Настоящее изобретение относится к новому производному 4-(4-фенил-1-пиперазинил)фенила, его фармацевтически приемлемой соли и к его стереоизомерной форме, а также к способу его получения и фармацевтической композиции, ингибирующей 5-липоксигеназу.
Известны производные 4-(4-фенил-1-пиперазинил)фенила в качестве промежуточных продуктов для получения соединений, обладающих фунгицидными и бактерицидными свойствами (Патенты США N 4267179 и 4619931; EP N 0228125).
Известен также N, N'-бис(4-оксифенил)пиперазин в качестве промежуточного продукта для получения соединений, обладающих фармакологическими свойствами, используемыми при лечении аллергических и аутоиммунных заболеваний.
Задачей настоящего изобретения является создание нового производного 4-(4-фенил-1-пиперазинил)фенила, обладающего свойством подавлять 5-липоксигеназу, а также способа получения таких соединений и новой фармацевтической композиции, ингибирующей 5-липоксигеназу, содержащей в качестве активного начала 4-(4-фенил-1-пиперазинил)фенилпроизводное.
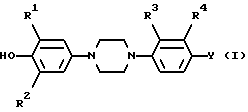
Поставленная задача достигается новым производным 4-(4-фенил-1-пиперазинил)фенила общей формулы: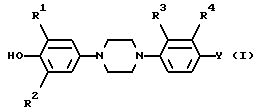
или его фармацевтически приемлемой соли присоединения кислоты или его стереоизомерной формой, где
R1 и R2 каждый независимо представляет водород, C1-6 алкил или галоид;
R3 представляет водород, галоид или нитро;
R4 представляет водород или трифторметил; Y представляет водород, нитро, C1-6 алкиламино, C1-6 алкилкарбониламино, C1-6 алкил, C1-6 алкилкарбонил, окси, галоид или гетероциклический радикал формулы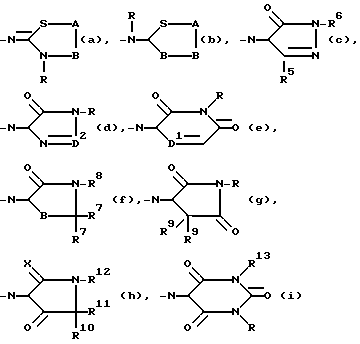
R представляет C1-6 алкил;
A - C/R14//R15/ и B -CH2- или -CH2-CH2 или A и B вместе взятые образуют двухвалентный радикал формулы -CH=CH-(j) или -CH=N(k), где атом C указанного последнего радикала (k) соединен с S; R14 и R15 каждый независимо представляет водород или C1-6 алкил; и в каждом из двухвалентных радикалов B, -CH=CH(j) и -CH=N(k) один или где возможно два атома водорода могут быть заменены на C1-6 алкил или фенил; и в двухвалентном радикале B два парных атома водорода могут быть замещены C4-6 алканедиилом;
R5 - водород или C1-6 алкил;
R6 - водород, C1-6 алкил, C3-7 циклоалкилтригало C1-6 алкил, (фенил) C1-6 алкил; причем C1-6 алкил, C3-7 циклоалкил и (фенил) C1-6 алкил могут быть замещены оксо- или окси- на любом атоме углерода C1-6 алкильной или C3-7 циклоалкильной части, при условии, что указанный атом C не присоединен к атому азота, имеющему указанный радикал R6;
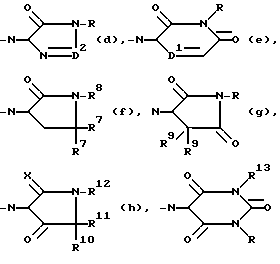
D2 - =N-, =CH- или =CH/CH3/;
D1 - -N= или -CH=;
каждый R7 - водород или C1-6 алкил;
R8 - C1-6 алкил, возможно замещенный оксо- на любом атоме углерода, при условии, что указанный атом углерода не находится рядом с атомом азота, имеющим указанный радикал R8;
каждый R9 - водород или C1-6 алкил;
X - O или S;
R10 - водород или C1-6 алкил;
R11 - водород или C1-6 алкил;
R12 - C1-6 алкил, не обязательно замещаемый оксо- на любом атоме углерода, при условии, что указанный атом углерода не прилегает к атому азота, имеющему указанный радикал R12; или R11 и R12 взятые вместе образуют радикал C3-5 алканедиил;
R13 - C1-6 алкил или (фенил) C1-6 алкил, причем указанный (фенил) C1-6 алкил может быть замещен оксо- или оксина любом из атомов углерода C1-6 алкильной части, при условии, что указанный атом углерода не прилегает к атому азота, имеющему указанный радикал R13;
одновременно фенил может быть замещен одним или двумя заместителями, выбранными из галоида, C1-6 алкила, C1-6 алкилокси, окси- или трифторметила,
при условии, что
a) по крайней мере один из R1 или R2 представляет C1-6 алкила или галоид, или
b) R3 представляет галоид или нитро; или
c) R4 представляет трифторметил; или
d) Y представляет C1-6 алкил, C1-6 алкиламино, C1-6 алкилкарбонил, галоид или гетероциклический радикал формулы (b); радикал (c), где R6 - C3-7 циклоалкил,
тригало C1-6 алкил, или C7 циклоалкил, замещенный оксо-; радикал (d) или (e); радикал (h), где R11 и R12 взятые вместе образуют C3-5 алканедиил радикал; или радикал формулы (i), где R13 - (фенил) C1-6 алкил, замещенный оксо- или гидрокси- на любом атоме углерода C1-6 алкильной части, при условии, что указанный атом углерода не присоединен к атому азота, имеющему указанный радикал R13; и
фенил возможно замещен одним или двумя радикалами, выбранными от галоида, C1-6 алкила, C1-6 алкилокси, окси- или трифторметила, в эффективном количестве.
В вышеприведенных определениях C1-6 алкил означает насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие 1-6 атомов углерода, например метил, этил, пропил, 1-метилэтил, четыре изомера бутила, изомеры пентила и гексила; C3-6 алкенил обозначает углеводородные радикалы с прямой и разветвленной цепью, содержащие одну двойную связь и от 3 до 6 атомов углерода, например 2-пропенил, 2-бутенил, 3-бутенил, 2-метил-2-пропенил, 2-пентенил, 3-пентенил, 4-пентенил, 3-метил-2-бутенил и изомеры гексенила; C3-6 алкинил обозначает углеводородные радикалы с прямой и разветвленной цепью, содержащие одну тройную связь и от 3 до 6 атомов углерода, например 2-пропинил, 2-бутинил, 3-бутинил, 2-пентинил, 3-пентинил или 4-пентинил и изомеры гексинила; и если сказано, что C3-6алкенил или C3-6 алкинил замещены на атоме азота, то атом углерода указанного C3-6 алкенила или C3-7 алкинила, связанный с указанным гетероатомом, предпочтительно является насыщенным; C3-5 циклоалкил обозначает циклопропил, циклобутил, циклопентил, циклогексил и циклогептил; C4-6 алкандиил и C1-6 алкандиил обозначают двувалентные насыщенные углеводородные радикалы, содержащие от 3 до 5 или, соответственно, от 4 до 6 атомов углерода, например 1,3-пропандиил, 1,4-бутандиил, 1,5-пентандиил и 1,6-гександиил; галоген обозначает фтор, хлор, бром или иод; термин "моно-, ди- и тригалоген C1-6 алкил", примененный выше, обозначает C1 алкильные радикалы, у которых один, два или три атома водорода замещены атомами галогена. В качестве примеров таких радикалов можно привести фторметил, дифторметил, трифторметил, трихлорметил, 2-хлорэтил, 2,2,2-трифторэтил и т.п.
В зависимости от природы различных заместителей соединения формулы (I) могут содержать асимметричный атом углерода. При отсутствии других указаний химическое название соединений обозначает смесь всех возможных стереохимических изомерных форм, причем указанные смеси содержат все диастереоизомеры и энантиомеры основной молекулярной структуры. Абсолютная конфигурация каждого хирального центра может быть указана стереохимическими обозначениями R и S, причем это обозначение с R и S соответствует правилам, приведенным в "Pure Appl. Chem.", 1976, 45, 11-30.
В некоторых соединениях стереохимическая конфигурация не определена экспериментально. В этих случаях решено условно обозначать стереохимический изомер, полученный первым, как "A", а стереоизомер, полученный вторым, как "В", без дальнейших ссылок на фактическую стереохимическую конфигурацию.
Чистые изомерные формы соединений формулы (I) могут быть выделены из смеси обычными методами разделения. Предпочтительно, если требуется получить конкретный стереоизомер, указанное соединение синтезируют стереоселективными методами получения. Для этих методов успешно применяют энантиомерно чистые исходные вещества.
Соединения формулы (I) обладают свойствами оснований и, следовательно, могут быть превращены в терапевтически активные нетоксичные кислотоаддитивные соли путем обработки соответствующими кислотами, например неорганическими кислотами, хлористоводородной, бромистоводородной и подобными кислотами, серной кислотой, азотной кислотой, фосфорной кислотой и т.п.; или органическими кислотами, например уксусной, пропановой, оксиуксусной, 2-оксипропановой, 2-оксопропановой, этандионовой, пропандионовой, бутадионовой, (Z)-2- бутендионовой, (E)-2-бутендионовой, 2-оксибутандионовой, 2,3-диоксибутандионовой, 2-окси-1,2,3-пропантрикарбоновой, метансульфоновой, этансульфоновой, бензолсульфоновой, 4- метилбензолсульфоновой, циклогексансульфаминовой, 2-оксибензойной, 4-амино-2-оксибензойной и подобными кислотами.
И наоборот, соли могут быть превращены в свободные основания путем обработки щелочами.
Используемый выше термин "кислотоаддитивная соль" включает в себя также сольваты, которые могут образовать соединения формулы (I), и указанные сольваты тоже входят в объем настоящего изобретения. Примерами таких сольватов являются, например, гидраты, алкоголяты и т.п.
Наиболее интересными новыми соединениями являются:
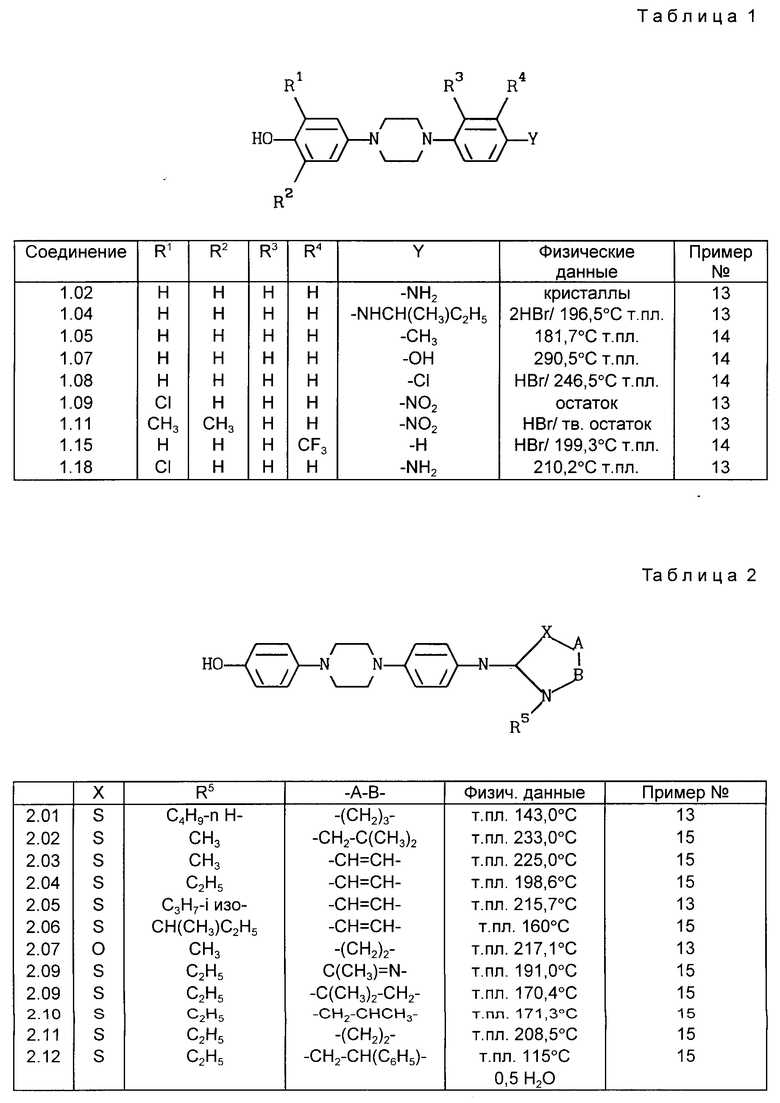
2,4-дигидро-4-/4-4-(4-окси-3,5-диметилфенил)-1-пиперазинил/фенил/-5 -метил-2-(1-метилпропил)-3H,-1,2,4-триазол-3-он,
2-/2-(4-бромфенил)-1-метил-2-оксоэтил)-2,4-дигидро-4- /4-/4-окси-3,5-диметилфенил)-1-пиперазинил/фенил/-5-метил-3H-1,2,4- триазол-3-он и
2-/2-(4-бромфенил)-2-окси-1-метилэтил/-2,4-дигидро-4-/4-/4-(окси-3,5- диметилфенил)-1-пиперазинил/фенил/-5-метил-3H-триазол-3-он.
Соединения общей формулы I получают диалкилированием алкоксипроизводного общей формулы II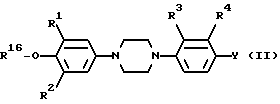
где
R1, R2, R3, R4 и Y имеют указанные в п. 1 значения, и R16-C1-6 алкил, в кислой среде или при помощи сильного нуклеофила, и при необходимости, превращением целевого продукта, где R6 или R13 представляет (фенил) C1-6 алкил, замещенный оксо- в C1-6 алкильной части, путем восстановления при помощи тетраборгидрата натрия в C1-4 алканоле, в целевой продукт формулы (I), где R6 или R13 представляет (фенил) C1-6 алкил, замещенный окси- в C1-6 алкильной части.
Соединения формулы (I) могут быть также превращены одно в другое известными способами функциональной перегруппировки. Ниже приведено несколько примеров.
Соединения формулы (I), содержащие нитрозаместитель, могут быть превращены в соответствующие амины путем перемешивания и, при желании, нагревания исходных нитросоединений в водородсодержащей среде в присутствии подходящего количества соответствующего катализатора, например платины на угле, палладия на угле, никеля Ренея и других подобных катализаторов. Пригодными растворителями являются, например, спирты, например метанол, этанол и т.п.
Атомы галогена - заместители на арильных группах - могут быть заменены водородом известными способами гидрогенолиза, то есть путем перемешивания и, при желании, нагревания исходных веществ в подходящем растворителе в атмосфере водорода в присутствии соответствующего катализатора, например палладия на угле или другого подобного катализатора.
Соединения формулы (I), в которой Y обозначает амин, могут быть также превращены в другие соединения, охватываемые формулой (I); например соединения, в которых Y обозначает (C4 алкил)карбониламино, могут быть получены при помощи реакции селективного N-ацилирования хлорангидридом или ангидридом карбоновой кислоты в пригодном растворителе, например ароматическом углеводороде, например бензоле, метилбензоле и т.п., диполярном апротонном растворителе, например N, N-диметилформамиде, диметилсульфоксиде и т.п., или в смеси таких растворителей, и в присутствии соответствующего основания, например N,N-диэтилэтанамина, пиридина и подобных оснований.
Многие промежуточные и исходные вещества, применяемые в вышеописанных реакциях, являются известными соединениями, которые могут быть получены в соответствии с известными методиками получения указанных или аналогичных соединений, а некоторые промежуточные соединения являются новыми. Некоторые способы получения таких соединений более подробно описаны ниже.
Стереохимически чистые изомерные формы соединений формулы (I) могут быть получены с применением известных методик. Диастереоизомеры могут быть выделены физическими методами разделения, например селективной кристаллизацией и хроматографией, например противоточного распределения, колоночной хроматографией или жидкостной хроматографией высокого разрешения, и энантиомеры могут быть разделены при помощи известных методов разделения, например методами селективной кристаллизации диастереомерных солей, полученных из оптически активных кислот. Чистые стереоизомеры могут также быть получены из соответствующих чистых стереохимических изомерных форм соответствующих исходных веществ при помощи стереоспецифических реакций.
Соединения формулы (I) являются сильными и селективными ингибиторами фермента 5-липоксигеназы как in vitro, так и in vivo. Подавление фермента 5-липоксигеназы эффективно блокирует обменные проводящие пути, ведущие от арахидиновой кислоты до лейкотриенов - веществ, которые, как известно, обладают рядом сильных физиологических действий и, как предполагают, участвуют в различных аллергических, анафилактических и воспалительных реакциях (Science, 220, 568-575, 1983).
Лейкотриены C4, D4 и E4 (LTC4, LTD4 и LTE4) интенсивно вызывают сокращение гладких мышц и, в частности, способны сильно сужать бронхи. Далее, указанные лейкотриены повышают проницаемость сосудов, в результате чего происходит вытекание внутрисосудистой жидкости и белков в ткани и образование эдем. Лейкотриен B2, сильный хемокинетический и хемотактический агент по отношению к лейкоцитам, предложено применять в качестве важного медиатора в реакциях непосредственной и подострой сверхчувствительности и при воспалительных процессах (The New England Journal of Medicine, 303, 822-825, 1980; "The Leukotrienes: Chemistry and Biology", ed. L.W. Chackin, D.M.Bailey, Academic Press, Orlando, 195-214, 1984). Все вышеупомянутые лейкотриены являются производными общего промежуточного соединения 5-гидропероксиэйкозатетраеновой кислоты (5-HPETE), образующейся из арахидоновой кислоты под действием 5-липоксигеназы. Другие липоксигеназы, например 12- и 15-липоксигеназа, превращают арахидоновую кислоту в другие моно- и диоксипроизводные с противоположной или синергической биологической активностью. Кроме того, известно о возросшем выделении продуктов ферментной активности 5-липоксигеназы и 12-липоксигеназы из поврежденной кожи пациентов, страдающих псориазом, а также атопическим дерматитом (Prostaglandins, 29, 611-619, 1985; J.Invest. Dermatol. 83, 70-73, 1983; Lancet; 222-223, 1984).
Следовательно, ингибиторы обусловленных действием липоксигеназ метаболических проводящих путей арахидоновой кислоты и обусловленных, в частности, действием фермента 5-липоксигеназы, считаются ценными терапевтическими лекарственными средствами для подавления вышеупомянутого вредного действия лейкотриенов. Сопутствующими болезнями и/или нарушениями являются, например, астма, аллергия, анафилаксия, псориаз и воспалительные реакции, например артриты и дерматиты. Значение настоящего изобретения заключается в том, что соединения формулы (I), рекомендуется для применения в соответствии с настоящим способом, являются сильными и селективными ингибиторами по отношению к ферменту 5-липоксигеназы. Большая часть других ингибиторов, о которых имеются сообщения, не обладают селективностью и одновременно подавляют другие липоксигеназы и/или циклооксигеназу - фермент, участвующий в метаболизме арахидоновой кислоты к простагландинам. Соединения формулы (I) незначительно подавляют соевую 15-дипоксигеназу, 12-липоксигеназу человеческого тромбоцита, циклогеназу тромбоцита человека и тромбоксан Ao синтетазу. Кроме того, соединения формулы (I) проявляют, как правило, только умеренные неспецифические противоокислительные свойства.
Соединения формулы (I)орально активны, как показал тест "Подавление вызванного декстраном образования эдемы в ушах у мышей" (пример 19).
Специалисты в данной области медицины легко могут определить эффективное количество ингибитора 5-липоксигеназы по результатам, представленным ниже. Обычно пригодная дневная доза для введения составляет приблизительно от 0,1 мг/кг до 10 мг/кг живого веса.
С учетом их подавляющей 5-липоксигеназы активности соединения согласно настоящему изобретению могут быть приготовлены для введения в различных фармацевтических формах. Для приготовления фармацевтической композиции согласно настоящему изобретению эффективное количество конкретного соединения или его кислотоаддитивной соли, взятые в качестве активного ингредиента, тщательно смешивают с фармацевтически приемлемым носителем, причем такой носитель может быть самого различного вида в зависимости от вида препарата, предназначенного для введения. Такие фармацевтические композиции желательно изготовлять в дозировке на один прием, пригодной предпочтительно для введения орально, ректально, подкожно или парентерально. Например, для приготовления композиции в виде оральной дозы может быть применен любой из обычных фармацевтических носителей, например вода, гликоли, масла, спирты и т.п. в случае приготовления жидких оральных составов, например суспензий, сиропов, эликсиров и растворов; или твердые носители, например крахмал, сахар, каолин, смазочные вещества, связующие вещества, расщепляющие агенты и т.п. для приготовления порошков, пилюль, капсул и таблеток. Наилучшей оральной формой дозы на один прием являются таблетки и капсулы вследствие удобства их приема, и в этом случае применяют твердые фармацевтические носители. Носитель для парентеральных композиций обычно включает в себя стерилизованную воду, по меньшей мере в большей части, но может содержать также и другие ингредиенты, например, для повышения растворимости активного ингредиента. Например, могут быть приготовлены растворы для инъекций, в которых носитель включает в себя солевой раствор, раствор глюкозы или смесь растворов соли и глюкозы. Могут быть приготовлены также суспензии для инъекций, и в этом случае могут быть применены соответствующие жидкие носители, суспендирующие агенты и т.п. В композициях для подкожного введения носитель не обязательно содержит агент, ускоряющий проникновение, и/или подходящий смачивающий агент, не обязательно в сочетании с минимальными количествами добавок любой природы, которые не оказывают значительного вредного действия на кожу. Указанные добавки могут облегчать введение в кожу и/или могут быть полезны при получении нужных композиций. Эти композиции могут быть введены различными способами, например трансдермальным путем в виде мази. Кислотоаддитивные соли соединений формулы (I) более пригодны для приготовления водных композиций вследствие их лучшей растворимости в воде по сравнению с соответствующими основаниями. Особенно целесообразно готовить вышеуказанные фармацевтические композиции с дозировкой на один прием для облегчения приема и достижения единообразия дозировки. Термин "дозировка на один прием", примененный здесь в описании и в формуле изобретения, относится к физически дискретным единицам, пригодным для применения в качестве одной дозы, причем каждая единица содержит заданное количество активного ингредиента, рассчитанное так, чтобы оказать желаемое терапевтическое действие в сочетании с требуемым фармацевтическим носителем. В качестве примеров таких дозировок на один прием можно привести таблетки (включая таблетки с надрезом или покрытием), капсулы, пилюли, порошки в пакетиках, облатки, растворы или суспензии для инъекций и т. п., а также их выделенные краткие количества.
Нижеследующие примеры иллюстрируют, но ни в коей мере не ограничивают настоящее изобретение во всех его особенностях. При условии других указаний все части даны по весу.
A. Получение промежуточных соединений
Пример 1.
a) К перемешиваемому раствору 20 ч. 1-(4-изотиоцианатфенил) -4-(4-метоксифенил)пиперазина в 325 ч. дихлорметана добавляют 40 ч. метанола, насыщенного аммиаком. Реакционную смесь перемешивают в течение 5 дней при комнатной температуре. Выпавший в осадок продукт отфильтровывают, промывают дихлорметаном и высушивают, получают 20,5 ч. (98,1 %) N-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/тиомочевины; т. пл. 265,2oC (промежуточное соединение 1).
b) Смесь 5 ч. N-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/мочевины, 3 ч. 2-хлор-1-фенилэтанона, 1,7 ч. ацетата натрия и 100 ч. уксусной кислоты перемешивают в течение 4 ч при температуре 80oC. После охлаждения реакционную смесь выпаривают в остаток перемешивают в 130 ч. дихлорметана. Все это нейтролизуют раствором бикарбоната натрия. Выпавший в осадок продукт отфильтровывают, промывают водой и дихлорметаном и кристаллизуют из 1,4-диоксана. Продукт отфильтровывают и высушивают, получают 4,5 ч. (69,6 %) N-/4-/4-метоксифенил)1-пиперазинил/фенил/-4-фенил-2-тиазоламина; т. пл. 269,7oC (промежуточное соединение 2).
c) Смесь 4,6 ч. N-/4-/4-(4-метоксифенил)-1-пиперазинил/- фенил/-4-фенил-2-тиазоламина, 2 ч. бромэтана, 1 ч. гидроксида натрия и 94 ч. N,N-диметилформамида перемешивают в течение 16 ч при комнатной температуре. Добавляют еще порцию 2 ч. бромэтана и 1 ч. гидроксида натрия и продолжают перемешивание в течение 4 ч при температуре 50oC. Реакционную смесь разбавляют водой. Выпавший в осадок продукт отфильтровывают и очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента трихлорметана. Чистые фракции собирают и элюент отгоняют. Остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и высушивают, получают 4,0 ч. (81,7 %) N-этил-/4-/4-(4-метоксифенил)-1-пиперазинил/ фенил/-4-фенил-2-тиазоламина; т.пл. 223,6oC (промежуточное соединение 3).
Пример 2.
a) Смесь 5,7 ч. 4-/4-(4-метоксифенил)-1-пиперазинил/бензоламина, 3 ч. 2-изотиоцианат-1,1-метоксиэтана и 100 ч. 1,4-диоксана перемешивают при кипении в течение 1 ч. Реакционную смесь выпаривают. Остаток очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента смеси трихлорметана и метанола (98:2 по объему). Чистые фракции собирают и элюент отгоняют. Остаток кристаллизуют из 4-метил-2-пентанона, получают 3,1 ч. (36%) N-(2,2- диметоксиэтил)-N'-4-(4-метоксифенил)-1-пиперазинил/фенил/- тиомочевины (промежуточное соединение 4).
b) Смесь 17,6 ч. N-(2,2-диметоксиэтил)-N'-4/-4/(4-метоксифенил)-1-пиперазинил/фенил/ тиомочевины и 120 ч. муравьиной кислоты перемешивают в течение 1 ч. при комнатной температуре. Реакционную смесь выпаривают в вакууме и остаток растворяют в 133 ч. дихлорметана. Смесь нейтрализуют раствором бикарбоната натрия. Выпавший в осадок продукт отфильтровывают, промывают водой и дихлорметаном и кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и высушивают, получают 8,5 ч. (52,0 %) 4,5-дигидро-5-метокси-N/4-/4-(4-метоксифенил)-1- пиперазинил/фенил/-2-тиазоламина; т. пл. 177,5oC (промежуточное соединение 5).
c) Смесь 15 ч. 4,5-дигидро-5-метокси-N-/4-/4-(4-метоксифенил)- 1-пиперазинил/фенил/-2-тиазоламина, 5,8 ч. бромэтана, 3 ч. гидроксида натрия в гранулах и 207 ч. N,N-диметилформамида перемешивают в течение 16 ч при комнатной температуре. Реакционную смесь разбавляют водой. Выпавший в осадок продукт отфильтровывают и очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента смеси трихлорметана, этилацетата, гексана и метанола (49:30:20:1 по объему). Вторую фракцию собирают и элюент выпаривают. Остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и сушат, получают 7,3 ч (44,4%) полугидрата N-этил-4,5-дигидро-5-метокси-N-/4-/4-(4-метоксифенил)-1-пипе- разинил/фенил/-2-тиазоламина; т.пл. 131,5oC (промежуточное соединение 6).
Пример 3.
a) Смесь 10 ч. 1-(4-изотиоцианатфенил)-4-(4-метоксифенил)- пиперазина, 3 ч. 2-амино-2-метил-1-пропанола и 260 ч. дихлорметана перемешивают в течение ночи при комнатной температуре. Выпавший в осадок продукт отфильтровывают, промывают дихлорметаном и 2-пропаноном и высушивают, получают 11,7 ч. (91,9%) N-(2-окси-1,1-диметилэтил)-N'-4/4-(4-метоксифенил)-1-пипе- разинил/фенил/тиомочевины; т.пл. 221,6oC (промежуточное соединение 7).
b) Смесь 74 ч. N-(2-окси-1,1-диметилэтил)-N'-4/-4/-(4- метоксифенил)-1-пиперазинил/фенил/тиомочевины и 360 ч. муравьиной кислоты перемешивают в течение 4 ч при температуре 70oC. Реакционную смесь выпаривают и остаток растворяют в 260 ч. дихлорметана. Все это нейтрализуют раствором бикарбоната натрия. Выпавший в осадок продукт отфильтровывают, промывают водой и дихлорметаном и очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента смеси трихлорметана и насыщенного аммиаком метанола (99: 1 по объему). Собирают чистые фракции и элюент отгоняют. Остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и высушивают, получают 44,1 ч. (62,4%) 4,5-дигидро-N-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-4,4- диметил-2-тиазоламина; т.пл. 232,0oC (промежуточное соединение 8).
c) Смесь 37 ч. 4,5-дигидро-N-/4-/4-(4-метоксифенил)- 1-пиперазинил/фенил/-4,4-диметил-2-тиазоламина, 5 ч. 50%-ной дисперсии гидрида натрия и 376 ч. N, N-диметилформамида перемешивают в течение 2 ч при температуре 70oC. После охлаждения до комнатной температуры к реакционной смеси медленно добавляют 14,1 ч. иодметана. Все это перемешивают в течение одного часа при комнатной температуре. Реакционную смесь разбавляют водой. Выпавший в осадок продукт отфильтровывают, промывают водой и 2-пропанолом и очищают методом колоночной хроматографии (жидкостная хроматография высокого разрешения) над силикагелем с применением в качестве элюента смеси дихлорметана и метанола (96 : 4 по объему). Первую фракцию собирают и элюент выпаривают. Остаток снова очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента смеси дихлорметана и метанола (98 : 2 по объему). Чистые фракции собирают и элюент выпаривают. Остаток кристаллизуют из 4-метил-2-пентанона, получают 4-/4-(4-метоксифенил)-1-пиперазинил/-N-(3,4,4-триметил-2- тиазолидинилиден)бензоламин (промежуточное соединение 9).
Вторую фракцию собирают и кипятят в 2-пропаноле. После охлаждения продукт отфильтровывают и сушат, получают 19,5 ч. (51,0%) 4,5-дигидро-N-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-N, 4,4- триметил-2-тиазоламина; т. пл. 166,6oC (промежуточное соединение 10).
Пример 4.
a) Смесь 47,8 ч. 1-(4-изотиоцианатофенил)-4-(4-метоксифенил) пиперазина, 100 ч. гидрата гидразина и 400 ч. 1,4-диоксана перемешивают при кипении в течение одного часа. Реакционную смесь охлаждают и выливают в воду. Выпавший в осадок продукт отфильтровывают, промывают водой и метанолом и высушивают, получают 46 ч. (89%) N-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/ гидразинкарботиоамида (промежуточное соединение 11).
b) Смесь 3,6 ч. N-/4-/4-(4-метоксифенил)-1-пиперазинил/- фенил/гидразинкарботиоамида, 1 ч. уксусного ангидрида и 150 ч. трихлорметана перемешивают в течение одного часа с рефлюксом. После охлаждения выпавший в осадок продукт отфильтровывают и высушивают, получают 3,6 ч. (90,1%) уксуснокислого 2-///4-/4-(4-метоксифенил)-1- пиперазинил/фенил/амино/тиоксометилгидразида; т.пл. 229,8oC (промежуточное соединение 12).
c) Смесь 2,6 ч. уксуснокислого 2-///4-/4-(4-метоксифенил)-1- пиперазинил/-фенил/амино/тиоксометилгидразида и 74 ч. метансульфокислоты перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь выливают при перемешивании в смесь гидроксида аммония с дробленым льдом. Выпавший в осадок продукт отфильтровывают, промывают водой и кристаллизуют из N,N-диметиоформамида. Продукт отфильтровывают и высушивают, получают 2,1 ч. (84,7%) N-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-5- метил-1,3,4-тиадиазол-2-амина; т.пл. 276,7oC (промежуточное соединение 13).
d) Смесь 10,6 ч. N/4-/4-(4-метоксифенил)-1-пиперазинил/-фенил-5- метил-1,3,4-тиадиазол-2-амина, 0,5 ч. бромэтана, 4 ч. гранулированного гидроксида натрия и 188 ч. N,N-диметилформамида перемешивают в течение 4 ч при температуре 40 - 50oC. После добавления воды кристаллизовавшийся продукт отфильтровывают и очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента смеси трихлорметана, метанола, этилацетата и гексана (48 : 2 : 30 : 20 по объему). Собирают первую фракцию и элюент отгоняют. Остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и сушат, получают 2,9 ч. (25,3%) N-(3-этил-5-метил-1,3,4-тиадиазол-2(3H(илиден)-4-/4-(4-метоксифенил) -1-пиперазинил/бензоламина; т.пл. 175,4oC (промежуточное соединение 14).
Собирают вторую фракцию и элюент отгоняют. Остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают, сушат и получают 6,5 ч. (56,7%) N-этил-N-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-5- метил-1,3,4-тиадиазол-2-амина; т.пл. 186,8oC (промежуточное соединение 15).
Пример 5.
К перемешиваемой смеси 17,2 ч. фенил-/4-/4-(4-метокси-3,5- диметилфенил)-1-пиперазинил/фенил/карбамата, 225 ч. N,N-диметилформамида и 9,1 ч. N,N-диэтилэтанамина добавляют 9,6 ч. хлортриметилсилана. Все это перемешивают сначала в течение 2 ч при комнатной температуре, потом в течение 2 ч при температуре 80oC. После охлаждения добавляют 10,1 ч. бромистоводородного 2-бромэтанамина и продолжают перемешивание в течение одного часа. Полученный раствор добавляют к перемешиваемой смеси 9,2 ч. 50%-ной дисперсии гидрида натрия и 45 ч. N,N-диметилформамида. После перемешивания в течение 2 ч при комнатной температуре добавляют по каплям 6,15 ч. 1-бромпропана. По окончании добавления продолжают перемешивание в течение ночи при комнатной температуре. Реакционную смесь выливают в воду. Выпавший в осадок продукт отфильтровывают и кристаллизуют из 2-пропанола, получают 5,6 ч. (33,1%) 1-/4-/4-(4-метокси-3,5- диметилфенил)-1-пиперазинил/-фенил/-3-пропил-2-имидазолидинона (промежуточное соединение 16).
Пример 6.
Смесь 50 ч. фенил-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/ карбамата, 22,7 ч. этил-2-пиперидинкарбоксилата, 4 ч. N,N-диметил-4-пиридинамина и 300 ч. 1,4-диоксана перемешивают в течение 5 ч при температуре кипения. После насыщения водой реакционную смесь нагревают в течение 30 мин. После охлаждения выпавший в осадок продукт отфильтровывают, промывают 2-пропанолом и очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента смеси трихлорметана и метанола (99 : 1 по объему). Чистые фракции собирают и элюент отгоняют. Остаток кристаллизуют из 1-бутанола. Продукт отфильтровывают и сушат, получают 24,8 ч. (47,5%) 5,6,7,8-тетрагидро-2-/4-/4-(метоксифенил)-1- пиперазинил/-фенил/имидазо/1,5-a/пиридин-1,3(2H, 8H)-диона; т.пл. 223,4oC (промежуточное соединение 17).
Пример 7.
a) К перемешиваемой и охлаждаемой на ледяной бане смеси 15 ч. 1-(4-нитрофенил)гидразина и 160 ч. абсолютного этанола добавляют 13,5 ч. хлористоводородного этилэтанимидата. После перемешивания в течение 3 ч при охлаждении реакционную смесь выливают в воду. Выпавший в осадок продукт отфильтровывают, промывают водой и высушивают, получают 19 ч. (85%) 1-(1-этоксиэтилиден)-2-(4- нитрофенил)гидразина; т. пл. 101,8oC; (промежуточное соединение 18).
b) Смесь 10 ч. 1-(1-этоксиэтилиден)-2-(4-нитрофенил) гидразина, 13 ч. морфолина и 135 ч. метилбензола перемешивают при температуре кипения в течение 72 ч. Реакционную смесь охлаждают. Выпавший в осадок продукт отфильтровывают, промывают метилбензолом и сушат, получают 8 ч. (67%) 1-/1-(4-морфоолинил)этилиден/-2-(4-нитрофенил)гидразина; т.пл. 175,9oC (промежуточное соединение 19).
c) Смесь 13 ч. 1-/1-(4-морфолинил)этилиден/-2-(4-нитрофенил) гидразина, 8,5 ч. 1-изоцианатпропана, 1 ч. N,N-диметил-4-пиридинамина и 39 ч. дихлорметана перемешивают при температуре кипения в течение 2 ч. Все это выпаривают и к остатку добавляют 90 ч. диметилбензола. Продолжают перемешивание при температуре кипения в течение 3 ч. Реакционную смесь охлаждают и фильтруют через диатомовую землю. Фильтрат насыщают петролейным эфиром. Выпавший в осадок продукт отфильтровывают и кристаллизуют из 2-пропанола, получают 8,5 ч. (65%) 2,4-дигидро-5-метил-2-(4-нитрофенил)-4-пропил-3H-1,2,4-триазол-3-она; т.пл. 125,4oC (промежуточное соединение 20).
d) Смесь 57 ч. 2,4-дигидро-5-метил-2-(4-нитрофенил)-4-пропил- 3H-1,2,4-триазол-3-она и 400 ч. метанола гидрируют при нормальном давлении при комнатной температуре в присутствии 5 ч. катализатора 20%-ного палладия на угле. После поглощения расчетного количества водорода катализатор отфильтровывают и фильтрат выпаривают. Остаток кристаллизуют из смеси 4-метил-2-пентанона и 2,2'-оксибиспропана. Продукт отфильтровывают и высушивают, получают 46 ч. (91%) 2-(4-аминофенил)-2,4-дигидро-5-метил-4-пропил-3H-1,2,4-триазол- 3-она; т.пл. 138,8oC (промежуточное соединение 21).
e) Смесь 25 ч. N,N-бис(2-хлорэтил)-4-метоксибензоламина, 23,2 ч. 2-(4-аминофенил)-2,4-аминофенил)-2,4-дигидро-5-метил-4-пропил- 3H-1,2,4-триазол-3-она, 2 ч. иодида калия и 200 ч. циклогексанона перемешивают при температуре кипения в течение 5 ч. с применением водоотделителя. Реакционную смесь охлаждают и нейтрализуют раствором бикарбоната натрия. Продукт отфильтровывают и растворяют в трихлорметане. Раствор фильтруют через силикагель и растворитель выпаривают. Остаток кристаллизуют из 1-бутанола и получают 18 ч. (44%) 2,4-дигидро-2-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-5- метил-4-пропил-3H-1,2,4-триазол-3-она; т.пл. 202,2oC (промежуточное соединение 22).
Пример 8.
a) К перемешиваемому раствору 42,8 ч. 50%-ной дисперсии гидрида натрия в 200 ч. диметилсульфоксида медленно добавляют по каплям раствор 50 ч. 2,4(1H, 3H)-пиримидиндиона в 800 ч. диметилсульфоксида, поддерживая температуру постоянной (20oC) путем охлаждения в водо-ледяной бане. Добавляют 62,9 ч. 1-фтор-4-нитробензола и реакционную смесь перемешивают в течение ночи при температуре 50oC. После охлаждения реакционную смесь выливают в 2500 ч. воды. Промывают дихлорметаном. pH водной фазы доводят до 5 - 6. Продукт отфильтровывают и высушивают в вакууме при температуре 60>C, получают 60 ч. (57,6%) 1-(4-нитрофенил)-2,4(1H,3H)-пиримидиндиона; т.пл. o 300oC (промежуточное соединение 23).
b) Смесь 3 ч. 1-(4-нитрофенил)-2,4(1H,3H)-пиримидиндиона, 1,4 ч. гидроксида калия и 67,5 ч. N,N-диметилацетамида перемешивают в течение одного часа при комнатной температуре в атмосфере азота. Добавляют 1,52 ч. бромэтана и продолжают перемешивание в течение ночи при комнатной температуре. Реакционную смесь выливают в 100 ч. ледяной воды. Продукт отфильтровывают и перемешивают в метаноле. Продукт отфильтровывают и сушат в вакууме при температуре 60oC, получают 2,4 ч. (65,6%) 3-этил-1-(4-нитрофенил)-2,4-(1H, 3H)-пиримидиндиона; т.пл. 182,5oC (промежуточное соединение 24).
c) Смесь 28,4 ч. 3-этил-1-(4-нитрофенил)-2,4(1H,3H)-пиримидиндиона, 5 ч. 4%-ного раствора тиофена в метаноле и 500 ч. 2-метоксиэтанола гидрируют при нормальном давлении и температуре 50oC в присутствии 3 ч. катализатора 5% платины на угле. После поглощения расчетного количества водорода катализатор отфильтровывают и фильтрат концентрируют до объема приблизительно 150 ч. После охлаждения продукт отфильтровывают (фильтрат отставляют в сторону) и сушат в вакууме при температуре 60oC, получают первую фракцию 16,5 ч. 1-(4-аминофенил)-3-этил-2,4(1H, 3H)-пиримидиндиона (промежуточное соединение 25). Отставленный в сторону фильтрат (см. выше) выпаривают. Остаток перемешивают в метаноле. Продукт отфильтровывают и сушат в вакууме при температуре 60oC, получают вторую фракцию 5,7 ч. промежуточного соединения 20. Общий выход: 22,2 ч. (88,3%) промежуточного соединения 25; т.пл. 190,8oC.
d) Смесь 17,47 ч. N,N-бис(2-хлорэтил)-4-метоксибензоламина, 16,3 ч. 1-(4-аминофенил)-3-этил-2,4(1H,3H)-пиримидиндиона, 11,83 ч. бикарбоната натрия и 240 ч. 1-бутанола перемешивают в течение 24 ч при температуре кипения. После охлаждения добавляют 150 ч. воды. Продукт отфильтровывают и кристаллизуют из метилбензола. Продукт отфильтровывают и сушат в вакууме при температуре 60oC, получают 10,6 ч. (37,0%) 3-этил-1/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-2,4(1H, 3H)- пиримидиндиона; т. пл. 210,2oC (промежуточное соединение 26).
Пример 9.
a) Смесь 40 ч. 2-(4-нитрофенил)-1,2,4-триазин-3,5(2H,4H)- диона, 25,7 ч. 1-бромбутана, 26,25 ч. карбоната калия и 720 ч. N,N-диметилформамида перемешивают в течение ночи при температуре 45oC. Реакционную смесь выливают в 2000 ч. ледяной воды. Выпавший в осадок продукт отфильтровывают, промывают водой и 2,2'-оксибиспропаном и перемешивают в метаноле. Продукт отфильтровывают и высушивают в вакууме при температуре 70oC, получают 33,9 ч. (68,6%) 4-бутил-2-(4-нитрофенил)-1,2,4-триазин- 3,5(2H, 4H)-диона (промежуточное соединение 27).
b) Смесь 33,9 ч. 4-бутил-2-(4-нитрофенил)-1,2,4-триазин-3,5(2H,4H)-диона, 2 ч. 4%-ного раствора тиофена в метаноле и 400 ч. 2-метоксиэтанола гидрируют при нормальном давлении и комнатной температуре в присутствии 3 ч. катализатора 10% палладия на угле. После поглощения расчетного количества водорода катализатор отфильтровывают, фильтрат выпаривают и получают 29 ч. (92,8%) 2-(4-аминофенил)-4-бутил-1,2,4-триазин- 3,5(2H, 4H)-диона в виде остатка (промежуточное соединение 28).
c) смесь 27,1 ч. N,N-бис(2-хлорэтил)-4-метоксибензамина, 29 ч. 2-(4-аминофенил)-4-бутил-1,2,4-триазин-3,5(2H, 4H)-диона, 18,4 ч. бикарбоната натрия и 350 ч. 2-метил-2-пропанола перемешивают в течение ночи при температуре кипения. После охлаждения прибавляют 200 ч. воды. Выпавший в осадок продукт отфильтровывают и кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и сушат в вакууме при температуре 75oC, получают 19,8 ч. (41,3%) 4-бутил-2-/4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-1,2,4- триазин-3,5(2H,4H)-диона; т.пл. 181,3oC; (промежуточное соединение 29).
Пример 10.
a) К 990 ч. охлажденного на ледяной бане тетрагидрофурана добавляют по частям 156 ч. хлорида алюминия и все это энергично перемешивают до полного растворения твердого вещества. Полученный раствор быстро добавляют к перемешиваемой суспензии 208 ч. азида натрия в 225 ч. тетрагидрофурана и продолжают перемешивание в течение 1 ч при температуре рефлюкса. После охлаждения до комнатной температуры добавляют по каплям раствор 54 ч. бутаноилхлорида, 225 ч. тетрагидрофурана при температуре ниже 30oC. Реакционную смесь нагревают медленно до температуры кипения и продолжают перемешивание в течение ночи при температуре рефлюкса. Реакционную смесь подкисляют при охлаждении 800 ч. 6 н. раствора хлористоводородной кислоты и смесь выпаривают. Остаток перемешивают в растворе бикарбоната натрия и промывают трихлорметаном. Водный слой подкисляют концентрированной соляной кислотой и смесь выпаривают. Остаток перемешивают в 2-пропаноле. Выпавший осадок отфильтровывают и фильтрат выпаривают, получают 32 ч. 1,4-дигидро-1-пропил-5H-тетразол-5-она в виде остатка (промежуточное соединение 30).
b) смесь 38 ч. 1-фтор-4-нитробензола, 32 ч. 1,4-дигидро-1-пропил-5H-тетразол-5-она, 14 ч. карбоната натрия и 200 ч. диметилсульфоксида перемешивают в нагревают в течение 4 ч при температуре 120oC. Реакционную смесь охлаждают и выливают в воду. Выпавший в осадок продукт отфильтровывают и кристаллизуют из 2-пропанола, получают 46 ч. (74%) 1,4-дигидро-1-(4-нитрофенил)-4-пропил-5H-тетразол-5-она; т.пл. 91,1oC (промежуточное соединение 31).
c) Смесь 43 ч. 1,4-дигидро-1-(4-нитрофенол)-4-пропил-5H- тетразол-5-она и 400 ч. метанола гидрируют при нормальном давлении и комнатной температуре в присутствии 4 ч. катализатора 10%-ного палладия на угле. После поглощения расчетного количества водорода катализатор отфильтровывают и фильтрат выпаривают. Остаток превращают в хлористоводородную соль в 2-пропаноле. Соль отфильтровывают и сушат, получают 39 ч. (90%) моногидрохлорида 1-(4-аминофенил)-1,4-дигидро-4-пропил-5H-тетразол-5-она; т. пл. 203,6oC (промежуточное соединение 32).
d) Смесь 25 ч. N,N-бис(2-хлорэтил)-4-метоксибензоламина, 25,5 ч. моногидрохлорида 1-(4-аминофенил)-1,4-дигидро-4-пропил-5H- тетразол-5-она, 2 ч. иодида калия и 200 ч. циклогексанола перемешивают при температуре кипения в течение 5 ч с применением водоотделителя. Реакционную смесь охлаждают и нейтрализуют раствором бикарбоната натрия. Продукт отфильтровывают и кристаллизуют из 1-бутанола, получают 15,5 ч. (39%) 1,4-дигидро-1-/4-/4-(4-метоксифенил)-1-пиперазинил/-фенил/-4- пропил-5H-тетразол-5-она; т. пл. 189,7oC (промежуточное соединение 33).
Пример 11.
a) К перемешиваемой смеси 54,3 ч. 3-бром-4-/4-(4-метоксифенил)- 1-пиперазинил/бензоламина и 189 ч. 1,1-диоксид тетрагидротиофена добавляют по частям при температуре 160oC в течение 30 мин 28,6 ч. этил-/(диметиламино)метилен/гидразинкарбоксилата. По окончании добавления продолжают перемешивание при температуре 170oC до полной отгонки этанола. После охлаждения до комнатной температуры добавляют 120 ч. 4-метил-2-пентанона для растворения клейкого остатка. Реакционную смесь нагревают до полного растворения. После охлаждения всплывшую фазу декантируют и остаток перемешивают в 2,2'-оксибиспропане. Продукт отфильтровывают и высушивают, получают 44.6 ч. 4-/3-бром-4-/4-(4-метоксифенил)-1- пиперазинил/фенил/-2,4-дигидро-3H-1,2,4-триазол-3-она (промежуточное соединение 34).
b) К перемешиваемой смесь 45 ч. 4-/3-бром-4-/4-(4-метоксифенил) -1-пиперазинил/фенил/-2,4-дигидро-3H-1,2,4-триазол-3-она и 90 ч. диметилсульфоксида добавляют 15 ч. измельченного в порошок гидроксида калия. Затем добавляют 6,2 ч. 2-бромбутана и реакционную смесь перемешивают в течение 20 ч при комнатной температуре. Реакционную смесь выливают в воду. Продукт экстрагируют трихлорметаном. Экстракт высушивают, фильтруют и выпаривают. Остаток очищают методом колоночной хроматографии над силикагелем с применением в качестве элюента смеси трихлорметана и метанола (99 : 1 по объему). Чистые фракции собирают и элюент выпаривают. Остаток кристаллизуют из смеси метилбензола и гексана (1 : 2 по объему). Выпавший в осадок продукт отфильтровывают и перекристаллизуют из 80 ч. метанола. Продукт отфильтровывают и сушат в вакууме при температуре 60oC, получают первую фракцию 14,7 (29,8%) 4-/3-бром-4-(4-метоксифенил)-1-пиперазинил/фенил/-2,4-дигидро-2- (1-метилпропил)-3H-1,2,4-триазол-3-она; т.пл. 144,6oC. Собирают менее чистые фракции и элюент отгоняют. Остаток дополнительно очищают методом колоночной хроматографии (жидкостная хроматография высокого разрешения) над силикагелем с применением смеси этилацетата и метанола (97:3 по объему) в качестве элюента. Собирают чистые фракции и элюент отгоняют. Остаток кристаллизуют из метанола, получают вторую фракцию 4-/3-бром-4-/4-(4-метоксифенил)-1-пиперазинил/фенил/-2,4-дигидро-2- (1-метилпропил)-3H-1,2,4-триазол-3-она; т.пл. 144,7oC (промежуточное соединение 35).
Пример 12.
a) К перемешиваемому раствору 25,0 ч. 2,2,2-трифторэтанола в 175 ч. N, N-диэтилэтанамина добавляют по частям 62,2 ч. 2-нафталинсульфонилхлорида. По окончании добавления добавляют в течение 20 мин смесь 1,5 ч. N,N-диметил-4-пиридинамина и 25 ч. этилацетата. По окончании добавления продолжают перемешивание в течение ночи при комнатной температуре. Реакционную смесь фильтруют и фильтрат выпаривают в вакууме.
Остаток перемешивают в воде и затвердевший продукт отфильтровывают при пониженном давлении. Выпавший в осадок продукт растворяют в дихлорметане. Органический слой высушивают, фильтруют и выпаривают в вакууме. Остаток обрабатывают петролейным эфиром. После фильтрования выпавший в осадок продукт кристаллизуют из 2-пропанола. Продукт отфильтровывают, сушат и получают 65,3 ч. (89%) 2,2,2-трифторэтил-2-нафталинсульфоната; т.пл. 72,7oC (промежуточное соединение 36).
b) Смесь 17,5 ч. 2,4-дигидро-4-/4-/4-(4-метоксифенил)-1- пиперазинил/фенил/-3H-1,2,4-триазол-3-она, полученного, как описано в примере XVII патента США N 4267179, 19,5 ч. 2,2,2-трифторэтил-2-нафталинсульфоната, 10,0 ч. карбоната калия и 135 ч. N,N-диметилформаамида 145oC. После охлаждения добавляют воду. Кристаллизовавшийся продукт отфильтровывают при пониженном давлении и растворяют в дихлорметане. Органический слой сушат, фильтруют и выпаривают в вакууме. Остаток очищают методом колоночной хроматографии над силикагелем с применением трихлорметана в качестве элюента. Собирают чистые фракции и элюент выпаривают. Остаток кристаллизуют из 2-бутанона. Продукт отфильтровывают и сушат, получают 9,2 ч. (42,4%) 2,4-дигидро-4-/4-/4-(4-метоксифенил)-1- пиперазинил/фенил/-2-(2,2,2-трифторэтил)-3H-1,2,4-триазол-3-она; т.пл. 208,0oC (промежуточное соединение 37).
B. Получение конечных соединений
Пример 13.
Смесь 16 ч. 4,5-дигидро-2-/4-/4-(4-метоксифенил)-1- пиперазинил/фенил/-5-метил-4-пропил-3H-1,2,4-триазол-3-она и 375 ч. 48%-ного раствора бромистоводородной кислоты в воде перемешивают при температуре кипения в течение 4 ч. После охлаждения выпавший в осадок продукт отфильтровывают и растворяют в смеси метанола и воды. Все это нейтрализуют раствором бикарбоната натрия. Выпавший в осадок продукт отфильтровывают и кристаллизуют из 1,4-диоксана, получают 13 ч. (85%) 4,5-дигидро-2-/4-/4-(4-оксифенил)-1-пиперазинил/фенил/-5-метил-4- пропил-3H-1,2,4-триазол-3-она; т. пл. 252,9oC (соединение 8.10).
Пример 14.
Смесь 2,2 ч. 1-этил 1-3-/4-/4-метоксифенил/-1-пиперазинил/фенил/- 5,5-диметил-2-тиоксо-4-имидазолидинона, 75 ч. раствора 48%-ной бромистоводородной кислоты и 30 ч. уксусной кислоты, насыщенной бромистым водородом, перемешивали в течение 4 ч при температуре дефлегмации. После охлаждения добавили 100 ч. воды. Осажденный продукт был отфильтрован и растворен в смеси метанола и воды. Затем раствор был нейтрализован при помощи углекислого натрия. Продукт был экстрагирован трихлорметаном. Экстракт выпаривают и остаток кристаллизуют из 1-бутанола. Продукт фильтруют, сушат, получают 1,4 ч. (66,0%) 1-/этил-3-/4-4-оксифенил/-1-пиперазинил/фенил/-5,5-диметил- 2-тиоксо-4-имидазолидинон; т.пл. 230,2oC.
Пример 15.
Смесь 6,3 ч. полугидрата N-этил-4,5-дигидро-5-метокси-N-/4-/4-(4- метоксифенил)-1-пиперазинил/фенил/-2-тиазоламина, 1 ч. сульфита натрия и 150 ч. 48%-ного водного раствора бромистоводородной кислоты перемешивают в течение 12 ч при температуре рефлекса. Реакционную смесь выпаривают в вакууме и остаток растворяют в смеси трихлорметана и воды. Раствор нейтрализуют раствором бикарбоната натрия и продукт экстрагируют 1500 ч. трихлорметана. Экстракт высушивают, фильтруют и выпаривают в вакууме. Остаток кристаллизуют из 2-пропанола. Продукт фильтруют и сушат, получают 4,5 ч. (81,5%) 4-/4-/4-/этил-(2-тиазолил)-амино/фенил/-1-пиперазинил/фенола; т. пл. 214,6oC (соединение 3.08).
Пример 16.
К перемешиваемой смеси 300 ч. 48%-ного водного раствора бромистоводородной кислоты, 100 ч. раствора бромистоводородной кислоты в уксусной кислоте и 2 ч. бисульфита натрия добавляют 18,9 ч. 4-бутил-2-/4-/4-(4-метоксифенил)-1-пиперазинил/-фенил/-1,2,4- триазин-3,5(2H,4H)-дона. Перемешивание продолжают в течение 5 ч при температуре кипения. После охлаждения выпавший в осадок продукт отфильтровывают и растворяют в смеси воды и метанола. Смесь нейтрализуют насыщенным раствором бикарбоната натрия. Выпавший в осадок продукт отфильтровывают и дважды кристаллизуют: сначала из 4-метил-2-пентанола и потом - из 1-пропанола. Продукт отфильтровывают и сушат в вакууме при температуре 75oC, получают 9,1 ч. (50, 2%) 4-бутил-2-/4-/4- (4-оксифенил)-1-пиперазинил/фенил/-1,2,4-триазин-3,5(2H, 4H)-диона; т. пл. 202,3oC (соединение 10.05).
Пример 17.
Смесь 6 ч. 2-/1-(4-хлорбензоил)пропил/-2,4-дигидро-4-/4-/4-/(4- оксифенил)-1-пиперазинил/фенил/-3H-1,2,4-триазол-3-она, 103 ч. 1,4-диоксана и 40 ч. метанола перемешивают при комнатной температуре, добавляя медленно по каплям раствор 1,5 ч. тетрагидробората натрия в 25 ч. воды. По окончании добавления продолжают перемешивание в течение 1 ч при комнатной температуре. Реакционную смесь выливают в 1500 ч. воды, к которой добавлено небольшое количество уксусной кислоты. После перемешивания в течение 30 мин выпавший в осадок продукт отфильтровывают, промывают водой и метанолом и высушивают, получают 5,7 ч. (95,3%) 2-/1-/4-хлорфенил)-оксиметил/пропил/-2,4- дигидро-4-/4-/4-(4-оксифенил)-1-пиперазинил/фенил/-3H-1,2,4- триазол-3-она; т.пл. 269,9•C (соединение 7.43).
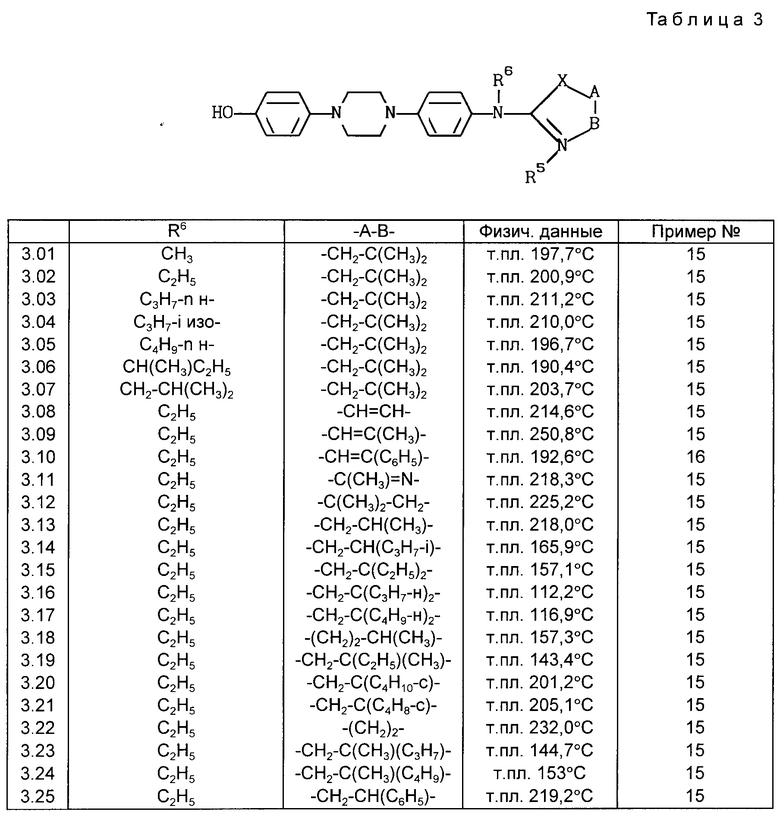
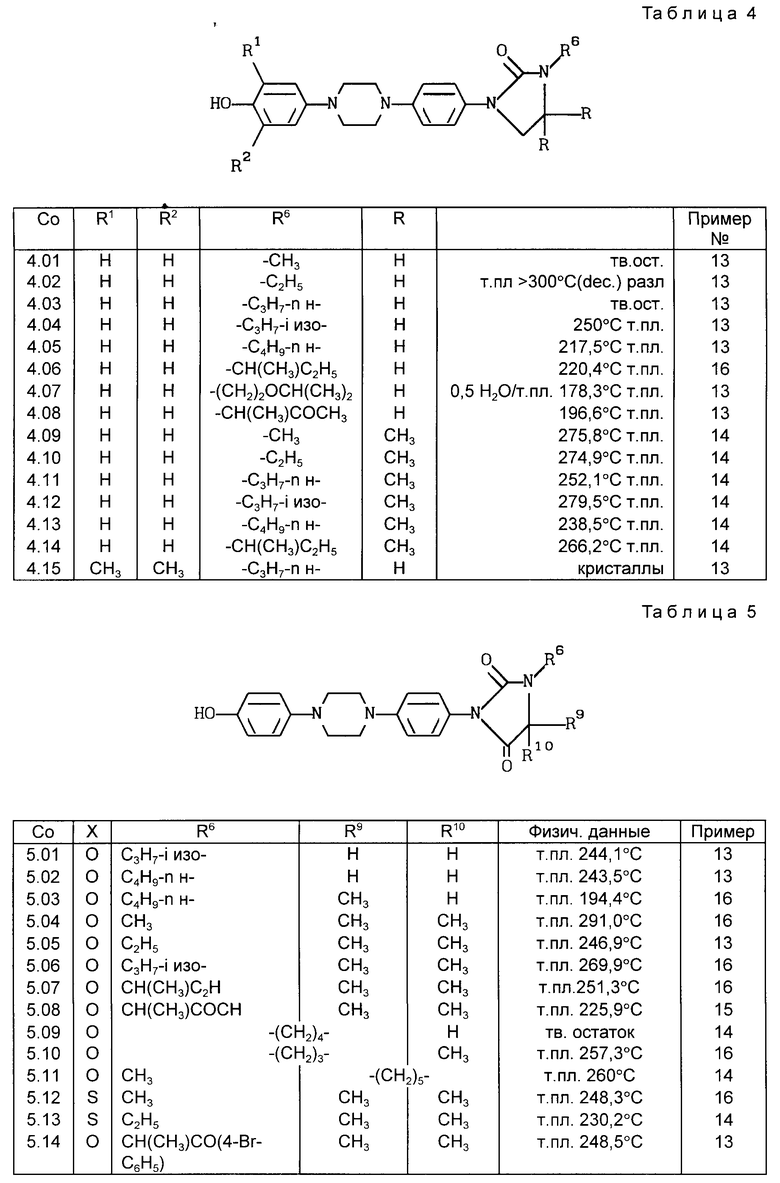
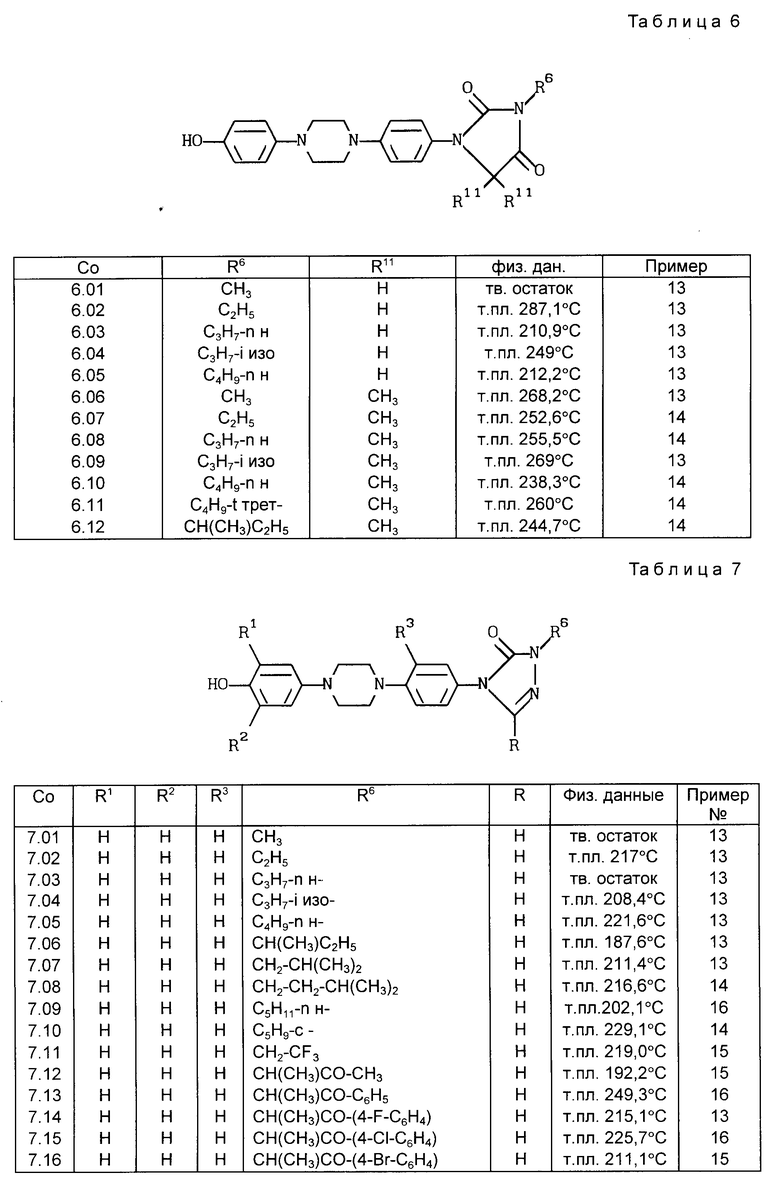
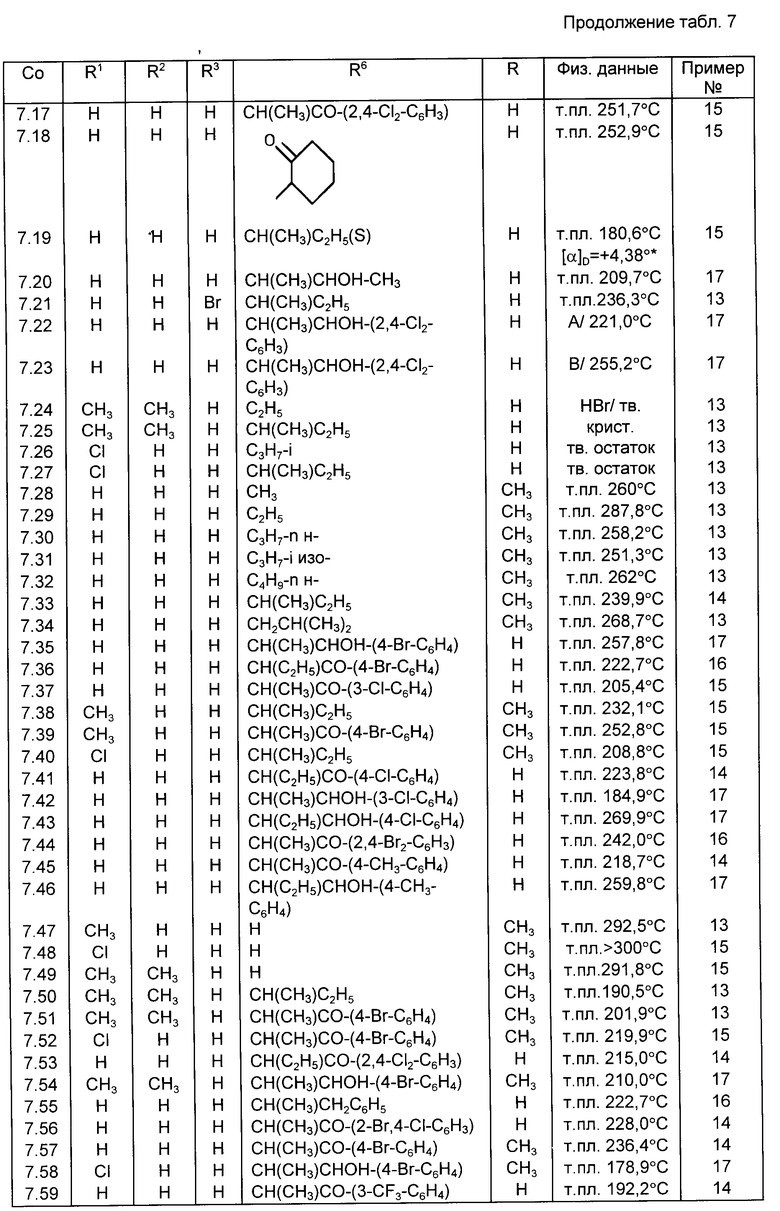
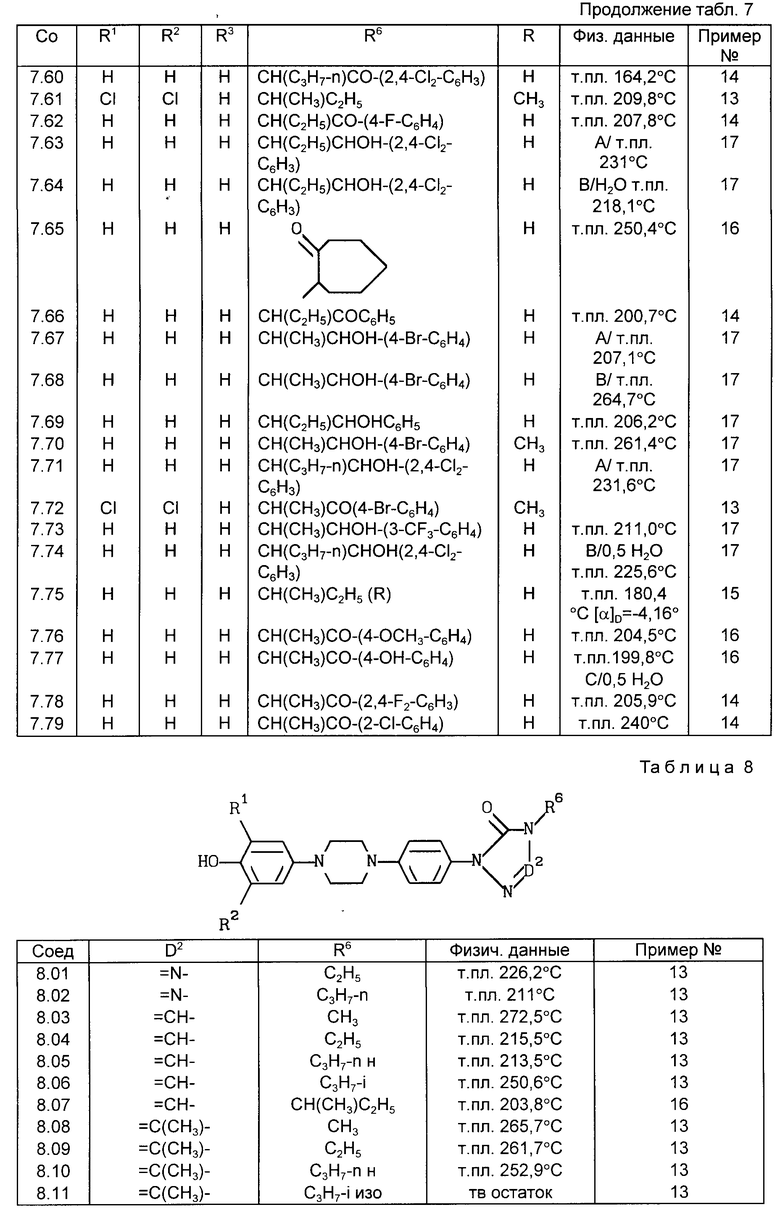
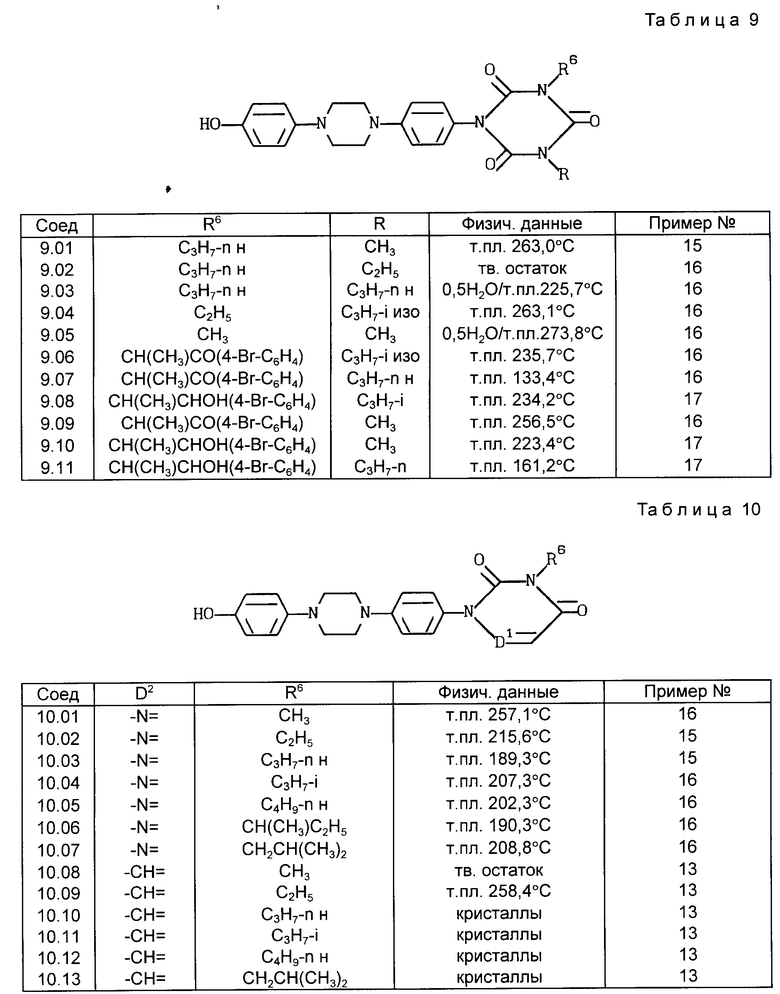
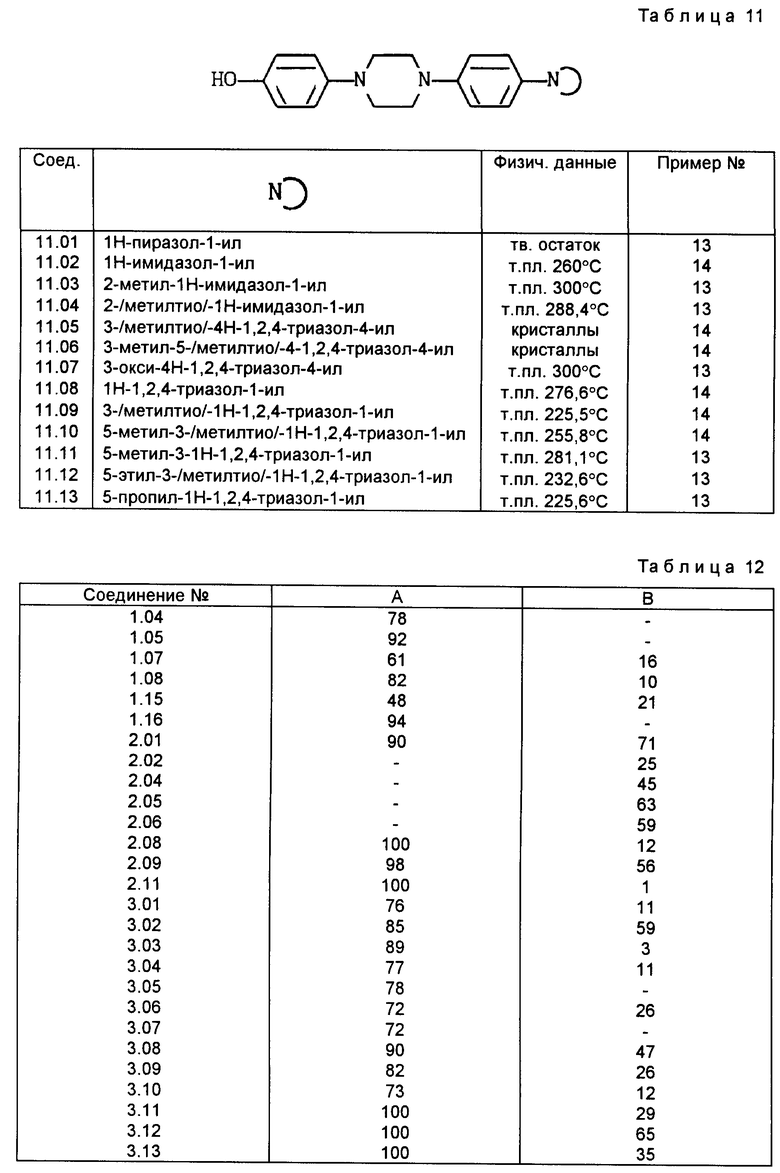
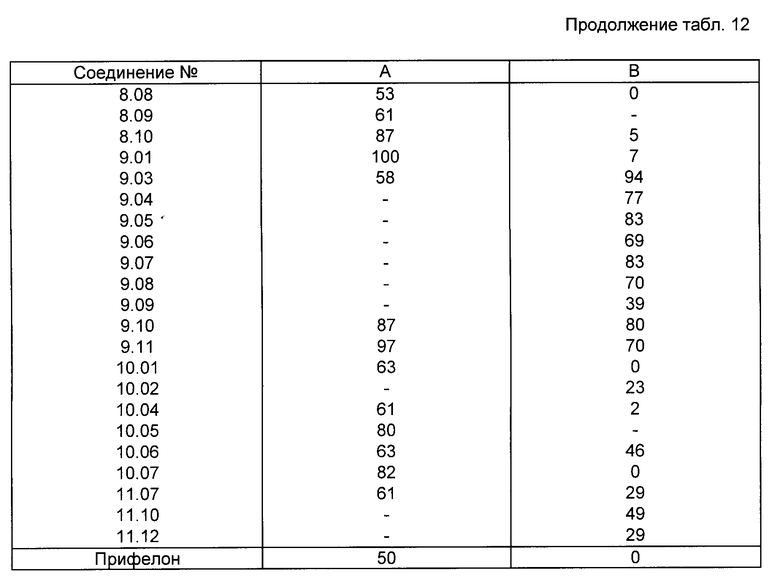
Все соединения, приведенные в табл. 1-11, получены по методике, описанной в примере, номер которого указан в столбце "Пример N".
C. Фармакологические примеры
Ценность ингибирующих липоксигеназу свойств соединений формулы (I) ясно доказана следующими тестами.
Пример 18.
Подавление арахидоната 5-липоксигеназы в всплывшем слое базофильных лейкемических клеток крысы
Крысиные базофильные лейкемические (КБЛ) клетки выращены согласно описанию (Adv. Prostaglandin Thromb. Leuk. Res., 11, 141-145). Их промывают и суспендируют в концентрации 5710o клеток/мл в 50-ммольном буферном растворе фосфата натрия (pH 7,4), содержащем 1 ммоль этилендиаминтетрауксусной кислоты (ЭДТК) и 0,1% желатина. Клетки гомогенизированы при помощи ультразвука, полученный при этом гомогенат центрифугирован при 1000 x g в течении 60 мин. Всплывший слой, сделанный аликвотным и хранящийся при температуре - 70oC, применен как источник 5-липоксигеназной активности.
Активность фермента проверена при температуре 37-5C в реакционной смеси (общий объем 0,4 мл), содержащей 50 ммоль буферного фосфата натрия (pH 7,4), 2 ммоль аденозинтрифосфата (АТФ), 2 ммоль хлорида кальция, 2 ммоль глютатиона, испытуемое соединение (10-8 -1014 моль) и фермент (60 мг белка). После пятиминутного предкубационного периода реакцию начинают путем добавления 0,1 мкюри 4C-арахидоновой кислоты и заканчивают через 15 мин путем добавления 0,3 мл ледяной смеси этиловый эфир:метанол:0,2 М лимонная кислота (30: 4: 1). После встряхивания и центрифугирования (300 x g, 5 мин) органический слой (N 150 мл) отделяют, сушат над безводным сульфатом натрия и экстрагируют 1 мл этилацетата. Затем экстракт выпаривают в вакууме, остаток растворяют в 20 мл этанола. Аликвотные пробы (20 - 30 000 отсчетов в минуту) наносят на покрытие слоем 0,25 мм диоксида кремния пленки (Merck) для тонкослойной хроматографии и элюируют смесью хлороформ: метанол:вода:уксусная кислота (90: 9: 0,05: 1). Радиоактивные пробы наносят методом авторадиографии, вырезают и их радиоактивность подсчитывают при помощи жидкого сцинтиллятора. Результаты подсчетов, полученные для проб, соответствующих продуктам арахидоновой кислоты и липоксигеназы, 5-HPETE и LTB50, суммированы и вычислен процент образования продуктов липоксигеназы. Для исследования подавления получены кривые ответа концентрации и значения ПК4 путем определения процента подавления образования продуктов липоксигеназы в присутствии испытуемого соединения по сравнению с неингибированным контрольным образцом. В первом столбце табл. 12 представлен процент подавления образования продуктов деятельности 5-липоксигеназы (то есть 5-HPETE и LTBo) в присутствии 2,5 ммоль соединения формулы (I).
Пример 19.
Подавление вызванного декстраном образования эдемы в ушах у мышей
Внутривенная инъекция мышам декстрана T500® (Pharmacia) и небесно-голубого красителя понтамина приводит к проведению сосудистой проницаемости и образованию эдемы, что характеризуется интенсивным посинением ушей. Предполагается, что определение количества вышедшего из сосудов красителя позволяет определить количественно ингибирующую 5-липоксигеназу активность испытуемых соединений (Drug. Dev. Res., 8, 213-218, 1986). Для экспериментов применены ослабленные самцы мышей Swiss весом 24-26 г. Эксперименты проведены между 13.00 и 17.00 часами пополудни при температуре окружающей среды 21+1oC. Мыши получили орально испытуемое соединение формулы (I), растворенное в 150 мл либо полиэтиленгликоля (PEG 200), либо оксипропилциклодекстрина в дозах, менявшихся от 1,25 до 40 мг/кг веса тела. В контрольных опытах мыши получили такое же количество одного только растворителя. Через час после обработки мышам был введен внутривенно изотонический солевой раствор, содержащий 60 мг/мл декстрина Т 500® и 13 мг/мл небесно-голубого красителя пентамина в количестве 0,1 мл на 10 г веса тела. Через 1 ч. 45 мин животных убили эфиром и отделили их уши. Экстракция и количественное определение вышедшего из сосудов красителя выполнены согласно описанию (Drug Dev. Res., 8, 213-218, 1986). Вычисленный процент подавления посинения уха вследствие введения соединения формулы (I) в дозе 10 мг/кг веса тела представлен во втором столбце табл. 12.
Столбец A: подавление 5-липоксигеназы во всплывшем слое крысиных базофильных лейкемических кислот, % подавления при дозе 2,5 ммоль.
Столбец B: подавление вызванного декстраном посинения ушей у мышей, % подавления при дозе 10 мг/кг веса тела - означает "не испытано".
D. Примеры композиций.
Нижеследующие составы служат примерами типичных фармацевтических композиций в дозировке на один прием, пригодных для системного введения животным и человеку в соответствии с настоящим изобретением.
Термин "активный ингредиент" (а. и.), примененный в этих примерах, относится к соединению формулы (I) или его фармацевтически приемлемой кислотоаддитивной соли.
Пример 20.
Капли для орального применения
Растворяют 500 ч. а.и. в 0,5 л 2-оксипропановой кислоты и 1,5 л полиэтиленгликоля при температуре 60-80oC. После охлаждения до температуры 30-40oC добавляют 35 л полиэтиленгликоля и смесь тщательно перемешивают. Затем к ней добавляют раствор 1750 ч. натриевого сахарина в 2,5 л очищенной воды и при перемешивании добавляют 2,5 л вкусового вещества какао и полиэтиленгликоль по потребности до объема 50 л, в результате получают раствор для капельного орального введения, содержащий 10 мг/мл а.и. Полученный раствор разливают в соответствующие контейнеры.
Пример 21.
Раствор для орального введения
Растворяют 9 ч. метил-4-оксибензоата и 1 ч. пропил-4-оксибензоата в 4 л кипящей очищенной воды. В 3 л этого раствора растворяют сначала 10 ч. 2,3-диоксибутандионовой кислоты, затем 20 ч. а.и. Последний раствор соединяют с оставшейся частью первого раствора и добавляют 12 л 1,2,3-пропентриола и 3 л 70%-ного раствора сорбита. Растворяют 40 ч. натриевого сахарина в 0,5 л воды и добавляют 2 мл малиновой и 2 мл крыжовной эссенции. Последний раствор соединяют с первым, добавляют необходимое количество воды до объема 20 л и получают раствор для орального приема, содержащий 5 мг активного ингредиента в чайной ложке (5 мл). Полученным раствором наполняют подходящие емкости.
Пример 22.
Капсулы
Энергично смешивают 20 ч. а.и., 6 ч. лаурилсульфата натрия, 56 ч. крахмала, 56 ч. лактозы, 0,8 ч. коллоидального диоксида кремния и 1,2 ч. стеарата магния. Затем полученной смесью наполняют 1000 пригодных твердых капсул, из которых каждая содержит 20 мг активного ингредиента.
Пример 23.
Покрытые пленкой таблетки
Приготовление сердцевины таблетки.
Смесь 100 ч. а.и., 570 ч. лактозы и 200 ч. крахмал тщательно перемешивают и затем увлажняют раствором, состоящим из 5 ч. додецилсульфата натрия и 10 ч. поливинилпирролидона (Kollidon-K 90 ®) приблизительно в 200 мл воды. Влажную порошкообразную смесь просеивают, сушат и снова просеивают. Затем добавляют 100 ч. микрокристаллической целлюлозы (Avicel ® ) и 15 ч. гидрированного растительного масла (Sterotex ®). Все это тщательно перемешивают и таблетируют, получают 10 000 таблеток, из которых каждая содержит 10 мг активного ингредиента.
Покрытие
К раствору 10 ч. метилцеллюлозы (Methocel 60 HG ®) в 75 мл денатурированного этанола добавляют раствор 5 ч. этилцеллюлозы (Ethocel 22 спз ®) в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. Расплавляют 10 ч. полиэтиленгликоля и растворяют в 75 мл дихлорметана. Последний раствор добавляют к первому и затем добавляют туда 2,5 ч. октадеканоата магния, 5 ч. поливинилпирролидона и 30 мл концентрированной суспензии красителя (Opa-spray K-1-2109 ®) и смесь гомогенизируют. Сердцевину таблетки покрывают полученной таким образом смесью в аппарате для нанесения покрытий на таблетки.
Пример 24.
Раствор для инъекций
Растворяют 1,8 ч. 4-оксибензоата и 0,2 ч. пропил-4-оксибензоата приблизительно в 0,5 л кипящей воды для инъекций. После охлаждения приблизительно до 50oC туда добавляют при перемешивании 4 ч. молочной кислоты, 0,05 ч. пропиленгликоля и 4 ч. а.и. Раствор охлаждают до комнатной температуры и добавляют воду для инъекций до объема 1 л, получают раствор, содержащий 4 мг/мл а.а. Раствор стерилизуют фильтрованием (Фармакопея США, ХУП, с. 811) и заполняют им стерильные контейнеры.
Пример 25.
Свечи
Растворяют 3 ч. а.и. в растворе 3 ч. 2,3-диоксибутандионовой кислоты в 25 мл полиэтиленгликоля 400. Сплавляют 12 ч. поверхностно-активного вещества (Span ®) и триглицериды в количестве, необходимом для доведения до 300 ч. Полученную смесь тщательно перемешивают с первым раствором. Полученную таким образом смесь выливают в формы при температуре 37-38C для получения 100 свечей, из которых каждая содержит 30 мг/мл а.и.
Сравнительные данные
Свойства ингибирования 5-липоксигеназы соединения 2,6-ди-третичн.-бутил-4-/2'-тиеноил/фенола, известного под давление прифелон и предложенного в патенте США N 4172082, анализировали в соответствии с процедурами испытаний, описанными в примерах 18 и 19.
Процент ингибирования образования продуктов 5-липоксигеназы в клетках крыс с Базофильной Лейкемией (КБЛ) при 2,5 μм составил 50%. Процент ингибирования посинения ушей в результате отека, индуцированного декстраном, в результате применения испытываемого соединения в дозе 10 мг/кг веса тела составил 0%.
Сравнение этих результатов для соединения-прототипа с биологическими данными для целевых соединений со всей очевидностью показывают, что соединение-прототип менее активное ин витро и ин виво.


Фармацевтическая композиция, ингибирующая 5-липоксигеназу, 4-(фенил-1-пиперазинил)фенилпроизводное и способ его получения. Сущность изобретения: 1. Фармацевтические композиции, ингибирующие 5-липоксигеназу, содержащие в качестве активного ингредиента 4-(4-фенил-1-пиперазинил) фенолы общей формулы I, приведенной в описании, где R1 и R3 каждый независимо H, C1 - 6 алкил или галоид: R3 - H, галоид, NO2; R4 - H или CF3, J - H, NO2, C1 - 6 алкиламино, С1 - 6 алкилкарбониламино, C1 - 6 алкил, C1 - 6 алкилкарбонил, окси, галоид или гетероциклический радикал, возможно замещенный, при условии, что по крайней мере один R1 и R2 представляет алкил или галоид, R3 - галоид или NO2 или R4 - трифторметил. 4-(4-фенил-1-пиперазинил)фенилпроизводное общей формулы I и способ его получения, заключающийся в том, что алкоксипроизводное общей формулы II, приведенный в описании, подвергают деалкилированию в кислой среде или при помощи сильного нуклеофила. 3 с.п.ф-лы, 12 табл.
или его фармацевтически приемлемую соль присоединения кислоты или его стереоизомерную форму,
где R1 и R2 каждый независимо представляет водород, C1 - C6 алкил или галоид;
R3 представляет водород, галоид или нитро;
R4 представляет водород или трифторметил;
Y представляет водород, нитро, C1 - C6 алкиламино, C1 - C6-алкилкарбониламино, C1 - C6 алкил, С1 - С6 алкилкарбонил, окси, галоид или гетероциклический радикал формулы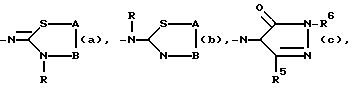
R представляет C1 - C6 алкил;
A - C/R1 4//R1 5/ и B - -CH2- или CH2 - CH2-, или А и В вместе взятые образуют двухвалентный радикал формулы -CH = CH - (j) или -CH = N - (K), где атом C указанного последнего радикала (K) соединен с S; R1 4 и R1 5 каждый независимо представляет водород или C1 - C6 алкил; и в каждом из двухвалентных радикалов B - CH = CH - (j) и -CH = N - (K) один или, где возможно, два атома водорода могут быть заменены на C1 - C6 алкил или фенил; и в двухвалентном радикале B два парных атома водорода могут быть замещены C4 - C6 алканедииломом;
R5 - водород или C1 - c6 алкил;
R6 - водород, C1 - C6 алкил, С3 - С7 циклоалкил, тригало С1 - С6 алкил, (фенил) С1 - С6 алкил; причем С1 - С6 алкил, С3 - С7 циклоалкил и (фенил) С1 - С6 алкил могут быть замещены оксо или окси на любом атоме углерода С1 - С6 алкильной или С3 - С7 циклоалкильной части, при условии, что указанный атом С не присоединен к атому азота, имеющему указанный радикал R6;
D2 - N-, CH- или =CH(CH3)-;
D1 - -N= или -CH=;
каждый R7 - водород или С1 - С6 алкил;
R8 - С1 - С6 алкил, возможно замещенный оксо на любом атоме углерода, при условии, чо указанный атом углерода не находится рядом с атомом азота, имеющим указанный радикал R8;
каждый R9 - водород или C1-6 алкил;
X - 0 или S;
R10 - водород или C1-6 алкил;
R11 - водород или C1-6 алкил;
R12 - C1-6 алкил, не обязательно замещаемый оксо на любом атоме углерода, при условии, что указанный атом углерода не прилегает к атому азота, имеющему указанный радикал R1 2; или R1 1 и R1 2 взятые вместе образуют радикал C3 - C5 алканедиил;
R1 3 - С1 - С6 алкил или (фенил) С1 - С6 алкил, причем указанный (фенил) С1 - С6 алкил может быть замещен оксо-или окси- на любом из атомов углерода С1 - С6 алкильной части, при условии, что указанный атом углерода не прилегает к атому азота, имеющему указанный радикал R1 3;
одновременно фенил-радикал может быть замещен одним или двумя заместителями, выбранными из галоида, С1 - С6 алкила, С1 - С6 алкилокси, окси или трифторметила,
при условии, что
a) по крайней мере один из R1 или R2 представляет С1 - С6 алкил или галоид, или
b) R3 представляет галоид или нитро; или
c) R4 представляет трифторметил; или
d) Y представляет С1 - С6 алкил, С1 - С6 алкиламино, С1 - С6 алкилкарбонил, галоид или гетероциклический радикал формулы (b); радикал (с), где R6 - С3 - С7 циклоалкил, тригало С1 - С6 алкил или С7-циклоалкил, замещенный оксо-; радикал (d) или (е); радикал (h), где R1 1 и R1 3, взятые вместе, образуют С3 - С5 алканедиил радикал; или радикал формулы (i), где R1 3 - (фенил) С1 - С6 алкил, замещенный оксо- или гидрокси- на любом атоме углерода С1 - С6 алкильной части, при условии, что указанный атом углерода не присоединен к атому азота, имеющему указанный радикал R1 3; и
фенил возможно замещен одним или двумя радикалами, выбранными от галоида, С1 - С6 алкила, С1 - С6 алкилокси, окси или трифторметила, в эффективном количестве.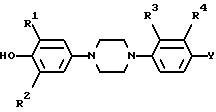
или его фармацевтически приемлемая соль присоединения кислоты или его стереоизомерная форма, где R1, R2, R3, R4 и Y имеют указанные в п.1 значения.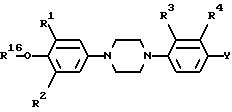
где R1, R2, R3, R4 и Y имеют указанные в п.1 значения и R1 6 - С1 - С6 алкил, в кислой среде или при помощи сильного нуклеофила и, при необходимости, превращают целевой продукт, где R6 или R1 3 представляет (фенил) С1 - С6 алкил, замещенный оксо в С1 - С6 алкильной части, путем восстановления при помощи тетраборгидрата натрия в С1 - С4 алканоле, в целевой продукт формулы (I), где R6 или R1 3 представляет (фенил) С1 - С6 алкил, замещенный окси в С1 - С6 алкильной части.
| US, патент, N 4619931, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент N 4267179, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| EP N 0228125, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| WO, 83/01897, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
