульфатом натрия и концентрирования в вакууме получали белое твердое вещество. Этот материал подвергали радиальной хроматографии на 22 мм-пластинах, элюируя 2%-ным (об./об.) метанолом в хлороформе. Таким образом получали 70 мг (58%) этилового сложного эфира 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты (24) в виде белого твердого вещества, т.пл. 224-226oC.
Пример 3. Метиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3- оксазолидинил)фенил)-1-пиперазинкарбоновой кислоты.
Целевое соединение получали в соответствии с методикой, описанной в примере 1, за исключением того, что вместо исходного материала (3,4-дифторонитробензола (2)) использовали 4-фторонитрбензол.
Пример 4. N-((2-Оксо-3-4-(4-фенилкарбонил)-1-пиперазин)-фенил)-5-оксазолидинон)метил)-ацетамид.
Целевое соединение получали в соответствии с методикой, описанной в примере 2, за исключением того, что исходный материал (3,4-дифторонитробензол (2)) заменяли на 4-фторонитробензол.
Пример 5. N-((3-(4-(3-фторо-4-(4-(2-цианоэтил)-1-пиперазинил))фенил)-2- оксо-5-оксазолидинон)метил)-ацетамид.
Раствор 75 мг (0,22 мМ) N-((3-(4-(3-фторо-4-(1-пиперазинил))фенил-2-оксо-5-оксазолидинил)метил)-ацетамида (22) (пример 1, часть k) в 5 мл метанола обрабатывали 13 мг (17 мкл, 0,25 мМ) акрилонитрила, а затем нагревали с обратным холодильником в течение 3 ч. После этого раствор охлаждали и концентрировали в вакууме. Образовавшийся остаток подвергали радиальной хроматографии на 4 мм-пластинах, элюируя 5 %-ным (об./об.)метанолом в хлороформе. В результате получали 84 мг (97%) нужного нитрила, N-((3-(4-(3-фторо-4-(4-(2-цианоэтил)-1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)-ацетамида в виде белого твердого продукта, т.пл. 125-130oC.
Пример 6. 2-Гидроксиэтиловый сложный эфир 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2-фторофенил)-1-пиперазинкарбоновой кислоты.
Раствор 208 мг (0,25 мМ) N-(93-(4-(3-фторо-4-(1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)-ацетамида (22) (продукт примера 1, часть k) в 3 мл ацетона и 2 мл воды обрабатывали бикарбонатом натрия (21 мг, 0,25 мМ), а затем охлаждали до 0oC. Полученную смесь обрабатывали 54 мг раствора (0,25 мМ) 2-бензилоксиэтилхлороформата в 2 мл ацетона. После этого раствор оставляли для нагревания до комнатной температуры на 22 ч, а затем разводили 30 мл этилацетата и экстрагировали водой (3х30 мл) и насыщенным раствором бикарбоната натрия (20 мл). Этот раствор осушали сульфатом натрия и концентрировали в вакууме с получением белого твердого вещества. Этот материал подвергали радиальной хроматографии на 4 мм-пластинах (элюент: 20%-ный (об. /об. ) ацетон в дихлорметане). В результате этих процедур получали 113 мг (прибл. 100%) хлороформата, 4-(4-(5-((ацетиламино)метил)2-оксо-3-оксазолидинил)-2-фторофенил)-1- пиперазинкарбоновой кислоты 2-гидроксиэтилового сложного эфира в виде белого твердого вещества, т.пл. 121-123oC. Раствор полученного материала в 5 мл метанола обрабатывали 35 мг 10 %-ного палладированного угля, после чего подвергали гидрогенолизу в течение 1 ч под давлением 1 атм. Полученную смесь фильтровали через целит, а образовавшийся осадок на фильтре промывали метанолом. Фильтрат концентрировали в вакууме и получали белое твердое вещество. Это вещество подвергали радиальной хроматографии на 2 мм-пластинах, элюируя 5%-ным (об./об.)метанолом в хлороформе, а затем 10%-ным (об./об.) метанолом в хлороформе. В результате получали 76 мг (82%) гидроксиэтилхлороформата, 2-гидроксэитилового сложного эфира 4-(4-(5-((ацетиламино)метил)-2-оксо-3-оксазолидинил)-2- фторофенил)-1-пиперазинкарбоновой кислоты в виде белого твердого продукта, т.пл. 203-206oC.
Пример 7. N-((3-(4-(3-фторо-4-((фенилкарбонил)-1-пиперазинил))фенил)-2-оксо-5- оксазолидинил)метил)-ацетамид.
В соответствии с методикой, описанной в примере 1, целевое соединение получали из 100 мг (0,297 мМ) N-((3-(4-(3-фторо-4-(1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)- метил)-ацетамида (22) (продукт примера 1, часть k) за исключением того, что метилхлороформат (исходный материал) заменяли на бензоилхлорид. В результате этой процедуры получали 73 мг (56%) N-((3-(4-(3-фторо-4-(фенилкарбонил)-1-пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)-ацетамида в виде мелкого белого порошка, т.пл.184-187oC.
Пример 8. 2-Метоксиэтиловый эфир 4-[4-[(5-[(ацетиламино)метил]-2-оксо-3-оксазолидинил]-2-фторофенил]-1-пиперазинкарбоновой кислоты.
Раствор 75 мг (0,22 мМ) пиперазинового производного 22 в 4 мл ацетона и 2 мл воды обрабатывали бикарбонатом натрия (2 мг, 0,25 мМ), а затем охлаждали до 0oC и добавляли раствор 35 мг (0,25 мМ) 2-метоксиэтилхлороформата в 0,5 мл тетрагидрофурана. Полученную смесь нагревали до комнатной температуры в течение 22 ч. После этого смесь разводили 30 мл этилацетата, а затем экстрагировали водой (3х20 мл) и насыщенным раствором бикарбоната натрия (20 мл). Объединенные водные слои экстрагировали этилацетатом (2х20 мл), а объединенные органические слои осушали сульфатом натрия и концентрировали в вакууме с получением белого твердого вещества. Полученное вещество подвергали радиальной хроматографии на 4 мм-пластинах, элюируя сначала 20%-ным (об./об.) ацетоном в дихлорметане, а затем 30%-ным (об./об.) ацетоном в дихлорметане. В результате получали 92 мг (96%) нужного соединения в виде белого твердого вещества.
Пример 9. 4-[4-[5(Ацетиламино)метил]-2-оксо-3-оксазолидинил]-2- фторофенил]-1-пиперазинацетонитрил.
Раствор 75 мг (0,22 мМ) пиперазинового производного 22 в 4 мл ацетона и 2 мл воды обрабатывали 21 мг (0,25 мМ) бикарбоната натрия, а затем охлаждали до 0oC. Полученную смесь обрабатывали 226 мг (190 мкл, 3,0 мМ) свежеприготовленного дистиллированного хлороацетонитрила, после чего эту смесь оставляли для нагревания до комнатной температуры на 60 ч. Образовавшийся раствор разводили 35 мл этилацетата, а затем экстрагировали водой (3х20 мл) и насыщенным раствором бикарбоната натрия (20 мл). Объединенные водные слои экстрагировали этилацетатом (3х20 мл), а объединенные органические слои осушали сульфатом натрия и концентрировали в вакууме с получением белого твердого вещества. Это вещество подвергали круговой хроматографии на 4 мм-пластинах (элюент: 5%-ный (об./об.) метанол в хлороформе). В результате этих процедур получали 80 мг (96%) нужного нитрила в виде блестящего белого твердого вещества.
Пример 10. (+/-)-N-[[3-[4-[4-(1,4-диоксопентил)-1-пиперазинил]-2-оксо-5-оксазолидинил]метил)ацетамид (Y=MeCO(CH2)2-CO-, моноF, рацемич.).
Соединение примера 1 (стадия k) (0,104 г) обрабатывали 0,041 г левулиновой кислоты, 0,083 г 1-этил-3-(3-диметиламинопропил)карбодиимида и 0,005 г N, N-диметиламинопиридина в 2 мл пиридина. Полученную смесь перемешивали 2 дня при 20oC. После водной экстракции, проведенной с использованием метиленхлорида, получали 0,139 г остатка. Этот материал очищали с помощью жидкостной хроматографии среднего давления (ЖХСД) на силикагеле (элюент: 5%-ный (об. /об.) метанол в этилацетате), в результате чего получали 0,118 г белого твердого продукта, т.пл. 148-150oC.
Пример 11. N-[[3-[4-[4-(1,4-диоксопентил)-1-пиперазинил]-3,5-дифторопентил]-2-оксо-5- оксазолидинил]метил)-ацетамид, (S)-(Y является таким, как он определен выше; diF, оптически активный)
В соответствии с общей методикой, описанной в примере 10, но с использованием трифтороуксусной соли пиперазина U-99472 (полученной, как описано ниже), получали 0,291 г соли. Эту соль подвергали жидкостной хроматографии среднего давления, а затем подвергали препаративной тонкослойной хроматографии (1000 мкм, элюент: 20%-ный ацетон/метиленхлорид (об./об.), в результате чего получали 0,103 г пенообразного белого твердого вещества, т.пл. 52-56oC.
Пример 12. (+/-)-N-[[3-[4-[4-[(1-оксо-6-окса-7-фенил)гептил]-1-пиперазинил]-3-фторофенил]-2- оксо-5-оксазолидинил]-метил)-ацетамид (Y= PhCH2O(CH2)4CO-).
Использовали общую методику, описанную в примере 10, за исключением того, что левулиновую кислоту заменяли на 5-бензилоксивалериановую кислоту (0,074 г), в результате чего получали 0,101 г соединения. Это соединение подвергали жидкостной хроматографии среднего давления (элюент: 10%-ный метанол в этилацетате) и получали 0,130 г целевого соединения, ТСХ: Rf = 0,24 (элюент: 10 %-ный метанол в этилацетате, об./об.).
Пример 13. (+/-)-N-[[3-[4-[4-(1-оксо-5-гидроксипентил)-1- пиперазинил] -3-фторопентил]-2-оксо-5-оксазолидинил]метил)- ацетамид (Y=HO(CH2)4CO-).
Соединение примера 12 (66 мг) растворяли в 5 мл метанола, реакционный сосуд продували и наполняли азотом (3 раза). К смеси добавляли 0,034 г палладиевой черни, и реакционный сосуд снова продували и наполняли водородом из баллона (3 раза). Полученную смесь перемешивали в атмосфере водорода в течение 3 ч, а затем фильтровали через диатомовую землю, промывали метанолом, и фильтрат выпаривали. Образовавшийся остаток растирали с хлороформом, и осажденный белый твердый осадок собирали и получали целевое соединение, т. пл. 171-172oC. ТСХ: Rf = 0,07 (10%-ный метанол в этилацетате, об./об.).
Пример 14. N-[[3-[3,5-дифторо-4-[4-[5-R, S-метил-[(1,3-ди-окса-2-оксо)циклопентил] ] ] -1-пиперазинил]фенил)-2-оксо-5-оксазолидинил] метил]ацетамид, -(5S) (Y=циклический карбонат: оптически активный при оксазолидиноне, но имеющий рацемат при циклическом карбонате, diF)
ВОС-пиперазин, diF, оптически активное соединение (0,094 г) обрабатывали 1,0 мл трифтороуксусной кислоты в 1,5 мл метиленхлорида при 0oC в течение 50 мин, а затем нагревали до 20oC. Образовавшиеся летучие вещества удаляли в вакууме и получали красное маслообразное вещество (трифтороацетат пиперазина U-99472). К этому веществу добавляли 0,036 г хлороэтилметиленкарбоната и 0,069 г карбоната калия в ацетонитриле, после чего смесь нагревали с обратным холодильником в течение 1 дня. Затем смесь фильтровали и выпаривали в вакууме с получением желтого маслообразного вещества. Полученный остаток очищали с помощью ЖХСД на силикагеле (элюируя градиентом 5-10% метанола в метиленхлориде, об./об.), а затем подвергали препаративной ТСХ (элюент: 7 %-ный метанол в метиленхлориде), в результате чего получали 0,022 г белого твердого вещества, т.пл. 106-111oC.
Пример 15. N-[[3-[3,5-дифторо-4-[4-(1-оксо-2-метоксиэтил)-1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид, (S) -(Y=MeOCH2CO-оптически активный, diF).
К раствору трифтороацетатной соли пиперазина (0,192 г) в 3 мл метиленхлорида и 1,0 мл триэтиламина в атмосфере азота и при 0oC добавляли 0,071 г метоксиацетилхлорида. Полученную смесь перемешивали при 0oC, а затем подвергали водной экстракции, проведенной с использованием метиленхлорида. Органический слой осушали сульфатом магния и концентрировали до объема 5-10 мл, после чего охлаждали, и образовавшийся твердый остаток собирали и перекристаллизовывали из этилацетата. В результате получали 44 мг белого твердого продукта, т.пл. 239-241oC.
Пример 16. (+/-)-N-[[3-[4-[4-(N-карбобензилокси)-2-амино-1-оксо-этил)-1-пиперазинил] ] -3-фторофенил] -2-оксо-5-оксазолидинил]метил]ацетамид (Y=Ph CH2O2CNHCH2CO-рацемич., моноF).
К соединению примера 1 k (0,115 г) добавляли 0,085 г N-карбобензилоксиглицина в 4 мл тетрагидрофурана и 2 мл воды, а затем pH доводили до значения 4 путем добавления 3 н. соляной кислоты и 0,203 г 1-этил-3-(3-диметиламинопропил)карбодиимида. После этого смесь перемешивали при 20oC в течение часа, добавляли еще 0,101 г N-карбобензилоксиглицина и 0,225 г 1-этил-3-(3-диметиламинопропил)карбодиимида, а pJ доводили до значений 3-4 и 5 путем добавления 2н гидроксида натрия. Полученную смесь перемешивали в течение ночи. После этого смесь подвергали водной экстракции, проведенной с использованием этилацетата, органические слои концентрировали, а остаток очищали путем концентрирования из метиленхлорида и метанола. В результате растирания с метанолом получали 0,042 мг белого твердого вещества, т.пл. 189-191oC.
Пример 17. (S)-N-[[3-[4-[4-(цианометил)-1-пиперазинил]-3-фторофенил]-2-оксо-5-оксазолидинил]метил-ацетамид (U-97665).
В последующих стадиях раскрывается получение моно-F-замещенного продукта настоящего изобретения.
(a) 1,1-Диметилэтиловый сложный эфир (S)-4-[4-[5-(гидроксиметил)-2-оксо-3-оксазолидинил]-2-фторофенил]-1-пиперазинкарбоновой кислоты (2):
в раствор 7,5 г (17,5 мМ) CBZ-производного 1 в 240 мл тетрагидрофурана при -78oC по капле примерно в течение 3 мин добавляли 12,0 мл (1,6 М, 19,25 мМ) н-бутиллития в гексане. Затем этот раствор перемешивали при -78oC в течение 30 мин; после этого приблизительно в течение 5 мин добавляли 2,78 г (2,73 мл, 19,25 мМ) чистого R-(-)-глицидилбутирата (по капле), и раствор нагревали до 0oC. Затем раствор нагревали до комнатной температуры в течение 18 ч. Полученную смесь разводили дихлорметаном и экстрагировали водой, а затем насыщенным водным раствором хлорида натрия. Органический слой осушали сульфатом натрия и концентрировали с получением смолообразного остатка. Этот остаток перекристаллизовывали из горячего этилацетата с добавлением некоторого количества гексана, в результате чего получали 6,4 г (93%) нужного продукта, т.пл. 130,5-133oC.
(b) 1,1-Диметилэтиловый сложный эфир (S)-4-[4-[5-(метансульфонилоксиметил)-2-оксо-3-оксазолидинил] -2-фторофенил] -1-пиперазинкарбоновой кислоты (3):
раствор 2,88 г (7,28 мМ) спирта (2) в 32 мл дихлорметана при 0oC обрабатывали 1,29 г (1,77 мл, 12,7 мМ) триэтиламина, а затем добавляли 1,04 г (0,70 мл, 9,10 мМ) метансульфонилхлорида. Полученный раствор перемешивали в течение 15 мин при 0oC, после чего разводили дихлорметаном и экстрагировали водой. Этот раствор осушали сульфатом натрия и концентрировали в вакууме с получением 3,4 г (98%) мезилата 3 в виде светло-розового твердого вещества (масс-спектроскопия высокого разрешения: для C20H28FN3O7S вычислено: 4731632, найдено: 4731631), достаточно чистого для использования в последующей стадии.
(c) 1,1-Диметилэтиловый сложный эфир (S)-4-[4-[5-(азидометил)-2-оксо-3-оксазолидинил]-2-фторофенил]-1-пиперазинкарбоновой кислоты (4):
раствор 16,8 г (35,5 мМ) мезилата 3 в 400 мл диметилформамида обрабатывали 11,5 г (177,5 мМ) азида натрия, а затем нагревали в течение 16 ч при 60oC. Полученный раствор разводили этилацетатом и экстрагировали водой. Органический слой осушали сульфатом натрия и концентрировали в вакууме, в результате чего получали 14,9 г (100%) азида 4 в виде светло-желтого твердого вещества (т.пл. 101-104oC), достаточно чистого для использования в последующей стадии.
(d) 1,1-Диметилэтиловый сложный эфир (S)-4-[4-[5-[(ацетиламино)метил]-2-оксо-3-оксазолидинил]-2-фторофенил]-1-пиперазинкарбоновой кислоты (5):
раствор 14,92 г (35,5 мМ) азида 4 в 200 мл этилацетата обрабатывали 2 г 10 %-ного палладия на углероде, а затем гидрогенизировали в течение 24 ч под давлением 1 атм. Реакционную колбу продували азотом, а затем добавляли 14,0 г (14,4 мл, 177,5 мМ) пиридина и 9,1 г (8,4 мл, 88,8 мМ) уксусного ангидрида. Полученную смесь перемешивали при комнатной температуре в течение 72 ч, после чего фильтровали через целит. Фильтрат экстрагировали водой, 1 н. раствором сульфата меди, осушали, концентрировали в вакууме и получали коричневатое твердое вещество. Это вещество очищали с помощью хроматографии на силикагеле; получали 12,7 г (82%) продукта 5 в виде порошкообразного белого твердого вещества, т.пл. 153-158oC.
(e) (S)-N-[[3-[4-[3-фторо-4-(1-пиперазинил)фенил] -2-оксо-5-оксазолидинил] метил]ацетамид:
35 мл трифтороуксусной кислоты при 0oC обрабатывали 5,0 г (11,46 мМ) ВОС-производного, а затем нагревали до комнатной температуры в течение часа. Полученный раствор концентрировали в вакууме и получали остаток, который растворяли в воде и перемешивали с 125 мл ионообменной смолы AGI-18 (OH- форма) в течение 2,5 ч. Затем смолу удаляли путем фильтрации, промывали водой, а объединенные фильтраты осушали путем замораживания, в результате чего получали 2,7 г (69%) нужного соединения в виде белых твердых хлопьев, т.пл. 73-76oC.
(f) (S)-N-[[3-[4-[4-(цианометил)-1-пиперазинил] -3-фторофенил]-2-оксо-5-оксазолидинил]метил]-ацетамид:
раствор 2,42 г (7,2 мМ) описанного соединения стадии (e) в 242 мл ацетона и 74 мл воды охлаждали до 0oC и обрабатывали 1,20 г (14,4 мМ) бикарбоната натрия, после чего добавляли 26,2 г (22,0 мл, 0,35 М) хлороацетонитрида. Полученный раствор нагревали до комнатной температуры в течение 36 ч. Смесь разводили этилацетатом и экстрагировали водой, а затем насыщенным раствором хлорида натрия. После осушки сульфатом натрия и концентрирования в вакууме получали беловатое твердое вещество, которое очищали с помощью хроматографии на силикагеле (элюент: система растворителей метанол/хлороформ). В результате получали 2,2 г (82%) целевого соединения в виде белого твердого хлопьеобразного вещества, т.пл. 166-167oC.
Пример 18. (S)-N-[[3-[4-[4-(цианометил)-1-пиперазинил]-3,5-дифторофенил] -2-оксо-5- оксазолидинил]метил]ацетамид.
В соответствии с методикой, описанной в примере 17 (за исключением того, что монофторо-производное примера 17 (e) заменяли на дифторопиперазиновое производное ((S)-N-[[3-[3,5-дифторо-4-(1-пиперазинил)фенил]-2-оксо-5,5-оксазолидинил] метил] -ацетамид)), получали целевое соединение в виде белого порошка с т.пл. 150-154oC.
Пример 19. (+)-N-[[3-[4-[4-(2-цианоэтил)-1-пиперазинил]-3-фторофенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Раствор 75 мг (0,22 мМ) рацемической смеси примера 17 (стадии e) в 5 мл метанола обрабатывали 13 мг (17 мкл, 0,25 мМ) акрилонитрила, а затем нагревали с обратным холодильником в течение 3 ч. Раствор концентрировали в вакууме. Образовавшийся остаток подвергали круговой хроматографии (элюент: 5%-ный (об. /об. ) метанол в хлороформе). В результате получали 84 мг (97%) целевого соединения в виде белого твердого вещества, т.пл. 125-130oC.
Пример 20. (+)-N-[[3-[4-[4-(2-циано-2-пропил)-1-пиперазидинил]-3-фторофенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Раствор 75 мг (0,22 мМ) рацемической смеси примера 17 (e) в 1 мл сухого ацетонитрила последовательно обрабатывали 5 мг (33 мкл, 0,45 мМ) сухого ацетона и 44 мг (59 мкл, 0,45 мМ) триметилсилилцианида. Полученный раствор нагревали с обратным холодильником в течение 18 ч, а затем разводили этилацетатом и экстрагировали водой. После осушки сульфатом натрия и концентрирования в вакууме получали коричневатое твердое вещество. Это вещество подвергали круговой хроматографии, элюируя 5%-ным (об./об.)метанолом в дихлорметане. В результате получали 36 мг (40%) целевого соединения в виде белого твердого вещества, т.пл.139-143oC.
Пример 21. (S)-N-[[3-[4-[4-(4-цианотетрагидропиран-4-ил)-1-пиперазинил] -3-фторофенил]-2-оксо-5-оксазолидинил]метил-ацетамид.
Раствор 50 мг (0,15 мМ) продукта примера 17(e) в 2 мл сухого ацетонитрила обрабатывали последовательно 3 мг (0,02 мМ) безводного хлорида цинка, 30 мг (28 мкл, 0,30 мМ) тетрагидропиран-4-он'а и 29 мг (40 мкл, 0,30 мМ) триметилсилилцианида. Полученный раствор нагревали с обратным холодильником в течение 30 ч, разводили этилацетатом и экстрагировали водой. После осушки сульфатом натрия и концентрирования в вакууме получали светло-желтое твердое вещество. Этот материал подвергали круговой хроматографии, элюируя метанолом/дихлорметаном. В результате получали 24 мг (36%) целевого соединения в виде белого твердого вещества, т.пл. 134-137oC.
Пример 22. (+)-N-[[3-[3-фторо-4-(4-формил-1-пиперазинил)-фенил]-2-оксо-5-оксазолидинил]метил]-ацетамид.
Раствор 0,250 г (0,267 мМ) рацемического продукта примера 17 (e) в 4 мл ТГФ и 2 мл воды обрабатывали 11 мг (9 мкл, 0,243 мМ) муравьиной кислоты и pH доводили до 4,5 путем добавления 0,1 н. водной соляной кислоты. После этого смесь добавляли к 153 мг (0,80 мМ) гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида в 1 мл воды, перемешивая при комнатной температуре. После этого смесь доводили до pH=4,5 путем добавления 2н.раствора гидроксида натрия и 0,1 н.раствора соляной кислоты. После приблизительно 1-часового перемешивания добавляли еще карбодиимид (100 мг, 0,54 мМ) и муравьиную кислоту (30 мг, 0,80 мМ), после чего смесь перемешивали при комнатной температуре в течение 16 ч. Эту смесь разбавляли водой и экстрагировали этилацетатом. Органический слой экстрагировали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, после чего осушали сульфатом натрия и концентрировали в вакууме. Образовавшееся белое твердое вещество подвергали хроматографии на силикагеле (элюент: метанол/метиленхлорид) и получали 0,094 г (97%) целевого соединения в виде белого твердого продукта, т.пл. 190-193,5oC.
Пример 23. Метиловый сложный эфир (S)-4-[4-[5-[(ацетиламино)метил]-2-оксо-3-оксазолидин)]-2-фторофенил]-1-пиперазинкарбоновой кислоты.
Раствор продукта примера 17 (стадии e) в 22 мл ацетона и 11 мл воды обрабатывали 150 мг (1,80 мМ) бикарбоната натрия и охлаждали до 0oC, а затем добавляли 0,170 г (0,14 мл, 1,80 мМ) метилхлороформата. После выдерживания в течение 2 ч смесь разводили этилацетатом и экстрагировали водой и насыщенным раствором хлорида натрия. После осушки сульфатом натрия и концентрирования в вакууме получали белое твердое вещество, которое очищали с помощью хроматографии на силикагеле (элюент: система ацетона и метиленхлорида). В результате получали 0,494 г (77%) целевого соединения в виде белого твердого вещества, т.пл.179,5-182oC.
Пример 24. Метиловый сложный эфир (S)-4-[4-[5-[(ацетиламино)-метил]-2-оксо-3-оксазолидин)]-2,6-дифторофенил]-1-пиперазинкарбоновой кислоты.
В соответствии с методикой, описанной в примере 23, за исключением того, что продукт примера 17(e) заменяли на дифторопиперазин, получали целевое соединение в виде белого порошка, т.пл.175-178oC.
Пример 25. (+)-N-[[3-[4-[3-фторо-4-[(фенилкарбонил)-1-пиперазинил]]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
В соответствии с методикой, описанной в примере 23 (используя рацемическую смесь примера 17(e), за исключением того, что метилхлороформат заменяли на бензоилхлорид, получали целевое соединение в виде белого порошка, т. пл. 184-187oC.
Пример 26. 2-Метоксиэтиловый сложный эфир (+)-4-[4-[5-[(ацетиламино) метил]-2-оксо-3-оксазолидин)]-2-фторофенил]-1-пиперазинкарбоновой кислоты.
Раствор 75 мг (0,22 мМ) монофторопиперазинового производного примера 25 в 4 мл ацетона и 2 мл воды обрабатывали 21 мг (0,25 мМ) бикарбоната натрия, а затем охлаждали до 0oC. Полученный раствор обрабатывали 35 мг (0,25 мМ) раствора 2-метоксиэтилхлороформата в 0,5 мл тетрагидрофурана. После этого раствор нагревали до комнатной температуры в течение 22 ч. Этот раствор разводили этилацетатом и экстрагировали водой, насыщенным раствором бикарбоната натрия, а затем насыщенным раствором хлорида натрия. Органический слой осушали сульфатом натрия и концентрировали в вакууме с получением белого твердого остатка. Этот остаток подвергали круговой хроматографии, элюируя 30%-ным (об./об.)ацетоном/дихлорметаном). В результате получали 92 мг (96%) целевого соединения в виде белого твердого вещества, т.пл.166-167oC.
Пример 27. 2-Метоксиэтиловый сложный эфир (S)-4-[4-[5-[(ацетиламино)метил] -2-оксо-3-оксазолидин)] -2,6-дифторофенил]-1-пиперазинкарбоновой кислоты.
В соответствии с методикой, описанной в примере 26, за исключением того, что продукт примера 17 (e) заменяли на дифторопиперазин 7, получали целевое соединение в виде белого твердого вещества, т.пл. 154,5-156oC.
Пример 28. 2-(Фенилметокси)этиловый сложный эфир (±)-4-[4-5-[(ацетиламино)метил]-2-оксо-3-оксазолинил-2-фторофенил]-1-пиперазинкарбоновой кислоты.
Раствор 208 мг (0,25 мМ) продукта примера 25 в 3 мл ацетона и 2 мл воды обрабатывали бикарбонатом натрия (21 мг, 0,25 мМ), а затем охлаждали до 0oC. После этого смесь снова обрабатывали 54 мг (0,25 мМ) раствора 2-(фенилметокси) этилхлорформата в 2 мл ацетона. Этот раствор оставляли для нагревания до комнатной температуры на 22 ч, после чего разводили этилацетатом и экстрагировали водой и насыщенным раствором бикарбоната натрия. В результате осушки сульфатом натрия и концентрирования в вакууме получали белое твердое вещество, которое подвергали круговой хроматографии (элюент: 20%-ный (об. /об. ) ацетон в дихлорметане). Таким образом получали 113 мг (100%) целевого соединения в виде белого твердого вещества с т.пл.121-123oC.
Пример 29. 2-(фенилметокси)этиловый сложный эфир (S)-4-[4-[5-[(ацетиламино)метил] -2-оксо-3-оксазолинил-2,6-дифторофенил]-1-пиперазинкарбоновой кислоты.
В соответствии с методикой, описанной в примере 28, за исключением того, что соединение примера 17 (e) заменяли на дифторопиперазин 7, получали целевое соединение в виде белого твердого вещества, т.пл. 108-110oC.
Пример 30. 2-Гидроксиэтиловый сложный эфир (±)-4-[4-[5-[(ацетиламино)метил] -2-оксо-3-оксазолинил-2,6-дифторофенил]-1-пиперазинкарбоновой кислоты.
Раствор 113 мг соединения примера 28 в 5 мл метанола обрабатывали 35 мг 10 %-ного палладированного угля, а затем гидрогенизировали при атмосферном давлении в течение часа. Полученную смесь фильтровали через целит, а осадок на фильтре промывали метанолом. Фильтрат концентрировали в вакууме и получали белое твердое вещество. Это вещество очищали с помощью круговой хроматографии, элюируя системой растворителей из метанола и хлороформа. В результате получали 76 мг (82%) целевого соединения в виде белого твердого вещества, т.пл. 203-206oC.
Пример 31. [S-(R)] -N-[[3-[3,5-дифторо-4-[4-[(тетрагидро-2-фуранил)карбонил]-1-пиперазинил]фенил]-2-оксо-5-оксазолидинил]- метил]ацетамид.
Раствор 100 мг (0,28 мМ) дифторопиперазинового производного ((S)-N-[[3-[3,5-дифторо-4-(1-пиперазинил)фенил] -2-оксо-5-5-оксазолидинил] метил]-ацетамида) и 144 мг (1,25 мМ) (R)-2-тетрагидрофурановой кислоты в 4 мл тетрагидрофурана и 2 мл воды доводили до pH=4,5 путем добавления 2 н.раствора NaOH. Этот раствор обрабатывали раствором 324 мг (1,69 мМ) гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида в 2 мл воды. Затем этот раствор поддерживали при pH=4,6 путем добавления 2 н.раствора NaOH, перемешивая при комнатной температуре в течение 1,5 ч. Полученный раствор разводили водой и экстрагировали этилацетатом. Объединенные органические слои осушали сульфатом натрия и концентрировали в вакууме, в результате чего получали коричневатое твердое вещество, которое подвергали круговой хроматографии (элюент: система растворителей из метанола/дихлорметана). В результате получали 106 мг (83%) целевого амида в виде белого твердого вещества, т.пл. 198-200oC.
Пример 32. (S)-N-[[3-[3,5-дифторо-4-[4-[2-(1-пиперидинил)-этил]-1-пиперазинил]фенил]-2-оксо-5-оксазолидинил]метил]-ацетамид.
В последующих стадиях раскрываются получения дифторопромежуточных соединений настоящего изобретения.
(a) 2,6-Дифторо-4-нитробензол(трифторометан)сульфонат.
2,6-Дифторо-4-нитрофенол (31,55 г, 180,19 мМ) объединяли с метиленхлоридом (300 мл) и пиридином (29,15 мл, 360,38 мМ). Полученную суспензию охлаждали до 0oC в ледяной бане и по капле обрабатывали ангидридом трифтороуксусной кислоты (31,8 мл, 189,2 мМ) в течение 45 мин. Реакционную смесь оставляли для перемешивания при 0oC на 2 ч, а затем хранили в течение ночи в холодильнике при 5oC. Завершение реакции контролировали с помощью ТСХ (15% EtOAc/гексан, коротковолновое УФ). После этого реакционную смесь концентрировали при пониженном давлении, а затем обрабатывали водой (50 мл) и этилацетатом (50 мл). Эту смесь переносили в делительную воронку со 100 мл этилацетата и промывали 1н. раствором соляной кислоты до тех пор, пока промывки не становились кислотными (2х100 мл). Водные фазы подвергали обратному экстрагированию этилацетатом (2х100 мл). Объединенные этилацетатные экстракты снова объединяли и промывали 1 н.раствором соляной кислоты (400 мл), после чего один раз промывали солевым раствором (400 мл). Органическую фазу осушали безводным сульфатом натрия, фильтровали и концентрировали с получением 54,092 г красно-золотистого маслообразного вещества. Хотя это вещество было чистым, о чем свидетельствовал ЯМР-анализ, его объединяли с неочищенными продуктами из других реакций и хроматографировали на колонке с силикагелем (550 г), упакованной 5 %-ным этилацетатом. После элюирования 2 л 5%-ного этилацетата и 2 л 10%-ного этилацетата получали целевое соединение (полный выход 95%) в виде бледно-желтого маслообразного вещества, ВРМС (М+) для C7H2F5NO5S: вычислено: 3069574; найдено: 3069590.
(b) 1-(трет-Бутоксикарбонил)-4-(2,6-дифторо-4-нитрофенил)-пиперазин.
Раствор 2,6-дифторо-4-нитробензола(трифторометан)сульфоната (55 г, 179 мМ) в сухом ДМФ (275 мл) обрабатывали 1-(трет-бутоксикарбонил)пиперазином (45,71 г, 250 мМ). Образовавшийся прозрачный желтый раствор становился оранжевым после добавления N,N-диизопропилэтиламина (47 мл, 269 мМ). Реакционную смесь нагревали с обратным холодильником в течение 15 ч в атмосфере азота. Завершение реакции контролировали с помощью ТСХ (30% EtOAc/гексан, коротковолновое УФ). Реакционную смесь концентрировали досуха и объединяли с неочищенным продуктом из другой реакции для очистки. Этот неочищенный материал растворяли в горячем метиленхлориде (420 мл; некоторые твердые вещества, не родственные данному продукту, не растворялись), а затем хроматографировали на 3 отдельных колонках (2 колонки: 750 г силикагеля; элюирование 1 л каждого из 1-5% EtOAc/CH2Cl2; одна колонка: 250 г силикагеля; упаковка метиленхлоридом; загрузка 60 мл соединения; элюирование 2,5 и 5 %-ным EtOAc/CH2Cl2), в результате чего получали целевое соединение (выход 87%) в виде оранжевого твердого продукта, ВРМС (М+) для C15H19F2N3O4: вычислено: 3431343, найдено: 3431358.
(c) 1-(трет-бутоксикарбонил)-4-[2,6-дифторо-4-(бензилокси-карбонил)аминофенил]пиперазин.
1-(трет-Бутоксикарбонил)-4-(2,6-дифторо-4-нитрофенил)- пиперазин (44,7 г, 130 мМ) растворяли в 20% ТГФ/MeOH (600 мл) в 2-литровой колбе. Затем к смеси порциями добавляли формат аммония (41 г, 651 мМ), после чего добавляли 10 %-ный палладий на углероде (1,12 г, 2,5 мас.%), охлаждая при этом на ледяной бане. После завершения добавления баню удаляли. Содержимое колбы слегка нагревали, в результате чего желтый цвет исчезал. Завершение реакции контролировали с помощью ТСХ (30% EtOAc/гексан, коротковолновое УФ) в течение 1,5 ч. После этого реакционную смесь фильтровали через целит, а осадок на фильтре промывали 500 мл MeOH. Фильтрат концентрировали при пониженном давлении и получали твердое вещество, которое обрабатывали 1 л EtOAc и 500 мл воды. Слои разделяли, и органический слой промывали водой (500 мл) и солевым раствором (500 мл). Водные порции подвергали обратному экстрагированию с дополнительным количеством EtOAc (2х300 мл). Объединенные органические экстракты осушали безводным сульфатом натрия, фильтровали и концентрировали с получением желтого твердого вещества (40,8 г), которое сразу растворяли в сухом ДМФ (500 мл) и охлаждали до -20oC (в бане из льда/MeOH) в атмосфере азота. Полученный раствор обрабатывали N,N-диметиланилином (20,6 мл, 163 мМ), а затем по каплям добавляли бензилхлороформат (21,5 мл, 143 мМ). Ледяную баню оставляли для таяния в течение ночи. Завершение реакции контролировали с помощью ТСХ (30% EtOAc/гекса, коротковолновое УФ). Смесь концентрировали до получения желтого маслообразного вещества, которое растворяли в 1 л EtOAc и промывали 500 мл солевого раствора. Водные части подвергали обратной экстракции дополнительным количеством EtOAc (2х 300 мл). Объединенные органические экстракты осушали безводным сульфатом натрия, фильтровали и концентрировали с получением желтого твердого вещества. Этот неочищенный материал перекристаллизовывали из горячего EtOAc/гексана и получали 39,11 г (67%) целевого соединения в виде бледно-желтого кристаллического твердого вещества, т.пл. 171-172oC.
(d) [3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метанол.
1-(трет-Бутоксикарбонил)-4-[2,6-дифторо-4-(бензилоксикарбонил)аминофенил]пиперазин (14,05 г, 31 мМ) растворяли в безводном ТГФ (158 мл), а затем охлаждали до -78oC (в бане из сухого льда/ацетона). После этого в реакционную смесь по капле в течение 25 мин добавляли н-бутиллитий (21,6 мл, 35 мМ). Эту реакционную смесь оставляли на 30 мин для перемешивания при -78oC, после чего по капле в течение 7 мин добавляли (R)-(-)-глицидилбутират (4,89 мл, 35 мМ). Реакционную смесь поддерживали при -78oC еще 15 мин, а затем баню удаляли, и медленно нагревали смесь до комнатной температуры в течение ночи. Завершение реакции контролировали с помощью ТСХ (5% MeOH/CHCl3, коротковолновое УФ). Полученную реакционную смесь разводили 500 мл метиленхлорида, а затем промывали водой (Хх300 мл) и солевым раствором (300 мл). Водные порции подвергали обратной экстракции дополнительным количеством CH2Cl2 (3х400 мл). Объединенные органические экстракты осушали сульфатом натрия, фильтровали и концентрировали, в результате чего образовывалось кремообразное желтое твердое вещество. Это неочищенное вещество очищали путем перекристаллизации из горячего EtOAc/гексана, и получали 11,063 г (85%) целевого соединения в виде белого твердого вещества, т.пл.164-166oC.
(e) [[3-[3,5-дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил]фенил]-2-оксо-5- оксазолидинил]метил]-р-толуолсульфонат.
[[3-3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] фенил] -2-оксо-5-оксазолидинил]метанол (24,2 г, 59 мМ) растворяли в пиридине (110 мл), а затем охлаждали до 0oC в ледяной бане. После этого добавляли только что перекристаллизованный п-толуолсульфонихлорид (13,4 г, 70 мМ), и реакционную смесь оставляли на 2,5 ч для перемешивания при 0oC в атмосфере азота. Затем реакционную колбу закрывали пробкой и хранили в холодильнике при 5oC в течение ночи. За это время, реакционная смесь становилась бледно-розовой суспензией. ТСХ обнаруживала присутствие некоторого количества спирта. После этого смесь обрабатывали еще 1,12 г п-толуолсульфонилхлорида (5,85 мМ), каталитическим количеством 4-(диметиламино)пиридина и 20 мл безводного метиленхлорида для облечения перемешивания. После выдерживания смеси в течение 4 ч при 0oC реакция была полностью завершена, о чем свидетельствовала ТСХ (5% MeOH/CH2Cl3, коротковолновое УФ). Затем к смеси добавляли 750 мл ледяной воды, и осажденный продукт выделяли с помощью вакуумной фильтрации, промывая 1 л воды и 500 мл эфира. После осушки продукта в вакууме получали 29,921 г (выход 90%) целевого соединения в виде белого твердого вещества, т.пл. 150,5-151,5oC.
(f) [[3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] -2-оксо-5- оксазолидинил]метил]метансульфонат.
[3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] фенил] -2-оксо-5-оксазолидинил метанол (3,831 г, 9,27 мМ) растворяли в метиленхлориде (40 мл), охлаждали до 0oC, а затем обрабатывали триэтиламином (1,74 г, 2,4 мл, 17,22 мМ) в атмосфере азота. Затем в течение одной минуты медленно добавляли метансульфонилхлорид (1,48 г, 1 мл, 12,9 мМ). Через полчаса ТСХ-анализ (20% ацетон/CH2Cl2) указывал на полное завершение реакции. Реакционную смесь разводили метиленхлоридом (200 мл), промывали водой (3х50 мл) и солевым раствором (50 мл), после чего осушали сульфатом натрия, фильтровали и концентрировали в вакууме, в результате чего получали целевое соединение в виде беловатого твердого продукта, ВРМС (М+) для C20H27F2N3O7S: вычислено: 4911538, найдено:4911543.
(g) [[3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил]фенил]-2-оксо-5-оксазолидинил]метил]азид.
[3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] фенил] -2-оксо-5-оксазолидинил] метил] -п-толуолсульфонат (29,661 г, 52 мМ) растворяли в сухом ДМФ (125 мл), а затем обрабатывали твердым NaN3 (10,19 г, 156 мМ) при комнатной температуре. После этого реакционную смесь нагревали до 60oC в течение 3 ч и оставляли для охлаждения до комнатной температуры в течение ночи в атмосфере азота. Завершение реакции контролировали с помощью ТСХ (30% EtOAc/гексан; два прогона; коротковолновое УФ). Эту реакционную смесь концентрировали в вакууме и получали кремообразное обесцвеченное твердое вещество. Этот неочищенный продукт растворяли в 600 мл EtOAc, а затем промывали водой (2х500 мл) и солевым раствором (500 мл). Водные фракции подвергали обратному экстрагированию дополнительным количеством EtOAc (2х400 мл). Объединенные органические экстракты осушали сульфатом натрия, фильтровали и концентрировали в вакууме, в результате чего получали 22,41 г (91%) целевого соединения в виде бледно-желтого твердого вещества, т.пл. 115-117oC.
Используя в основном идентичные условия, соответствующий мезилат превращали в тот же самый азид.
(h) N-[[3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил]фенил] -2-оксо-5-оксазолидинил метил ацетамид.
[[3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил]фенил]- 2-оксо-5-оксазолидинил] метил]азид (22,4 г, 51 мМ) растворяли в 1 л этилацетата, а затем три раза дегазировали азотом. После этого добавляли 10 %-ный палладированный уголь (4,48 г, 20% по массе), и раствор снова три раза дегазировали азотом, после чего азот заменяли на водород (баллонный). Через 3 ч завершение реакции контролировали с помощью ТСХ (20% MeOH/CHCl3, коротковолновое УФ). Затем добавляли пиридин (8,26 мл, 102 мМ), после чего смесь обрабатывали уксусным ангидридом (9,64 мл, 102 мМ). Полученную реакционную смесь оставляли в течение ночи при комнатной температуре для перемешивания. Завершение реакции контролировали с помощью тонкослойной хроматографии (20% MeOH/CHCl3, коротковолновое УФ-излучение). Затем реакционную смесь фильтровали через целит, а образовавшийся на фильтре осадок промывали 500 мл этилацетата. Фильтрат концентрировали до получения объема приблизительно 600 мл, а затем промывали водой (2х500 мл) и солевым раствором (500 мл). Водные части подвергали обратному экстрагированию дополнительным количеством этилацетата (2х500 мл). Объединенные органические экстракты осушали безводным сульфатом натрия, фильтровали и концентрировали с получением желтого твердого вещества. Этот неочищенный продукт перекристаллизовывали из горячего CHCl3 и гексана, в результате чего получали 19,167 г (83%) целевого соединения в виде белого твердого продукта, т.пл.177-179oC.
(i) N-[[3-[3,5-Дифторо-4-(1-пиперазинил)фенил] -2-оксо-5-оксазолидинил] метил] ацетамид.
N-[[3-[3,5-Дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] фенил]-2-оксо-5- оксазолидинил]метил]ацетамид (1,00 г, 2,20 мМ) растворяли в метиленхлориде (6 мл) и охлаждали до 0oC в ледяной бане. После добавления 20 мл трифтороуксусной кислоты охлаждающую баню удаляли, и реакционную смесь оставляли для нагревания до комнатной температуры на 1 ч. После этого реакционную смесь концентрировали в вакууме, а образовавшийся остаток растворяли в воде (15 мл). Полученный раствор добавляли к ионообменной смоле AG-I-X8 (Bio Pad) (12 мл; OH- форма; промытая водой до нейтрализации). После добавления 5 мл воды смесь перемешивали в течение 10 мин. Затем смесь фильтровали, а смолу промывали дополнительным количеством воды (3х5 мл). Этот водный фильтрат лиофизиловали и получали 0,559 г (72%) целевого соединения в виде белого твердого вещества, т.пл. 108-112oC (с разл.).
(j) (S)-N-[[3-[3,5-Дифторо-4-[4-[2-(1-пиперидинил)этил] -1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Смесь (S)-N-[[3-[3,5-дифторо-4-(1-пиперазинил)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамида (0,200 г, 0,565 мМ), моногидрохлорида 1-(2-хлороэтил)пиперидина (0,125 г, 0,678 мМ) и карбоната калия (0,478 г, 3,39 мМ) в ацетонитриле (10 мл) нагревали с обратным холодильником в течение полутора часов. Полученную реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Образовавшийся остаток растирали с дихлорметаном, твердые вещества отфильтровывали, а фильтрат концентрировали в вакууме, в результате чего получали беловатое твердое вещество (0,248 г). Этот неочищенный материал хроматографировали на силикагеле (5 г), элюируя 5 %-ным метанолом/хлороформом, а затем 10 %-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,137 г (52%) целевого соединения в виде беловатого твердого вещества, т.пл.198-200oC.
Пример 33. (S)-N-[[3-[3-фторо-4-[4-[2-(1-пиперидинил)этил]-1-пиперидинил)этил]-1- пиперазинил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Смесь (S)-N-[[3-[3-фторо-4-(1-пиперазинил)фенил]-2-оксо-5-оксазолидинил] метил] ацетамид (0,200 г, 0,595 мМ), моногидрохлорида 1-(2-хлороэтил)пиперидина (0,131 г, 0,714 мМ) и карбоната калия (0,493 г, 3,57 мМ) в ацетонитриле (12 мл) нагревали с обратным холодильником в течение 1 ч. Образовавшийся остаток растирали с гексаном, охлаждали до комнатной температуры и концентрировали в вакууме. Твердые вещества отфильтровывали, а фильтрат концентрировали в вакууме с получением неочищенного продукта (0,308 г). Этот неочищенный материал хроматографировали на 5 г силикагеля, элюируя 5%-ным метанолом/хлороформом, а затем 10 %-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,192 г (72%) целевого соединения в виде беловатого твердого вещества, т.пл. 169-170oC.
Пример 34. (S)-N-[[3-[3-фторо-4-[4-[2-(4-морфолинил)этил]-1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Смесь (S)-N-[[3-[3-фторо-4-(1-пиперазинил)фенил]-2-оксо-5-оксазолидинил] метил] ацетамида (0,200 г, 0,595 мМ), гидрохлорида 4-(2-хлороэтил)морфолина (0,133 г, 0,714 мМ) и карбоната калия (0,493 г, 3,57 мМ) в ацетонитриле (12 мл) нагревали с обратным холодильником в течение часа. Полученную реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток растирали с дихлорметаном, твердые вещества отфильтровывали, а фильтрат концентрировали в вакууме с получением смолы янтарного цвета (0,201 г). Этот неочищенный материал хроматографировали на силикагеле (5 г), элюируя 5 %-ным метанолом/хлороформом, а затем 10%-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,129 г (48%) целевого соединения в виде беловатого твердого вещества, т.пл.150-151,5oC.
Пример 35. (S)-N-[[3-[4-[2-(диэтиламино)этил]-1-пиперазинил]-3-фторофенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Смесь (S)-N-[[3-[3-фторо-4-(1-пиперазинил)фенил]-2-оксо-5-оксазолидинил] метил] ацетамида (0,200 г, 0,595 мМ), гидрохлорида 2-диэтиламиноэтилхлорида (0,123 г, 0,714 мМ) и карбоната калия (0,493 г, 3,57 мМ) в ацетонитриле (12 мл) нагревали с обратным холодильником в течение часа. Полученную реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Образовавшийся остаток растирали с дихлорметаном, твердые вещества отфильтровывали, а фильтрат концентрировали в вакууме с получением 0,241 г беловатого смолообразного твердого вещества. Этот неочищенный материал хроматографировали на силикагеле (5 г), элюируя 5%-ным метанолом/хлороформом, а затем 10%-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,159 г (61%) целевого соединения в виде беловатого твердого вещества, т.пл. 131-133oC.
Пример 36. (S)-N-[[3-[3-фторо-4-[4-(2-метоксиэтил)-1-пиперазинил]фенил] -2-оксо-5-оксазолидинил]метил]ацетамид.
Смесь (S)-N-[[3-[3-фторо-4-(1-пиперазинил)фенил]-2-оксо-5-оксазолидинил] метил] ацетамида (0,450 г, 1,34 мМ), 2-хлороэтилметилового эфира (1,220 мл, 13,40 мМ) и карбоната калия (1,110 г, 8,04 мМ) в ацетонитриле (25 мл) нагревали с обратным холодильником в течение 24 ч. Полученную реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Образовавшийся остаток растирали с дихлорметаном, твердые вещества отфильтровывали, а фильтрат концентрировали в вакууме с получением желтого пенообразного твердого вещества (0,326 г). Этот неочищенный материал хроматографировали на силикагеле (25 г), элюируя 1%-ным метанолом/хлороформом, 3%-ным метанолом/хлороформом, а затем 5%-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,296 г (56%) целевого соединения в виде беловатого твердого вещества, т.пл.144,5-146oC.
Пример 37. (S)-N-[[3-[3,5-Дифторо-4-[4-(2-метоксиэтил)-1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Раствор (S)-N-[[3-[3,5-дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] фенил] -2-оксо-5-оксазолидинил] метил] ацетамида (0,200 г, 0,441 мМ) в дихлорметане (1 мл) обрабатывали трифтороуксусной кислотой (4 мл) при комнатной температуре в течение часа. Реакционную смесь концентрировали в вакууме, а полученный остаток объединяли с 2-хлороэтилметиловым эфиром (403 мкл, 4,41 мМ), карбонатом калия (0,730 г, 5,28 мМ) и ацетонитрилом (9 мл), и полученную смесь нагревали с обратным холодильником в течение 15 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток растирали с дихлорметаном, твердые вещества отфильтровывали, а фильтрат концентрировали в вакууме с получением неочищенного продукта. Этот неочищенный материал хроматографировали на силикагеле (10 г), элюируя 1%-ным метанолом/хлороформом, 3%-ным метанолом/хлороформом, а затем 5%-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,097 г (53%) целевого соединения в виде беловатого твердого вещества, т.пл. 162-164oC.
Пример 38. (S)-N-[[3-[3-фторо-4-[4-(3-гидроксипропил)-1-пиперазинил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Смесь (S)-N-[[3-[3-фторо-4-(1-пиперазинил)фенил] -2-оксо-5- оксазолидинил] метил] ацетамида (0,200 г, 0,595 мМ) 3-хлоро-1-пропанола (299 мкл, 3,57 мМ) и карбоната калия (0,493 г, 3,57 мМ) в ацетонитриле (12 мл) нагревали с обратным холодильником в течение 7 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Полученный неочищенный материал растворял и в 10%-ном метаноле/хлороформе и адсорбировали на слой силикагеля (2 г). Образовавшийся остаток хроматографировали на 10 г силикагеля, элюируя 1%-ным метанолом/хлороформом, 3%-ным метанолом/хлороформом, а затем 6%-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,096 г (41%) целевого соединения в виде белого твердого вещества, т. пл. 154-155,5oC.
Пример 39. (S)-N-[[3-[3,5-дифторо-4-[4-(2-гидроксиэтил)-1-пиперазинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид.
Раствор (S)-N-[[3-[3,5-дифторо-4-[4-(трет-бутоксикарбонил)-1-пиперазинил] фенил] -2-оксо-5-оксазолидинил] метил] ацетамида (0,400 г, 0,881 мМ) в дихлорметане (3 мл) при комнатной температуре и в течение часа обрабатывали 7 мл трифтороуксусной кислоты. Полученную реакционную смесь концентрировали в вакууме, а образовавшийся сироп янтарного цвета объединяли с 2-хлороэтанолом (354 мкл, 5,27 мМ), карбонатом калия (0,730 г, 5,27 мМ) и ацетонитрилом (20 мл). Полученную смесь нагревали с обратным холодильником в течение 24 ч. После этого смесь охлаждали до комнатной температуры и концентрировали в вакууме. Образовавшийся неочищенный продукт хроматографировали на силикагеле (10 г), элюируя 1 %-ным метанолом/хлороформом, 3%-ным метанолом/хлороформом, а затем 6%-ным метанолом/хлороформом. После концентрирования соответствующих фракций получали 0,067 г (выход 19%) целевого соединения в виде беловатого твердого вещества, т.пл.172-174oC.
Пример 40. (S)-N-[[3-[3-фторо-4-[4-[3-(4-морфолинил)-1-оксопропил]-1-пиперазинил]фенил]-2-оксо-5-оксазолидинил]-метил] ацетамид.
3-(4-Морфолинил)пропионовую кислоту (0,600 г, 2,11 мМ), полученную путем конденсирования морфолина с этилакрилатом (3 эквивалента) в кипящем этаноле, а затем путем дистилляции, омыления (1 н.водный раствор гидроксида натрия; кипящий тетрагидрофуран), нейтрализации (1 н. HCl) и лиофилизации, объединяли с 1,3-дициклогексилкарбодиимидом (0,434 г, 2,11 мМ), 4-(диметиламино)пиридином (13 мг, 0,11 мМ), (S)-N-[[3[3-фторо-4-(1- пиперазинил)фенил] -2-оксо-5-оксазолидинил]-метил]ацетамидом (0,354 г, 1,05 мМ) и тетрагидрофураном/дихлорметаном (1:1) (50 мл) при комнатной температуре. Через 3 дня реакционную смесь фильтровали для удаления осажденной 1,3-дициклогексилмочевины, а фильтрат концентрировали в вакууме. Полученный неочищенный продукт хроматографировали на силикагеле (20 г), элюируя градиентом 1-6%-ного метанола/хлороформа, в результате чего получали 0,446 г (95%) целевого соединения в виде белого твердого вещества, т.пл. 209-210oC.
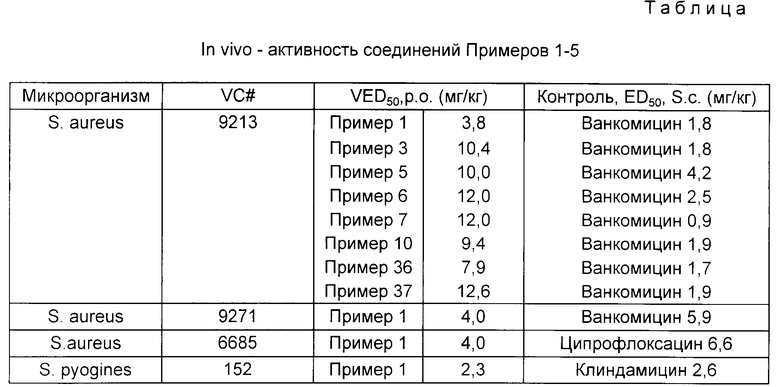
Использование: в химии гетероциклических соединений, проявляющих противомикробную активность. Сущность изобретения: производное оксазолидинона формулы I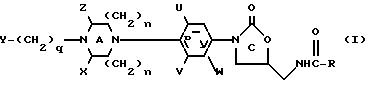
или его фармацевтически приемлемая соль, где n = 1; A - пиперазиновое кольцо; Y представляет собой: - C1-6-алкил; -C(O)-C1-6-алкил; -C(O)-)-C1-6-алкил; бензоил; 2-бензилоксиэтоксикарбонил, 1,4-диоксопентил; N(C1-4)2, пиперидил, морфолинил, группа формулы
X и Z независимо представляют собой C1-6-алкил или водород; причем в каждом случае указанный C1-6-алкил может быть замещен одним или несколькими заместителями, выбранными из F, Cl, Br, CN или OR1, где R1 представляет водород или C1-4-алкил; U, V и W независимо представляют собой F или водород; R - низший алкил; q равно 0-4 включительно. Предложен также способ лечения микробных инфекций у теплокровных животных путем введения 0,1-100 мг/кг веса тела в день (лучше 3,0-50 мг/кг веса в день) соединения формулы I. 2 с. и 11 з.п. ф-лы, 1 табл.
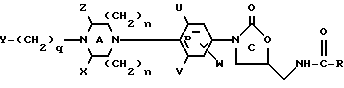
или его фармацевтически приемлемая соль, где n 1;
кольцо А представляет собой пиперазиновое кольцо;
Y -С1 - 6-алкил;
-С(О)-С1 - 6-алкил; -С(О)-О-С1 - 6-алкил; бензоил, 2-бензилоксиэтоксикарбонил; 1,4-диоксопентил; -N(С1 - 4-алкил)2, пиперидил, морфолинил, группа формулы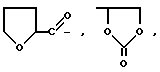
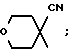
X и Z независимо С1 - 6-алкил или водород,
причем в каждом случае указанный С1 - 6-алкил может быть замещен одним или несколькими заместителями, выбранными из F, Cl, Br, CN или OR1, где R1 водород или С1 - 4-алкил;
U, V и W независимо фтор или водород;
R низший алкил;
q 0 4 включительно.
(а) 4-(4-(5-((ацетиламино)метил) -2-оксо-3-оксазолидинил)-2-фторфенил)-1-пиперазинкарбоновой кислоты метиловый сложный эфир;
(b) 4-(4-(5-((ацетиламино)метил)- 2-оксо-3-оксазолидинил)-2-фторфенил)-1-пиперазинкарбоновой кислоты этиловый сложный эфир;
(с) 4-(4-(5-((ацетиламино)метил)- 2-оксо-3-оксазолидинил)фенил)-1-пиперазинкарбоновой кислоты метиловый сложный эфир;
(d) N-((2-оксо-3-(4-(фенилкарбонил) -1-пиперазинил)фенил)-5-оксазолидинил)метил)ацетамид;
(е) N-((3-(4-(3-фтор-4-(2-цианоэтил)-1-пиперазинил))фенил) -2-оксо-5-оксазолидинил)метил)ацетамид;
(f) N-((3-(4-(3-фтор-4-(4-(2- гидроксиэтил)карбонил-1- пиперазинил))фенил)-2-оксо-5-оксазолидинил)метил)ацетамид;
(g) N-((3-(4-(3-фтор-4-((фенилкарбонил)-1-пиперазинил))фенил)-2- оксо-5-оксазолидинил)метил)ацетамид;
(h) 4-[4-[5-[(ацетиламино)метил] -2-оксо-3-оксазолидинил] -2-фторфенил] -1-пиперазинкарбоновой кислоты 2-метоксиэтиловый сложный эфир;
(i) 4-[4-[5-(ацетиламино)метил]-2- оксо-3-оксазолидинил]-2-фторфенил]-1-пиперазинацетонитрил;
(j) (±)-N-[[3-[4-[4-(1,4-диоксопентил)-1-пиперазинил]-3-фторфенил] -2-оксо-5-оксазолидинил]метил]ацетамид;
(k) (S)-N-[[3-[3-фтор-4-[4-(2-метоксиэтил)-1-пиперазинил] фенил] -2-оксо-5- оксазолидинил]метил]ацетамид или
(l) (S)-N-[[3-[3,5-дифтор-4-[4-(2-метоксиэтил)-1-пиперазинил] фенил]-2- оксо-5-оксазолидинил]метил]ацетамид.
| EP, патент, 352781, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 316594, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 312000, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

