Изобретение относится к способу получения 2-карбокси-3-[2-(диметиламино)этил] -N-метил-1H-индол-5- метансульфонамида и его сложных низших алкиловых эфиров, соответствующих общей формуле (I)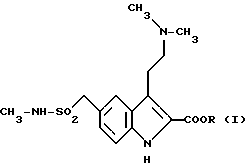
где
R может быть водородом или низшим алкилом. Эти синтетические соединения находят свое применение в качестве промежуточных соединений при синтезе 3-[2-(диметиламино)этил] - N-метил-1H-индол-5-метансульфонамида, продукта, обладающего ценными терапевтическими свойствами, позволяющими использовать его для лечения мигрени.
Продукты изобретения, а также промежуточные соединения для их получения ранее не описывались.
В патенте США 1189064 (1968) раскрывается получение 2-карбоалкокси-3-(галоидэтил)индолов путем индолизации Фишера соответствующих фенилгидразонов в спиртовой насыщенной хлористым водородом среде.
Получение фенилгидразонов из α -кето- δ -валеролактона с помощью реакции указанного валеролактона с соответствующим фенилгидрозином описано J. Lehmann (Arch. Pharm., 320, 22 - 29 (1987)), а также в вышеупомянутом патенте.
В ES 523039 раскрывается получение 4-гидразино-N-метилбензолметансульфонамида, который является исходным продуктом изобретения.
В изобретении описывается способ получения 2-карбокси-3-[2-(диметиламино)этил] -N-метил-1H-индол-5- метансульфонамида (формула (I)) и его низших алкиловых сложных эфиров, где Alk является низшей алкильной группой, предпочтительно метилом или этилом.
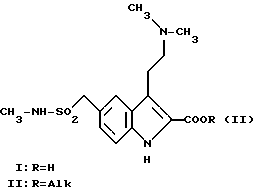
Соединения изобретения могут быть получены из соединений общей формулы (III)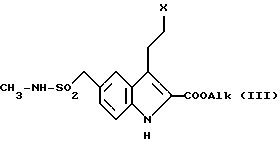
где
Alk является таким же, как он был определен для формулы (II), а X является уходящей группой, такой как атом галогена, мезильная или тозильная группа, предпочтительно тозильная группа, с помощью реакции указанных соединений (III) со спиртовым раствором диэтиламина при температуре в пределах от 0 до 100oC, предпочтительно при около 75oC. Затем полученные сложные диметиламиноэфиры II очищают путем экстракции в водной среде с последующим подщелачиванием.
Диметиламинокислоту I, в свою очередь, получают путем реакции омыления сложных диметиламиноэфиров II (Alk = метил или этил) в щелочной среде, например, в этанольном 5%-ном растворе гидроокиси натрия, при температуре в пределах от 20 до 100oC, предпочтительно при около 75oC. Затем продукт выделяют традиционным способом и очищают путем перекристаллизации. Соединения (III) могут быть получены согласно следующей схеме: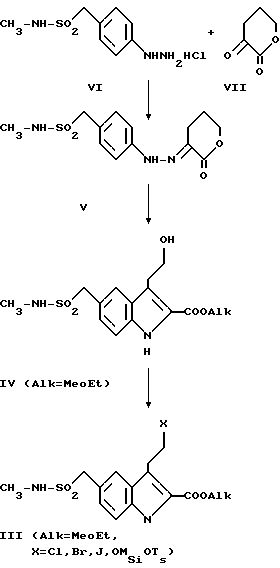
После этого гидрохлорид гидразина VI подвергают реакции с α -кето- δ -валеролактоном VII в водной среде при температуре 70oC и pH 2 в соответствии с процедурами, описанными выше. В результате получают гидразон V, который затем выделяют путем фильтрации, при этом нет необходимости в дополнительной очистке.
Сложные гидроксиэфиры (IV) (например, Alk = Me или Et) получают путем индолизации Фишера гидразона V с последующей переэтерификацией в той же самой реакционной среде. Эту реакцию проводят в безводном растворе хлорводорода в соответствующем спирте (например, метаноле или этаноле), при концентрации кислоты от 3 до 10 н., предпочтительно 5 н., в случае этанола. Указанная реакция может быть осуществлена при температуре в диапазоне 0 - 80oC, предпочтительно примерно при 20oC. Продукты IV выделяют стандартными способами, причем последующей очистки не требуется.
Соединения (III) получают из сложных гидроксиэфиров путем замещения гидроксильной группы атомом галогена с использованием стандартных реагентов галоидирования либо путем их реакции с мезилхлоридом или тозилхлоридом.
В случае тозилатов реакцию замещения уходящей группы соединения (III) диметиламином проводят в особенно мягких условиях. Указанные соединения обычно получают с помощью реакции сложных гидроксиэфиров IV с тозилхлоридом в присутствии пиридина в качестве растворителя и 5 - 10%-ном М (в отношении к IV) 4-диметиламинопиридина в качестве катализатора. Эта реакция может быть проведена при температуре от 0 до 50oC, предпочтительно около 20oC.
Пример 1. Получение гидразона V. Раствор 0,371 М α -кето- δ -валеролактона, полученного путем декарбоксилирования 69 г (0,371 М) α -этоксиаллил- γ -бутиролактона в 150 мл H2SO4, нагретого до температуры перегонки, добавляли по капле в предварительно охлажденный при 5oC раствор 95 г (0,378 М) гидрохлорида 4-гидразин-N-метилбензолметансульфонамида и 19,4 г КОН в 600 мл воды. После завершения добавления значение pH доводили до 2 с помощью 2 н. водного раствора NaOH. Эту смесь нагревали, интенсивно размешивая в течение 15 мин при 60oC, затем оставляли для доведения ее до комнатной температуры. Образовавшийся бледно-желтый осадок отфильтровывали и промывали водой. Полученное твердое вещество сушили при 45oC, в результате получали 111 г (96% по отношению к гидразину) гидразона V.
Т. пл.: 190 - 191oC.
ИК (KBr), см-1: 3419, 3309, 3277, 2947, 1690, 1614, 1582, 1529, 1387, 1308, 1250, 1152, 1124, 1073, 1052, 840, 578.
ЯМР (DMCO-d6), млн. дол.: 1,65 (м, 2H, протоны γ -лактона); 2,55 (д, J = 7, 3H, CH3NH-); 3,45 (т, J = 7, 2H, протоны β- -лактона), 4,10 (широкий сигнал; 2H, протоны δ- -лактона); 4,20 (с, 2H, -SO2CH2-); 6,85 (кв, J = 7, 1H, CH3NH-); 7,30 (с, 4H, ароматич.); 10,00 (с, 1H, -NH-N=).
Анализ: C10H17N3O4S (M.W.: 311,36).
Вычислено, %: C 50,15; H 5,50; N 13,50; S 10,30.
Найдено, %: C 50,19; H 5,48; N 13,53; S 10,31.
Пример 2. Получение 2-карбэтокси-3-(2-гидроксиэтил)-N- метил-1H-индол-5-метансульфонамида (IV, Alk = Et). 70,0 г (0,255 М) гидразина V добавляли в 700 мл перемешанного раствора 10 н. сухого хлороводорода в безводном этаноле. Затем эту смесь размешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь разбавляли 700 мл безводного этанола и перемешивали еще 15 ч при комнатной температуре, а затем выливали на 700 г льда и доводили до значения pH 8 - 9 безводным раствором K2CO3. Этанол отгоняли в вакууме, а остаток экстрагировали четыре раза с использованием 250 мл этилацетата каждый раз. Объединенные органические слои промывали 250 мл насыщенного водного раствора хлорида натрия и высушивали безводным сульфатом магния. Этот растворитель выпаривали досуха, в результате получали 47,4 г (62%) продукта в виде пены. Полученный сырой продукт очищали путем фильтрации на силикагеле, элюируя смесью Cl2CH2Ac = Et (1 : 1) с последующей перекристаллизацией из этанола/воды.
Т. пл.: 153,5 - 155,5oC.
ИК (KBr), см-1: 3336, 2928, 1694, 1553, 1445, 1378, 1317, 1253, 1155, 1119, 1090, 1040, 897, 846, 782, 531.
ЯМР(DMCO-d6), млн. под. : 1,35 (т, J= 7,3H; -COOCH2CH3); 2,55 (c, 3H, CH3NH-); 3,25 (т, J=7,2H; CH2CH2-OH); 5,55 (т. J=7, 2H, -CH2CH2-OH); 4,29 (кв, J= 7, 2H, -COOCH2CH3); 4,31 (c,1H, OH); 4,35 (c, 2H, -SO2CH2-); 6,75 (широкий сигнал, 1H, CH3NH-); 7,30 (AB-система, J=8, 2H; ароматич. в С6 и C7); 7,60 (с. 1H; ароматич. протон в C4); 13,2 (с. 1H, NH-индол).
Анализ: C15H20N2O5S (M.W.: 340,39).
Вычислено,(%): C 52,93; H 5,92; N 8,23; S 9,42.
Найдено,%: C 52,97; H 5,90; N 8,26; S 9,39.
Пример 3. Получение 2-карбэтокси-3-(2-тозилоксиэтил)-N-метил-1H-индол-5-метансульфонамида (III, Alk+Et, X=OTs). К перемешанному раствору 44,4 г (0,13 М) соединения IV (Alk=Et) в 256 мл пиридина добавляли 38 г (1,5 экв.) тозилхлорида и 1,7 г (0,1 экв.) 4-диметиламинопиридина и перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь выливали в 1 л предварительно охлажденного 3н. HCl-раствора при 0oC. Экстракцию осуществляли три раза с использованием 400 мл (каждый раз) дихлорметана. Объединенные органические слои последовательно промывали насыщенным водным раствором бикарбоната натрия и хлорида натрия. После осушки безводным сульфатом натрия растворитель отгоняли досуха и получали 55 г (86%) тозилата продукта III (Alk= этил) в виде белого твердого вещества, которое может быть затем очищено (но необязательно) путем фильтрации на силикагеле с использованием Cl2CH2/AcOEt в качестве элюента.
Т.пл.: 130-131oC.
ИК (KBr) см-1: 3301, 2968, 2960, 1545, 1358, 1316, 1256, 1177, 1122, 1001, 949, 911, 783, 662, 578, 554, 533.
ЯМР (CDCl3) млн. под. : 1,30 (т, J = 7, 3H, -COOCH2CH3); 2,30 (c. 3H, CH3NH-); 2,65 (c. 3H,CH3-Ph-So2-O-), 3,32 (т, J=8, 2H, -CH2CH2-OTs); 4,00-4,60 (комплексная система, 6H, -SO2CH2-, -COOCH2CH3 и -CH2CH2-OTs); 7,30 (AB-система, J=10, 4H, ароматич. из тозильной группы); 7,35 (АВ-система, J= 8, 2H, ароматич. в С6 и С7); 7,52 (с. 1H, ароматич. в C4); 9,05 (c, 1H, NH-индол).
Анализ: C22H26N2O7S2(M.W: 494,58).
Вычислено,% C 53,43; H 5,30; N 5,66; S 12,96.
Найдено,% C 53,36; H 5,33; N 5,61; S 13,00.
Пример 4. Получение 2-карбоэтокси-3-[2-(диметиламино)-этил]-N-метил-1H-индол-метансульфонамида (II, Alk= Et). 49,4 г (0,1М) соединения (III) (Alk=Et, X=OTs) растворяли в 200 мл 33%-ного раствора диметиламина в спирте. Полученный раствор размешивали при комнатной температуре в течение 15 ч, а затем нагревали с обратным холодильником 30 мин. После этого растворитель выпаривали досуха, а остаток растворяли в 200 мл 3 н, HCl и три раза промывали 80 мл (каждый раз) Cl2CH2. Промытый водный слой охлаждали, доводили pH до 12 с помощью 10 н. NaOH и три раза экстрагировали 100 мл (каждый раз) Cl2CH2. Органические объединенные слои промывали 100 мл NaCl-насыщенного раствора и осушали безводным сульфатом натрия. Растворитель выпаривали досуха и получали 30 г (83%) соединения (II) (Alk=Et). Затем сырой продукт перекристаллизовывали из этанола.
Т. пл.: 153-155oC.
ИК (KBr) см-1: 3343, 2945, 2790, 1674, 1545, 1446, 1322, 1264, 1116, 1012, 781, 736.
ЯМР (CDCl3) млн. под. : 1,40 (т, J=7, 3H, -COOCH2CH3); 2,30 (c,6H, -N(CH3)2); 2,50 (м, 2H, -CH2CH2-N); 2,70 (c, 3H, CH3NH-); 3,00 (м, 2H, -CH2CH2-N), 4,25 (c, 2H -SO2CH2-); 4,30 (кв., J=7, 2H, -COOCH2CH3); 7,25 (c, 2H, ароматич. в C6 и C7); 7,55 (с, 1H, ароматич. в С4); 9,30 (с, 1H, NH-индол).
Анализ: C17H25N3O4S (М.W.: 367, 46).
Вычислено,%: C 55,57; H 6,86; N 11,44; S 8,72.
Найдено %: C 56,00, H 6,90, N 11,41; S 8,69.
Пример 5. Получение 2-карбокси-3-[2-(диметиламино)этил]-N-метил-1H-индол-5- метансульфонамида (1). 14,3 г (0,04 М) соединения II (Alk=этил) растворяли в 140 мл 5%-ного раствора KOH в этаноле. Полученный раствор нагревали с обратным холодильником в течение 5 ч, затем охлаждали, а растворитель отгоняли досуха. Остаток растворяли в 100 мл воды и три раза промывали 70 мл (каждый раз) Cl2 CH2. Затем водный раствор охлаждали до 5oC, а pH доводили до 6 с помощью ледяной AcOH. Размешивание продолжали в течение 1 ч при 5oC, осажденное твердое вещество отфильтровывали и осушали при 45oC, в результате получали 12,6 г (96%) продукта 1.
Т.пл.: 245-250oC.
ИК(KBr) см-1: 3405, 3275, 1600, 1343, 1296, 1120, 972.
ЯМР (DMCO-d6) млн. дол.: 2,65 (шир. синглет, 9H, CH3NH-, и -N(CH3)2), 3,05 (м, 2H, -CH2CH2 -N); 3,35 (м, 2H, -CH2CH2-N), 4,35 (c,2H,- SO2CH2-), 5,10 (шир. с. , 1H, протон аминокислоты); 6,85 (шир. с. 1H, CH3NH-); 7,25 (AB-система J= 8, 2H, ароматич. в C6 и C7); 7,55 (с. 1H, ароматич. протон в C4); 12,15 (с. 1H, nH-индол).
Анализ: C15H21N3O4S (M.W.: 339,41).
Вычислено,%: C 53,08; H 6,24; N 12,38; S 9,45.
Найдено,%: C 53,10; H 6,28; N 12,36; S 9,44.
Пример 6. Получение 3-[2-(диметиламино)этил]-N-метил-1H-индол-5-метансульфонамида. 2 г (6 ммоль) 2-карбокси-3-[2-(диметиламино)этил]-N-метил-1H-индол-5-метансульфонамида растворяются в 20 мл сухого хинолина. Добавляется 40 мг (0,3 ммоль) окиси меди (1), перемешиваемая суспензия нагревается с помощью тока сухого азота до 205oC. Реакционная смесь поддерживается при данной температуре до тех пор, пока не закончится выделение CO2 (примерно 30-40 мин). Смесь охлаждается до комнатной температуры и реакционная смесь фильтруется через декалит. Фильтрат концентрируется с помощью вакуумной отгонки растворителя, давая остаток, который суспендируется в 40 мл дихлорметана, а затем фильтруется. Новый фильтрат промывается три раза 15 мл водного раствора 10%-ного аммиака, сушится над безводным сульфатом натрия и упаривается досуха. Полученный твердый остаток промывается 20 мл гексана и сушится при 40oC, давая 1,4 г (80%) продукта в виде твердого вещества цвета чистой охры, который очищается с помощью перекристаллизации из изопропанола.
Т.пл.: 170-171oC.
ИК(KBr), см-1: 3360, 3040, 2779, 1485, 1315, 1152, 1124, 1092, 826, 801, 628, 533, 513.
ЯМР (CDCl3), млн. дол. : 2,31 (с., 6H, N(CH3)2), 2,61 (c.3H, CH3-NH), 2,60-2,90 (м., 4H, CH2-CH2-N), 2,80 (широкий сигнал, 1H, NH-SO2), 4,25 (c., 2H, SO2-CH2), 7,00-7,50 (широкий сигнал, 4H, ароматич. протон), 9,5 (широкий сигнал, 1H, NH индол).
Анализ: C14H21N3O2S(M.W.: 295,40).
Вычислено,%: C 56,92; H 7,17; N 14,22; S 10,83.
Найдено,%: C 56,88; H 7,20; N 14,19; S 10,80.
2-Карбокси-3-/2-(диметиламино)этил-N-метил-1Н-индол-5-метансульфонамид получают взаимодействием 2-карбоалкокси-3-(2-тозилоксиэтил)-N-метил-1Н-индол-5-метансульфонамида со спиртовым раствором диметиламина с последующим омылением полученного продукта. Указанное соединение может быть использовано в качестве промежуточного продукта при синтезе 3-/2-(диметиламино)-этил/-N-метил-1Н-индол-5-метансульфонамида, обладающего ценными терапевтическими свойствами, позволяющими использовать его для лечения мигрени. 5 з.п.ф-лы.
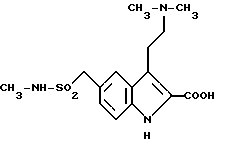
или его сложных эфиров, отличающийся тем, что соединение общей формулы III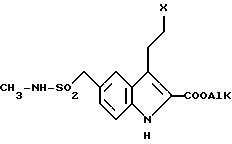
в которой Х представляет уходящую группу;
AlK - низшая алкильная группа,
подвергают взаимодействию с диметиламином в присутствии подходящего растворителя с получением сложных эфиров общей формулы (II)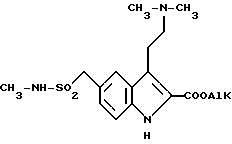
где AlK - низшая алкильная группа,
с последующим, в случае необходимости, их омылением.
| GB, 2124210, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| GB, 2162522, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| GB, 2150932, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

