Изобретение относится к новому способу получения [4S-(4 альфа, 12a альфа)] -4-(диметиламино)-7-(замещенный)-9-[[(замещенный амино) замещенный] -амино] -1, 4, 4a, 5, 5a, 6, 11, 12a-октагидро-3, 10, 12, 12a-тетрагидрокси-1, 11-диоксо-2-нафтаценкарбоксамидов, называемых в последующем 7-(замещенный)-9[(замещенный глицил)-амидо]-6-деметил-6-деокситетрациклинами, которые используют в качестве антибиотиков.
Изобретение также касается получения новых промежуточных соединений типа прямого или разветвленного 9-[(галоидацил) амидо]-7-(замещенный)-6-деметил-6-деокситетрациклина, которые используют для получения новых соединений настоящего изобретения.
Данное изобретение касается нового способа получения 7-(замещенный)-9-[(замещенный глицил)амидо] -6-деметил-6-деокситетрациклинов, представленных формулой I: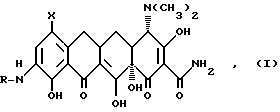
в которой
X - группа -NR1R2, в которой R1 - метил или этил; R2 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил или R1 н-пропил, и R2 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, или R1 - 1-метилэтил, и R2 - н-бутил, 1-метилпропил, 2-метилпропил, или R1 - н-бутил, и R2 - н-бутил, 1-метилпропил, 2-метилпропил; или R1 - 1-метилпропил, и R2 -2-метилпропил;
R - группа R4(CH2)nCO-, в которой n = 1-4; R4 представляет прямой или разветвленный моно(C1-C6)алкиламино с алкильными группами, выбранными из метила, этила, н-пропила, 1-метилэтила, н-бутила, 1-метилпропила, 2-метилпропила, 1,1-диметилэтила, н-пентила, 2-метилбутила, 1,1-диметилпропила, 2,2-диметилпропила, 3-метилбутила, н-гексила, 1-метилпентила, 1,1-диметилбутила, 2,2-диметилбутила, 3-метилпентила, 1,2-диметилбутила, 1,3-диметилбутила и 1-метил-2-этилпропила; или циклопропиламино; циклобутиламино; или ди(C1-C4)алкиламино, выбранный из диметиламино, диэтиламино, метил(бутил)амино и этил(1-метил)амино, и их солей.
Согласно изобретению, способ включает взаимодействие галоидацилгалогенида формулы: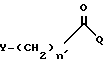
в которой
Q - галоген, выбранный из брома, хлора, фтора и иода;
n' = O,
Y - прямая или разветвленная альфагалоид(C2-C4)алкильная группа, выбранная из бромметила, хлорметила, иодметила, альфа-бромэтила, альфа-хлорэтила, альфа-бромбутила и альфа-хлоризобутила,
или
n' = 1-4
Y - галоген, выбранный из брома, хлора, иода и фтора;
с 9-амино-7-(замещенный)-6-деметил-6-деокситетрациклином формулы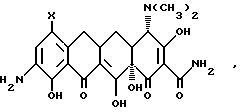
в которой X имеет значения, определенные выше, или его формакологически приемлемых органических и неорганических солей для получения промежуточного продукта 9[(галоидацил)амидо]-7-(замещенный) -6-деметил-6-деокситетрациклина формулы: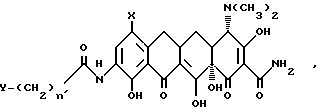
в которой
X, Y и n' имеют значения, определенные выше, или его органических или неорганических солей, и взаимодействие промежуточного продукта, 9-[(галоидацил)амидо] -7-(замещенный)-6-деметил-6-деокситетрациклина или его фармакологически приемлемых органических или неорганических солей, с нуклеофилом формулы R4H, в которой R4 имеет значения, определенные выше, с получением 7-(замещенный)-9-[(замещенный глицил) амидо-6-деметил-6-деокситетрациклина в соответствии с формулой I или его органических и неорганических солей.
Данный новый способ является эффективным путем получения 7-(замещенный)-9-[(замещенный глицил) амидо] -6-деметил-6-деокситетрациклина или его фармакологически приемлемых органических и неорганических солей. Новый способ допускает возможность этих соединений в двух реакциях.
Первая реакция приводит к образованию обычного промежуточного продукта, 9-[(галоидацил)амидо]-7-(замещенный) 6-деметил-6-деокситетрациклина, или его фармакологически приемлемых органических или неорганических солей. Вторая реакция обеспечивает возможность взаимодействия обычного промежуточного продукта с широким множеством аминов и приводит к широкому спектру 7-(замещенный)-9-[(замещенный глицил) амино]-6-деметил-6-деокситетрациклинов или их фармакологически приемлемых органических или неорганических солей. Использование сложных защитных групп исключается, что позволяет получать конечные продукты всего лишь в двух реакциях.
Предпочтительным является способ получения соединений в соответствии с вышеуказанной формулой I,
в которой X - группа -NR1R2, в которой R1 = метил или этил и R2 = метил, этил, н-пропил, 1-метилэтил, н-бутил;
R - R4(CH2)nCO-, в которой n = 1 - 4, и R4 - прямая или разветвленная моно (C1-C6) алкиламино группа, в которой алкил выбран из метила, этила, н-пропила, 1-метилэтила, н-бутила, 1-метилпропила, 2-метилпропила, 1,1-диметилэтила, н-пентила, 2-метилбутила, 1,1-диметилпропила, 2,2-диметилпропила, 3-метилбутана, н-гексила, 1-метилпентила, 1,1-диметилбутила, 2,2-диметилбутила, 3-метилпентила, 1,2-диметилбутила, 1,3-диметилбутила и 1-метил-2-этил-пропила; или циклопропиламино; циклобутиламино; или дизамещенная аминогруппа, выбранная из диметиламинодиэтиламино, метил(бутил) амино и этил (1-метилэтил) амино, а также фармакологически приемлемых органических и неорганических солей.
В настоящее изобретение также включен способ получения нового промежуточного продукта, прямого или разветвленного 9-[(галоидацил) амидо]-7-(замещенный)-6-деметил-6-деокситетрациклина, используемого для получения вышеуказанных соединений формулы I. Данный промежуточный продукт включает соединения, имеющие формулу II: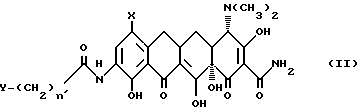
в которой X - группа -NR1R2, в которой R1 - метил или этил, R2 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, или R1 - н-пропил, R2 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, или R1 - 1-метилэтил, R2 - н-бутил, 1-метилпропил, 2-метилпропил, или R1 - н-бутил, R2 - н-бутил, 1-метилпропил, 2-метилпропил; или R1 - 1-метилпропил, R2 - 2-метилпропил;
n' = 0,
Y - прямая или разветвленная альфа-галоид(C1-C4)алкильная группа, выбранная из бромметила, хлорметила, иодметила, альфа-бромэтила, альфа-хлорэтила, альфа-бромбутила, альфа-хлоризобутила,
или
n' = 1 - 4,
Y - галоген, выбранный из брома, хлора, фтора и иода, а также и фармакологически приемлемых органических и неорганических солей.
Новый способ получения промежуточного соединения формулы II включает взаимодействие 9-амино-7-(замещенный)-6-деметил-6-деокситетрациклина с соединением формулы: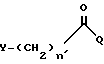
в которой
Y, n' и Q имеют определенные значения.
Предпочтительным является способ получения соединений в соответствии с вышеприведенной формулой II,
в которой
X - группа - NR1R2, в которой R1 = метил или этил, R2 = метил, этил, н-пропил, 1-метилэтил, н-бутил;
n' = 0,
Y - прямая или разветвленная альфа-галоид (C1-C4) алкильная группа, выбранная из бромметила, хлорметила, йодметила, альфа-бромэтила, альфа-хлорэтила, альфа-бромбутила и альфа-хлоризобутила,
или
n' = 1 - 4,
Y - галоген, выбранный из брома, хлора, йода и фтора, а также фармакологически приемлемых органических и неорганических солей.
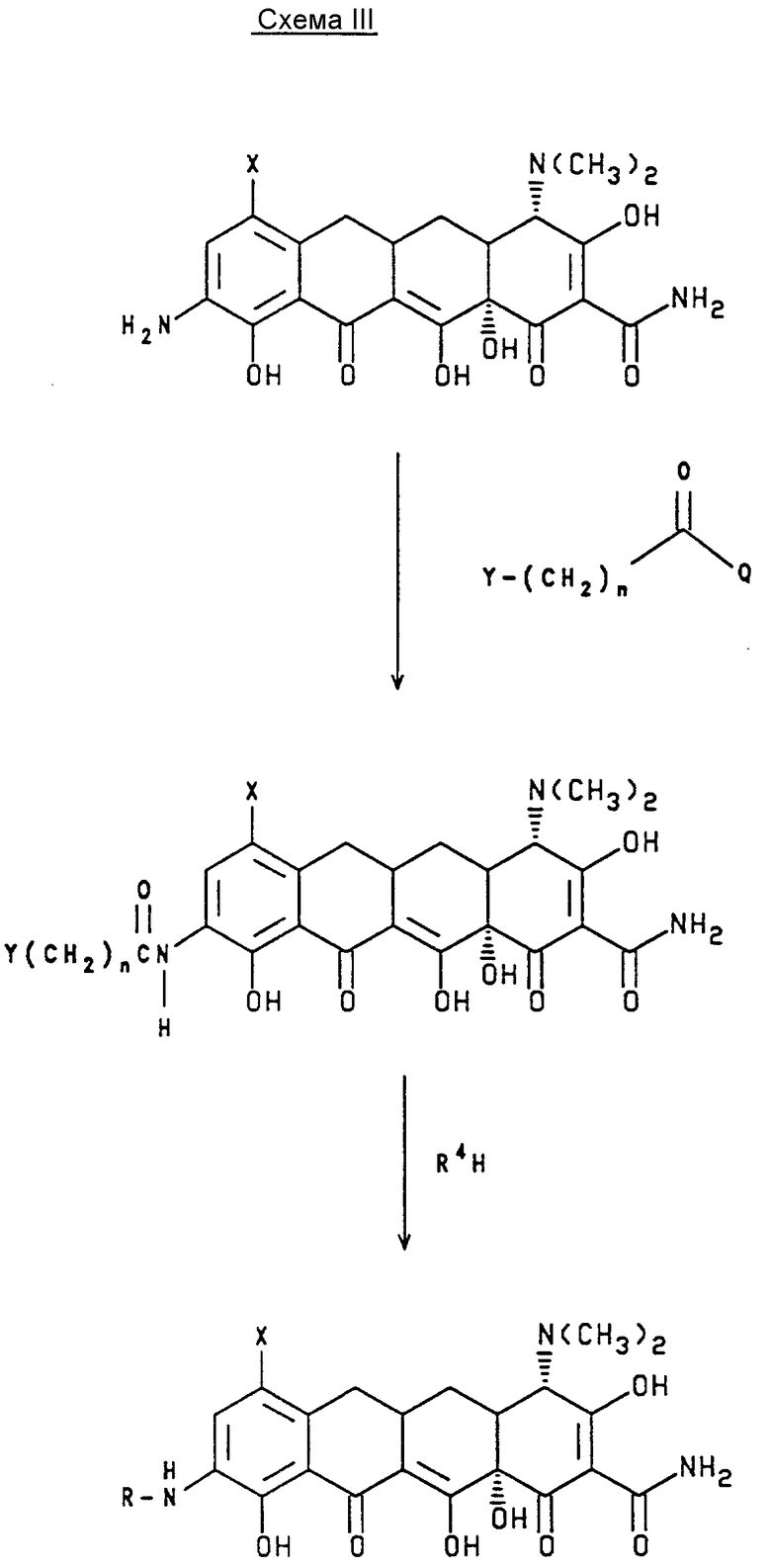
Новый способ настоящего изобретения, схема III, предусматривает более легкий путь получения 7-(замещенный)-9-[(замещенный глицил) амидо]-6-деметил-6-деокситетрациклинов или их фармакологически приемлемых органических и неорганических солей. Данный новый способ предусматривает путь получения некоторых из 7-(замещенный)-9-[(замещенный глицил) амидо]-6-деметил-6-деокситетрациклинов или их фармакологически приемлемых органических и неорганических солей, которые было бы очень трудно получить, используя любой из способов, имеющихся на предшествующем уровне развития техники, показанных на схеме I или II.
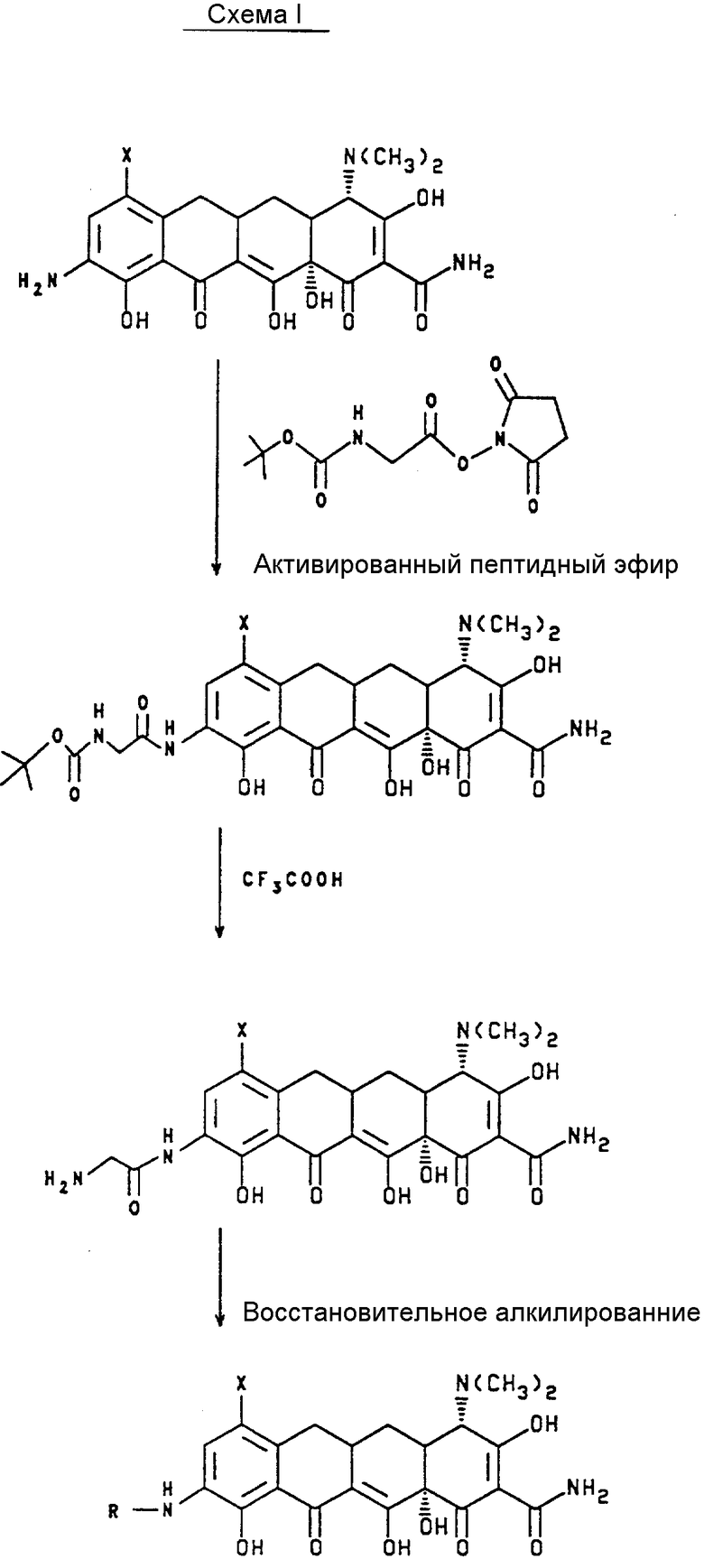
Способ, показанный на схеме I, основан на гидро N- алкилирования-9-(глициламидо)-7-(замещенный)-6-деметил-6- деокситетрациклина. Этот способ возможно использовать, лишь когда на азот вводят два идентичных заместителя. Было бы невыполнимо ввести последовательно два различных заместителя на азот, поскольку условия гидроалкилирования таковы, что оба азота замещают в один и тот же момент. Таким образом, используя способ схемы I, было бы невозможно ввести эффективно единственный заместитель. В дополнение, начальная реакция трет-бутилового эфира (сукцинилоксикарбонил) метил карбаминовой кислоты с соответствующим 9-амино-7-(замещенный)-6-деметил-6-деокситетрациклином дает лишь умеренные выходы.
Способ, показанный на схеме II, основан на образовании кислого хлорида из моно- или дизамещенной (C1-C6) амино замещенный ацил кислоты и взаимодействии таким образом образованного кислого хлорида с амином в положении 9 9-амино-7-(замещенный)-6-деметил-6-деокситетрациклина. Обычно, кислый хлорид образуют взаимодействием подходящего моно- или дизамещенного (C1-C6) амина либо с галогенуксусными кислотами (или сложными эфирами), либо с их синтетическими эквивалентами, например n-толуолсульфонилокси-уксусной кислотой или метансульфонилоксиуксусной кислотой. В случае N-(монозамещенных) амино кислот способ, показанный на схеме II, можно использовать только через использование азотных защитных групп. Однако защитные группы должны уцелеть в реакциях образования кислого хлорида, но также должны легко удаляться из конечных продуктов без ущерба для присоединенного ядра тетрациклина. Включение защитных групп в этот процесс влечет за собой дополнительные стадии и является эксплуатационно сложным. При использовании способа, показанного на схеме II, для каждой новой структурной единицы, например, 9-[(замещенный глицил)амидо] -7-(замещенный)-6-деметил-6-деокситетрациклина., потребовалось бы минимум 4 стадии синтеза и даже до 8 стадий синтеза.
В противоположность, новый способ настоящего изобретения позволяет получение конечного продукта только в две стадии синтеза. В соответствии с новым способом на схеме III, введение монозамещенных (С1-С6) аминов или дизамещенных (C1-C6) аминов на 9-[(галоидацил)амино]-7-(замещенный)-6-деметил-6-деокситетрациклины не требует использования азотных защитных групп. Таким образом, данный процесс позволяет использовать структурно однородные или химически чувствительные амины, например, амины, которые могут разлагаться благодаря избытку кислоты. Эти особые амины можно было бы использовать в процессе с эксплуатационной эффективностью. Поскольку многие амины являются летучими, их удаление из реакционной смеси вакуумной перегородкой будет минимизировать побочные продукты, которые могут усложнять процесс очистки. В заключение, амины можно также было бы восстанавливать для дальнейшего использования. Наиболее важно то, что можно получить более широкое разнообразие структурных единиц не более чем двумя стадиями синтеза.
В соответствии с новым способом настоящего изобретения, схемой III, исходный 9-амино-7-(замещенный)-6-деметил-6-деокситетрациклин или фармакологически приемлемую органическую и неорганическую соль, полученный(ую) процедурой, описанной в патентной заявке США N 771576, поданной 4 октября 1991 г., смешивают с
a) полярным апротонным растворителем, таким как 1,3-диметил-3, 4,5,6-тетрагидро-2(IH)-пиримидон, в последующем называемый DMPU, гексаметилфосфорамид, в последующем называемый HMPA, 1,3-диметил-2-имидазолидинон, диметилформамид, диметилацетамид, N-метилпирролидон, 1,2-диметоксиэтан или их эквиваленты;
b) инертным растворителем, таким как ацетонитрил, метилен хлорид, тетрагидрофуран, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, тетерахлорэтан, диэтиловый эфир, трет-бутилметиловый эфир, изопропиловый эфир или их эквиваленты;
c) основанием, таким как карбонат натрия, бикарбонат натрия, ацетат натрия, карбонат калия, бикарбонат калия, триэтиламин, карбонат цезия, эквиваленты карбоната или бикарбоната лития; и
d) прямым или разветвленным галогенацил галогенидом формулы: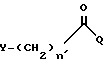
в которой
Y, n и Q имеют значения, определенные выше; таким как бромацетил бромид, хлорацетил хлорид или 2-бромпропионил бромид; галоген и галоген в галоидацилгалогениде могут быть одинаковыми или различными, и выбираются из хлора, брома, йода и фтора;
e) в течение 0,5-5 ч при от комнатной температуры до температуры флегмы реакции; для образования соответствующего 9-[(галоидацил)амидо] -7-(замещенный)-6-деметил-6-деокситетрациклина или его фармакологически приемлемой органической или неорганической соли.
Для получения 7-(замещенный)-9-[(замещенный глицил)амидо] -6-деметил-6-деокситетрациклина или его фармакологически приемлемой органической или неорганической солей, 9-[(галоидацил)] амидо)-7-(замещенный)-6-деметил-6-деокситетрациклин или его фармакологически приемлемые органические и неорганические соли обрабатывают, в атмосфере аргона, азота или гелия,
a) нуклеофилом R4H, где R4 имеет вышеопределенное значение, таким как амин или замещенный амин, например, метиламин, диметиламин, этиламин, н-бутиламин, пропиламин или н-гексиламин;
b) в полярном апротонном растворителе, таком как DMPU, HMPA, диметилформамид, диметилацетамид, N-метилпирролидон, 1,2-диметоксиэтан, тетрагидрофуран, или полярном протонном растворителе, таком как вода, метанол или их эквиваленты;
c) в течение 0,5-2 ч при комнатной температуре или температуре флегмы для получения желаемого-7(замещенный)-9-[(замещенный глицил)амидо]-6-деметил-6-деокситетрациклина, или его фармакологически приемлемых органических и неорганических солей.
В том случае, когда желательными являются формы неорганической и органической соли, 7-(замещенный)-9-[(замещенный глицил)амидо]-6-деметил-6-деокситетрациклины можно получить в виде неорганических и органических солей, используя методы, известные специалистам [Richard C. Larock, Comprehensive Organic Transformations (Всесторонние органические предобразования), VCH Publishers, 411 - 415, 1989]. Для специалиста хорошо известно, что подходящая форма соли выбирается, исходя из физической и химической устойчивости, подвижности, гигроскопичности и растворимости. Предпочтительно, 7-(замещенный)-9-[(замещенный глицил) амидо] -6-деметил-6-деокситетрациклины получают в виде неорганической соли, такой как хлористоводородная, бромистоводородная, йиодистоводородная, фосфорная, азотная или сульфат; или органической соли, такой как ацетат, бензоат, цитрат, цистеин или другие аминокислоты, фумарат, гликолят, малеат, сукцинат, тартрат, алкилсульфонат или арилсульфонат. В зависимости от стехиометрии используемых кислот образование соли имеет место с C(4)-диметиламино группой (I эквивалент кислоты) или с обеими C(4)-диметиламино группой и заместителем на R4 группе (2 эквивалента кислоты). Соли являются предпочтительными для перорального и парентерального приема.
Некоторые из соединений вышеописанной схемы III имеют центры асимметрии на углероде, несущем R4 заместитель. Соединения могут, следовательно, существовать в по крайней мере двух (2) стереоизомерных формах. Настоящее изобретение заключает в себе способ получения рацемической смеси стереоизомеров, а также всех стереоизомеров соединений, либо свободных от других стереизомеров, либо смешанных с стереизомерами в любой пропорции энантиомеров. Абсолютная конфигурация любого соединения может быть определена с помощью обычной рентгеновской кристаллографии. Стереохимия центров на единице тетрациклина (т.е. C-4, C-4a, C-5a и C-12a) остается нетронутой в течение последовательности реакций.
Данное изобретение будет описано более подробно следующими неограничивающими примерами.
Пример 1.
Терт-бутиловый эфир (сукцинилоксикарбонил) метил карбаминовой кислоты
К 5oC раствору 8,76 г N-(тертбутоксикарбонил) глицина и 5,75 г N-гидроксисукцинимида в 100 мл диоксана и 160 мл 1,2-диметоксиэтана добавляли 10,3 г дициклогексилкарбодиимида. Смесь держали при 0oC в течение 24 ч. Реакционную смесь отфильтровывали, промывали диоксаном, и фильтрат выпаривали в вакууме до получения твердого тела. Твердое тело тритуратировали диэтиловым эфиром, собирали и высушивали с получением 12 г желаемого промежуточного продукта.
Вышеописанный эксперимент представляет литературную процедуру, найденную в JACS, T.86, 1839 (1939).
Пример 2
1,1-Диметилэтиловый эфир [7,S-(7-альфа, 10a-альфа)]-2-[[[9-(аминокарбонил)4, 7-бис(диметиламино)-5,5a, 6, 6a, 7, 10, 10a, 12-октагидро-1,8,10a, 11-тетрагидрокси-10,12 -диоксо-2-нафтаценил] амино]-2-оксоэтил] карбаминовой кислоты.
Смесь 0,850 г 9-амино-4,7-бис(диметиламино)-6-деметил -6-деокситетрациклина, 0,680 г ацетата натрия в 25 мл тетрагидрофурана и 5 мл воды перемешивали при 25oC в течение 5 мин. Раствор обрабатывали, 0,359 г продукта из примера 1, перемешивали в течение 2 ч. и экстрагировали хлороформом. Органический слой выпаривали в вакууме с получением 0,50 г желаемого продукта.
MC(FAB) : m/Z 630 (M+H)
Пример 3
[4S-(4 альфа, 12 альфа)]-9-[(аминоацетил)амино]-4,7-бис- (диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12, 12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид моно(трифторацетат).
Раствор 0,030 г продукта из примера 2 и 1,0 мл трифторуксусной кислоты выдерживали при комнатной температуре в течение 24 ч с последующим выпариванием in vacuo. Остаток растирали с метиловым спиртом и собирали твердое тело с выходом 0,024 г желаемого продукта.
MC(FAB) : m/z 530(M+H).
Пример 4
Диметиламиноацетил хлорид гидрохлорид
Смесь 15 г N,N-диметилглицин гидрохлорида (измельченного и высушенного в вакуумной печи при 45-50oC в течение 24 ч) и 13,85 мл тионилхлорида нагревали на песочной бане до 78oC и держали при этой температуре в течение 1,5 ч. К смеси добавляли толуол, и избыток жидкости удаляли пипеткой. Эту операцию повторяли несколько раз. Твердое тело затем переносили в воронку Бюхнера, промывали метилен хлоридом и высушивали под вакуумом при 50oC в течение 24 ч с выходом 14,2 г желаемого промежуточного продукта.
Пример 5
[4S-(4 альфа, 12 альфа)]-4, 7-бис (диметиламино)-9-[[(диметиламино) ацетил] амино] -1,4,4a, 5,5a,6, 11,12a - октагидро-3,10,12,12a-тетрагидрокси-1, 11-диоксо-2-нафтаценкарбоксамид дигидрохлорид
К смеси 6,68 г 9-амино-4,7-бис(диметиламино)-6-деметил-6 -деокситетрациклин дисульфата в 120 мл (DMPU) и ацетонитрила добавляли 6,57 г карбоната натрия. Смесь перемешивали в течение 5 мин, с последующим добавлением 2,83 г продукта из примера 4. Реакцию перемешивали в течение 1 ч., отфильтровывали, и фильтрат медленно добавляли к смеси метиленхлорид/диэтиловый эфир (1200 мл/400 мл).
Твердое вещество собирали, растворяли в 250 мл метилового спирта и медленно добавляли к 1600 мл метиленхлорида. Осадок собирали, промывали диэтиловым эфиром и высушивали с получением 5,75 г желаемого продукта.
MC(FAB) : m/z 558(M+H).
Пример 6
[4S-(4 альфа, 12a альфа)]-9-[хлорацетил)амино]-4,7-бис-(диметиламино)-1,4,4a, 5,5a, 6, 11, 12a-октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2- нафтаценкарбоксамид дигидрохлорид.
К находящемуся при комнатной температуре раствору 0,334 г 9-амино-4,7-бис(диметиламино)-6-деметил-6-деокситетрациклин дисульфата, 6 мл (1,3-диметил-3,4,5, 6-тетрагидро-2-(1H)-пиримидинона, называемого в последующем DMPU и 2 мл ацетонитрила добавляли 0,318 г карбоната натрия. Смесь перемешивали в течение 5 мин с последующим добавлением 0,068 г хлорацетилхлорида. Реакцию перемешивали в течение 30 мин, отфильтровывали и фильтрат добавляли по каплям к 100 мл диэтилового эфира, содержащего 1 мл 1М соляной кислоты в диэтиловом эфире. Полученное твердое вещество собирали и высушивали с получением 0,340 г желаемого промежуточного продукта.
MC (FAB) : m/z 549 (M+H).
Пример 6A
[4S-(4 альфа, 12a альфа)]-9-[(хлорацетил)амино]-4,7-бис(диметиламино)-1, 4,4a, 5,5a, 6,11,12a-октагидро-3,10,12,12a- тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид (свободное основание).
Указанное соединение получали процедурой примера 6, используя 0,51 г 9-амино-4,7-бис(диметиламино)-6-деметил-6-деокситетрациклин гидрохлорида, 50 мл DMPU, 5 мл ацетонитрила, 0,668 г карбоната натрия и 0,452 г хлорацетил хлорида с получением 0,52 г желаемого продукта как свободного основания.
1H ЯМР (ДМСО-d6): δ 9,3 (с, 1H); 7,9 (с, 1H); 4,45 (с, 2H).
Пример 7
[4S-(4 альфа, 12a альфа]-9-[(бромацетил)амино]-4,7-бис (диметиламино)-1,4,4a, 5,5a,6,11,12a-октагидро-3,10,12,12a-тетрагидрокси-1,11- диоксо-2-нафтаценкарбоксамид моногидробромид.
К раствору 5,01 г 9-амино-4,7-бис(диметиламино)-6-деметил -6-деокситетрациклин дисульфата, 100 мл DMPU и 25 мл ацетонитрила добавляли 5,0 г карбоната натрия. Реакцию перемешивали в атмосфере аргона при комнатной температуре в течение 5 мин, с последующим добавлением 3,03 г бромацетил бромида. Перемешивание продолжали в течение дополнительного часа. Твердое вещество собирали, и фильтрат медленно добавляли в изопропиловому спирту /диэтиловому эфиру (200 мл/750 мл). Желтое твердое вещество собирали, промывали изопропанолом и диэтиловым эфиром с получением 5,77 г желаемого промежуточного продукта.
MC(FAB) : m/z 593(M+H)
Пример 7A
[4S-(4 альфа, 12a альфа)]-9-[(бромацетил)амино]- 4,7-бис(диметиламино)1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12,12a- тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид (свободное основание)
К 0,20 г продукта из примера 7 в 3 мл 1,3-диметил-2- имидазолидинона добавляли 0,30 г бикарбоната натрия. Реакцию перемешивали при комнатной температуре в течение 15 мин и отфильтровывали. Фильтрат добавляли к 15 мл диэтилового эфира и полученный осадок собирали с получением 0,150 г желаемого промежуточного продукта как свободного основания
MC(FAB):m/z 593(M+H).
Пример 8
[4S-(4 альфа, 12a альфа)]-9-[(бромацетил)амино]-4,7-бис (диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12,12a-тетрагидрокси- 1,11-диоксо-2-нафтаценкарбоксамид дигидрохлорид
Указанное соединение получали процедурой примера 6, используя 0,668 г 9-амино-4,7-бис(диметиламино)-6-деметил-6- деокситетрациклиндисульфата, 6 мл DMPU, 2 мл ацетонитрила, 0,636 г карбоната натрия и 0,215 г бромацетилхлорида. Получили семь десятых грамма желаемого промежуточного продукта
MC(FAB):m/z 593(M+H).
Пример 9
[4S-(4 альфа, 12a альфа)] -9-[(2-бром-1-оксопропил)амино] - 4,7-бис(диметиламино)-1,4,4a,5,5a,6,11,12a-октагидро-3,10,12,12a- тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид гидробромид
Указанное соединение получали процедурой примера 6, используя 1,00 г 9-амино-4,7-бис-(диметиламино)-6-деметил-6- деокситетрациклиндисульфата, 1,0 г карбоната натрия и 0,648 г 2-бромпропионил бромида с получением 0,981 г желаемого промежуточного продукта
MC(FAB):m/z 607(M+H).
Пример 10
[4S-(4 альфа, 12a альфа)]-9-[(4-бром-1-оксобутил)амино]- 4,7-бис(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12,12a- тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид дигидрохлорид
Указанное соединение получали процедурой примера 6, используя 1,34 г 9-амино-4,7-бис(диметиламино)-6-деметил-6- деокситетрациклиндисульфата, 1,3 г карбоната натрия, 24 мл DMPU, 8 мл ацетонитрила и 0,389 г 4-бромбутирил хлорида с получением 1,45 г желаемого продукта.
Пример 11
[4S-(4 альфа, 12a альфа)]-4,7-бис(диметиламино)-9- [[(диметиламино)ацетил] амино] -1, 4, 4a, 5, 5a, 6, 11, 12a-октагидро-3, 10, 12, 12a-тетрагидрокси-1, 11-диоксо-2-нафтаценкарбоксамид дигидрохлорид
К раствору 0,15 г продукта из примера 6 в 4 мл DMPU добавляли 0,85 г диметиламина (40% в воде). Реакцию перемешивали в течение 20 мин с последующим выпариванием в вакууме для удаления избытка диметиламина. Смесь отфильтровывали, и фильтрат добавляли по каплям к 70 мл изопропилового спирта /диэтилового эфира (1:1). К этому раствору добавляли 1 мл 1М соляная кислота/ диэтиловый эфир. Полученный осадок собирали, промывали изопропиловым спиртом и диэтиловым эфиром, и высушивали с получением 0,11 г желаемого продукта.
MC)FAB):m/z 558 (M+H).
Пример 12
[4S-(4 альфа, 12a альфа)]-4,7-Бис(диметиламино)-1,4,4a,5,5a, 6,11,12a-октагидро-3,10,12,12a-тетрагидрокси-9-[[(метиламино) ацетил] амино] -1,11-диоксо-2-нафтаценкарбоксамид дигидрохлорид
Смесь 0,1258 г продукта из примера 7, 5 мл 40% метиламина в воде и 5 мл метилового спирта в атмосфере аргона перемешивали при комнатной температуре в течение 30 мин. Избыток метиламина удаляли в вакууме и остаток разбавляли небольшим объемом метилового спирта. Разбавленные реакционный раствор добавляли по каплям к 100 мл диэтилового эфира, содержащего 1 мл 1М соляной кислоты в диэтиловом эфире, и 10 мл изопропилового спирта. Полученное твердое вещество собирали и высушивали с получением 0,106 г желаемого продукта
MC(FAB):m/z 554(M+H).
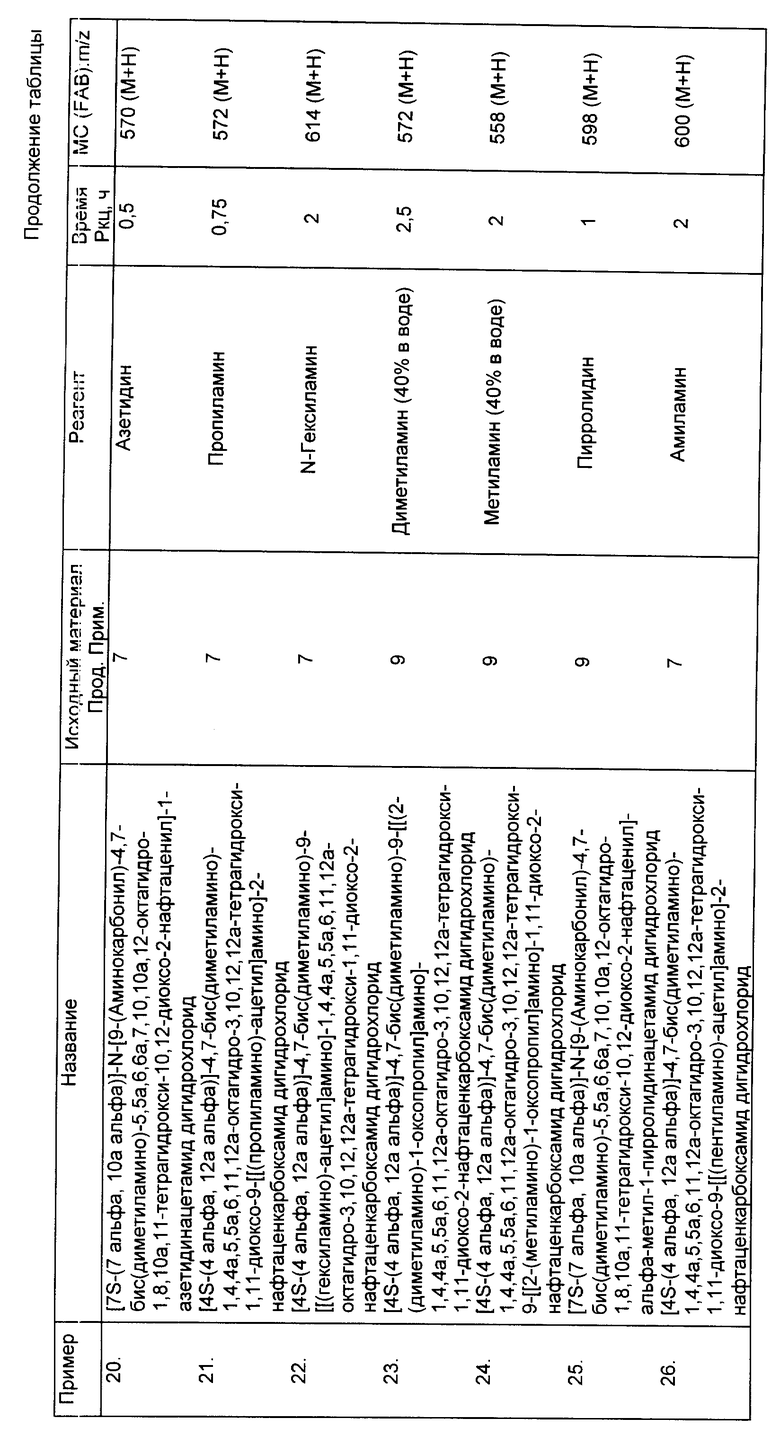
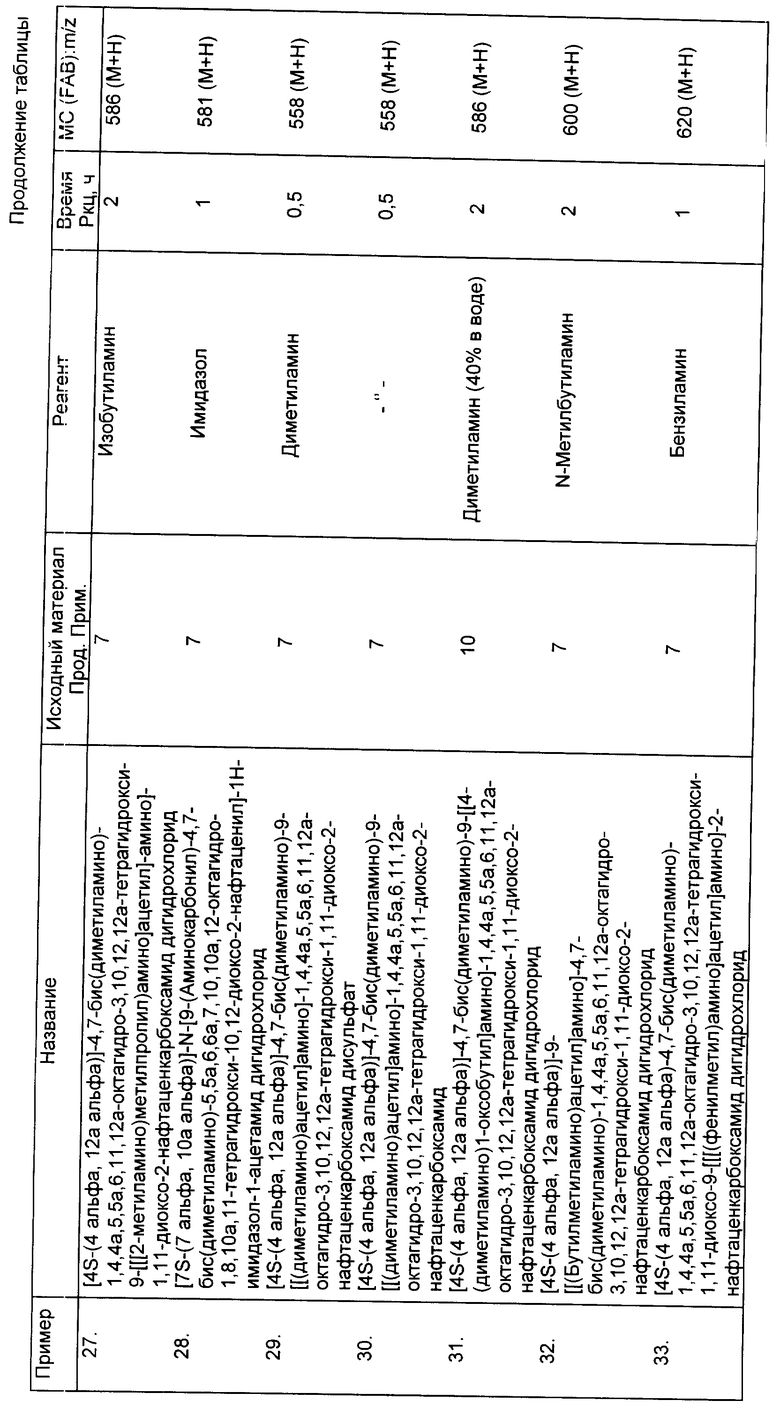
Соединения данного изобретения, перечисленные ниже в примерах 13-33 (см. таблицу), получены по существу теми же методами, которые детально описаны выше в примере 12.
Пример 34
[7S-(7 альфа, 10a альфа)]-N-[2-[[9-(аминокарбонил)-4,7-бис (диметиламино-5,5a,6,7,10a,12-октагидро-1,8,10a,11-тетрагидрокси- 10,12-диоксо-2-нафтаценил]амино]-2-оксоэтил]глицин фенилметиловый эфир
К 0,30 г бензилглицин гидрохлорида в 3 мл 1,3-диметил-2- имидазолидинона добавляли 0,60 г бикарбоната натрия. Смесь перемешивали при комнатной температуре в течение 15 мин и отфильтровывали. К фильтрату добавляли 0,20 г продукта из примера 7A. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем добавляли к диэтиловому эфиру. Полученное твердое вещество собирали.
Пример 43
[7S-(7 альфа, 10a альфа)]-N-[2-[[9-(аминокарбонил)4,7-бис (диметил-амино)-5,5a, 6,7,10a, 12-октагидро-1,8,10a,11-тетрагидрокси- 10,12-диоксо-2-нафтаценил]амино]-2-окоэтил]глицин
Одну десятую грамма продукта из примера 34 в 10 мл 2-метоксиэтана восстанавливали каталитически, в вибраторе Парра, с помощью 0,10 г 10% палладия на углероде, при 30 фунтов/кв. дюйм водорода, в течение 2 ч. Реакционную смесь отфильтровывали и фильтрат выпаривали с получением 0,050 г желаемого продукта.
FAB - MC:m/z 588 (M+H).

Изобретение относится к новому способу получения соединений формулы I, приведенной в описании. Изобретение также относится к способу получения промежуточных соединений, используемых для получения соединений формулы I. Используя обычное промежуточное соединение, с помощью нового способа можно эффективно получать соединения формулы I. 2 с. и 3 з.п.ф-лы, 1 табл.
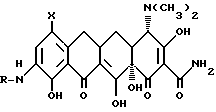
в которой X - группа -NR1R2, в которой R1 - метил или этил, R2 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, или R1 - н-пропил и R 2 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, или R1 - 1-метилэтил и R2 - н-бутил, 1-метилпропил, 2-метилпропил, или R1 - н-бутил и R2 - н-бутил, 1-метилпропил, 2-метилпропил, или R1 - 1-метилпропил и R2 - 2-метилпропил;
R - группа R4(CH2)nCO-, в которой n = 1 - 4, R4 - прямой или разветвленный моно (C1 - C6)-алкиламино с алкильными группами, выбранными из метила, этила, н-пропила, 1-метилэтила, н-бутила, 1-метилпропила, 2-метилпропила, 1,1-диметилэтила, н-пентила, 2-метилбутила, 1,1-диметилпропила, 2,2-диметилпропила, 3-метилбутила, н-гексила, 1-метилпентила, 1,1-диметилбутила, 2,2-диметилбутила, 3-метилпентила, 1,2-диметилбутила, 1,3-диметилбутила и 1-метил-2-этилпропила, или циклопропиламино, циклобутиламино, или ди(C1 - C6)алкиламино, выбранный из диметиламино, диэтиламино, метил(бутил)амино и этил(1-метилэтил)амино, отличающийся тем, что 9-амино-7-(замещенный)-6-диметил-6- деокситетрациклин или его фармакологически приемлемую неограническую соль смешивают с полярным апротонным растворителем, инертным растворителем, основанием и подвергают взаимодействию с прямым или разветвленным галоидацилгалогенидом общей формулы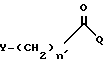
в которой Q - галоген, выбранный из брома, хлора, фтора и иода;
n' = 0;
Y - прямая или разветвленная альфа-галоид(C1 - С4)алкильная группа, выбранная из бромметила, хлорметила, иодметила, альфа-бромэтила, альфа-хлорэтила, альфа-бромбутила и альфа-хлориз обутила, или n' = 1 - 4 и Y - галоген, выбранный из брома, хлора, иода и фтора,
в течение 0.5 - 5.0 ч при температуре от комнатной до температуры дефлегмации реакционной смеси, полученный 9-[(галоидацил]амидо]-7-(замещенный)-6-диметил-6-деокситетрациклин или его фармакологически приемлемую неорганическую соль выделяют и подвергают взаимодействию в полярном растворителе в инертной атмосфере гелия, азота или аргона с нуклеофилом формулы R4H, в которой R4 имеет указанные значения, в течение 0,5 - 2,0 ч при температуре от комнатной до температуры дефлегмации реакционной смеси, и соединение формулы I или его фармакологически приемлемую неорганическую соль выделяют.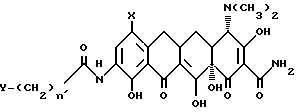
в которой X - группа - NR1R2, в которой R1 - метил или этил и R2 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, или R1 - н-пропил и R2 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, или R1 - 1-метилэтил и R2 - н-бутил, 1-метилпропил, 2-метилпропил, или R1 - н-бутил и R2 - н-бутил, 1-метилпропил, 2-метилпропил, или R1 - 1-метилпропил и R2 - 2-метилпропил;
n' = 0;
Y - прямая или разветвленная альфагалоид(C1 - C4)алкильная группа, выбранная из бромметила, хлорметила, иодметила, альфа-бромэтила, альфа-хлорэтила, альфа-бромбутила, альфа-хлоризобутила,
или n' = 1 - 4 и Y - галоген, выбранный из брома, хлора, фтора и иода,
отличающийся тем, что 9-амино-7-(замещенный)-6-диметил-6-деокситетрациклин или его фармакологически приемлемую неорганическую соль смешивают с полярным апротонным растворителем, инертным растворителем, основанием и подвергают взаимодействию с прямым или разветвленным галоидацилгалогенидом общей формулы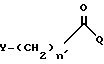
в котором Q - галоген, выбранный из брома, хлора, фтора и иода;
n' и Y имеют указанные значения,
в течение 0,5 - 5,0 ч при температуре от комнатной до температуры дефлегмации реакционной смеси и соединение формулы II или его фармакологически приемлемую неорганическую соль выделяют.
| Richard C | |||
| Larock, Comprehersive Organic Transformatins | |||
| VCH Publishers, 411 - 415, 1989 | |||
| EP, патент, 535346, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

