Настоящее изобретение относится к новым производным тетрациклинов, далее называемым 7, -8, -9(замещенными)-6-деметил-6-дегидрокситетрациклинами, которые пригодны для применения в качестве антибиотиков и проявляют антибактериальную активность против широкого спектра организмов, включая организмы, устойчивые к тетрациклинам.
Изобретение относится также к новым промежуточным соединениям 7-9(замещенным-6)-деметил-6-дегидрокситетрациклинам, пригодным для получения вышеуказанных новых соединений в соответствии с настоящим изобретением, к новым способам получения новых соединений и промежуточных соединений, а также к антибактериальной фармацевтической композиции.
В частности, настоящее изобретение качается новых 7-(замещенный)-8-(замещенный)-9-(замещенный)-6-деметил- 6-дегидрокситетрациклинов, представленных общей формулой 1, которые проявляют антибактериальную активность; фармацевтических препаратов, содержащих эти соединения; новых промежуточных соединений и способов получения этих соединений, которые проявляют "ин витро" и "ин виво" антибиотическую активность против стойких к тетрациклинам штаммов, а также высокий уровень активности против штаммов, которые обычно чувствительным к тетрациклинам.
В наиболее предпочтительном варианте изобретение относится к производным 7, -8, -9(замещенных)-6-деметил-6-дегидрокситетрациклинов общей формулы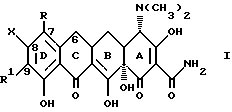
где
X представляет галоген, выбранный из брома, фтора или йода;
R и R1 являются одинаковыми или различными и представляют собой водород, амино, галоген (выбранный из хлора, брома, фтора или йода) или NR2R3; и когда R или R1 представляют -NR2R3, а R2 = водород, то
R3 представляет собой метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил;
а когда R2 = метил или этил,
R3 представляет собой метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-пропил,
R3 представляет собой н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = 1-метилэтил,
R3 представляет собой н-бутил, 1-метилпропил, 2-метилпропил;
когда R2 = н-бутил,
R3 представляет собой н-бутил, 1-метилпропил, 2-метилпропил;
когда R2 = 1-метилпропил,
R3 представляет собой 2-метилпропил;
когда R и R1 = - NR2R3 и R2 = водород,
R3 представляет собой -COR4, где R4 - водород; амино; прямая или разветвленная моно (C1-C6) алкиламиногруппа, выбранная из: метил-, этил-, н-пропил-, 1-метилэтил-, н-бутил-, 1-метилпропил-, 2-метилпропил-, 1,1-диметилэтил-, н-пентил-, 2-метилбутил-, 1,1-диметилпропил-1, 2,2-диметилпропил-2, 3-метилбутил-, н-гексил-, 1-метилпентил-, 1,1-диметилбутил-, 2,2-диметилбутил-, 3-метилпентил-, 1,2-диметилбутил-, 1,3-диметилбутил-, 1-метил-1-этилпропил-амино-группы и диастереоизомеров и энантиомеров указанной прямой или разветвленной моно (C1-C6)алкиламино-группы; либо R3 ди(C1-C6)алкиламино-группа, с прямой или разветвленной цепью, выбранная из диметиламино-, диэтиламино-, метил(этил)амино-, этил(1-метилэтил)аминогрупп и диастереомеров и энантиомеров указанной ди(C1-C6)-алкиламино-группы.
В соответствии с настоящим изобретением предлагаются пригодные в качестве промежуточных соединений для получения соединений формулы I, особенно предпочтительны из которых производные 7-9(замещенного)-6-деметил-6-дегидрокситетрациклина формулы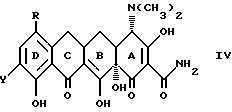
где Y = -N2 +Cl- или -N3; R = -NR2R3
и когда R2 = метил, этил, R3 = метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = 1-метилэтил, R3 = н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-бутил, R3 = н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = -1-метилпропил, R3 = 2-метилпропил.
Кроме того, в соответствии с изобретением предложены: способ получения производного 7,-8, -9(защемленного)-6-деметил-6-дегидрокситетрациклина формулы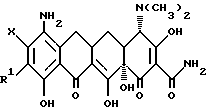
где X - галоген, выбранный из брома, хлора, фтора, йода;
R1 - галоген, выбранный из брома, хлора, фтора, йода, заключающийся в том, что азид формулы: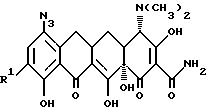
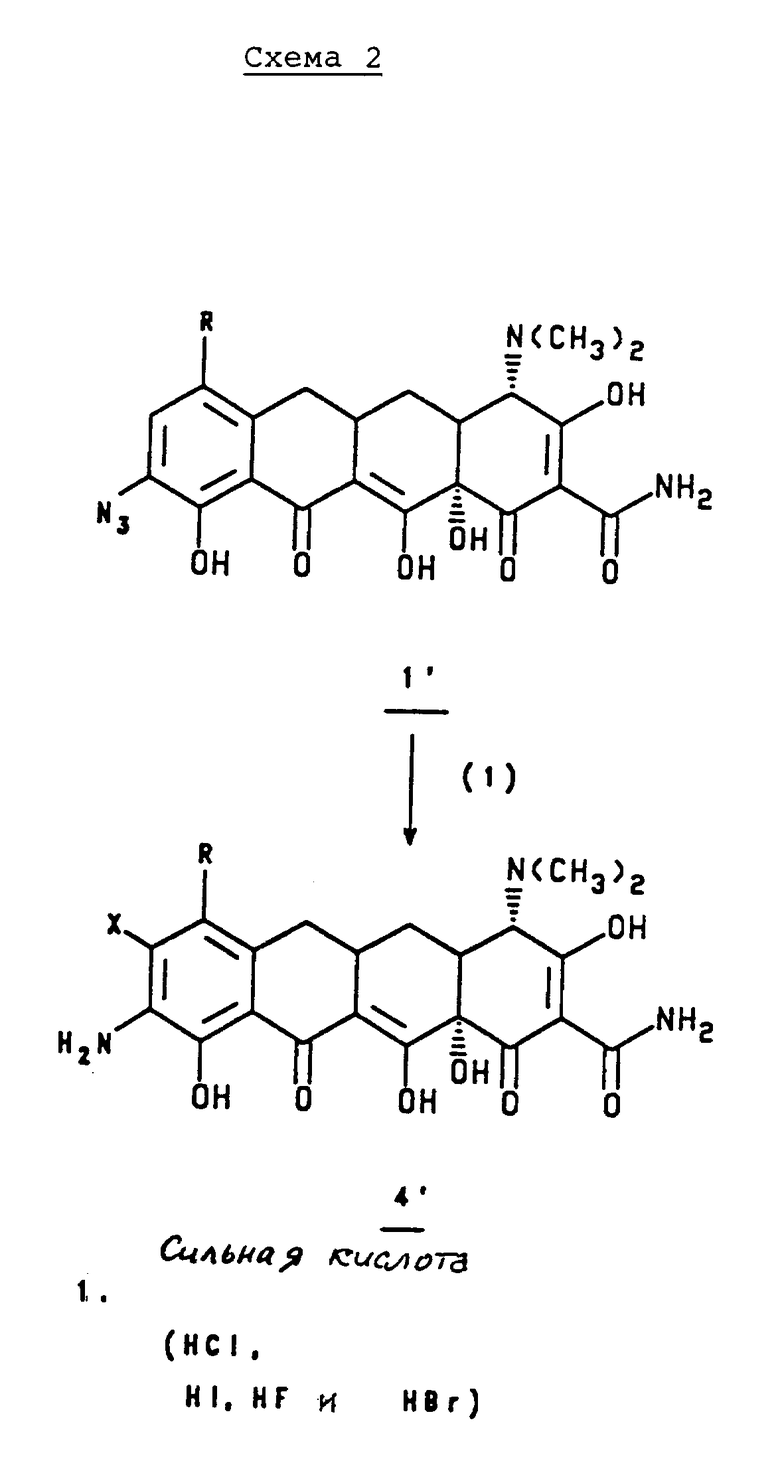
подвергают взаимодействию с сильной кислотой формулы HX, где X - галоген;
способ получения 7,-8-,-9(замещенного)-6-деметил-6-дегидрокситетрациклина формулы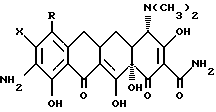
где X - галоген, выбранный из брома, хлора, фтора, йода;
R - водород, или - NR2R3
и когда R представляет собой -NR2R3, то
R2 = метил, этил, R3 = метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-пропил, R3 = н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-пропил, R3 = н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-бутил, R3 = н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = 1-метилпропил, R3 = 2-метилпропил, заключающийся в том, что соединение формулы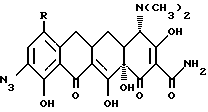
подвергают взаимодействию с сильной кислотой HX, где X - галоген и
способ получения 7,-8,-9(замещенного)-6-деметил-6-дегидрокситетрациклина формулы
где X - галоген, выбранный из хлора, брома, фтора, йода;
R - водород или -NR2R3 и когда R2 = метил, этил, то R3 = метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-пропил, R3 = н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = н-бутил, R3 = н-бутил, 1-метилпропил, 2-метилпропил;
а когда R2 = 1-метилпропил, R3 = 2-метилпропил;
заключающийся в том, что проводят реакцию соединения формулы
с уксусным ангидридом.
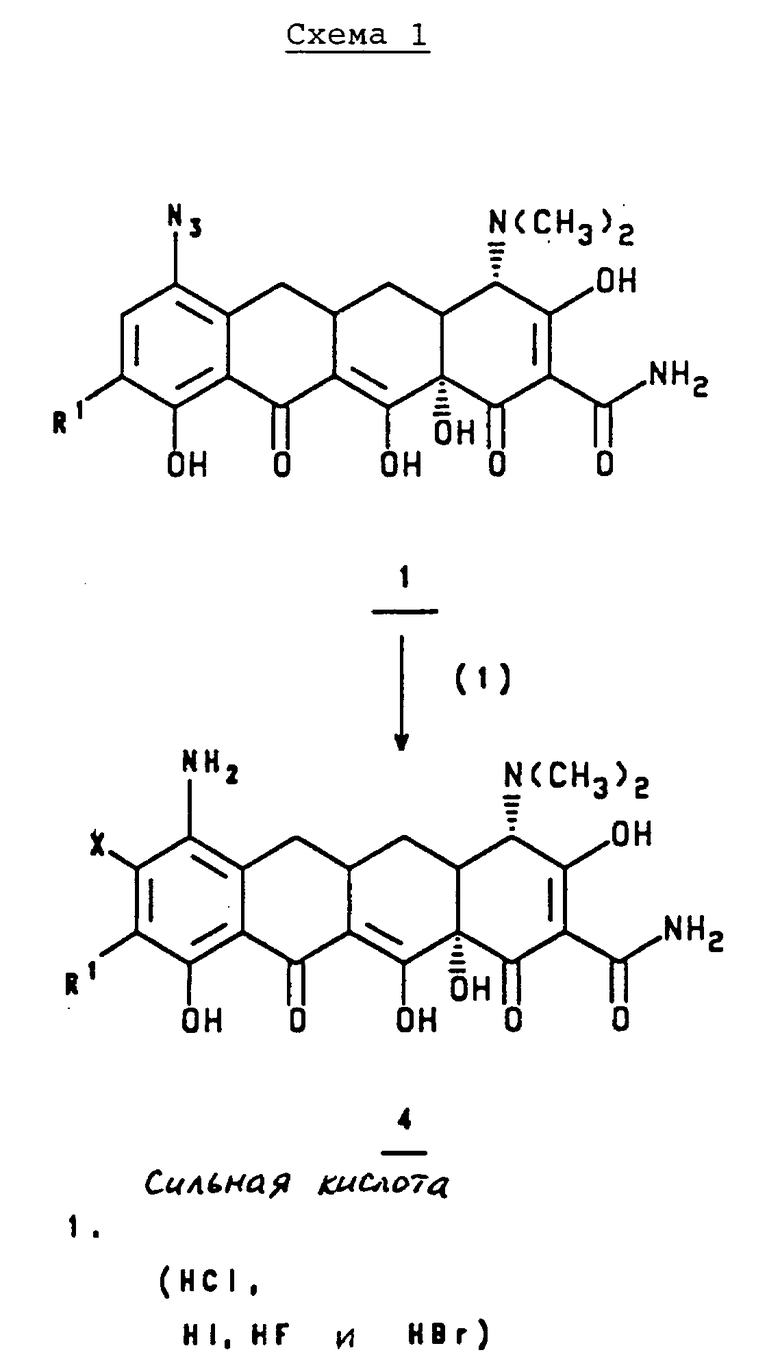
Более конкретно, в соответствии со схемами 1 или 2, приведенными в конце описания, 7-азидо-9-(замещенный)-6-деметил-6-дегидрокситетрациклин, 1, или 9-азидо-7-(замещенный)-6-деметил-6-дегидрокситетрациклин, 1', или их соль минеральной кислоты или галоидную соль обрабатывают сильной кислотой, такой как соляная кислота, бромистоводородная кислота, иодистоводородная кислота, или фтористый водород и получают 7-амино, 8-(замещенный)-9-(замещенный)-6-деметил-6-дегидрокситетрациклин, 4, или 9-амино-8-(замещенный)-7-(замещенный)-6-деметил-6- дегидрокситетрациклин, 4', или его соль минеральной кислоты или галоидную соль.
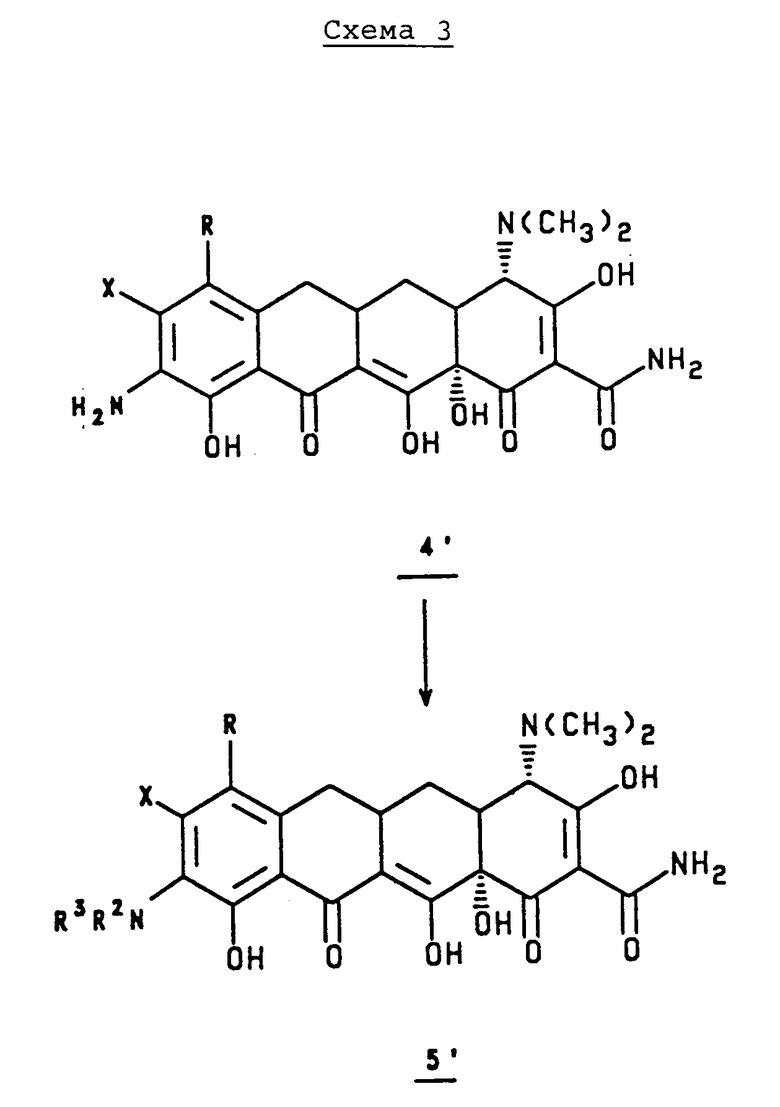
В соответствии со схемой 3, приведенной в конце описания, 9-амино-8-(замещенный)-7-(замещенный)-6-деметил-6-дегидрокситетрациклин, 4', или его минеральную или галоидную соль обрабатывают ацилхлоридом, ангидридом кислоты, смешанным ангидридом кислоты в присутствии подходящего акцептора кислоты в различных растворителях и получают соответствующий 9-(ацил)-8-(замещенный)-7-(замещенный)-6-деметил- дегидрокситетрациклин, 5', или его минеральную или галоидную соль. Акцептор кислоты выбирают из бикарбоната натрия, ацетата натрия, пиридина, триэтиламина, N,O-бис-(триметилсилил)ацетамида, N,O-бис(триметилсилил)трифторацетамида, карбоната калия или основной ионообменной смолы. Растворители выбирают из ТГФ, N-метилпирролидона, 1,3-диметил-2-имидазолидинона, гексаметилфосфороамида, 1,3-диметил-3,4,5,6-тетрагидро-2-(1H)пиримидинона или 1,2-диметоксиэтана.
Предпочтительно 7-(замещенный)-8-(замещенный)-9-(замещенный)-6-деметил-6-дегидрокситетрациклины получают в виде неорганических солей, таких как гидрохлорид, гидробромид, гидроиодид, фосфат, нитрат или сульфат; или органических солей, таких как ацетат, бензоат, цитрат, соль цистеина или другой аминокислоты, фумарат, малеат, сукцинат, тартрат, алкилсульфонат или арилсульфонат. Во всех случаях солеобразование происходит по C(4)-диметиламино-группе. Соли являются предпочтительными для орального или парентерального введения.
Определение биологической активности.
Способ оценки антибактериальной активности in vitro (табл. 1).
Минимальную ингибирующую концентрацию (МИК) - самую низкую концентрацию антибиотика, которая ингибирует рост тест-организма, определяют методом разбавления на агаре, используя агар Мюллера-Хинтона II (Baltimore Biological Laboratories). Используют плотность инокулята 1 - 5•105 KOE (колониеобразующая единица)/мл и концентрацию антибиотика 32-≤0,015 мкг/мл. Чашки Петри инкубируют 18 ч при 35oC в аэрируемом инкубаторе. Тест-организмы представляют собой штаммы, чувствительные к тетрациклину и генетически определенные штаммы, которые являются устойчивыми к тетрациклину благодаря неспособности связывать бактериальные рибосомы (tetM).
Система трансляции протеина E.coli in vitro (табл. II)
In vitro, свободная от клеток система трансляции протеина с использованием экстрактов из E.coli штаммa MPE 600 (чувствительный к тетрациклину) и производного MRE 600, содержащего tetM детерминанту, проводилась на основе литературных методик (J.M.Pratt Coupled Transcription-translation in Procaryotic Cell-free Systems, Transcription and Translation, Practical Approach (B.D.Hames and S.J.Higgins, eds.) стр. 179 - 209, IRL Press Oxford-Washington, 1984).
При использовании описанной выше системы испытывают тетрациклиновые соединения настоящего изобретения на их способность ингибировать синтез протеина in vitro. Вкратце каждые 10 мкл реакционной смеси содержат S30 экстракт (цельный экстракт), полученный или из чувствительных к тетрациклину клеток, или из изогенного устойчивого к тетрациклину (tetM) штамма, низкомолекулярные компоненты, необходимые для транскрипции и трансляции (например, АТФ и ГТФ), смесь 19 аминокислот (без метионина), 35S меченый метионин, ДНК матрицу (или pBR22 или pUC119), и/или DMCO (контроль), или новое тетрациклиновое соединение, которое должно быть испытано ("новый ТЦ"), растворенное в DMCO.
Реакционные смеси инкубируют в течение 30 мин при 37oC. Отсчет времени начинают с добавления S30 экстракта - последнего добавляемого компонента. Через 30 мин отбирают 2,5 мкл реакционной смеси и смешивают с 0,5 мл 1 н NaOH для разрушения PHK, и тPHK. Добавляют 2 - 3 мл 25%-ной трихлоруксусной кислоты и инкубируют смесь при комнатной температуре в течение 15 мин. Собирают осажденный трихлоруксусной кислотой материал на фильтрах Ватман GF/C и промывают раствором 10%-ной трихлоруксусной кислоты. Фильтраты сушат и подсчитывают удержанную радиоактивность, представляющую включение 35S- метионина в полипептиды, используя стандартные методики жидкостной сцинцилляции.
Процент ингибирования (П.И.) синтеза протеина определяют как
In vivo антибактериальная активность (табл. III).
Терапевтическое действие тетрациклинов определяли против острой летальной инфекции, вызванной штаммом Staphylococcus aureus штамм Смита (чувствительный к тетрациклину). Самок мышей штамма CD-1 (Charles River Laboratories) весом 20 ± 2 г отбирают для интраперитонеальной инъекции достаточным количеством бактерий (суспендированных в слизи свиньи), чтобы убить необработанные контроли в течение 24 - 48 ч. Антибактериальные агенты, содержащиеся в 0,5 мл 0,2%-ного водного агара, вводят подкожно или орально через 30 мин после заражения. При использовании методики орального дозирования животных не кормят за 5 ч до и 2 ч после заражения. Пять мышей обрабатывают каждым уровнем доз. Соотношения выживания через 7 дней для 3 отдельных тестов объединяют, чтобы рассчитать среднюю эффективную дозу (ED50).
Результаты испытаний.
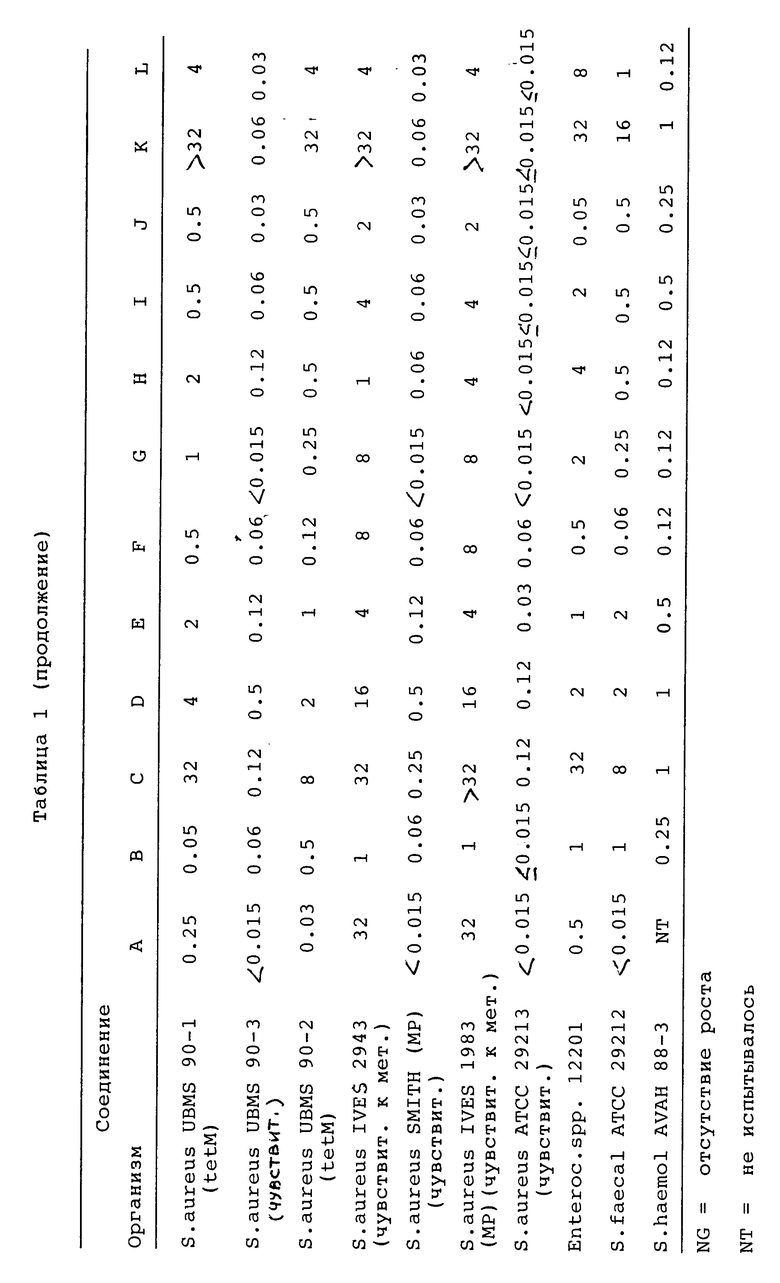
Заявленные соединения проявляют антибактериальную активность против спектpа чувствительных к тетрациклину и устойчивых грамположительных и грамотрицательных бактерий, особенно против штаммов E.coli, S.aureus и E.faecalis, содержащих tetM детерминанты устойчивости (табл. I). Заметным является 8-хлор-9-(формиламино)-4-(диметиламино)-6-деметил-6-дегидрокситетрациклин (соединение A в табл. I), который показал хорошую активность in vitro против устойчивых к тетрациклину штаммов, содержащих tetM детерминанту устойчивости (таких как S.aureus UBMS 88 - 5 и S.aureus UBMS 90 - 1 и 90 - 2) и является эффективным так же, как миноциклин против чувствительных к тетрациклину штаммов.
Синтез протеина, определенный при использовании не содержащих клеток экстрактов из чувствительного к тетрациклину штамма MRE600, ингибируется тетрациклином, миноциклином и 8-хлор-9-(формиламино)-4-(диметиламино)-6-деметил-6 -дегидрокситетрациклином по настоящему изобретению (табл. II). Синтез протеина, определенный при использовании не содержащих клеток экстрактов из штамма MRE600 (tetM), является устойчивым к тетрациклину и миноциклину, поскольку менее 20% ингибирования достигается даже при уровне концентрации 1 мг/мл миноциклина относительно 90% ингибирования 0,3 мг/мл чувствительных к тетрациклину рибосомальных экстрактов, полученных из штамма MRE600 (табл. II). В противоположность этому 8-хлор-9-(формиламино)-4-(диметиламино)-6-деметил-6-дегидрокситетрациклин эффективно ингибирует синтез протеина в экстрактах, полученных или из MRF600, или MRE600 (tetM) (табл. II). Предоставленное доказательство показывает, что 8-хлор-9-(формиламино)-4-(диметиламино)-6-деметил-6- дегидрокситетрациклин является ингибитором синтеза протеина на рибосомальном уровне. Способность 8-хлор-9-(формиламино)-4-(диметиламино)-6- деметил-6-дегидрокситетрациклина ингибировать рост бактерий почти определенно отражает непосредственное ингибирование синтеза бактериального протеина. Следовательно, ожидаемым является проявление бактериостатического эффекта против чувствительных бактерий, как это происходит в случае других тетрациклинов.
Антибактериальная активность 8-хлор-9-(формиламино)-4-(диметиламино)-6-деметил-6-дегидрокситетрациклина также была показана in vivo на животных, зараженных S.aureus Смита (табл. III).
Повышенная эффективность 8-хлор-9-(формиламино)-4-(диметиламино)-6-деметил-6-дегидрокситетрациклина была продемонстрирована на активности in vitro против изогенных штаммов, в которые была клонирована детерминанта устойчивости, такая как tetM, (табл. I); ингибированием синтеза протеина tetM рибосомами (табл. II); и антивностью in vivo против экспериментальных инфекций (табл. III).
Легенда для соединений.
A 8-Хлор-4-(диметиламино)-9-(формиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10, 12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид гидрохлорид или сульфат
B 8-хлор-4,7-бис(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10, 12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид сульфат
C 7-амино-8-хлор-4-(диметиламино)-1,4,4a, 5,5a,6,11,12a-октагидро-3,10, 12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид гидрохлорид
D 8-(аминокарбонил)-2-хлор-10-(диметиламино)-5,6a, 10,10a,11,11a,12-октагидро-5,7-диоксо-1-нафтацендиазонийхлорид
E 8-хлор-4-(диметиламино)-1,4,4a,5,5a,6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид сульфат
F 8-хлор-4,7-бис(диметиламино)-9-(формиламино)-1,4,4a,5,5a,6,11,12a -октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид сульфат
G 9-амино-8-хлор-4-(диметиламино)-1,4,4a, 5,5a, 6,11,12a -октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид
H 9-амино-8-хлор-4,7-(диметиламино)-1,4,4a, 5,5a, 6,11,12a -октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид сульфат
I метиловый эфир [7S-(7альфа, 10a альфа)]-[9-(аминокарбонил)-3-хлор-7-(диметиламино)-5,5a, 6,6a, 7,10,10a,12 -октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксо-2-нафтаценил]карбаминовой кислоты
J [4S(4альфа, 12a альфа)]-8-хлор-4-(диметиламино)-9-гидразино-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид моногидрохлорид
K Тетрациклина гидрохлорид
L Миноциклина гидрохлорид
M [4S(4 альфа, 12a альфа)]-9-амино-8-хлор-7-(диэтиламино)-4-диметиламино-1,4,4a, 5,5a,6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид
При использовании соединений в качестве антибактериальных агентов они могут быть объединены с одним или более из фармацевтически приемлемых носителей, например растворителей, разбавителей и т.п., и они могут быть введены орально в таком виде, как таблетки, капсулы, диспергирующиеся порошки, гранулы или суспензии, содержащие, например, от примерно 0,05 до 5% суспендирующего агента, сиропов, содержащих, например, от примерно 10 до 50% сахара, и эликсиров, содержащих, например, от примерно 20 до 50% этанола, и т.п., или парентерально в виде стерильных растворов для инъекций или суспензий, содержащих от примерно 0,05 до 5% суспендирующего агента в изотонической среде. Такие фармацевтические препараты могут содержать, например, от примерно 25 до примерно 90% активного ингредиента в сочетании с носителем, обычно между примерно 5 и 60 мас.%.
Эффективное количество соединения от 2,0 до 100,0 мг/кг массы тела должно быть введено за 1 - 5 раз в день любым обычным путем введения, включая, но не ограничиваясь оральным, парентеральным (включая подкожные, внутривенные, внутримышечные, внутригрудинные инъекции или инфузию), местным или ректальным в виде единичных дозированных форм, содержащих традиционные нетоксичные фармацевтически приемлемые носители, адъюванты и разбавители. Однако должно быть понятно, что конкретный уровень дозы и частота введения для любого конкретного пациента могут варьировать и будут зависеть от разнообразных факторов, включая активность конкретного использованного соединения, стабильность метаболизма и продолжительность действия такого соединения, возраст, массу тела, состояние здоровья, пол, диету, способ и время введения и тяжесть конкретного состояния.
Эти активные соединения могут быть введены орально, а также внутривенно, внутримышечно или подкожно. Твердые носители включают крахмал, лактозу, дикальцийфосфат, микрокристаллическую целлюлозу, сахарозу и каолин, тогда как жидкие носители включают стерильную воду, полиэтиленгликоли, неионные поверхностно-активные вещества и пищевые масла, такие как кукурузное, арахисовое и кунжутное масла, в соответствии с природой активного ингредиента и конкретной желаемой формой введения. Адъюванты, обычно применяемые для приготовления фармацевтических композиций, могут быть такими, как ароматизирующие агенты, красители, консерванты и антиоксиданты, например, витамин E, аскорбиновая кислота, BHT, и BHA.
Предпочтительными фармацевтическими композициями с точки зрения легкости приготовления и введения являются твердые композиции, в частности, таблетки и капсулы, заполненные жидкостью или твердым веществом. Предпочтительным является оральное ведение соединений.
Эти активные соединения также могут быть введены парентерально или интраперитонеально. Растворы или суспензии этих активных соединений в виде свободного основания или фармакологически приемлемой соли могут быть приготовлены в воде и смеси с подходящим поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Также могут быть приготовлены дисперсии в глицерине, жидкости, полиэтиленгльколях и их смесях с маслами. В обычных условиях хранения и применения эти препараты содержит консервант для предотвращения роста микроорганизмов.
Фармацевтические формы, приемлемые для инъекционного использования, включают стерильные водные растворы или дисперсии и стерильные порошки для импровизированного приготовления стерильных растворов или дисперсий для инъекций. Во всех случаях форма должна быть стерильной и жидкой. Она должна быть стабильной в условиях производства и хранения и должна быть предохранена против загрязнения микроорганизмами, такими как бактерии и грибы. Носитель может быть растворителем или диспергирующей средой, содержащим, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящими смесями и растительным маслом.
Изобретение будет более полно описано в сочетании со следующими конкретными примерами, которые не должны рассматриваться как ограничивающие область изобретения.
Пример 1
[7S-(7 альфа, 10 альфа)]-9-(аминокарбонил)-4,7-бис(диметиламино)-5,5a, 6,6a, 7,10,10a, 12 -октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксо-2-нафтацендиазоний хлорид сульфат (1:1)
К охлажденному до 0oC раствору 3,0 г 9-амино-4,7-бис(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12,12a- тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида сульфата в 100 мл 0,1 н раствора хлористого водорода в метаноле прибавляют по каплям 6,6 мл бутилнитрита.
Реакционную смесь перемешивают при 0oC в течение 1 часа, выливают в 400 мл диэтилового эфира, собирают и сушат, получают 2,64 г целевого продукта, MC(FAB): m/z 484 (M + H).
Пример 2
[4S-(4альфа, 12a альфа)] -9-азидо-4,7-бис(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3, 10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид (1:1)
К раствору 2,64 г продукта примера 1 при комнатной температуре, растворенным в 84 мл 0,1 н метанольного хлористого водорода, прибавляют 0,353 г азида натрия. Смесь перемешивают при комнатной температуре в течение 4 часов, выливают в 500 мл диэтилового эфира и собирают, получают 2,5 г целевого продукта. ИК(КВг): 2080-1.
Пример 3
[4S -(4α, 12aα)] -9-амино-8-хлор-4,7-бис(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10, 12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида сульфат
Прибавляют 1 г продукта примера 2 к 10 мл концентрированной серной кислоты при 0oC. Реакционную смесь перемешивают при 0oC в течение 1,5 часов, выливают в 500 мл диэтилового эфира, собирают и сушат, получают 1,1 г целевого продукта. MC (FAB): m/z 507 (M + H).
Пример 4
[4S -(4α, 12aα)] -8-хлор-4,7-бис(диметиламино)-9-(формиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро -3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида сульфат (1:1)
К раствору 0,092 г продукта примера 3, растворенного в 5,0 мл 98%-ной муравьиной кислоты, при 0oC прибавляют 0,0164 г ацетата натрия. Полученную в результате смесь перемешивают при 0oC в течение 10 минут, затем прибавляют 0,23 мл уксусного ангидрида. Реакционную смесь перемешивают при комнатной температуре 1 час, выливают в диэтиловый эфир и собирают, получают 0,045 г твердого продукта. Собранный твердый продукт растирают с 50 мл этилацетата и фильтруют. Фильтрат концентрируют в вакууме, получают 0,019 г целевого продукта. MC (FAB): m/z 535 (M + H).
Пример 5
[4S -(4α, 12aα)]- 8-хлор-4,7-бис(диметиламино)-1,4,4a, 5,5a,6,11,12a-октагидро-3,10, 12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида сульфат (1:1)
При 0oC к раствору 0,090 г продукта примера 3, растворенного в 35 мл 0,1 н метанольного хлористого водорода, прибавляют 0,2 мл бутилнитрита. Реакционную смесь перемешивают при комнатной температуре 1 час, выливают в 70 мл диэтилового эфира и собирают, получают 0,070 г целевого промежуточного диазонийхлорида.
Раствор 0,070 г вышеупомянутого промежуточного продукта в 20 мл метилового спирта кипятят с обратным холодильником в течение 45 минут, выливают в диэтиловый эфир и собирают, получают 0,056 г целевого продукта. MC (FAB): m/z 491 (M+).
Пример 6
[4S -(4α, 12aα)]- 7-амино-8-хлор-(диметиламино)-1,4,4a,5,5a,6,11,12a-октагидро -3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид (1:1)
Прибавляют 3 г 7-азидо-6-деметил-6-деокситетрациклина гидрохлорида, полученного по методике, описанной в J.Am.Chem. Soc., 84, 1426 - 1430, к 120 мл холодной концентрированной соляной кислоты и перемешивают 1 3/4 часа на ледяной бане. Реакционную смесь концентрируют в вакууме, получают 2,9 г целевого продукта. MC (FAB): m/z 535 (M + H).
Пример 7
[6aS -(6aα, 10aα)]- 8-(аминокарбонил)-2-хлор-10-(диметиламино)-5,6a, 10,10a,11,11a,12-октагидро- 5,7-диоксо-1-нафтацендиазонийхлорида гидрохлорид
К раствору 0,50 г продута примере 6, в 15 мл 0,1 н метанольного хлористого водорода при 0oC прибавляют 1,0 мл бутилнитрита. Реакционную смесь перемешивают при 0oC в течение 1 часа, выливают в 500 мл диэтилового эфира и собирают, получают 0,48 г целевого продукта. ИК (КВг): 2200 см-1.
Пример 8
[4S-(4 альфа, 12a альфа)]-7-азидо-8-хлор-4-(диметиламино)-1,4,4a,5,5a, 6,11,12a-октагидро- 3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид
При комнатной температуре к раствору 0,48 г продукта примера 7 в 20 мл 0,1 н метанольного хлористого водорода прибавляют 0,055 г азида натрия. Реакционную смесь перемешивают при комнатной температуре в течение 4 часов, выливают в 100 мл диэтилового эфира и собирают, получают 0,366 г целевого продукта. MC (FSB): m/z 490 (M + H).
Пример 9
[4S -(4α, 12aα)]- 8-хлор-4-(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10, 12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид (1:1)
При 0oC к раствору 0,095 г продукта примера 6 в 5 мл 0,1 и метанольного раствора хлористого водорода, прибавляют 0,3 мл бутилнитрита. Реакционную смесь перемешивают при 0oC в течение 1 часа, выливают в диэтиловый эфир и собирают, получают 0,070 г целевого промежуточного продукта.
Раствор 0,050 г указанного выше промежуточного продукта в 15 мл метилового спирта кипятят с обратным холодильником 1 час и концентрируют в вакууме, получают 0,035 г целевого продукта. MC (FAB): m/z 449 (M + H).
Пример 10
[4S -(4α, 12aα)]- 9-амино-8-хлор-4-(диметиламино)-1,4,4a,5,5a, 6,11,12a-октагидро -3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид (1:1)
К 10 мл концентрированной соляной кислоты при 0oC прибавляют 0,20 г 9-азидо-6-деметил-6-дегидрокситетрациклина гидрохлорида, полученного по методике, описанной в J.Am.Chem.Soc., 84, 1426 - 1430. Реакционную смесь перемешивают при 0oC в течение 1,5 часов и концентрируют в вакууме, получают 0,195 г целевого продукта. MC (FAB): m/z 464 (M + H).
Пример 11
[4S -(4α, 12aα)]- 8-хлор-4-(диметиламино)-9-(формиламино)-1,4,4a,5,5a, 6,11,12a-октагидро -3,10,12,12a-тетрагидрокси-1,11- диоксо-2-нафтаценкарбоксамида гидрохлорид (1:1)
При 0oC к раствору 0,103 г продукта примера 10 в виде гидрохлорида в 6 мл 98%-ной муравьиной кислоты прибавляют 0,23 мл уксусного ангидрида. Полученную смесь перемешивают 5 минут при 0oC, затем 1 час при комнатной температуре. Реакционную смесь выливают в 500 мл диэтилового эфира и собирают, получают 0,090 г целевого продукта. MC (FAB): m/z 492 (M + H).
Пример 12
[4S -(4α, 12aα)]- 7-амино-4-(диметиламино)-1,4,4a, 5,5a,6,11,12a-октагидро -3,10,12,12a-тетрагидрокси-9-иод-1,11-диоксо-2-нафтаценкарбоксамида сульфат (1:1)
При 0oC к раствору 0,285 г 7-амино-6-деметил-6-деокситетрациклина в 5 мл концентрированной серной кислоты прибавляют 1,2 эквивалента N-иодсукцинимида. Реакционную смесь перемешивают при 0oC в течение 15 минут, затем выливают в 400 мл диэтилового эфира. Полученный твердый продукт собирают и сушат, получают 0,23 г целевого продукта.
1H-ЯМР-спектр (DMCO-d6) :δ 8,0 (C-8 H).
Пример 13
[6aS-(6a альфа, 10a альфа)]-8-(аминокарбонил)-]0-(диметиламино)- 5,6a, 7,10,10a, 11,11a,12-октагидро-4,6,6a,9-тетрагидрокси-3-иод-5,7-диоксо-1- нафтацендиазонийхлорида сульфат (1:1:1)
При 0oC к раствору 0,15 г продукта примера 12 в достаточном количестве 0,1 н метанольного хлористого водорода, чтобы получить раствор, прибавляют по каплям 0,143 мл н-бутилнитрита. Реакционную смесь перемешивают при 0oC в течение 30 - 45 минут, затем выливают в холодный диэтиловый эфир при перемешивании. Полученный твердый продукт собирают, промывают диэтиловым эфиром и сушат, получают 0,12 г целевого продукта.
1H-ЯМР (DMCO-d6) :δ 8,52 (C-8 H).
Пример 14
[4S-(4 альфа, 12a альфа)] -7-азидо-4-(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12,12a- тетрагидрокси-9-иод-1,11-диоксо-2-нафтаценкарбоксамида сульфат (1:1)
Целевое соединение получают по методике примера 8, используя 2,2 г продукта примера 13, 60 мл 0,1 н метанольного хлористого водорода и 0,203 г азида натрия, получают (после очистки) 0,65 г целевого продукта. ИК-спектр (КВг): 2100 см-1. MC (FAB): m/z 582 (M + H)
Пример 15
[4S-(4 альфа, 12a альфа)]-7-амино-8-хлор-4-(диметиламино)-1,4,4a,5,5a, 6,11,12a-октагидро -3,10,12,12a-тетрагидрокси-9-иод-1,11-диоксо-2-нафтаценкарбоксамида сульфат (1:1)
Смесь 0,2 г продукта примера 14 и 1 мл концентрированной соляной кислоты перемешивают при комнатной температуре 2 часа. Реакционную смесь растирают с изопропанолом и эфиром, собирают и сушат, получают 0,18 г целевого продукта. MC (FAB): m/z 590 (M + H).
Пример 16
[7S-(7 альфа, 10a альфа)] -9-(аминокарбонил)-4-(диэтиламино)-7-(диметиламино)-5,5a, 6,6a,7,10,10a, 12-октагидро-1,8,10a, 11-тетрагидрокси-10,12-диоксо-2-нафтацендиазонийхлорид сульфат (2:1)
При 0oC к раствору 1,85 г 9-амино-7-(диэтиламино)-6-деметил-6-дегидрокситетрациклина, полученного по методике, описанной в заявке на патент в США серийный N 07/771697, поданной 4 октября 1991 г, в 40 мл 0,1 н метанольного хлористого водорода прибавляют 1,85 мл н-бутилнитрита. Реакционную смесь перемешивают при 0oC в течение 2 часов, выливают в диэтиловый эфир, собирают твердый продукт и промывают диэтиловым эфиром, получают 2,1 г целевого продукта.
1H-ЯМР-спектр (DMCO-d6) :δ 7,9 (C-8 H).
Пример 17
[4S-(4 альфа, 12a альфа)] -9-азидо-7-(диэтиламино)-4-(диметиламино)-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида дисульфат
При комнатной температуре прибавляют к раствору 1,192 г продукта примера 16 в 75 мл 0,1 н метанольного хлористого водорода 0,104 г азида натрия. Реакционную смесь перемешивают при комнатной температуре 2 часа, медленно выливают в диэтиловый эфир и собирают, получают 0,8 г целевого продукта.
1H-ЯМР-спектр (DMCO-d5) :δ (C-8 H).
Пример 18
[4S-(4 альфа, 12a альфа)] -8-азидо-7-(диэтиламино)-4-(диметиламино)-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида дигидрохлорид
При комнатной температуре к раствору 0,6 г продукта примера 17 в воде прибавляют твердый ацетат натрия до достижения pH 5. Смесь экстрагируют хлороформом 2 раза, органический слой сушат над сульфатом натрия и концентрируют в вакууме. Остаток повторно растворяют в 5 мл метанола и прибавляют 2 капли концентрированной соляной кислоты. Затем реакционный раствор прибавляют по каплям к 120 мл диэтилового эфира. Полученный твердый собирают, получают 0,4 г целевого продукта. ИК-спектр (КВг): 2100 см-1.
Пример 19
[4S-(4 альфа, 12a альфа)]-9-амино-8-хлор-7-(диэтиламино)-4-(диметиламино)-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида сульфат
Смесь 0,23 г продукта примера 18 и 5 мл концентрированной соляной кислоты перемешивают при комнатной температуре 2 часа. Полученный твердый продукт растирают с изопропанолом и диэтиловым эфиром. Собирают твердый продукт, промывают диэтиловым эфиром и сушат, получают 0,21 г целевого продукта. MC (FAB): m/z 535 (M + H).
Пример 20
[7S-(7 альфа, 10a альфа)]-9-(аминокарбонил)-3-хлор-7-(диметиламино)-5,5a, 6,6a, 7,10,10a,12- октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксо-2-нафтаценил]карбаминовой кислоты метиловый эфир
При комнатной температуре к раствору 0,20 г продукта примера 10 в 4 мл 1-метил-пирролидинона прибавляют 0,30 г бикарбоната натрия. Смесь перемешивают 5 минут, затем прибавляют 34 мкл метилхлорформата. Реакционную смесь перемешивают при комнатной температуре 1 час, фильтруют в 200 мл диэтилового эфира и собирают, получают 0,066 г целевого продукта. MC (FAB): m/z 522 (M + H).
Пример 21
[4S-(4 альфа, 12a альфа)]-8-хлор-4-(диметиламино)-9-гидразино-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида моногидрохлорид
К 0,30 г продукта примера 10, растворенным в 8 мл 0,10 н метанольного хлористого водорода, прибавляют 0,60 мл н-бутилнитрита. Реакционную смесь перемешивают на ледяной бане в течение 1 часа, выливают в 200 мл диэтилового эфира и собирают, получают 0,260 г. Прибавляют 50 мг собранного материала к 3 мл 6%-ной серной кислоты, перемешивают при комнатной температуре 1 час и концентрируют в вакууме, получают 0,037 г целевого продукта.
CI-MS: m/z 479 (M + H).
Пример 22
[4S-(4 альфа, 12a альфа)]-9-амино-4,7-бис(диметиламино)-8-фтор-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид
Целевое соединение получают по методике примера 3, используя продукт примера 2 и жидкий фтористый водород.
Пример 23
[6aS-(6a альфа, 10 альфа)]-3-амино-8-(аминокарбонил)-10-(диметиламино)-5,6a, 7,10,10a,11,11a,12- октагидро-4,6,6a,9- тетрагидрокси-5,7-диоксо-2-нафтацениловый эфир трифторметан-сульфоновой кислоты
Целевое соединение получают по методике примера 3, используя 9-азидо-6-деметил-6-деокситетрациклина, полученный по методике, описанной в J.Am. Chem.Soc., 84, 1426 - 1430, и трифторметансульфоновой кислоты.
Пример 24
[4S-(4 альфа, 12a альфа)]-9-амино-4-(диметиламино)-8-фтор-1,4,4a,5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид
Целевое соединение получают по методике примера 22, используя 9-азидо-6-деметил-6-деокситетрациклин, полученный по методике, описанной в приведенной выше ссылке.
Пример 25
[4S-(4 альфа, 12a альфа)] -4-(диметиламино)-8-фтор-9-(формиламино)-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид
Целевое соединение получают по методике примера 4, используя продукт примера 24.
Пример 26
[4S-(4 альфа, 12a альфа)]-4-(диметиламино)-8-фтор-1,4,4a,5,5a,6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид
Целевое соединение получают по методике, описанной в примерах 1 и 5, используя продукт примера 24.
Пример 27
[4S-(4 альфа, 12a альфа)] -4,7-бис(диметиламино)-8-фтор-1,4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид
Целевое соединение получают по методике примера 5, используя продукт примера 22.
Пример 28
[7S-(7 альфа, 10a альфа)]-9-(аминокарбонил)-7-(диметиламино)-3-фтор-5,5a, 6,6a, 7,10,10a,12- октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксо-2-нафтаценил]карбаминовой кислоты метиловый эфир
Целевое соединение получают по методике примера 20, используя продукт примера 24.
Пример 29
[6aS-(6a альфа, 10 альфа)] -3-амино-8-(аминокарбонил)-1,10-бис-(диметиламино)-5,6a, 7,10,10a, 11, 11a,12-октагидро-4,6,6a,9-тетрагидрокси-5,7-диоксо-2-нафтацениловый эфир трифторметансульфоновой кислоты
Целевое соединение получают по методике примера 3,используя продукт примера 3 и трифторметансульфоновую кислоту.
Пример 30
[4S-(4 альфа, 12a альфа)]-7-амино-4-(диметиламино)-8-фтор-1,4,4a,5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорида
Целевое соединение получают по методике примера 3, используя 7-азидо-6-деметил-6-деокситетрациклин, полученный по методике, описанной в J.Am. Chem.Soc., 84: 1426 - 1430, в жидкий фтористый водород при -30oC.
Пример 31
[7S-(7 альфа, 10a альфа)]-9-(аминокарбонил)-3-хлор-4,7-бис-(диметиламино)-5,5a, 6,6a, 7,10,10a,12- октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксо-2-нафтаценил] карбаминовой кислоты 2-(диметиламино)-этиловый эфир
Целевое соединение получают по методике примера 27, используя продукт примера 3 и бета-диметиламиноэтилхлороформиат
Пример 32
[4S-(4 альфа, 12a альфа)]-8-хлор-9-[[[(диэтиламино)окси]карбонил]амино] -4,7-бис(диметиламино)- 1,4,4a, 5,5a,6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид
Получают целевое соединение по методике примера 27, используя продукт примера 3 и диэтиламинооксихлороформиат
Пример 33
[4S-(4 альфа, 12a альфа)]-8-хлор-7-(диэтиламино)-4-(диметиламино)-9-(формиламино)-1,4,4a, 5, 5a,6,11,12a-октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2 -нафтаценкарбоксамида сульфат
Целевое соединение получают по методике примера 4, используя продукт примера 19.
Пример 34
[6aS-(6a альфа, 10 альфа)]-8-(аминокарбонил)-2-хлор-10-(диметиламино)-5,6a, 7,10,10a, 11,11a, 12- октагидро-4,6,6a,9-тетрагидрокси-5,7-диоксо-1-нафтаценил]карбаминовой кислоты метиловый эфир
Целевое соединение получают по методике примера 20, используя продукт примера 6.
Пример 35
[4S (4α, 12aα)]- 4-(диметиламино)-8-фтор-7-(формиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12, 12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидрохлорид
Целевое соединение получают по методике примера 4, используя продукт примера 8.
Пример 36
[7S-(7альфа, 10a альфа)] -[9-(аминокарбонил)-7-(диметиламино)-3-фтор-5,5a, 6,6a, 7,10,10a,12- октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксо-2-нафтаценил]карбаминовой кислоты метиловый эфир
Целевое соединение получают по методике примера 20, используя соединение примера 24.
Пример 37
[7S-(7 альфа, 10a альфа)]-9-(аминокарбонил)-7-(диметиламино)-3-фтор-5,5a, 6,6a, 7,10,10a,12- октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксокарбаминовая кислота
Целевое соединение получают по методике примера 33, используя продукт примера 24.
Пример 38
[6S-(6a альфа, 10 альфа)] -8-(аминокарбонил)-10-(диметиламино)-5,6a, 7,10,10a, 11,11a, 12- октагидро-4,6,6a,9-тетрагидрокси-3-(метокикарбонил)-амино-5,7-диоксо-2- нафтацениловый эфир трифторметансульфоновой кислоты
Целевое соединение получают по методике примера 20, используя продукт примера 23.
Пример 39
[4S-(4альфа, 12a альфа)]-8-хлор-4-(диметиламино)-9-[[[(диметиламино)окси]карбонил]-1,4,4a,5,5a, 6,11,12a-октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид
Целевое соединение получают по методике примера 20, используя продукт примера 10 и диметиламиноксихлорформиат.
Пример 40
[4S-(4альфа, 12a альфа)]-8-хлор-9-[[[(диэтиламино)окси]-карбонил]амино] -4-(диметиламино)-1, 4,4a, 5,5a, 6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид
Целевое соединение получают по методике примера 20, используя продукт примера 10 и диэтиламинохлороформиат.
Пример 41
[4S -(4α, 12αa)]- 9-амино-8-бром-4-(диметиламино)-1,4,4a,5,5a,6,11,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид гидробромид (1:1)
Целевое соединение получают по методике примера 3, используя 9-азидо-6-деметил-6-дегидрокситетрациклин, полученный по методике, приведенной в J. Am. Chem. Soc., 84: 1426 - 1430, и 30%-ный раствор бромистого водорода в уксусной кислоте.
Пример 42
[4S -(4α, 12aα)]- 9-амино-8-бром-4,7-бис(диметиламино)-1,4,4a, 5,5a, 6,11,12a-октагидро-3,10, 12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамид гидробромид
Целевое соединение получают по методике примера 3, используя продукт примера 2 и раствор бромистого водорода в уксусной кислоте (30 мас.%).
Пример 43
[4S-(4альфа, 12a альфа)] -9-амино-4,7-бис(диметиламино)-8-иод, 1,4,4a, 5,5a, 6,11,12a-октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2 -нафтаценкарбоксамид гидроиодид
Целевое соединение получают по методике примера 3, используя продукт примера 2 и иодистоводородную кислоту.
Пример 44
[4S-(4альфа, 12a альфа)] -9-амино-4-(диметиламино)-8-иодо-1,4,4a,5,5a, 11,12,12a- октагидро-3,10,12,12a-тетрагидрокси-1,11-диоксо-2-нафтаценкарбоксамида гидроиодид
Целевое соединение получают по методике примера 3, используя 9-азидо-6-деметил-6-деокситетрациклин, полученный по методике, описанной в J.Am. Chem.Soc., 84: 1426 - 1430, и иодистоводородную кислоту.
Пример 45
Метиловый эфир [7S-(7альфа, 10a альфа)]-[9-(аминокарбонил)-7-(диметиламино)-3-иодо-5,5a,6,7,10,10a,12- октагидро-1,8,10a,11-тетрагидрокси-10,12-диоксо-2-нафтаценил]карбаминовой кислоты
Целевое соединение получают по методике примера 20, используя продукт примера 44.
Пример 46
Метиловый эфир [7S-(7 альфа, 10a альфа)]-[9-(аминокарбонил)-3-бром-7-(диметиламино)-5,5a, 6,7,10,10a, 12- октагидро-1,8,10a, 11-тетрагидрокси-10,12-диоксо-2-нафтаценил]карбаминовой кислоты
Целевое соединение получают по методике примера 20, используя продукт примера 41.
Следующие примеры готовых препаративных форм могут включать в качестве активного ингредиента любое из соединений формулы 1. Примеры только иллюстративные и никоим образом не предназначены для ограничения объема изобретения.
Пример 47
Твердые желатиновые капсулы получают с использованием следующих ингредиентов, мг/капсула:
Соединение примера 19 - 50
Высушенный крахмал - 100
Оксид магния - 10
Указанные выше ингредиенты смешивают и смесью заполняют твердые желатиновые капсулы в количестве 160 мг на капсулу.
Пример 48
Таблетированную форму получают, используя приведенные ниже ингредиенты, мг/таблеткa:
Соединение примера 21 - 25
Целлюлоза микрокристаллическая - 200
Коллоидный диоксид кремния - 10
Стеариновая кислота - 5
Компоненты смешивают и прессуют для образования таблеток весом 240 мг каждая.
Пример 49
Капсулы, каждая из которых содержит 80 мг лекарственного средства, готовят следующим образом, мг:
Соединение примера 20 - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Всего - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито меш N 45 шкалы США и вводят в твердые желатиновые капсулы в количестве 200 мг на капсулу.

Производные 7, -8,-9 (замещенных)-6-деметил-6-дегидрокситетрациклинов, где X - галоген; R и R1 - одинаковые или различные и означают H, NH2, галоген, NR2R3 и когда R и R1 = -NR2R3, а R2 = H, то R3 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил; а когда R2 - метил, этил, то R3 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилбутил, 2-метилпропил; а когда R2 - н-пропил, то R3 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил; а когда R2 - 1-метилэтил, то R3 - н-бутил, 1-метилпропил, 2-метилпропил; когда R2 - н-бутил, то R3 - н-бутил, 1-метилпропил, 2-метилпропил; когда R2 - метилпропил, то R3 - 2-метилпропил; когда R и R1 - = -NR2R3 и R2 - Н, то R3 - COR4, где R4 - Н, NH2, моно(C1-C6)-алкиламиногруппа, диастереоизомеры и энантиомеры моно(C1-C6)-алкиламиногруппы; либо R3 - ди(C1-C6)-алкиламиногруппа. Используют в качестве фармацевтической композиции с антибактериальной активностью. 6 с. п. ф-лы, 3 табл.
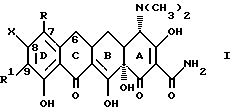
где Х представляет галоген, выбранный из брома, фтора или йода;
R и R1 являются одинаковыми или различными и представляют собой водород, амино, галоген (выбранный из хлора, брома, фтора или йода) или NR2R3, и когда R или R1 представляют -NR2R3, а R2 - водород, то R3 представляет собой метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил; а когда R2 - метил или этил, R3 представляет собой метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, а когда R2 - н-пропил, R3 представляет собой н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, а когда R2 - 1-метилэтил, R3 представляет собой н-бутил, 1-метилпропил, 2-метилпропил; когда R2 - н-бутил, R3 представляет собой н-бутил, 1-метилпропил, 2-метилпропил; когда R2 - 1-метилпропил, R3 представляет собой 2-метилпропил; когда R и R1 - NR2R3 и R2 - водород, R3 представляет собой -COR4, где R4 - водород, амино, прямая или разветвленная моно -(С1-С6)-алкиламиногруппа, выбранная из метил-, этил, н-пропил-, 1-метилэтил-, н-бутил-, 1-метилпропил-, 2-метилпропил-, 1,1-диметилэтил-, н-пентил-, 2-метилбутил-, 1,1-диметилпропил-, 2,2-диметилпропил-, 3-метилбутил, н-гексил-, 1-метилпентил-, 1,1-диметилбутил-, 2,2-диметилбутил-, 3-метилпентил-, 1,2-диметилбутил-, 1,3-диметилбутил-, 1-метил-1-этилпропил-аминогруппы и диастереоизомеров и энантомеров указанной прямой или разветвленной моно (С1-С6)-алкиламиногруппы; либо R3-ди-(С1-С6)-алкиламиногруппа, с прямой или разветвленной цепью, выбранная из диметиламино-, диэтиламино-, метил(этил)-амино-, этил(1-метилэтил)-аминогрупп и диастереомеров и энантиомеров указанной ди-(С1-С6)-алкиламиногруппы.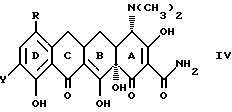
Y - N2 +Cl- или N3;
R - NR2R3, и когда R2 - метил, этил, R3 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, в когда R2 - н-пропил, R3 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил; а когда R2 - 1-метилэтил, R3 - н-бутил, 1-метилпропил, 2-метилпропил; а когда R2 - н-бутил, R3 н-бутил, 1-метилпропил, 2-метилпропил; а когда R2 - 1-метилпропил, R3 - 2-метилпропил.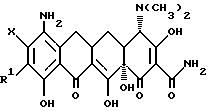
где X - галоген, выбранный из брома, хлора, фтора, йода;
R1 - галоген, выбранный из брома, хлора, фтора, йода,
отличающийся тем, что азид формулы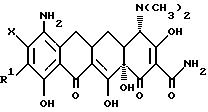
подвергают взаимодействию с сильной кислотой формулы
HX,
где Х - галоген.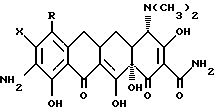
где Х - галоген, выбранный из брома, хлора, фтора, йода;
R - водород, или - NR2R3, и когда R представляет собой - NR2R3, то R2 - метил, этил, R3 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил; а когда R2 - н-пропил, R3 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил; 2-метилпропил; а когда R2 - н-пропил, R3 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил; а когда R2 - н-бутил, R3 - н-бутил, 1-метилпропил, 2-метилпропил; а когда R2 - 1-метилпропил, R3 - 2-метилпропил,
отличающийся тем, что соединение формулы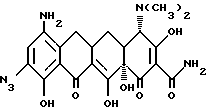
подвергают взаимодействию с сильной кислотой формулы
НХ,
где Х - галоген.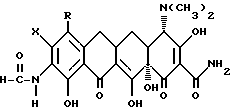
где Х - галоген, выбранный из хлора, брома, фтора, йода%
R - водород или NR2R3, и когда R2 - метил, этил, то R3 - метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метил-пропил, 2-метилпропил; а когда R2 - н-пропил, R2 - н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, а когда R2 - н-бутил, R3 - н-бутил, 1-метилпропил, 2-метилпропил, а когда R2 - 1-метилпропил, R3 - 2-метилпропил,
отличающийся тем, что проводят реакцию соединения формулы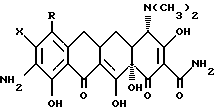
с уксусным ангидридом.
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, т.2, с.223 - 229. |

