Изобретение относится к физиологии и биохимии растений, но может быть применено в надмолекулярной биологии индивидуального развития при морфо-, онтогенетической оценке параметрической регуляции на уровне интерфазных ядер.
Известен способ определения углеводов в зародышах злаков [1], в котором содержание глюкозы, мальтозы, ксилозы тестируют в целых тканях зародышей. Недостаток этого метода заключается в том, что углеводы определяются в клетках зародышей без дифференциации клеточных органелл.
Известен способ определения гексоз [2], основанный на их способности в растворах концентрированной серной кислоты взаимодействовать с L-цистеином, образуя окрашенные продукты. Недостаток этого метода заключается в том, что в качестве объектов, содержащих гексозы, были использованы химически синтезированные реагенты - гексозы, а не рассматриваемый объект - клеточные ядра растений.
Известен способ определения гексоз [3], основанный на способности в присутствии концентрированной серной кислоты давать с антроном окрашенные продукты. Недостаток этого метода заключается так же, как и в способе [2], в том, что в качестве объектов, содержащих гексозы, используют химически синтезированные реагенты - гексозы с довольно высокой концентрацией 100-200 мкг/мл.
Известен способ определения углеводных компонентов ядерного матрикса печени и гепатомы крыс [4]. Недостаток этого метода заключается в том, что клеточные ядра из животных тканей выделяли без применения их градиентной очистки [5] , что связано с загрязнением ядер цитоплазматическими компонентами.
Известен способ определения содержания мукополисахаридов в клеточных ядрах в различных тканях и органах животных организмов [6]. Недостаток этого метода заключается в том, что клеточные ядра из животных тканей выделяли без применения градиентной очистки [5], что связано с загрязнением ядер цитоплазматическими компонентами.
За прототип принят способ определения [6], в котором для определения содержания углеводов используют клеточные ядра из замороженных животных тканей с последующим их гидролизом 2,5 н. соляной кислотой. Недостатком этого способа является то, что не указан температурный фактор замораживания животных тканей, а также то, что клеточные ядра не были очищены через градиентную систему для освобождения от цитоплазматического загрязнения и не было проведено фракционирование структурно-функциональных элементов ядер для выяснения преимущественной локализации компонентов внутри интерфазного ядра.
Использование прототипа, где в качестве объекта исследования используют органеллы представителей других эукариотических царств, широко применяется в практической биохимии [7].
Цель изобретения - обеспечение максимального выхода углеводсодержащего материала из клеточных ядер с одновременной реализацией возможности определения его внутриядерной локализации. Указанная цель достигается тем, что углеводсодержащий материал (компоненты) извлекают из клеточных ядер (предварительно законсервированных в тканях проростков пшеницы глицериновой средой), впоследствии выделенных и очищенных в градиенте плотности глицерина с последующей последовательной экстракцией ядерных фракций: 0,14 М (нуклеоплазма), 0,35 М (хроматин I), 2 М хлористым натрием (хроматин II), 6 М гуанидин-гидрохлоридом в присутствии 0,1%-ного β -меркаптоэтанола (ядерный матрикс), 0,5 н. NaOH (ядерно-цитоплазматический остаток) и анализом (определением) в них содержания углеводов.
Пример 1. Опыты проводили на покоящихся - воздушно-сухих зародышах, 1-2-дневных проростках, 3-4-дневных тканях колеоптилей, листьев и корневой системы с междоузлием проростков пшеницы Московской-35 и Мироновской озимой и яровой. Проростки выращивали при температуре 25o-27oC в термостатированной камере между слоями фильтровальной бумаги, смоченной дистиллированной водой. Отделенные от эндосперма воздушно-сухие зародыши, проростки, колеоптили, листья и корневую систему с междоузлием в течение 3 с промывали холодным эфиром и многократно дистиллированной водой. Проростки и ткани консервировали при -20 - -25oC в 80-90% глицерине. Авторы считают, что подобная не замораживающая ткани консервация сохраняет внутриклеточную, внутриядерную архитектуру объекта и предохраняет от стягивания фибриллярных внутриклеточных структур на поверхность и внутренние системы ядер. Отклонения от вышеизложенных условий консервации могут привести к стягиванию внутриклеточных структур, например, под действием более высоких температурных факторов или других детергентов и в связи с этим количественному изменению истинных показаний содержания углеводных компонентов во внутриядерных структурах (см. табл. 4). Затем зародыши, проростки и ткани в количестве примерно 10 г заливали 25 мл гомогенизационной средой: 20% глицерин; 0,005 М MgCl2; 0,025 M KCl; 0,003 M CaCl2; 0,005 M NaCl; 0,004 M β -меркаптоэтанол; 0,01% триэтаноламин (ТЭА)•HCl pH 6,8; 0,004 M н-октиловый спирт. Гомогенизацию проростков проводили 4 с (чтобы не повредить клеточные ядра) при 15 000 об/мин (гомогенизатор МПВ-302, Польша). Гомогенат фильтровали через 1 слой фланели, флюзелина и 3 слоя капрона (размер пор 70 мкм). С грубым остатком растительного материала гомогенизацию и фильтрование проводили еще 2 раза 14 с (20 мл гомогенизационной среды) и 25 с (17 мл гомогенизационной среды) при 15 000 об/мин. Такая мягкая гомогенизационная обработка растительных тканей увеличивает процентный выход неразрушенных клеточных ядер и способствует последовательному извлечению максимально сохраненных внутриядерных структур с адекватным определением в них внутриядерной локализации углеводов. Объединенные гомогенаты центрифугировали при 500 об/мин (ЦЛР-1, СССР) в течение 5 мин с целью освобождения от грубых неразрушенных тканей и целых клеток, затем надосадок центрифугировали при 2700 об/мин (К-23, ГДР) в течение 20 мин. Осадок ядер собирали суспендированием средой гомогенизации (50 мл) и наслаивали на прерывистый глицериновый градиент, состоящий из 5 слоев (по 30 мл) возрастающей концентрации глицерина (50%, 60%, 70%, 80%, 90% вес/объем), приготовленном на ТЭА•HCl pH 6,8 буфере со всеми вышеперечисленными компонентами гомогенизационной среды, исключая 20% глицерин. Градиентное центрифугирование проводили при 2000 об/мин в течение часа (ЦЛР-1, СССР). Осадок ядер промывали 0,5% тритоном Х-100 на среде гомогенизации, но без глицерина, с последующим центрифугированием при 2700 об/мин (К-23, ГДР) в течение 15 мин, после чего ядра промывали трижды в среде следующего состава: 0,005 M MgCl2; 0,003 M CaCl2; 0,025 M KCl; 0,005 M NaCl; 0,01 M трис-HCl pH 6,8 с последующим центрифугированием при вышеуказанных условиях. 1% тритоном Х-100 ядра промывать нельзя, т.к. они набухают и разваливаются.
Степень чистоты выделенных препаратов клеточных ядер определяли микроскопически, спектрофотометрически и по компонентному составу клеточных ядер (белок/ДНК, РНК/ДНК) (см. табл. 1). Сравнение процентного выхода ядер и их удельной ферментативной активности при использовании сахарозно-гуммиарабикового и глицеринового методов изоляции ядер представлены в табл. 2.
Из очищенных препаратов ядер нуклеоплазму экстрагировали при низкой ионной силе 0,14 M NaCl, 0,01 M трис-HCl pH 6,8 буфером. Хроматин I выделяли путем трех последовательных экстракций 0,35 M NaCl на том же буфере.
Далее осадок фракционировали суспендированием в трис-HCl буфере с 2 M NaCl (хроматин II). В осадке оставалась фракция, содержащая ядерный матрикс. Последующую экстракцию проводили 6 М гуанидин-гидрохлоридом с 0,1% β -меркаптоэтанолом на трис-HCl буфере pH 6,8 (ядерный матрикс), остаточные компоненты экстрагировали 0,5 н. NaOH (ядерный остаток).
Расчет экстракционных объемов обработок ядер зависел от выхода клеточных ядер. Так, если выход клеточных ядер в осадке был в пределах от 1,5•106 до 5•106, то экстракционного раствора брали 2 мл; если выход ядер в осадке был в пределах 5•106 - 10•106, то экстракционного раствора брали 3 мл; если выход ядер в осадке был в пределах ≥10•106, то экстракционного раствора брали 5 мл.
Все фракции ядерных компонентов получали путем трех последовательных экстракций. Достаточность экстракций контролировали по выходу белка. Ядерные фракции хранили при -196o в азоте.
Количество белка определяли по связыванию белка с кумасси ярко-синим Г (Loba, Австрия) [8]. Метод использовался в случае микронаноколичественного определения белка: 0,3 мл исследуемого раствора смешивали с 2,6 мл 0,05 н. NaOH и 0,4 мл кумасси (1 мг кумасси растворяли в 5 мл 85% H3PO4, доводили до 15 мл бидистиллированной водой и фильтровали для удаления нерастворившегося красителя); оптическую плотность раствора измеряли при 596 нм. Количественную оценку белка давали в соответствии с калибровочной кривой по бычьему сывороточному альбумину (марка Б, Реахим).
Для определения ДНК и РНК использовали метод Спирина А.С. [9].
Анализ полноты выделения ядерных фракций (см. табл. 3) показал, что все фракции представляют собой не чистый белок, а комплексы белка и нуклеиновых кислот.
В определении углеводов ядерных фракций за основу приняты реакции с L-цистеин гидрохлоридом [2, 3] (пример 1) и антроном [3] (пример 2).
Сущность метода определения углеводов цистеиновым методом заключается в том, что большинство углеводов, содержащих пять или более углеродных атомов, образуют фурфурол или его гомологи в растворах концентрированной серной кислоты. Производные фурфурола вступают во взаимодействие с L-цистеином или другими соединениями, образуя окрашенные продукты.
Вследствие того что продажный препарат L-цистеин гидрохлорида (Reanal) в контрольных опытах дает высокое поглощение при 415 нм и имеет tпл.-210o, что свидетельствует о наличии в нем примесей и было подтверждено хроматографией на Silufol UW 254 (Reanal) в хлороформе и метаноле (10:1), возникла необходимость очистки этого препарата. Из-за наличия реакционно способных SH-групп L-цистени легко переходит в цистеин и поэтому плохо очищается.
Очистку L-цистеина проводили перекристаллизацией из абсолютного спирта (99,8%). Абсолютный спирт получали из 96% ректификата [10].
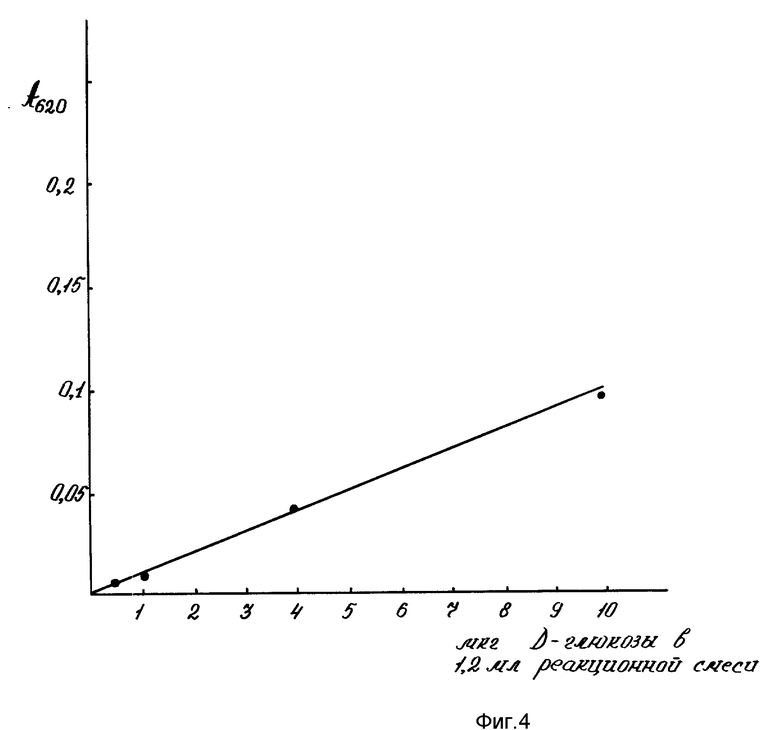
Чувствительность метода отрабатывали в пределе от 0,5-20 мкг/мл D-глюкозы (Serva) при построении стандартной калибровочной кривой (фиг. 1).
Реагенты:
1. Реагент A: охлажденная до 0o 86% (по объему) серная кислота (охч, удельный вес 1,831). Готовится осторожным добавлением к 14 мл дистиллированной H2O + 86 мл конц. серной кислоты. Каждую вновь поступившую партию серной кислоты проверяли на качество: к 0,2 мл дистиллированной H2O прибавляли 1 мл 86% H2SO4. Оптическую плотность измеряли при A415 против воды до и после кипячения. Низкая оптическая плотность (близкая к нулю) свидетельствует о хорошем качестве кислоты.
2. Реагент B: свежеприготовленный раствор L-цистеин гидрохлорида (700 мг/л) в реагенте A (охладить до 0oC).
Ход определения:
а. В стеклянные пробирки вносят по 0,2 мл опытной пробы или контроля. Пробирки нельзя оставлять открытыми, так как определению могут помещать карбогидраты, содержащиеся в воздухе.
б. Пробы выдерживают в течение 5 мин в ледяной бане, после чего добавляется 1 мл реагента B (0oC). Перемешать каждую пробу в течение 5 с.
в. Пробирки, закрытые металлической фольгой, помещали в кипящую водяную баню (100oC). Через 3 мин охлаждали до комнатной температуры.
г. Оптическую плотность проб измеряли против воды при A415 на спектрофотометре СФ-26 (ЛОМО, СССР).
Контроль 1:0,2 мл 0,05 М трис-HCl буфера pH 6,8 + 1 мл реагента B.
Контроль 2: 0,2 мл опытной пробы + 1 мл 86% H2SO4.
Общий объем реакционной смеси 1,2 мл.
Количество углеводов в общем объеме реакционной смеси находили по стандартной калибровочной кривой, построенной по первичной реакции D-глюкозы (Serva) (фиг. 1) и выражали в мкг на 100 зародышей или проростков.
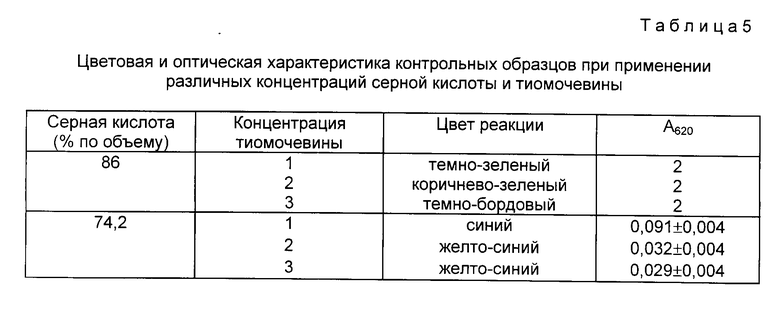
Эффективность чувствительности цистеиновой реакции для качественной оценки (≈10 мкг в пробе): пентоз (арабиноза, ксилоза); гексоз (глюкоза, галактоза, манноза, N-ацетилглюкозамин); гликопротеидов (РНК-аза); полисахаридов (крахмал - линейный полимер глюкозы, альгиновая кислота - линейный полимер D-глюкуроновой, D-манноуроновой кислот), протеогликанов (гепарин - содержит одну полисахаридную цепь глюкуроновой кислоты, глюкозамина); РНК; ДНК по первичной реакции с L-цистеином и серной кислотой) - представлена на фиг. 2.
Из всех исследованных веществ по первичной реакции с L-цистеином и серной кислотой (фиг. 2) для построения калибровочной кривой была выбрана D-глюкоза (фиг. 1). Соответственно этому чувствительность реакции приобретала следующую зависимость: глюкоза > гликопротеины (РНК-аза) > линейный полисахарид - альгиновая кислота ≥галактоза ≥ манноза≥крахмал > ксилоза ≥ арабиноза ≥ РНК > гепарин ≥ ДНК > N-ацетилглюкозамин (фиг. 2).
Присутствие гептоз в растворе делает определение гексоз невозможным, так как гептозы сразу дают оранжевую окраску при A415, быстро переходящую в пурпурную. Влияние других белков кроме РНК-азы гликопротеида и протеогликана - гепарина на первичную реакцию с L-цистеином и серной кислотой изучалось на примерах с трипсином (СПОФА), протаминсульфатом (СПОФА), сывороточным бычьим альбумином (реахим). Практически эти белки не оказывали влияния на определение содержания углеводов в пробах. Показания были на уровне контролей, за исключением протаминсульфата, который только в количестве 40-60 мкг в пробе мог давать дополнительное слабое окрашивание.
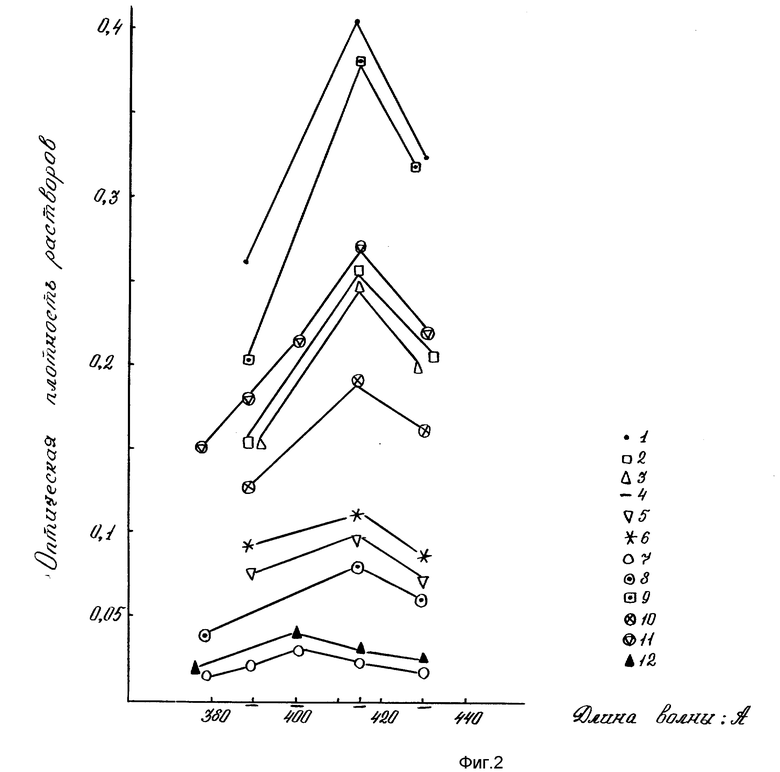
Эффективность последовательной ступенчатой экстракции ядерных фракций из растительных тканей с последующим определением в них углеводов по первичной реакции с L-цистеином и серной кислотой представлена на фиг. 3.
Метод мало устойчив, иногда дает резкие колебания. Интенсивность окраски нестабильна в течение 1 ч.
Пример 2. Работа по выращиванию проростков, их консервированию и выделению из них клеточных ядер и их фракций проводилась точно так же, как в примере 1.
Определение углеводов в ядерных фракциях проводили антроновым методом.
Антроновый метод [3] является одним из распространенных. Сущность метода заключается в том, что при кипячении в присутствии концентрированной серной кислоты от пептидов отщепляются гликозильные группировки, легко распадающиеся на моносахара. При действии на моносахара соединений, содержащих SH-группы, в кислой среде образуются тиоацетали, которые при взаимодействии с антроном дают продукты, окрашенные в синий цвет, имеющий оптимальное поглощение (особенно для гексоз) при A620.
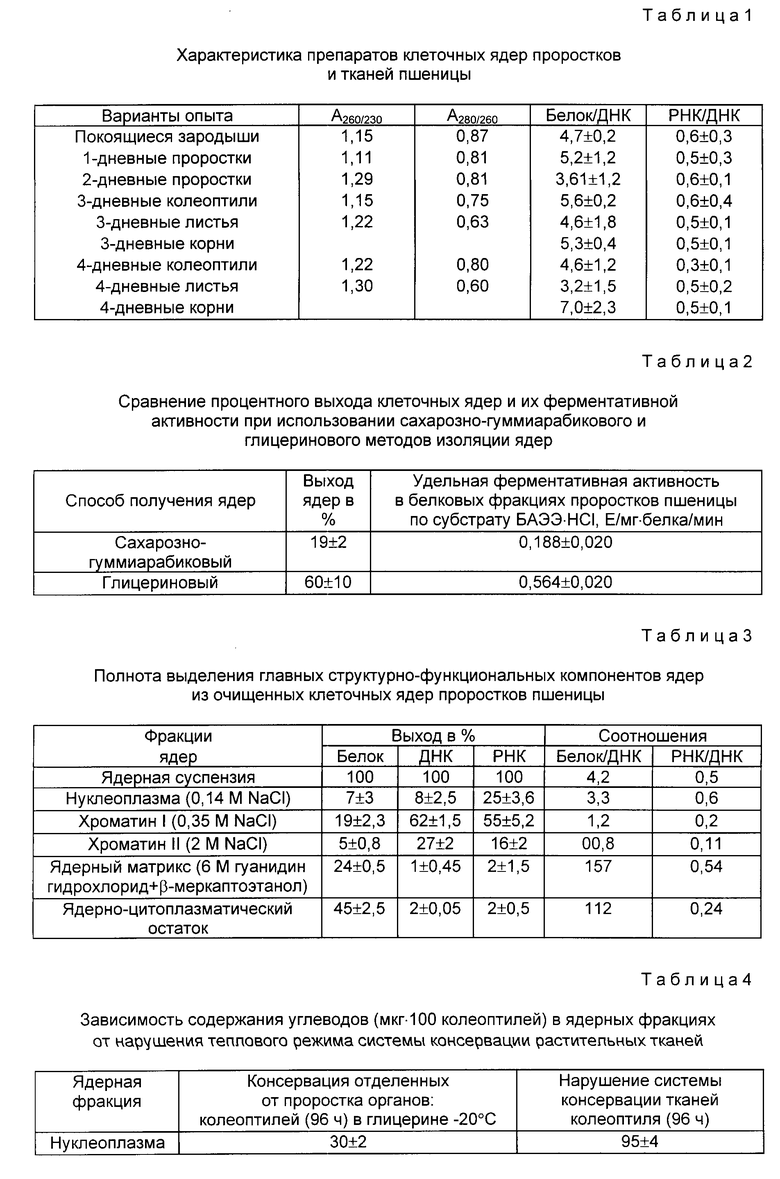
Прямое применение исходной методики [3] не дало результатов. Поэтому были поставлены эксперименты, выявляющие оптимальные условия проведения реакции. По литературным данным, чистый антрон имеет температуру плавления 163oC (бесцветные иглы), 154oC [11, 12]. Продажный препарат антрона (реахим, Ереван, 1987), по-видимому, вследствие большого количества примесей вместо белых игл имеет кристаллы желтого цвета с температурой плавления 105-110oC. Тонкослойная хроматография на силуфоле (Silufol UW 254, Reanal) дает 2 полосы. Перекристаллизационная очистка антрона позволила получить кристаллы с tпл 145oC, которые оказались стойкими при хранении. Перекристаллизованная тиомочевина имела tпл. 180oC. Концентрацию серной кислоты и тиомочевины подбирали эмпирически (см. табл. 5), руководствуясь тем, что антроновая реакция является избирательной для гексоз с максимумом поглощения при A620, и тем, что ароматические аминокислоты, белки, пентозы (рибоза, ксилоза), гексозамины, уроновые кислоты не проявляют максимума поглощения при A620 [3] .
На основании данных, приведенных в табл. 5, для постановки метода были выбраны концентрации тиомочевины - 2% и серной кислоты - 74,2%. Серная кислота должна быть достаточно концентрированной, так как в случае дальнейшего разбавления выпадают кристаллы тиомочевины. Для работы подходит продажная серная кислота 92% с плотностью 1,83.
Для определения микроколичеств - гексоз (0,5-10 мкг) антроновым методом построена стандартная калибровочная кривая по D-глюкозе (Serva, 1988), (фиг. 4).
Приготовление антронового реактива: 72 мл серной кислоты (по объему) осторожно добавляется к 25 мл дистиллированной воды. Смесь охлаждают до 35oC и добавляют 50 мг антрона, перемешивая до растворения, затем добавляют 2 г тиомочевины, также перемешивая до растворения. Реактив готовится перед опытом.
Ход реакции: к 0,2 мл исследуемого раствора прибавляют 1 мл антронового реактива, быстро перемешивают и охлаждают на ледяной бане до 0oC. Затем ставят в кипящую водяную баню на 15 мин. Быстро охлаждают до комнатной температуры t - 20oC. Развивающуюся синюю окраску колориметрируют на СФ-26 (ЛОМО, СССР) против дистиллированной воды при A620.
Содержание углеводов в анализируемых пробах рассчитывали на 100 зародышей или проростков.
Метод устойчив, не дает резких колебаний. Интенсивность окраски проб сохраняется в течение 1,5 ч, затем понижается.
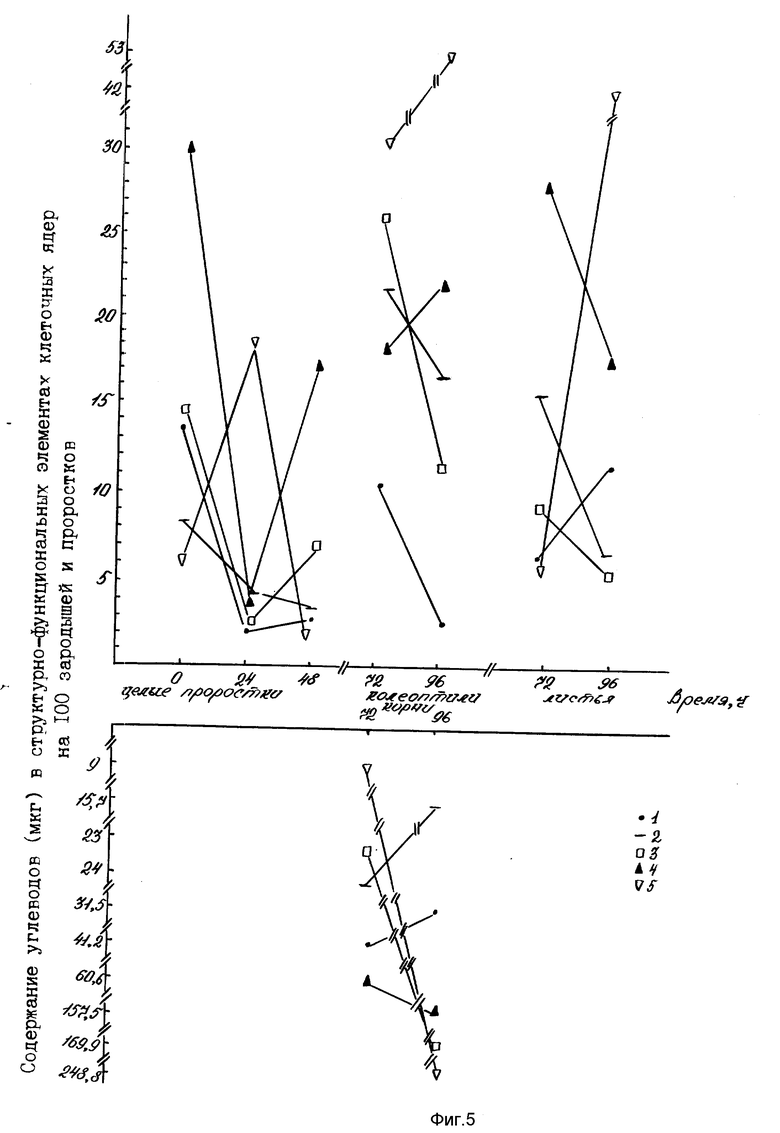
Эффективность последовательной ступенчатой экстракции ядерных фракций из растительных тканей с последующим определением в них углеводов по антроновой реакции представлено на фиг. 5.
Исследование ядерных фракций на содержание углеводов включает предварительную консервацию растительных тканей и выделение из них клеточных ядер в глицериновой среде.
Табл. 1 показывает, что структура клеточных ядер по содержанию генетического материала не нарушена; табл. 2 иллюстрирует сохранность ферментативных метаболитических систем в ядрах; табл. 3 - полноту выделения ядерных фракций; табл. 4 - зависимость содержания углеводов от консервации тканей; табл. 5 - эффективность концентраций серной кислоты и тиомочевины на антроновую реакцию.
На фиг. 1 показана чувствительность для определения микронаноколичеств углеводов цистеиновым методом; на фиг. 2 - эффективность цистеиновой реакции для определения пентоз, гексоз, полисахаридов, гликопротеидов, протеогликанов, ДНК, РНК. 1-глюкоза; 2-галактоза; 3-манноза; 4-N-ацетилглюкозамин; 5-арабиноза; 6-ксилоза; 7-ДНК; 8-РНК; 9-РНК-аза; 10-крахмал; 11-альгиновая кислота; 12-гепарин; на фиг. 3 - влияние ступенчатой последовательности экстракций ядерных фракций на выход углеводов цистеиновым методом. 1-нуклеоплазма; 2-хроматин I; 3-хроматин II; 4-ядерный матрикс; 5-ядерно-цитоплазматический остаток; на фиг. 4 - чувствительность для определения микронаноколичеств углеводов антроновым методом; на фиг. 5 - влияние ступенчатой последовательности экстракций ядерных фракций на выход углеводов антроновым методом. 1-нуклеоплазма; 2-хроматин I; 3-хроматин II; 4-ядерный матрикс; 5-ядерно-цитоплазматический остаток.
Предложенный способ рекомендуется в исследовании надмолекулярно-генетических основ организации генетических, морфогенетических программ индивидуального развития организма на разных онтогенетических стадиях дифференцированного роста растений.
Источники информации.
1. Osman M. , Doas H.A., El-Sakr A.S. Carbohydrates in cereal germs // Grasas y acotes. 1988. V. 39. N 4-5. P. 203-209.
2. Chaplin M. F. Monosacharides // Carbohydrate analysis. A practical approach. 1986. IRL Press Limited. Oxford. England. P. 1.
3. Дише З. Цветные реакции гексоз // Методы химии углеводов. М.: Мир, 1967. С. 30.
4. Вокуркова Н., Бульдяева Т.В., Троицкая Л.П., Збарский И.Б. Углеводные компоненты ядерного матрикса печени и гепатомы крысы // Бюл. экспер. биол. 1984. Т. 97. С. 46-48.
5. Збарский И.Б., Перевощикова К.А. Сократительные свойства белков клеточного ядра // Биохимия. 1951. Т.16. N 2. С. 112-124.
6. Бычков С. М. , Збарский И.Б., Хазанова А.И., Фомина В.А. Содержание мукополисахаридов и мукопротеидов в клеточных ядрах // ДАН СССР. 1951. Т. 78. N 1. С. 99-101.
7. Методы практической биохимии. М.: Мир. 1978. С. 33.
8. Иванова Э.А., Вафина Г.Х. Методика выделения протеиназ и их ингибиторов из клеточных ядер проростков пшеницы // Физиология растений. 1990. Т. 37. N 3. С. 609-615.
9. Спирин А.С. Спектрофотометрическое определение суммарного количества нуклеиновых кислот // Биохимия. 1968. Т. 23. N 5. С. 656.
10. Воскресенский П.И. Техника лабораторных работ. М.-Л.: Химия. 1964. С. 433.
11. Чичибабин А.И. Основные начала органической химии. М.: Госхимиздат. 1962.
12. Справочник химика. Л.: Химия. 1971. Т. 2. С. 444-445.
Использование: изобретение относится к физиологии и биохимии растений и может быть применено в надмолекулярной биологии индивидуального развития при морфо-, онтогенетической оценке параметрической регуляции на уровне интерфазных ядер. Сущность изобретения: углеводсодержащий материал (компоненты) извлекают из клеточных ядер, предварительно законсервированных в тканях проростков пшеницы глицериновой средой, затем выделенных и очищенных в градиенте плотности глицерина с последующей последовательной экстракцией ядерных фракций 0,14 М (нуклеоплазма), 0,35 М (хроматин I) и 2 М (хроматин II) хлористым натрием, 6 М гуанидингидрохлоридом в присутствии 0,1%-ного β -меткаптоэтанола (ядерный матрикс); 0,5 н. NaOH (ядерно-цитоплазматический остаток) и анализом (определением) в них содержания углеводов. 5 табл., 5 ил.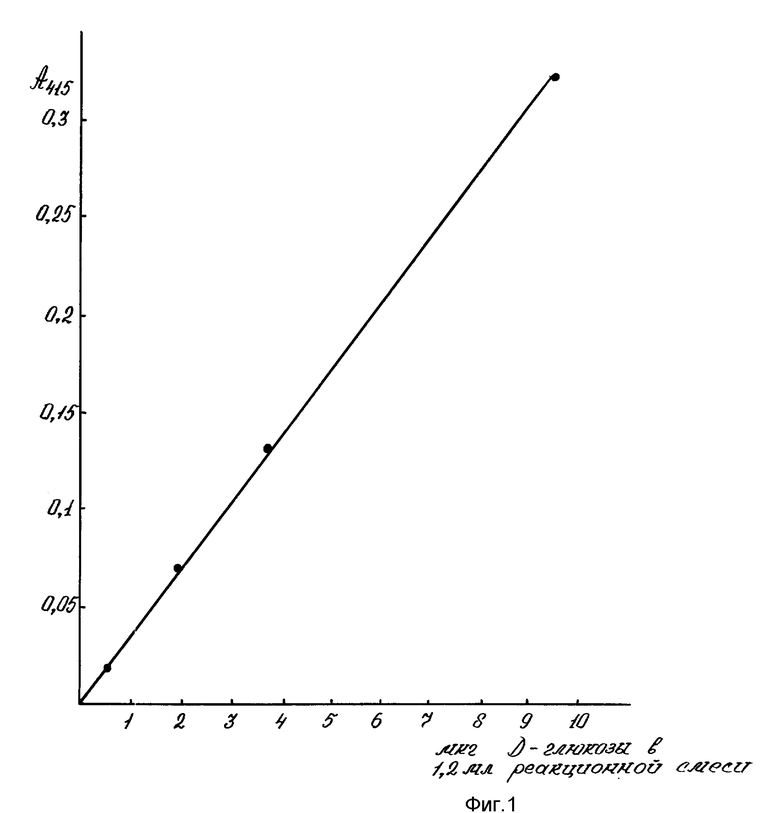
Способ определения углеводных компонентов в клеточных ядрах, включающий консервирование образцов животной ткани путем замораживания, гомогенизацию их в буферном растворе, выделение ядер, кислотный гидролиз ядерного материала и оценку содержания углеводных компонентов в гидролизате по результатам специфической цветной реакции, отличающийся тем, что в качестве исследуемых образцов используют воздушно-сухие зародыши, 1 - 2-дневные проростки и/или ткани колеоптиля, листьев и корневой системы с междоузлием 3 - 4-дневных проростков пшеницы, которые предварительно консервируют без замораживания при (-20) - (-25)oС в 80 - 90%-ном глицерине: выделенные ядра подвергают дополнительной очистке при помощи центрифугирования в градиенте плотности глицерина, затем осуществляют последовательную экстракцию ядерного материала 0,14; 0,35; 2 М хлористым натрием, 6 М гуанидин - гидрохлоридом с 0,1%-ным β- меркаптоэтанолом и 0,5 н. NaOH с получением соответствующих фракций (нуклеоплазмы, хроматина I, II, ядерного матрикса, ядерно-цитоплазматического остатка), в которых одновременно проводят кислотный гидролиз и цветную реакцию с антроном или цистеином.
| Бычков С.М., Збарский И.Б., Хазанова А.И., Фомина В.А | |||
| Содержание мукополисахаридов и мукопротеидов в изолированных клеточных ядрах | |||
| ДАН СССР, 1951, т.78, N 1, с.99-101. |
