Изобретением даются новые соединения, новые препараты, способы их применения и способы получения, причем такие соединения, как правило, фармакологически применимы в качестве средств, препятствующих агрегации тромбоцитов при разнообразной сосудистой патологии. Указанная фармакологическая активность может быть использована для лечения млекопитающих. Более конкретно, сульфонамидные соединения изобретения действуют путем блокирования молекулярного участка рецептора белка фибриногена. Фибриноген представляет собой гликопротеин, циркулирующий в плазме крови, участок тромбоцитного рецептора которого представлен гликопротеином 11b/111a. Блокированием действия фибриногена у рецептора (гликопротеин 11b/111a) соединения изобретения влияют на агрегацию тромбоцитов, являющуюся причиной многочисленных сосудистых патологий. В настоящее время существует необходимость в области сосудистой терапии в таких блокирующих рецептор фибриногена средствах. Влиянием на гемостаз подобный вид лечения мог бы уменьшить заболеваемость и смертность от тромботических болезней.
Гемостаз - это спонтанный процесс прекращения кровотечения из поврежденного кровеносного сосуда. Предкапиллярные сосуды сжимаются сразу же при их разрезе. В течение секунд тромбоциты или кровяные пластинки связываются с обнаженной матрицей поврежденного сосуда в процессе, называемом адгезией тромбоцитов. Кроме того, тромбоциты слипаются друг с другом в процессе, известном как агрегация тромбоцитов, с образованием пробки тромбоцитов. Такая пробка тромбоцитов способна быстро остановить кровотечение, но она должна усиливаться белковым фибрином для длительной эффективности, пока разрыв в кровеносном сосуде не будет устойчиво восстановлен за счет фибробластов, являющихся специализированными клетками восстановления ткани.
Внутрисосудистый тромб (сгусток) возникает в результате патологического нарушения гемостаза. Тромб может вырасти до размера, достаточного для закупорки артериальных кровеносных сосудов. Тромбы могут также образоваться в областях стасиса или замедленного кровотока в венах. От венозных тромбов легко отрываются их кусочки, называемые эмболями, движение которых в системе кровообращения может привести к закупорке других сосудов, например легочных артерий. Таким образом, артериальные тромбы вызывают серьезные заболевания за счет местной блокады, в то время как венозные тромбы вызывают заболевания за счет дистанционной блокады или эмболизации. Такие заболевания включают венозный тромбоз, тромбофлебит, артериальную эмболию, коронарный и церебральный артериальный тромбоз и инфаркт миокарда, шок, церебральную эмболию, эмболию почек и легочную эмболию.
В области сердечно-сосудистых и церебрально-сосудистых заболеваний существует необходимость в средстве, которое может быть использовано для профилактики и лечения тромбоза с минимальными побочными эффектами, включая нежелательное длительное кровотечение в других частях системы кровообращения во время профилактики или ликвидации целевого тромба. Соединения изобретения отвечают такой необходимости, представляя собой лечебные средства для профилактики и лечения тромбоза.
Соединения изобретения проявляют активность в качестве противотромбозных средств за счет своей способности блокировать действие фибриногена в его участке (сайте) рецептора тромбоцитов с предотвращением тем самым агрегации тромбоцитов.
Изобретение относится к новым соединениям общей структурной формулы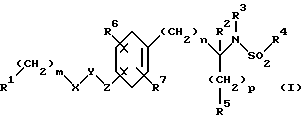
и их фармацевтически приемлемым солям,
где
R1 представляет
четырех-восьмичленный гетероцикл, содержащий 1, 2, 3 или 4 гетероатома, выбранных из N, O или S, и гетероцикл необязательно замещен по любому атому водородом, R6 или R7, NR6R7, 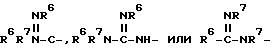
где
R6 и R7 независимо представляют водород и незамещенный или замещенный C0-10-алкил и циклоалкил, где заместители включают
C1-C10-алкокси,
C1-C10-алкоксиалкил,
C1-C10-алкоксиалкилокси,
C1-C10-алкоксикарбонил,
C1-C10-алкилкарбонил,
C4-C10-аралкилкарбонил,
C1-C10-алкилтиокарбонил,
C1-C10-аралкилтиокарбонил,
тиокарбонил,
C1-C10-алкокситиокарбонил,
арил,
пяти-шестичленное насыщенное гетероциклическое кольцо, содержащее 1, 2, 3 или 4 гетероатома, выбранных из группы, включающей N, O и S,
C1-C10-алканоиламино,
C1-C6-алкоксикарбонил-C0-C6-алкиламино,
C1-C10-алкилсульфониламино,
C4-C10-аралкилсульфониламино,
C4-C10-аралкил,
C1-C10-алкарил,
C1-C10-алкилтио,
C4-C10-аралкилтио,
C1-C10-алкилсульфинил,
C4-C10-аралкилсульфинил,
C1-C10-алкильсульфонил,
C4-C10-аралкилсульфонил,
аминосульфонил,
C1-C10-алкиламиносульфонил,
C4-C10-аралкилсульфониламино,
оксо,
тио,
незамещенный или моно- или дизамещенный 1-этенил, 2-этенил, 3-пропенил, заместители которых выбирают из группы, включающей
водород, C1-C10-алкил и C4-C10-аралкил,
карбокси,
гидрокси,
амино,
C1-C6-алкиламино,
C1-С6-диалкиламино,
галоген, где галоген определяется как F, Cl, Br или J,
нитро и
циано,
и, кроме того, N может быть дополнительно замещен с образованием иона четвертичного аммония заместителем, определенным ранее для R6 и R7;
R2 и R3 независимо представляют
водород
арил и
незамещенный или замещенный C0-С10-алкил или циклоалкил, где заместителем является C1-C10-алкоксиалкил,
четырех-восьмичленную насыщенную гетероциклическую систему, содержащую 1, 2, 3 или 4 гетероатома, выбранных из группы, включающей N, O и S,
C4-C10-аралкил,
C1-C10-алкарил,
C1-C10-алкилтио,
C4-C10-аралкилтио,
C1-C10-алкилсульфинил,
C4-C10-аралкилсульфинил,
C1-C10-алкилсульфонил,
C4-C10-аралкилсульфонил,
карбокси,
C1-C10-алкилкарбонил,
C1-C10-алкилтиокарбонил,
C4-C10-аралкилкарбонил,
C4-C10-аралкилтиокарбонил,
C1-C6-алкоксикарбонил,
C4-C10-аралкоксикарбонил,
C1-C6-алкокси,
C1-C6-алкоксикарбонил-C1-C4-алкил,
C4-C10-аралкоксикарбонил-C1-C4-алкил,
C4-C10-аралалкокси,
C1-C6-алкиламино,
C1-C12-диалкиламино,
C1-C6-алканоиламино,
C4-C10-аралканоиламино,
C4-C10-аралкиламино;
R4 представляет
арил,
C1-C10-алкил или циклоалкил,
C4-C10-аралкил,
C1-C10-алкоксиалкил,
C1-C10-алкарил,
C1-C10-алкилтиоалкил,
C1-C10-алкокситиоалкил,
C1-C10-алкиламино,
C4-C10-аралкиламино,
C1-C10-алканоиламино,
C4-C10-аралканоиламино,
C1-C10-алканоил,
C4-C10-аралканоил и
незамещенный или замещенный C1-C10-карбоксиалкил, где заместитель представлен арилом или C1-C10-аралкилом, кроме того, любой из заместителей для R4 может быть замещен заместителями, выбранными из группы, определенной для R6;
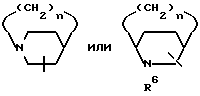
R5 представляет
четырех-восьмичленный насыщенный или ненасыщенный гетероцикл, содержащий 1, 2, 3 и 4 гетероатома, выбранных из N, O или S, и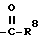 ,
,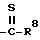 ,
,
где
R8 представляет
гидрокси,
C1-C10-алкилокси,
C1-C10-алкарилокси,
C4-C10-аралкилокси,
C4-C10-аралкилкарбонилокси,
C1-C10-алкоксиалкилокси,
C1-C10-алкоксиалкилкарбонилокси,
C1-C10-алкоксикарбонилалкил,
C1-C10-алкилкарбонилоксиалкилокси,
L- или D-аминокислоту, присоединенную амидной связью, или L- или D-аминокислоту, присоединенную амидной связью и в которой карбоксильная группа аминокислоты этерифицирована C1-C6-алкилом или C4-C10-аралкилом,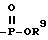 ,
,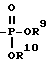 ,
,
где R9 и R10 выбраны из группы, включающей водород, C1-C10 - алкил и C4-C10-аралкил;
X и Y независимо представляют
NR6,
O,
S, SO, 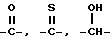 ,
,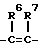 ,
,
-C≡C- ,
четырех-восьмичленный цикл, содержащий 0, 1, 2, 3 или 4 гетероатома, выбранных из N, O и S, причем цикл независимо замещен при любом атоме группой R6 ,
арил,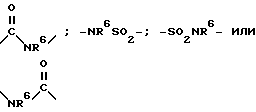
представляет возможный заместитель, который в случае присутствия имеет значения, независимо выбранные и приведенные для X и Y;
m представляет целое число от нуля до десяти;
n представляет целое число от нуля до десяти;
p представляет целое число от нуля до трех.
Предпочтительная группа соединений изобретения включает соединения, определяемые общей структурной формулой
где R1 представляет
пяти-шестичленный гетероцикл, в котором гетероатомы выбраны из N, O и S, и где гетероцикл необязательно замещен C1-C5-алкилом,
или NR6R7,
где R6 и R7 независимо представляют
водород,
незамещенный или замещенный C1-C10-алкил, заместители для которого выбраны из C1-C10-алкоксикарбонила, арила, C0-C5-алкиламино-С1-С10-алкила,
C4-C10-аралкила,
и, кроме того, атом N может быть дополнительно замещен с образованием иона четвертичного аммония, в котором дополнительный заместитель имеет значения, указанные для R6 и R7;
R2 и R3 представляют
водород,
C1-C4-алкил, C4-C10-аралкил;
R4 представляет
арил,
C1-C10-алкил или циклоалкил,
C4-C10-аралкил,
C1-C10-алкоксиалкил,
C1-C10-алкарил,
незамещенный или замещенный C1-C10-карбоксиалкил, где заместитель выбран из арила, C1-C6-алкила или C4-C10-аралкила;
R11 представляет
водород или C1-C10-алкил;
X и Y независимо представляют
арил,
O, SO2,
-CH=CH-, 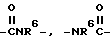 , -SO2NR6- или -NR6SO2,
, -SO2NR6- или -NR6SO2,
пяти-шестичленный цикл, содержащий 0, 1 или 2 гетероатома, выбранных из N или O;
Z представляет возможный заместитель, который в случае присутствия представляет O, SO2 , -NR6CO-, -CONR6-, прямой или разветвленный C1-C10-алкил;
m представляет целое число от нуля до восьми;
n представляет целое число от нуля до двух;
p представляет целое число от нуля до двух.
Более предпочтительная группа соединений изобретения включает соединения, определяемые общей структурной формулой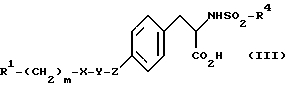
где R1 представляет
пяти-шестичленный гетероцикл с гетероатомами, выбранными из N и O, причем гетероцикл необязательно замещен C1-C5-алкилом,
NR6R7,
где R6 и R7 независимо представляют C1-C10-алкил или C4-C10-аралкил,
и далее атом N может быть дополнительно замещен с образованием иона четвертичного аммония, в котором дополнительный заместитель имеет значения, указанные ранее для R6 и R7;
R4 представляет
арил,
C1-C10-алкил или циклоалкил, или
C4-C10-аралкил;
X и Y независимо представляют
фенил,
O, SO2,  или
или
пяти-шестичленный цикл, содержащий 0 или 1 гетероатом, выбранный из N или O;
Z представляет возможный заместитель, который в случае присутствия представляет O, SO2 , -NR6CO-, -CONR6- или -CH2-;
m представляет целое число от нуля до шести.
Термин "фармацевтически приемлемые соли" означает неядовитые соли соединений изобретения, которые обычно получают реакцией свободного основания с приемлемой органической или неорганической кислотой. Примеры характерных представителей солей включают ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат кальция, камзилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдизилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликоллиларзанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, памеат, пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид, валерат.
Термин "физиологически эффективное количество" означает такое количество лекарственного или фармацевтического средства, которое будет создавать медицинскую или биологическую реакцию ткани, системы или животного, ожидаемую исследователем или клиницистом. Термин "противокоагулирующее средство" включает аспирин, гепарин и варфарин. Термин "фибринолитическое средство" выключает стрептокиназу и активатор тканевого плазминогена.
Термин "арил" означает моно- или полициклическую систему, состоящую из пяти- шестичленных ароматических циклов, содержащих 0, 1, 2, 3 и 4 гетероатома, выбранных из N, O и S, и которые незамещены или замещены группой R6.
Термин "алкил" означает нормальный или разветвленный алкан, алкен или алкин.
Термин "алкоксигруппа" будет рассматриваться как включающий алкильную часть, где алкил имеет вышеуказанные значения.
Термины "аралкил" и "алкарил" следует рассматривать как включающие алкильную часть, где алкил имеет вышеуказанные значения, и арильную часть, где арил имеет вышеуказанные значения.
Термин "галоген" включает фтор, хлор, йод и бром.
Термин "оксо" означает радикал = 0.
Термин "тио" означает радикал = S.
Соединения изобретения могут быть введены больным, если хотят предотвратить тромбоз ингибированием связывания фибриногена с рецептором гликопротеинового комплекса 11b/111a мембраны тромбоцита. Соединения применимы в хирургии на периферийных артериях (артериальные трансплантаты, каротидная эндартерэктомия) и в сердечно-сосудистой хирургии в случаях, когда операции на сосудах и органах и/или контактирование тромбоцитов с искусственными поверхностями приводит к агрегации тромбоцитов и их расходу. Агрегированные тромбоциты могут образовывать тромбы и тромбоэмболии. Соединения могут быть введены таким послеоперационным больным для предотвращения образования тромбов и тромбоэмболий.
Экстракорпоральная система кровообращения обычно применяется в сердечно-сосудистой хирургии для насыщения крови кислородом. При этом происходит прилипание тромбоцитов к поверхностям экстракорпоральной цепи. Налипание зависит от взаимодействия между GP 11b/111a на мембранах тромбоцитов и фибриногеном, адсорбированным поверхностью цепи (Gluszko и др. , Amer. J. Physiol. , 1987, 252:H, с. 615-621). Отделившиеся от искусственных поверхностей тромбоциты характеризуются ухудшенным функционированием. Для предотвращения налипания могут быть введены соединения изобретения.
Другие области применения данных соединений включают предотвращение тромбоза кровяных пластинок, тромбоэмболии и реокклюзии в ходе и после тромболитической терапии, а также предотвращение тромбоза кровяных пластинок, тромбоэмболии и реокклюзии после пластических операций на коронарных и других артериях и после процедур обхода коронарной артерии. Соединения могут быть также использованы для профилактики инфаркта миокарда.
Соединения изобретения могут быть введены в таких пероральных дозированных формах, как таблетки, капсулы, пилюли, порошки, гранулы, эликсиры, тинктуры, суспензии, сиропы, эмульсии. Точно также соединения могут быть введены в виде внутривенной, внутрибрюшинной, подкожной или внутримышечной инъекции, и каждая такая применяемая форма хорошо известна специалисту в фармацевтической области. Эффективное, но нетоксичное количество целевого соединения может быть использовано в качестве противоагрегационного средства.
Режим дозировок с применением соединений изобретения подбирают с учетом разнообразных факторов, в том числе типа, вида, возраста, веса, пола и медицинского состояния больного, тяжести подлежащих лечению симптомов, пути введения, функционирования почек и печени пациента и конкретного применяемого соединения или его соли. Любой опытный лечащий врач или ветеринар способен легко определить и прописать эффективное количество лекарственного средства, необходимое для профилактики, лечения или приостановки развития симптомов.
Пероральные дозировки изобретения при их использовании для достижения целевого эффекта будут находиться в интервале от 0,01 мг кг массы тела в день (мг/кг/день) до 100 мг/кг/день, предпочтительно 1-100 мг/кг/день и наиболее предпочтительно 1-50 мг/кг/день. Внутривенные, наиболее предпочтительные дозировки находятся в интервале 1-10 мг/кг мин при вливании с постоянной скоростью. Соединения изобретения могут с успехом назначаться в виде единственной ежедневной дозы или общая дневная доза может быть введена дробными дозами один, два, три или четыре раза ежедневно. Кроме того, предпочтительные соединения изобретения могут назначаться через нос с применением приемлемых интраназальных носителей или могут назначаться трансдермальным путем с применением хорошо известных специалистам в данной области кожных пластырей. При введении в виде трансдермальной системы доставки дозированное введение, разумеется, будет скорее непрерывным, чем прерывистым во всем дозировочном режиме.
В способах изобретения подробно охарактеризованные здесь соединения являются активными компонентами, которые обычно вводят в смеси с фармацевтически приемлемыми разбавителями, наполнителями или носителями (обобщенно называемые здесь "носителями"), которые подбирают соответствующим образом в зависимости от намеченной формы введения, т.е. пероральных таблеток, капсул, эликсиров, сиропов и т.п. в соответствии с обычной фармацевтической практикой.
К примеру, для перорального введения в форме таблетки или капсулы активный лекарственный компонент может быть смешан с пероральным неядовитым фармацевтически приемлемым инертным носителем, например лактозой, крахмалом, сахарозой, глюкозой, метилцеллюлозой, стеаратом магния, дикальцийфосфатом, сульфатом кальция, маннитом, сорбитом и т.п.; для перорального введения в жидкой форме лекарственный компонент может быть смешан с любым пероральным неядовитым инертным фармацевтически приемлемым носителем, например этанолом, глицерином, водой и т.п. Кроме того, при желании или необходимости в смесь могут быть введены приемлемые связующие средства, смазки, рыхлители и красители. Приемлемые связующие средства включают крахмал, желатин, природные сахара, например глюкозу или бета-лактозу, кукурузные подслащивающие вещества, природные и синтетические камеди, например камедь акации, трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и т.п. Применяемые в дозированных формах смазки включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Рыхлители или раздробляющие агенты включают (но без ограничения только ими) крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п.
Соединения изобретения могут быть также введены в форме липосомной системы доставки, например, в виде небольших однослойных пузырьков, крупных однослойных пузырьков и многослойных пузырьков. Липосомы могут быть получены из самых разнообразных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолин.
Соединения изобретения могут быть также поставлены с использованием моноклональных антител в качестве индивидуальных носителей, к которым присоединены молекулы соединения. Соединения изобретения могут быть также присоединены к растворимым полимерам в качестве целевых носителей лекарственного средства. Подобные полимеры могут включать поливинилпирролидон, сополимеры пирана, полигидроксипропилметакриламин-фенол, полигидроксиэтиласпартамид-фенол или полиэтиленоксидполилизин, замещенный пальмитоильными остатками. Кроме того, соединения изобретения могут быть присоединены к ряду биоразлагаемых полимеров, полезных при достижении регулируемого выделения лекарственного средства, например полимолочной кислоты, полигликолевой кислоты, сополимеров полимолочной кислоты и полигликолевой кислоты, поли-эпсилон-капролактона, полигидроксимасляной кислоты, полиортоэфиров, полиацеталей, полидигидропиранов, полицианоакрилатов и сшитых или амфипатических блок-сополимеров гидрогелей.
Соединения изобретения могут быть также введены совместно с приемлемыми противокоагулятными средствами или тромболитическими средствами с получением синергических эффектов при лечении разнообразной сосудистой патологии.
Соединения формулы (I) могут быть легко получены согласно нижеследующим схемам реакций и примерам или их модификаций с применением легко доступных исходных продуктов, реактивов и обычных методик синтеза. В таких реакциях возможно применение вариантов, которые сами по себе известны специалисту, но которые не были упомянуты с большими подробностями.
К наиболее предпочтительным соединениям изобретения относятся любое соединение или все соединения, конкретно описанные в примерах. Однако эти соединения не следует рассматривать как образующие единственный род соединений, считающихся изобретением, и любые сочетания соединений или их фрагментов могут сами образовать род или группу соединений. Нижеследующие примеры дополнительно иллюстрируют подробности получения соединений изобретения. Специалисту понятно, что при получении этих соединений возможны известные вариации условий и способов осуществления приемов получения этих соединений. Все температуры даны в градусах Цельсия, если нет особых указаний.
Условные обозначения реагентов имеют следующие значения:
Boc - трет-бутоксикарбонил,
Pd-C - палладий на активированном угле в качестве катализатора,
ДМФА или DMF - диметилформамид,
ДМСО - диметилсульфоксид,
CBZ - карбобензилокси,
CH2Cl2 - хлористый метилен,
CHCl3 - хлороформ,
EtOH - этанол,
MeOH - метанол,
EtOAc - этилацетат,
HOAc - уксусная кислота,
ТГФ - тетрагидрофуран.
Ниже указаны источники следующих соединений: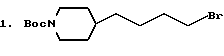
описан ниже,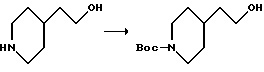
2-(4-N-трет-бутилоксикарбонилпиперидинил)этанол.
В 700 мл диоксана растворяют 4-пиперидин-2-этанол (производство фирмы Олдрич) (130 г, 1 моль), охлаждают до 0oC и обрабатывают 3 н. NaOH (336 мл, 1 моль) и ди-трет-бутил-карбонатом(221,8 г, 1 моль). Ледяную баню удаляют и реакционную смесь перемешивают в течение ночи. Затем реакционную смесь концентрируют, разбавляют водой и экстрагируют эфиром. Эфирные слои объединяют, промывают рассолом, сушат над MgSO4, фильтруют и после испарения получают 225,8 г (98%) продукта.
Rf=0,37 в смеси EtOAc-гексан (1:1), окрашивание нингидрином.
1H-ЯМР (300 МГц, CDCl3) δ : 4,07 (ш.с, 2H), 3,7 (ш.с, 2H), 2,7 (т, J = 12,5 Гц, 2H), 1,8-1,6 (м, 6H), 1,51 (c, 9H), 1,1 (ддд, J= 4,3, 12,5 и 12 Гц, 2H).

Метиловый эфир 4-(4-N-трет-бутилоксикарбонилпиперидинил)-бут-2-еновой кислоты.
В 1 л CH2Cl2 растворяют оксалилхлорид (55,8 мл, 0,64 моля) и в атмосфере N2 охлаждают до -78oC. Затем по каплям прибавляют ДМСО (54,2 мл, 0,76 моля). После прекращения выделения газа в течение 20 мин прибавляют 2-(4-N-трет-бутилоксикарбонилпиперидинил)этанол (102,5 г, 0,45 моля), растворенный в 200 мл CH2Cl2. После перемешивания еще 20 мин по каплям прибавляют триэтиламин (213 мл, 1,53 моля) и охлаждающую баню удаляют. Спустя 1,5 ч, ТСХ показывает отсутствие исходных продуктов. Добавляют карбометокситрифенилфосфаран (179 г, 0,536 моля) и реакционную смесь перемешивают в течение ночи. Раствор разбавляют 300 мл Et2O, экстрагируют один раз 800 мл H2O, дважды 300 мл 10%-го раствора KHSO4, затем один раз рассолом. Органический слой сушат над MgSO4, фильтруют и испаряют. Колоночной хроматографией (SiO2, 5% EtOAc в гексане) получают 78,4 г (62%) чистого метилового эфира 4-(4-N-трет-бутилоксикарбонилпиперидинил)бут-2-еновой кислоты.
1H-ЯМР (300 МГц, CDCl3) δ : 6,9 (ддд, J=15,6, 7,6 и 7,6 Гц, 1H), 5,8 (д, J= 15,6 Гц, 1H), 4 (ш.с, 2H), 3,7 (с, 3H), 2,6 (т, J = 12,6 Гц, 2H), 2,1 (т, J = 7,4 Гц, 2H): 1,7-1,4 (с, 9H), 1,1 (м, 2H).
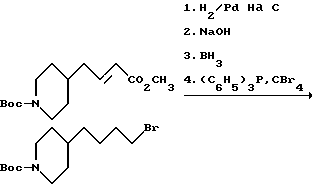
4-(4-N-трет-бутилоксикарбонилпиперидинил)бутилбромид.
В 500 мл EtOAc растворяют метиловый эфир 4-(4-N-трет-бутилоксикарбонилпиперидинил)бут-2-еновой кислоты. К раствору добавляют 10% палладия на угле (10 г) в виде взвеси в EtOAc и реакционную смесь выдерживают в течение ночи в атмосфере H2 (из баллона). Затем реакционную смесь фильтруют через Солкафлок, фильтровальный пирог промывают EtOAc и после испарения этилацетата получают 34,7 г (90%) метилового эфира 4-(4-N-трет-бутилоксикарбонилпиперидин-4-ил)бутановой кислоты. ТСХ
Rf = 0,69 в 30% EtOAc в гексане.
1H-ЯМР (300 Мгц, CDCl3) δ : 4 (ш.с, 2H), 3,6 (с, 3H), 2,6 (т, J = 12,3 Гц, 2H), 2,2 (т, J = 7,4 Гц, 2H), 1,6 (м, 4H), 1,4 (с, 9H), 1,4 (м, 1H), 1,2 (м, 2H), 1 (м, 2H).
Эфир бутановой кислоты (45,3 г, 0,159 моля) растворяют в CH3OH и выдерживают в течение ночи с 1 н. NaOH (500 мл, 0,5 моля). Растворитель удаляют в вакууме, добавляют воду и раствор промывают эфиром, после чего подкисляют 10%-ным раствором KHSO4. Водный слой промывают эфиром, эфирные слои объединяют, промывают рассолом, сушат над MgSO4 и концентрированием получают в виде прозрачного масла соответствующую кислоту (41,85 г, выход 97%).
1H-ЯМР (300 МГц, CDCl3) δ : 4 (ш.с, 2H), 2,6 (м, 2H), 2,25 (м, 2H), 1,6 (ш.с, 4H), 1,4 (с, 9H), 1,3-0,9 (9H).
Полученную кислоту (20,4 г, 0,077 моля) выдерживают 1 ч с бораном (BF3-ТГФ, 235 мл, 235 ммоля) при 0oC в ТГФ, по каплям прибавляют NaOH (1 н., 250 мл) и раствор перемешивают в течение ночи. Образовавшуюся реакционную смесь концентрируют с удалением ТГФ и экстрагируют эфиром. Эфирные экстракты объединяют, сушат над MgSO4, фильтруют и после испарения получают в виде бесцветного масла (19,7 г) соответствующего спирта.
Rf = 0,7 в смеси этилацетат-гексан (2:1).
1H-ЯМР (300 МГц, CDCl3) δ : 4,1 (ш.с, 2H), 3,6 (т, 2H), 2,65 (т, 2H), 2,1 (ш.с, 1H), 1,65 (ш.с, 2H), 1,55 (м, 2H), 1,4 (с, 9H), 1,35 (м, 3H), 1,25 (м, 2H), 1,1 (м, 2H).
Полученный спирт (19,7 г, 76,5 ммоля) растворяют в ТГФ, обрабатывают трифенилфосфином (23,1 г, 88 ммолей) и охлаждают до 0oC. Одной порцией добавляют четырехбромистый углерод (29,8 г, 89,9 ммоля), охлаждающую баню удаляют и реакционную смесь перемешивают в течение ночи. Для завершения реакции добавляют дополнительное количество трифенилфосфина (11,71 г) и четырехбромистого углерода (14,9 г). Смесь фильтруют, жидкость разбавляют эфиром и снова фильтруют. После удаления растворителя полученную жидкость адсорбируют на SiO2 и алюированием 5% EtOAc в гексане получают в виде прозрачного бесцветного масла 4-(4-N-трет-бутилоксикарбонилпиперидин-4-ил)бутилбромид (20,7 г, 85% выход).
Rf = 0,6 в смеси этилацетат-гексан (1:4).
1H-ЯМР (300 МГц, CDCl3) δ : 4,1 (ш.с, 2H), 3,4 (т, 2H), 2,65 (т, 2H), 1,85 (м, 2H), 1,65 (ш.д, 2H), 1,4 (с, 9H), 1,35 (м, 2H), 1,3 (м, 3H), 1,1 (м, 2H).
2. BocNH(CH2)6Br.
По стандартной методике промышленный H2N(CH2)5CH2OH защищался в виде N-Boc производного и с помощью Ph3P-CBr4 в ТГФ превращался в бромид. Использование исходных аминоспиртов с различной длиной цепи дает по схеме 1, приведенной в конце описания, аналогичные галогениды: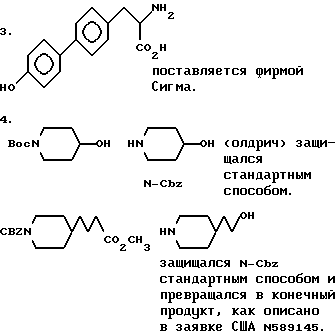
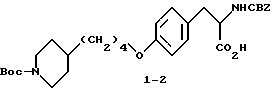
2-s-(бензилоксикарбониламино)-3-/4-(N-трет-бутилоксикарбонил-пиперидин- 4-ил)бутилоксифенил/пропионовая кислота (1-2).
В ДМФА (75 мл) растворяют N-CBZ-L-тирозин (1-1) (17,58 г, 0,055 моля), охлаждают до 0-10oC и обрабатывают гидридом натрия (2,88 г, 0,12 моля). Полученную суспензию перемешивают 1 ч при 0-10oC и затем по каплям в течение 15 мин прибавляют N-трет-бутилоксикарбонилпиперидин-4-илбромид (17,7 г, 0,055 моля) в 25 мл ДМФА. После этого реакционную смесь перемешивают 16 ч при комнатной температуре. Растворитель удаляют в вакууме, а остаток переносят в смесь 500 мл EtOAc и 100 мл 10% KHSO4. Органическую фазу промывают рассолом, сушат (Na2SO4) и после отгонки растворителя получают вязкое масло. Очисткой полученного масла вытеснительной хроматографией на силикагеле с элюированием смесью CHCl3-CH3-OH-HOAc (98:2:0,5) получают чистое соединение 1-2 (23,75 г), Rf = 0,35, в виде бледно-желтого масла.
1H-ЯМР (300 МГц, CDCl3) δ : 1-1,15 (2H, м), 1,2-1,8 (16H, м), 2,62 (2H, т), 3,1 (2H, м), 3,91 (2H, т), 4,04 (2H, м), 5,1 (2H, м), 5,22 (1H, д), 6,78 (2H, д), 7,04 (2H, д), 7,35 (5H, м).
Пример 2.
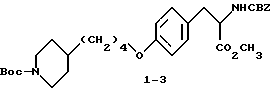
Метиловый эфир 2-s-(бензилоксикарбониламино)-3-/4-(N-трет- бутилоксикарбонилпиперидин-4-ил)бутилоксифенил/пропионовой кислоты (1-3).
В 150 мл CH3OH растворяют при комнатной температуре соединение 1-2 (10,05 г, 18,1 ммоля), добавляют карбонат цезия (2,95 г, 9,06 ммоля) и после перемешивания полученной смеси 15 мин получают прозрачный раствор. Метанол удаляют при пониженном давлении, остаток растворяют в ДМФА (150 мл) и обрабатывают прибавлением по каплям йодистого метила (2,57 мл, 18,1 ммоля). Полученный раствор перемешивают в течение ночи при комнатной температуре. Растворитель удаляют в вакууме, остаток переносят в 400 мл эфира, промывают порциями 3 x 50 мл H2O, 50 мл рассола и сушат (Na2SO4). Удалением растворителя получают в виде масла соединение 1-3.
1H-ЯМР (300 МГц, CDCl3) δ : 1-1,15 (2H, м), 1,3-1,7 (16H, м), 2,68 (2H, дт), 3,05 (2H, м), 3,72 (3H, с), 3,91 (2H, т), 4,08 (2H, д), 4,61 (1H, м), 5,1 (2H, м), 5,18 (1H, м), 6,79 (2H, д), 6,98 (2H, д), 7,35 (5H, м).
Пример 3.

Метиловый эфир 2-s-амино-3-/4-(N-трет-бутилоксикарбонил-пиперидин- 4-ил)бутилоксифенил/пропионовой кислоты (1-4).
К соединению 1-3 (5 г, 8,79 ммоля), растворенному в абсолютном этаноле (150 мл), добавляют 10% Pd-C (0,5 г) и образовавшуюся суспензию гидрируют 12 ч под давлением водорода из баллона. Катализатор отфильтровывают и удалением растворителя в вакууме получают в виде масла соединение 1-4 (3,6 г).
1H-ЯМР (300 МГц, CDCl3) δ : 1-1,2 (2H, м), 1,22-1,55 (12H, м), 1,6-1,75 (4H, м), 2 (2H, ш.с), 2,68 (2H, т), 2,87 (1H, дд), 3,05 (1H, дд), 3,72 (3H, с), 3,93 (2H, т), 4,09 (2H, м), 6,82 (2H, д), 7,1 (2H, д).
Пример 4.
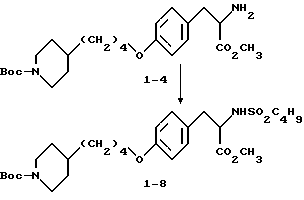
Метиловый эфир 2-s-(н-бутилсульфониламино)-3-/4-(N-трет- бутилоксикарбонилпиперидин-4-ил)-бутилоксифенил/пропионовой кислоты (1-8).
В 10 мл этилацетата растворяют соединение 1-4 (0,59 г, 1,36 ммоля), добавляют NaHCO3(0,7 г, 8,68 ммоля) при перемешивании и при комнатной температуре с последующим прибавлением бутансульфонилхлорида (0,36 мл, 2,76 ммоля) и полученную смесь кипятят 26 ч. Охлажденную реакционную смесь фильтруют, концентрируют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (4: 1) получают чистое соединение 1-8 (0,305 г).
Rf = 0,7 в смеси гексан-EtOAc (1:1), окрашивание нингидрином.
1H-ЯМР (300 МГц, CDCl3) δ: 0,82 (3H, т), 1,05 (2H, ддд), 1,45 (9H,с), 1,1-1,6 (1H, м), 1,7 (4H, м), 2,6 (2H,т), 2,6-2,8 (2H, м), 2,78 (1H, дд), 3,05 (1H, дд), 3,7 (3H, с), 3,93 (2H, т), 4,05 (2H, ш.д.), 4,15 (1H, дд), 6,85 (2H, д), 7,15 (2H, д).
Пример 5.
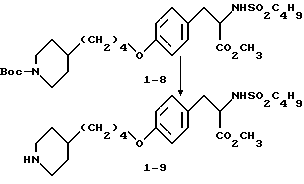
Гидрохлорид 2-s-(н-бутилсульфониламино)3-/4-(пиперидин-4-ил) бутилоксифенил/пропионовой кислоты (1-9).
В смеси CH3OH-H2O-ТГФ (1:1:1) растворяют соединение 1:8 (0,325 г, 0,59 ммоля) и к раствору добавляют LiOH•H2O(0,157 г, 3,76 ммоля). Полученный раствор перемешивают 3 ч при комнатной температуре, затем концентрируют, разбавляют 10% KHSO4 и экстрагируют EtOAc. В результате получают 2-s-(н-бутилсульфониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин -4-ил)бутилоксифенил/пропионовую кислоту. Полученную кислоту (0,315 г, 0,59 ммоля) растворяют в EtOAc (20 мл) и обрабатывают 15 мин при -20oC газообразным HCl. Затем реакционную смесь перемешивают 1 ч при -5oC, и за это время происходит полное исчезновение исходного продукта. Через реакционную смесь пробулькивают аргон и после удаления растворителя получают остаток, переосаждением которого из эфира получают в виде бледно-желтого твердого вещества соединение 1-9.
1H-ЯМР (300 МГц, CD3OD) 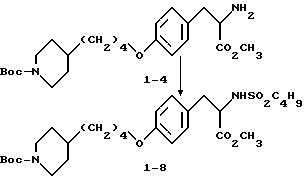 : 0,85 (3H, т), 1,2 (2H, дд), 1,2-1,7 (9H, м), 1,7 (2H, м), 1,95 (2H, ш.с.), 2,65 (2H, т), 2,8 (1H, дд), 2,95 (2H, ш.т.), 3,1 (1H, дд), 3,83, (2H, ш.с.), 3,95 (2H, т), 4,1 (1H, дд), 6,85 (2H, д), 7,2 (2H, д).
: 0,85 (3H, т), 1,2 (2H, дд), 1,2-1,7 (9H, м), 1,7 (2H, м), 1,95 (2H, ш.с.), 2,65 (2H, т), 2,8 (1H, дд), 2,95 (2H, ш.т.), 3,1 (1H, дд), 3,83, (2H, ш.с.), 3,95 (2H, т), 4,1 (1H, дд), 6,85 (2H, д), 7,2 (2H, д).
Анализ для C22 H36N2O5S•HCl•0,8H2O.
Вычислено: C 53,76; H 7,92; N 5,70.
Найдено: C 53,76; H 7,66; N 5,44.
Пример 6.
δ
Метиловый эфир 2-s-(бензилсульфониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин -4-ил)бутилоксифенил/пропионовой кислоты (1-10).
Соединение 1-4 (0,59 г, 1,36 ммоля) обрабатывают бензилсульфонилхлоридом (0,263 г, 1,38 ммоля) по вышеприведенной методике получения соединения 1-8. Сырой продукт реакции очищают вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (3: 1) и получением в виде масла чистого соединения 1-10 (0,35 г).
1H-ЯМР (300 МГц, CD3OD) 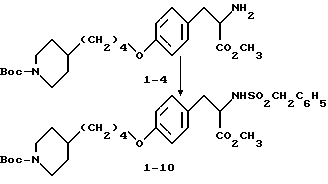 : 0,85-1,1 (2H, м), 1,1-1,23 (2H, м), 1,35-1,52 (11H, м), 1,61-1,8 (4H, м), 2,65-3 (4H, м), 3,65 (3H,c), 3,9-4,14 (5H, м), 6,85 (2H, д), 7,08 (2H, д), 7,22 (2H, м), 7,3 (3H, м).
: 0,85-1,1 (2H, м), 1,1-1,23 (2H, м), 1,35-1,52 (11H, м), 1,61-1,8 (4H, м), 2,65-3 (4H, м), 3,65 (3H,c), 3,9-4,14 (5H, м), 6,85 (2H, д), 7,08 (2H, д), 7,22 (2H, м), 7,3 (3H, м).
Пример 7.
δ
Гидрохлорид 2-s-(бензилсульфониламино)-3-/4-(пиперидин-4-ил)бутилоксифенил/пропионовой кислоты (1-11).
Обработкой соединения 1-10 (0,354 г, 0,6 ммоля) LiOH (0,15 г, 3,7 ммоля) по методике, приведенной для получения соединения 1-8, получают в виде вязкого масла 2-s-(бензилсульфониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил) бутилоксифенил/пропионовую кислоту (0,35 г).
1H-ЯМР (300 МГц, CD3OD) 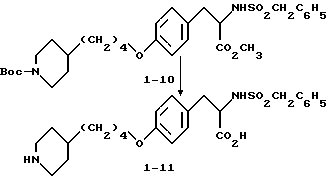 : 0,84-1,06 (3H, м), 1,23 (4H, м), 1,34-1,5 (11H, м), 1,6-1,78 (5H, м), 2,65 (2H, ш. т. ), 2,82 (1H, м), 3,02 (1H, м), 3,91 (2H, м), 3,96-4,12 (5H, м), 6,83 (2H, д), 7,15 (2H, д), 7,22 (2H, м), 7,29 (3H, м).
: 0,84-1,06 (3H, м), 1,23 (4H, м), 1,34-1,5 (11H, м), 1,6-1,78 (5H, м), 2,65 (2H, ш. т. ), 2,82 (1H, м), 3,02 (1H, м), 3,91 (2H, м), 3,96-4,12 (5H, м), 6,83 (2H, д), 7,15 (2H, д), 7,22 (2H, м), 7,29 (3H, м).
Полученную кислоту (0,35 г, 0,6 ммоля) растворяют в 20 мл EtOAc и обрабатывают газообразным HCl по методике, приведенной для синтеза соединения 1-9, с получением чистого соединения 1-11 (0,3 г) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD) δ: 1,32 (4H, м), 1,4-1,65 (3H, м), 1,72 (2H, м), 1,92 (2H, д), 2,77-3,08 (4H, м), 3,33 (3H, м), 3,95-4,14 (5H, м), 6,86 (2H, д), 7,17 (2H, д), 7,28 (2H, м), 7,31 (3H, м).
δ
Метиловый эфир 2-s-(2-стирилсульфониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил) бутилоксифенил/пропионовой кислоты (1-12).
Соединение 1-4 (0,647 г, 15 ммолей) растворяют в этилацетате (20 мл), добавляют NaHCO3 (0,454 г, 5,4 ммоля) с последующим прибавлением  -стиролсульфонилхлорида (0,365 г, 18 ммолей) и полученную реакционную смесь кипятят с перемешиванием 16 ч. Охлажденную реакционную смесь фильтруют, растворитель удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-этилацетат (3:1) получают чистое соединение 1-12.
-стиролсульфонилхлорида (0,365 г, 18 ммолей) и полученную реакционную смесь кипятят с перемешиванием 16 ч. Охлажденную реакционную смесь фильтруют, растворитель удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-этилацетат (3:1) получают чистое соединение 1-12.
1H-ЯМР (300 МГц, CDCl3) β: 1,1 (2H, м), 1,3-1,55 (14H, м), 1,65-1,8 (4H, м), 2,68 (2H, т), 3,01 (2H, дт), 3,62 (3H, с), 3,88 (2H, т), 4,09 (2H, м), 4,22 (1H, м), 4,98 (1H, д), 6,45 (1H, д), 6,8 (2H, д), 7,06 (2H, д), 7,4 (4H, с).
Гидрохлорид 2-s-(2-стирилсульфониламино)-3-/4-(пиперидин-4-ил) бутилоксифенил/пропионовой кислоты (1-13).
Соединение 1-12 (0,58 г, 0,97 ммоля) растворяют в 15 мл смеси ТГФ-H2O-МeOH (1:1:1), добавляют гидроксид лития (0,12 г, 5 ммолей) и полученный прозрачный раствор перемешивают в течение ночи при комнатной температуре.
Реакционную смесь разбавляют 75 мл H2O, подкисляют до pH 2-3 добавлением 10%-ного раствора KHSO4 и затем экстрагируют EtOAc (3•50 мл). Органический экстракт сушат, растворитель удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью CHCl3-MeOH-HOAc (97:3:1) получают целевую кислоту (Rf = 0,2).
Полученную кислоту растворяют в EtOAc и обрабатывают газообразным HCl по методике синтеза соединения 1-9 с получением соединения 1-13.
1H-ЯМР (300 МГц, CD3OD) δ: 1,15-1,7 (10H, м), 1,7-1,82 (2H, т), 1,97 (2H, т), 2,78-3,12 (5H, м), 3,35 (3H, м), 3,87 (2H, т), 4,03 (1H, м), 6,5 (1H, д), 6,69 (2H, м), 7,18 (3H, м), 7,41 (5H, ш.с.).
δ
2-s-(2-фенилэтилсульфониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил) бутилоксифенил/пропионовая кислота (1-14).
Соединение 1-12a (0,21 г) растворяют в 20 мл абсолютного этанола, добавляют 0,1 г 10% Pd-C и перемешиваемую суспензию гидрируют под давлением водорода из баллона. Спустя 4 ч, реакцию прекращают и удалением растворителя получают целевое соединение 1-14 (0,194 г).
1H-ЯМР (300 МГц, CD3OD) 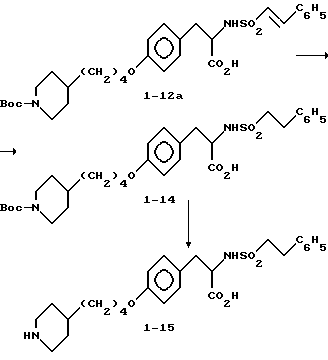 : 1,05 (2H, м), 1,3-1,4 (3H, м), 1,47 (14H, м), 1,72 (5H, м), 2,67-2,93 (8H, м), 3,13 (1H, м), 3,31 (2H, м), 3,82 (2H, м), 4-4,2 (4H, м), 6,82 (2H, д) 7,07 (2H, д), 7,21 (5H, м).
: 1,05 (2H, м), 1,3-1,4 (3H, м), 1,47 (14H, м), 1,72 (5H, м), 2,67-2,93 (8H, м), 3,13 (1H, м), 3,31 (2H, м), 3,82 (2H, м), 4-4,2 (4H, м), 6,82 (2H, д) 7,07 (2H, д), 7,21 (5H, м).
2-s-(2-фенилэтилсульфониламино)-3-/4-(пиперидин-4-ил)бутилоксифенил/пропионовой кислоты гидрохлорид (1-15).
Соединение 1-14 (0,194 г) растворяют в EtOAc и обрабатывают по методике синтеза соединения 1-9 газообразным HCl с получением чистого соединения 1-15 (0,145 г).
1H-ЯМР (300 МГц, CD3OD) δ : 1,25 -1,68 (8H, м), 1,73 (2H, м) 1,93 (2H, м), 2,78 (3H, м), 2,91 (4H, м), 3,13 (1H, м), 3,33 (4H,м), 3,86 (2H, м), 4,18 (1H, м), 6,8 (2H, д), 7,09 (2H, д), 7,22 (5H, м).
δ
метиловый эфир 2-s-(фенилсульфониламино)3-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил) бутилоксифенил/пропионовой кислоты (1 - 16).
По методике получения соединения 1 - 8 проводят реакцию между соединением 1 - 4 (0,647 г, 1,5 ммоля) и фенилсульфонилхлоридом (0,318 г, 1,8 ммоля). Очисткой сырого продукта на селикагеле с элюированием смесью CHCl3-МeOH (98:2) получают чистое соединение 1 - 16 (0,67 г).
1H-ЯМР (300 МГц, CDCl3)  : 1,09 (2H, м), 1,25 - 1,4 (3H, м), 1,42 (9H, ш.с.), 1,6 - 1,85 (6H, м), 2,66 (2H, м), 2,96 (2H, д), 3,55 (3H, с), 3,89 (2H, т), 4,09 (4H, м), 5,12 (1H, д), 6,72 (2H, д), 6,95 (2H, д), 7,4 - 7,65 (3H, м), 7,75 (2H, м).
: 1,09 (2H, м), 1,25 - 1,4 (3H, м), 1,42 (9H, ш.с.), 1,6 - 1,85 (6H, м), 2,66 (2H, м), 2,96 (2H, д), 3,55 (3H, с), 3,89 (2H, т), 4,09 (4H, м), 5,12 (1H, д), 6,72 (2H, д), 6,95 (2H, д), 7,4 - 7,65 (3H, м), 7,75 (2H, м).
Гидрохлорид 2-s-(фенилсульфониламино)-3-(4-пиперидин-4-ил-бутилоксифенил)пропионовой кислоты (1 - 17).
Соединение 1 - 16 (0,525 г) обрабатывают по методике получения соединения 1 - 8 гидроксидом лития с получением сырого продукта, очисткой которого вытеснительной хроматографией на силикагеле с элюированием смесью CHCl3-МeOH-HOAc (97:3:1) получают чистую кислоту (Rf= 0,2).
Обработкой полученной кислоты по методике синтеза соединения 1 - 9 газообразным HCl в EtOAc получают чистое соединение 1 - 17.
1H-ЯМР (300 МГц, CD3OD) δ : 1,28 - 1,47 (6H, м), 1,5 - 1,7 (3H, м), 1,75 (2H, м), 1,97 (2H, д), 2,77 (1H, м), 2,95 (3H, м), 3,35 (4H, м), 3,93 (3H, м), 6,72 (2H, д), 7,02 (2H, д), 7,41 (2H, м), 7,52 (1H, м), 7,67 (2H, м).
δ
Метиловый эфир 2-s-(2-тиенилсульфониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил)бутилоксифенил/пропионовой кислоты (1 - 18).
Соединение 1 - 4 (0,304 г, 0,7 ммоля) обрабатывают 2-тиенилсульфонилхлоридом (0,155 г, 0,85 ммоля) по методике синтеза соединения 1 - 8 с получением сырого продукта. Полученный продукт очищают вытеснительной хроматографией на силикагеле с элюированием смесью CHCl3-CH3OH (98:2) с получением в виде вязкого масла чистого соединения 1 - 18.
Rf = 0,3 (силикагель, CHCl3-CH3OH (98:2)).
1H-ЯМР (300 МГц, CDCl3) 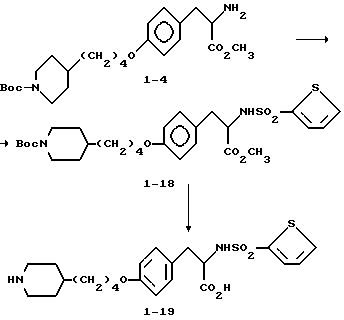 : 1,1 (2H, м), 1,31 (4H, м), 1,36 - 1,8 (16H, м), 2,68 (2H, ш.т.), 3,03 (2H, д), 3,57 (3H, с), 3,91 (2H, т), 4,08 (2H, м), 4,29 (1H, м), 5,16 (1H, д), 6,78 (2H, д), 7 (4H, м), 7,55 (2H, дд).
: 1,1 (2H, м), 1,31 (4H, м), 1,36 - 1,8 (16H, м), 2,68 (2H, ш.т.), 3,03 (2H, д), 3,57 (3H, с), 3,91 (2H, т), 4,08 (2H, м), 4,29 (1H, м), 5,16 (1H, д), 6,78 (2H, д), 7 (4H, м), 7,55 (2H, дд).
2-s-(2-тиенилсульфониламино)-3-/4-(пиперидин-4-ил)бутилоксифенил/пропионовой кислоты гидрохлорид (1 - 19).
Обработкой соединения 1 - 18 (0,22 г, 0,38 ммоля) LiOH (0,045 г, 1,89 ммоля) по методике синтеза соединения 1 - 8 получают целевую кислоту, которую очищают вытеснительной хроматографией на силикагеле с элюированием смесью CHCl3-CH3OH-HOAc (97:3:1).
1H-ЯМР (300 МГц, CD3OD) δ : 1,05 (2H, дт), 1,2 - 1,4 (5H, м), 1,4 - 1,6 (12H, м), 1,65 - 1,8 (5H, м), 2,65 - 1,82 (4H, м), 2,98 (1H, дд), 3,3 (1H, м), 3,92 (2H, т), 4 - 4,13 (5H, м), 6,75 (2H, д), 7,02 (3H, м), 7,39 (1H, д), 7,67 (1H, д).
Обработкой полученной кислоты по методике синтеза соединения 1 - 9 газообразным HCl получают соединение 1 - 19 в виде белого твердого вещества после переосаждения.
Анализ для C22H30N2O5S2 •HCl•0,5H2O.
Вычислено: C 51,60; H 6,3; N 5,47.
Найдено: C 51,57; H 6,2; N 5,51.
1H-ЯМР (300 МГц, CD3OD) δ : 1,29 - 1,45 (4H, м), 1,47 - 1,7 (3H, м), 1,71 - 1,83 (2H, м), 1,92 - 2 (2H, ш.д.), 2,79 (1H, м), 2,9 - 3,04 (3H, м), 3,95 (2H, т), 4,04 (1H, м), 6,76 (2H, д), 7,05 (3H, м), 7,4 (1H, м), 7,79 (1H, м).
δ
2-s-(данзиламино)-3-[4-(N-трет-бутилоксикарбонилпиперидин-4-ил)бутилоксифенил]пропионат (1 - 20).
Соединение 1 - 4 (0,304 г, 0,7 ммоля) обрабатывают по методике синтеза соединения 1 - 8 данзилхлоридом (0,208 г, 0,77 ммоля) с получением сырого продукта, очисткой которого вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (75:25) получают чистое соединение 1 - 20).
Rf = 0,25 (силикагель, элюирование смесью гексан-EtOAc (75:25)).
1H-ЯМР (300 МГц, CDCl3) 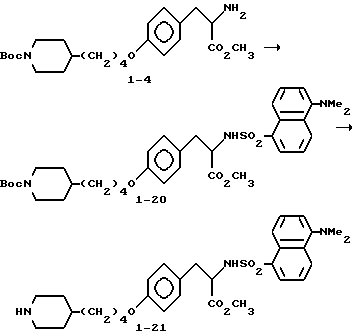 : 1,1 (2H, М), 1,21 - 1,38 (6H, м), 1,4 - 1,53 (11H, м), 1,6 - 1,8 (6H, м), 2,68 (2H, ш.т.), 2,89 (6H, с), 3,33 (2H, с), 3,89 (2H, т), 4,05 - 4,19 (4H, м), 5,24 (1H, м), 6,62 (2H, д). 6,82 (2H, д), 7,18 (1H, д), 7,5 (2H, м), 8,19 (2H, т), 8,51 (1H, д).
: 1,1 (2H, М), 1,21 - 1,38 (6H, м), 1,4 - 1,53 (11H, м), 1,6 - 1,8 (6H, м), 2,68 (2H, ш.т.), 2,89 (6H, с), 3,33 (2H, с), 3,89 (2H, т), 4,05 - 4,19 (4H, м), 5,24 (1H, м), 6,62 (2H, д). 6,82 (2H, д), 7,18 (1H, д), 7,5 (2H, м), 8,19 (2H, т), 8,51 (1H, д).
2-s-(данзиламино)-3/4-(пиперидин-4-ил)бутилоксифенил/пропионовой кислоты гидрохлорид (1 - 21).
Обработкой соединения 1 - 20 (0,275 г, 0,412 ммоля) LiOH по методике синтеза соединения 1 - 8 получают целевую кислоту в виде высокофлуоресцентного вязкого остатка.
1H-ЯМР (300 МГц, CD3OD) δ : 1,09 (2H, м), 1,22 - 1,4 (3H, м), 1,4 - 1,57 (12H, м), 1,65 - 1,8 (3H, м), 2,6 - 2,8 (3H, м), 2,9 (6H, с), 3,31 (3H, м), 3,8 (2H, т), 3,9 (1H, м), 4,01 - 4,15 (4H, м), 6,47 (2H, д), 7,21 (1H, д), 7,42 (2H, м), 7,98 (1H, д), 8,2 (2H, д), 8,46 (1H, д).
Обработкой полученной кислоты по методике синтеза соединения 1 - 9 газообразным HCl получают соединение 1 - 21 в виде белого твердого вещества после переосаждения из этилацетата.
Анализ для C30H39N3O5S•1,8HCl •H2O.
Вычислено: C 56,53; H 6,77; N 6,59; C1 10,01.
Найдено: C 56,48; H 6,66; N 6,36; C1 10,21.
1H-ЯМР (300 МГц, CD3OD) δ : 1,3 - 1,51 (7H, м), 1,52 - 1,8 (4H, м), 1,95 (2H, ш. т. ), 2,65 (1H, м), 2,95 (3H, м), 3,3 - 3,4 (4H, м), 3,45 (6H, с), 3,84 - 3,97 (3H, м), 6,45 (2H, д), 6,77 (2H, д), 7,71 (2H, м), 8 (1H, д), 8,16 (2H, д), 8,55 (1H, д), 8,7 (1H, д).
Схема 2 приведена в конце описания.
Пример 8.
δ
2-s-(бензилоксикарбониламино)-3-/4-(6-N-трет- бутилоксикарбониламиногексилокси)фенил/пропионовая кислота (2 - 1).
В 75 мл ДМФА растворяют N-CBZ-L-тирозин (15 г, 0,045 моля) и полученный раствор прибавляют при 0 - 10oC к суспензии гидрида натрия (2,16 г, 0,09 моля) в 25 мл ДМФА. Образовавшуюся суспензию перемешивают 1 ч при 0 - 10oC, после чего по каплям при 0 - 5oC прибавляют 6-(трет-бутилоксикарбониламино)гексилбромид (12,6 г, 0,045 моля) и прозрачную темную реакционную смесь перемешивают в течение ночи при комнатной температуре.
После удаления растворителя остаток переносят в EtOAc и подкисляют 10%-ным раствором KHSO4. Органическую фазу отделяют, промывают рассолом, сушат (Na2SO4) и после испарения растворителя получают масло. Очисткой масла колоночной хроматографией на силикагеле с элюированием смесью CHCl3-CH3OH-HOAc (98:2:1) получают в виде прозрачного масла соединение 2 - 1.
1H-ЯМР (300 МГц, CD3OD) 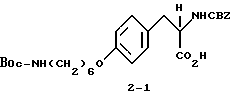 : 1,45 (15H, м), 1,75 (2H, м), 2,8 - 3,15 (6H, м), 3,91 (2H, т), 4,38 (1H, м), 4,95 (6H, м), 6,85 (2H, д), 7,06 (2H, д).
: 1,45 (15H, м), 1,75 (2H, м), 2,8 - 3,15 (6H, м), 3,91 (2H, т), 4,38 (1H, м), 4,95 (6H, м), 6,85 (2H, д), 7,06 (2H, д).
Пример 9.
δ
Метиловый эфир 2-s-(бензилоксикарбониламино)-3-/4-(6-N-трет-бутилоксикарбониламиногексилокси)фенил/пропионовой кислоты (2-2).
Соединение 2-1 (10 г, 19,43 ммоля) перемешивают 1,9 ч при комнатной температуре с карбонатом цезия (3,16 г, 9,72 ммоля). Затем по каплям добавляют йодистый метил (2,76 г, 19,43 ммоля) и реакционную смесь перемешивают в течение ночи при комнатной температуре. Растворитель удаляют в высоком вакууме (30oC), остаток переносят в 300 мл EtOAc, промывают порциями 2 • 40 мл насыщенного раствора NaHCO3, рассолом и сушат (Na2SO4). Удалением растворителя получают соединение 2-2 (8,5 г, 83%) в виде прозрачного масла.
1H-ЯМР (300 МГц, CDCl3) 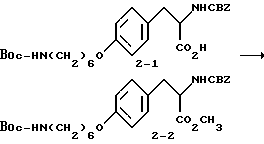 : 1,25-1,53 (16H, м), 1,76 (2H, м), 2,96-3,17 (4H, м), 3,71 (3H, с), 3,9 (2H, мт), 4,61 (1H, м), 5,1 (2H, м), 5,19 (1H, м), 6,88 (2H, д), 6,98 (2H, д), 7,32 (5H, м).
: 1,25-1,53 (16H, м), 1,76 (2H, м), 2,96-3,17 (4H, м), 3,71 (3H, с), 3,9 (2H, мт), 4,61 (1H, м), 5,1 (2H, м), 5,19 (1H, м), 6,88 (2H, д), 6,98 (2H, д), 7,32 (5H, м).
Пример 10.
δ
Метиловый эфир 2-s-амино-3-/4-(6-N-трет- бутилоксикарбониламиногексилокси)фенил/пропионовой кислоты (2-3).
Соединение 2-2 (8 г, 15,1 ммоля) растворяют в 150 мл абсолютного этанола и к раствору добавляют 1 г 10% Pd-C. Полученную суспензию гидрируют 3,5 ч в аппарате Парра под давлением 50 фунт/кв.дюйм (3,5 ат). Затем катализатор отфильтровывают и удалением растворителя в роторном испарителе получают в виде прозрачного масла чистое соединение 2-3 (5,56 г).
Rf = 0,4 на SiO2 • CHCl3 - CH3OH (95:5).
1H-ЯМР (300 МГц, CDCl3) 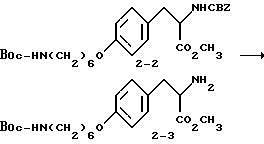 : 1,3-1,55 (16H, м), 1,7 (2H, м), 2,8 (1H, м), 3-3,17 (3H, м), 3,71 (3H, с), 3,93 (2H, т), 6,82 (2H, т), 7,09 (2H, д).
: 1,3-1,55 (16H, м), 1,7 (2H, м), 2,8 (1H, м), 3-3,17 (3H, м), 3,71 (3H, с), 3,93 (2H, т), 6,82 (2H, т), 7,09 (2H, д).
Пример 11.
δ
2-s-(метилсульфониламино)-3-/4-(6-N-трет-бутилоксикарбонил-аминогексилокси)фенил/пропионовая кислота (2-4).
Соединение 2-3 (0,4 г, 1,01 ммоля) обрабатывают метансульфонилхлоридом (0,116 г, 1,01 ммоля) и NaHCO3 (0,25 г, 3 ммоля) по методике получения соединения 1-8. Очисткой сырого продукта реакции вытеснительной хроматографией на силикагеле с элюированием 30% EtOAc в гексане получают в виде прозрачного масла чистое соединение 2-4 (0,1 г).
1H-ЯМР (300 МГц, CDCl3) 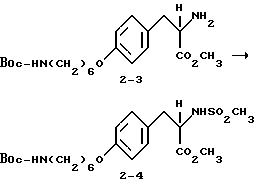 : 1,36-1,56 (15H, м), 1,77 (2H, м), 2,7 (3H, с), 3,78 (3H, с), 3,92 (2H, т), 4,36 (1H, м), 4,9 (1H, д), 6,82 (2H, д), 7,09 (2H, д).
: 1,36-1,56 (15H, м), 1,77 (2H, м), 2,7 (3H, с), 3,78 (3H, с), 3,92 (2H, т), 4,36 (1H, м), 4,9 (1H, д), 6,82 (2H, д), 7,09 (2H, д).
Пример 12.
δ
2-s-(метилсульфониламино)-3-/4-(6-аминогексилокси)фенил/-пропионовой кислоты гидрохлорид (2-5).
Соединение 2-4 (0,1 г, 0,212 ммоля) обрабатывают по методике синтеза соединения 1-8 LiOH (0,026 г, 1,06 ммоля) и получают в виде вязкого масла 2-s-(метилсульфониламино)-3-/4-(6-N-трет-бутилоксикарбониламиногексилокси)фенил/пропиновую кислоту (0,125 г).
1H-ЯМР (300 МГц, CD3OD)  : 1,3-1,55 (16H, м), 1,75 (2H, м), 2,63 (3H, с), 2,85 (1H, дд), 3-3,13 (3H, м), 3,93 (2H, т), 4,17 (1H, м), 6,83 (2H, д), 7,2 (2H, д).
: 1,3-1,55 (16H, м), 1,75 (2H, м), 2,63 (3H, с), 2,85 (1H, дд), 3-3,13 (3H, м), 3,93 (2H, т), 4,17 (1H, м), 6,83 (2H, д), 7,2 (2H, д).
Полученную кислоту растворяют в EtOAc (20 мл) и обрабатывают по методике получения соединения 1-9 газообразным HCl. Удалением растворителя получают остаток, переосаждением которого из 30 мл Et2O получают чистое соединение 2-5 (0,09 г) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD) δ : 1,4-1,6 (4H, м), 1,6 (2H, м), 1,69 (2H, м), 2,68 (3H, с), 2,82 (1H, дд), 2,92 (2H, т), 3,1 (1H, дд), 3,3 (2H, м), 3,97 (2H, т), 4,18 (1H, м), 6,83 (2H, д), 7,19 (2H, д).
Анализ для C16H26N2O5S • HCl • 0,25 H2O.
Вычислено: C 48,11; H 6,94; N 7,01.
Найдено: C 48,16; H 6,82; N 6,98.
Пример 13.
δ
Метиловый эфир 2-s-(бутилсульфониламино)-3-4-(6-N-трет- бутилоксикарбониламиногексилокси)фенил/пропионовой кислоты (2-6).
Соединение 2-3 (0,4 г, 1,01 ммоля) обрабатывают бутилсульфонилхлоридом (0,47 г, 3,03 ммоля) и NaHCO3 (0,5 г, 6 ммолей) по методике получения соединения 1-8. Очисткой сырого продукта реакции вытеснительной хроматографией на силикагеле с элюированием 30% EtOAc в гексане получают в виде прозрачного масла чистое соединение 2-6 (0,22 г).
1H-ЯМР (300 МГц, CDCl3) 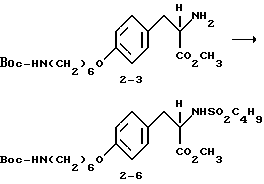 : 0,87 (3H, т), 1,35-1,54 (18H, м), 1,61 (2H, м), 1,77 (2H, м), 2,74 (2H, т), 2,95 (1H, дд), 3,05-3,18 (3H, м), 3,9 (2H, т), 4,32 (1H, м), 4,72 (1H, м), 6,82 (2H, д), 7,07 (2H, д).
: 0,87 (3H, т), 1,35-1,54 (18H, м), 1,61 (2H, м), 1,77 (2H, м), 2,74 (2H, т), 2,95 (1H, дд), 3,05-3,18 (3H, м), 3,9 (2H, т), 4,32 (1H, м), 4,72 (1H, м), 6,82 (2H, д), 7,07 (2H, д).
Пример 14.
δ
2-s-(бутилсульфониламино)-3-/4-(6-аминогексилокси)фенил/-пропионовой кислоты гидрохлорид (2-7).
Соединение 2-6 (0,2 г, 0,39 ммоля) в смеси ТГФ-H2O-CH3OH (1:1:1) обрабатывают LiOH (0,05 г, 2,12 ммоля) по методике синтеза 1-8 и получают 2-s-(бутилсульфониламино-3-/4-(6-N-трет- бутилоксикарбониламиногексилокси)фенил/пропионовую кислоту (0,235 г) в виде вязкого масла.
1H-ЯМР (300 МГц, CD3OD) 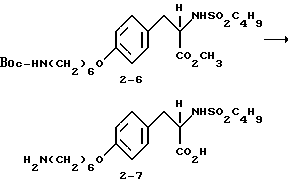 : 0,83 (3H, т), 1,35-1,56 (16H, м), 1,76 (2H, м), 2,61 (2H, т), 2,79 (1H, ддд), 3-3,14 (3H, м), 3,92 (2H, т), 4,11 (1H, м), 6,82 (2H, д), 7,18 (2H, д).
: 0,83 (3H, т), 1,35-1,56 (16H, м), 1,76 (2H, м), 2,61 (2H, т), 2,79 (1H, ддд), 3-3,14 (3H, м), 3,92 (2H, т), 4,11 (1H, м), 6,82 (2H, д), 7,18 (2H, д).
Полученную кислоту (0,235 г, 0,7 ммоля) растворяют в EtOAc (30 мл) и обрабатывают газообразным HCl по методике получения соединения 1-9. Переосаждением остатка из смеси эфира (40 мл) и EtOAc (10 мл) получают соединение 1-7 (0,17 г) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD) δ : 0,85 (3H, т), 1,24 (2H, м), 1,35-1,6 (6H, м), 1,7 (2H, м), 1,8 (2H, м), 2,66 (2H, т), 2,78 (1H, дд), 2,92 (2H, т), 3,1 (1H, дд), 3,3 (1H, м), 6,85 (2H, д), 7,2 (2H, д).
Анализ для C19H32N2O5S • HCl.
Вычислено: C 52,22; H 7,61; N 6,41.
Найдено: C 51,80; H 7,61; N 6,33.
Пример 14A.
δ
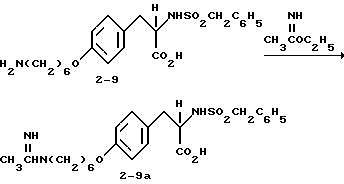
2-s-(бутилсульфониламино)-3-/4-(6-ацетамидиногексилокси)-фенил/пропионовая кислота (2-7a).
Раствор соединения 2-7 (1 г, 2,29 ммоля) в ТГФ (30 мл) обрабатывают этилацетимидатом (0,2 г, 2,29 ммоля) и полученную реакционную смесь перемешивают 16 ч при комнатной температуре. Растворитель затем удаляют и перекристаллизацией из этилацетата получают чистое соединение 2-7a.
Пример 14B.
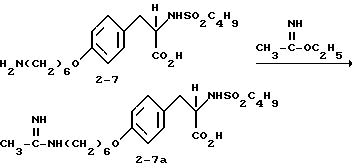
2-s-(бутилсульфониламино)-3-/4-(6-бензамидиногексилокси)-фенил/пропионовая кислота (2-7b).
Раствор соединения 2-7 (1 г, 2,29 ммоля) в ТГФ (30 мл) обрабатывают этилбензимидатом (0,34 г, 2,29 ммоля) и полученный раствор перемешивают 20 ч при комнатной температуре. Растворитель удаляют, остаток переносят в EtOAc, фильтруют и после перекристаллизации получают чистое соединение 2-7b.
Пример 14C.
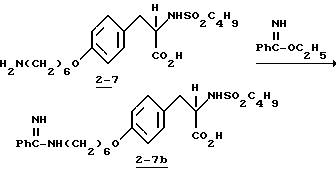
2-s-(бутилсульфониламино)-3-/4-(6-гуанидиногексилокси) фенил/пропионовая кислота (2-7c).
Смесь соединения 2-7 (1 г, 2,29 ммоля) и N-нитрозометилтиогуанидина (0,32 г, 2,29 ммоля) нагревают 5 мин при 40oC в абсолютном EtOH (15 мл) и затем оставляют на 1 день при комнатной температуре. Растворитель удаляют в вакууме и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью CHCl3CH3OH-HOAc (95:5:2) получают целевое нитрогуанидинопромежуточное соединение.
Полученное промежуточное соединение растворяют в 10% HCl-CH3OH (20 мл) и встряхивают 8 ч при комнатной температуре в аппарате Парра под давлением 50 фунт/кв.дюйм (3,5 ат) в присутствии 10% Pd-C (100 мг). Катализатор отфильтровывают, растворитель удаляют в вакууме, остаток растворяют в 10%-ной водной соляной кислоте и кипятят 2 ч. Растворитель удаляют в вакууме и очисткой остатка на колонке Дауэкс 1-X2 с элюированием водой получают чистое соединение 2-7c.
Пример 15.
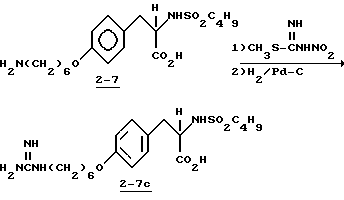
Метиловый эфир 2-s-(бензилсульфониламино)-3-/4-(6-N-трет-бутилоксикарбониламиногексилокси)фенил/пропионовой кислоты (2-8).
Соединение 2-3 (0,29 г, 0,735 ммоля) обрабатывают бензилсульфонилхлоридом (0,14 г, 0,735 ммоля) и NaHCO3 (0,185 г, 2,2 ммоля) по методике получения соединения 1-8. Очисткой сырого продукта вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (1:1) получают в виде прозрачного масла соединение 2-8 (0,27 г).
1H-ЯМР (300 МГц, CDCl3)  : 1,47-1,69 (15H, м), 1,9 (2H, м), 2,18 (2H, с), 3,08 (2H, д), 3,25 (2H, м), 3,85 (3H, с), 4,05 (2H, т), 4,19-4,2 (4H, м), 4,8 (1H, д), 6,83 (2H, д), 7,12 (2H, д), 7,47 (5H, м).
: 1,47-1,69 (15H, м), 1,9 (2H, м), 2,18 (2H, с), 3,08 (2H, д), 3,25 (2H, м), 3,85 (3H, с), 4,05 (2H, т), 4,19-4,2 (4H, м), 4,8 (1H, д), 6,83 (2H, д), 7,12 (2H, д), 7,47 (5H, м).
Пример 16.
δ
2-s-(бензилсульфониламино)-3-/4-(6-аминогексилокси)фенил/-пропионовой кислоты гидрохлорид (2-9).
Соединение 2-8 (0,48 г, 0,875 ммоля) обрабатывают LiOH (0,105 г, 4,37 ммоля) по методике синтеза соединения 1-8 и получают в виде пены 2-s-(бензилсульфониламино)-3-/4-(6-N-трет-бутилоксикарбониламиногексилокси)фенил/пропионовую кислоту (0,4 г).
1H-ЯМР (300 МГц, CD3OD) 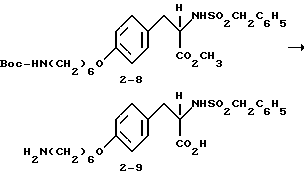 : 1,3-1,52 (15H, м), 1,72 (2H, м), 2,81 (1H, дд), 3 (3H, м), 3,93 (2H, м), 4,04 (2H, м), 6,81 (2H, д), 7,13 (2H, д), 7,2-7,32 (5H, м).
: 1,3-1,52 (15H, м), 1,72 (2H, м), 2,81 (1H, дд), 3 (3H, м), 3,93 (2H, м), 4,04 (2H, м), 6,81 (2H, д), 7,13 (2H, д), 7,2-7,32 (5H, м).
Полученную кислоту (0,4 г, 0,75 ммоля) растворяют в EtOAc (30 мл) и обрабатывают по методике получения соединения 1-9 газообразным HCl. Переосаждением сырого продукта реакции из эфира получают чистое соединение 2-9 (0,35 г) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD) δ : 1,38-1,57 (4H, м), 1,65 (2H, м), 1,73 (2H, м), 2,71 (1H, дд), 2,89 (2H, т), 3,02 (1H, дд), 3,3 (3H, м), 3,94-4,15 (5H, м), 6,83 (2H, д), 7,15 (2H, д), 7,29 (5H, м).
Пример 16A.
δ
2-s-(бензилсульфониламино)-3-/4-(6-ацетамидиногексилокси)-фенил/пропионовая кислота (2-9a).
Раствор соединения 2-9 (1 г, 2,1 ммоля) в ТГФ (30 мл) обрабатывают этилацетимидатом (0,18 г, 2,1 ммоля) по методике примера 14A и получают после перекристаллизации из этилацетата чистое соединение 2-9a.
Пример 16B.
2-s-(бензилсульфониламино)-3-/4-(6-гуанидиногексилокси) фенил/пропионовая кислота (2-9b).
Смесь соединения 2-9 (1 г, 2,1 ммоля) и N-нитрозометилтиогуанидино (0,29 г, 2,1 ммоля) обрабатывают по методике примера 14C и получают соединение 2-9b.
Схема 3 приведена в конце описания.
Метиловый эфир 2-s-амино-3-/4-(4-гидроксифенил)оксифенил/-пропионовой кислоты (3-2).
Метанол (100 мл) охлаждают до 0oC и обрабатывают 15 мин при 0oC и перемешивании SOCl2 (47 ммолей), затем добавляют соединение 3-1 (1,5 г, 5,49 ммоля) и перемешивание продолжают 16 ч с повышением температуры до комнатной.
Реакционную смесь фильтруют и удалением растворителя получают масло, из которого промыванием эфиром получают соединение 3-2 (1,57 г).
1H-ЯМР (300 МГц, CD3OD)  : 3,1-3,3 (2H, м), 3,81 (3H, с), 6,76-6,90 (6H, м), 7,2 (2H, д).
: 3,1-3,3 (2H, м), 3,81 (3H, с), 6,76-6,90 (6H, м), 7,2 (2H, д).
Метиловый эфир 2-s-(N-бензилоксикарбониламино)-3-/4-(4-гидроксифенил)оксифенил/пропионовой кислоты (3-3).
Раствор соединения 3-2 (0,2 г, 0,62 ммоля) в смеси воды с диоксаном (1: 1) охлаждают до 0oC и обрабатывают Na2CO3 (0,131 г, 1,23 ммоля) и бензилхлорформатом (0,619 ммоля). Спустя 1,5 интенсивного перемешивания, диоксан удаляют при пониженном давлении, остаток разбавляют H2O и экстрагируют EtOAc. Органический экстракт промывают рассолом, сушат (Na2SO4)и удалением растворителя получают в виде масла соединение 3-3.
1H-ЯМР (300 МГц, CDCl3) δ : 3,06 (2H, м), 3,75 (3H, с), 4,64 (1H, м), 5,1 (2H, м), 5,36 (1H, м), 6,83 (6H, м), 7 (2H, д), 7,37 (5H, ш.с).
Метиловый эфир 2-s-(N-бензилоксикарбониламино)-3-/4-(4-N-трет-бутилоксикарбонилпиперидин-4-ил)оксифенилокси/ фенилпропионовой кислоты (3-4).
Раствор соединения 3-3 (0,5 г, 1,18 ммоля) в бензоле (40 мл) обрабатывают при комнатной температуре, перемешивании и постоянном пропускании N2N-трет-бутилоксикарбонилпиперидин-1-олом (0,24 г, 1,18 ммоля) и Ph3P (0,31 г, 1,18 ммоля). Добавляют диэтилазодикарбоксилат (1,18 ммоля) и полученный раствор перемешивают 16 ч при комнатной температуре.
Затем растворитель удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (70:30) получают чистое соединение 3-4.
1H-ЯМР (300 МГц, CDCl3) δ : 1,48 (9H, с), 1,8 (2H, м), 1,95 (2H, м), 3,08 (2H, м), 3,36 (2H, м), 3,76 (3H, с), 4,4 (1H, м), 4,63 (1H, м), 5,1 (1H, м), 5,25 (1H, м), 6,8-7,04 (8H, м), 7,36 (5H, ш.с).
Метиловый эфир 2-s-(бутилсульфониламино)-3-/4-(4-N-трет-бутилоксикарбонилпиперидин-4-ил)оксифенилокси/фенилпропионовой кислоты (3-5).
К раствору соединения 3-4 (0,5 г, 0,082 ммоля) в EtOH (40 мл) добавляют 10% Pd-C (125 мг) и полученную суспензию гидрируют 1,5 ч в колбе Парра под давлением 50 фунт/кв.дюйм (3,5 ат). Катализатор отфильтровывают и отгонкой растворителя получают в виде прозрачного масла целевой аминоэфир.
1H-ЯМР (300 МГц, CDCl3) δ : 1,48 (9H, с), 1,5-1,8 (8H, м), 1,91 (2H, м), 2,82 (1H, м), 3,04 (1H, м), 3,34 (2H, м), 3,76 (3H, с), 4,2 (1H, м), 7,9 (8H, м), 8,11 (2H, д).
Полученный аминоэфир (0,36 г, 0,77 ммоля) растворяют в EtAOc (10 мл), добавляют NaHCO3 (0,386 г, 4,6 ммоля) и н-бутилсульфонилхлорид (1,53 ммоля) и кипятят 48 ч. Растворитель удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (65:35) получают в виде масла чистое соединение 3-5.
1H-ЯМР (300 МГц, CDCl3) δ : 0,88-1,02 (4H, м), 1,25-1,45 (3H, м), 1,5 (9H, с), 1,51-1,8 (2H, м), 1,93 (2H, м), 2,8 (2H, м), 2,95-3,2 (2H, м), 3,21-3,4 (2H, м), 3,72 (2H, м), 3,74 (3H, с), 4,38 (2H, м), 4,8 (1H, д), 6,9 (6H, м), 7,1-7,27 (2H, м).
2-s-(бутилсульфониламино)-3-/4-(пиперидин-4-ил)оксифенилокси/пропионовой кислоты гидрохлорид (3-6).
Раствор соединения 3-5 (0,2 г, 0,34 ммоля) в смеси ТГФ-H2O-CH3OH (1:1:1) обрабатывают 8 ч при комнатной температуре LiOH (0,075 г, 1,78 ммоля). Растворитель удаляют, остаток подкисляют 10%-ным раствором KHSO4 и несколько раз экстрагируют EtOAc. Органические экстракты объединяют, промывают рассолом, сушат (Na2SO4) и удалением растворителя получают целевую кислоту.
Rf = 0,3 (силикагель, CHCl3-CH3OH-HOAc (97:3:1).
1H-ЯМР (300 МГц, CDCl3) δ : 0,85 (3H, т), 1,2-1,3 (3H, м), 1,46 (9H, с), 1,5-2 (6H, м), 2,75 (2H, м), 2,97 (1H, м), 3,18 (1H, м), 3,33 (2H, м), 3,76 (2H, м), 4,35 (2H, м), 5,07 (1H, м), 6,89 (6H, м), 7,13 (2H, м).
Полученную кислоту (0,15 г, 0,26 ммоля) растворяют в EtOAc и обрабатывают газообразным HCl по методике синтеза соединения 1-9 с получением чистого соединения 3-6 в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD) δ : 0,89 (3H, т), 1,32 (2H, м), 1,53 (2H, м), 1,97-2,21 (4H, м), 2,75 (2H, м), 2,63 (1H, м), 3,2 (3H, м), 3,4 (2H, м), 4,14 (1H, м), 6,82-7,05 (6H, м), 7,23 (2H, м).
Схема 4 приведена в конце описания.
4-/4-(N-бензилоксикарбонилпиперидин-4-ил)-2-метил/пентан-2-ол (4-2).
Метиловый эфир 4-(N-бензилоксикарбонилпиперидин-4-ил)-бутановой кислоты (4-1) (10,07 г, 0,032 моля) в ТГФ (200 мл) охлаждают до 0oC и обрабатывают 3 ч CH3MgI (0,095 моля). Реакционную смесь переносят на лед, подкисляют 10% KHSO4 и экстрагируют 3 раза EtOAc. Объединенные органические экстракты промывают рассолом, сушат (MgSO4) и растворитель удаляют. Очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (7:3) получают чистое соединение 4-2.
Rf = 0,3 (силикагель, гексан-EtOAc (7:3)).
Метиловый эфир 2-s-(бутилсульфониламино)-3-/4-(N- бензилоксикарбонилпиперидин-4-ил)-2,2- диметил/бутилоксифенилпропионовой кислоты (4-3).
Метиловый эфир N-н-бутилсульфонил-L-тирозина (7,21 г, 0,023 моля) растворяют в смеси соединения 4-2 (1 г), CH2Cl2 (30 мл) и бензола (250 мл). Добавляют трифенилфосфин (5,97 г, 0,023 моля) и после пропускания N2 прибавляют диэтилазодикарбоксилат (3,6 мл, 0,023 моля), при этом реакционная смесь приобретает красно-оранжевую окраску. Реакционную смесь перемешивают 7 дней при комнатной температуре. Растворитель удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (60:40) получают чистое соединение (4-3).
1H-ЯМР (300 МГц, CDCl3) δ : 0,88 (6H, т), 1,1-1,4 (12H, м), 1,43-1,78 (8H, м), 2,7-2,82 (4H, м), 2,95-3,1 (3H, м), 3,75 (3H, с), 4,18 (2H, м), 4,32 (1H, м), 5,13 (2H, с), 6,88 (2H, д), 7,06 (2H, д), 7,38 (5H, м).
2-s-(бутилсульфониламино)-3-/4-(N-бензилоксикарбонилпиперидин-4-ил)-2,2-диметил/бутилоксифенилпропионовая кислота (4-4).
Соединение 4-3 (0,64 г, 0,001 моля) растворяют в смеси ТГФ-H2O-CH3OH и обрабатывают LiOH (0,26 г, 0,0062 моля) 8 ч при комнатной температуре. Удалением растворителя, подкислением раствором KHSO4 и экстрагированием EtOAc получают сырое соединение 4-4, очисткой которого вытеснительной хромотографией на силикагеле с элюированием смесью CHCl3-CH3OH-HOAc (97:3:1) получают чистое соединение 4-4.
1H-ЯМР (300 МГц, CDCl3) δ : 0,86 (6H, с), 1,05-1,5 (13H, м), 1,55-1,8 (5H, м), 2,77 (4H, м), 3,04 (2H, м), 4,1 (2H, ш.д), 4,17 (1H, м), 4,85 (1H, д), 5,14 (2H, с), 6,88 (2H, д), 7,13 (2H, д), 7,39 (5H, м).
2-s-(бутилсульфониламино)-3-/4-(пиперидин-4-ил)-2,2- диметил/бутилоксифенилпропионовая кислота (4-5).
К формату аммония (0,23 г, 3,65 ммоля) в CH3OH (5 мл) добавляют соединение 4-4 (0,22 г, 3,65 ммоля) в 10 мл CH3OH и затем при комнатной температуре добавляют 10% Pd-C (100 мг). Через 15 мин реакционную смесь пропускают через слой Солка Флок и растворитель удаляют. Очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью EtOH-H2O-NH4OH (9:1:1) получают чистое соединение 4-5.
1H-ЯМР (300 МГц, CD3OD) δ : 0,88 (6H, с), 1,15-1,4 (12H, м), 1,42-1,7 (7H, м), 1,9 (2H, д), 2,78-3 (6H, м), 3,06 (1H, дд), 3,35 (3H, м), 3,93 (1H, м), 6,86 (2H, д), 7,2 (2H, д).
δ
Схема 5 приведена в конце описания.
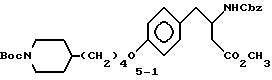
Метиловый эфир 3-s-(бензилоксикарбониламино)-4-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил)бутилоксифенил/масляной кислоты (5-1).
Раствор соединения 1-2 (1 г, 1,8 ммоля) и N-метилморфолина (0,21 мл, 1,9 ммоля) в EtOAc (10 мл) перемешивают при 15oC и обрабатывают изобутилхлорформатом. Спустя 15 мин, гетерогенную смесь обрабатывают добавлением порциями эфирного раствора диазометана (0,5 г, 10 мл, 5 ммолей) с последующим непрерывным перемешиванием 1 ч при 0oC. Затем для удаления избытка диазометана реакционную смесь 10 мин продувают аргоном. Органическую фазу промывают порциями 2 • 5 мл H2O, рассолом, сушат (MgSO4) и испаряют. Остаток растворяют в CH3OH (15 мл) и последовательно добавляют триэтиламин (0,7 мл, 5 ммолей) и AgO2CPh (110 мг, 0,5 ммоля) при перемешивании и комнатной температуре с интенсивным выделением газа. Через 30 мин растворитель испаряют и последующей очисткой сырого продукта реакции вытеснительной хроматографией на силикагеле с элюированием смесью гексана с EtOAc (4:1) получают в виде масла соединение 5-1 (0,52 г).
ТСХ Rf = 0,23 (30% EtOAc в гексане).
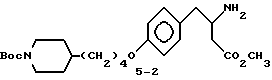
Метиловый эфир 3-s-амино-4-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил)бутилоксифенил/масляной кислоты (5-2).
К раствору соединения 5-1 (0,52 г, 0,9 ммоля) в абсолютном этаноле (20 мл) добавляют 10% Pd-C (0,25 г) и полученную суспензию гидрируют 12 ч под давлением водорода из баллона. Затем катализатор отфильтровывают и удалением растворителя в вакууме получают в виде масла соединение 5-2 (0,35 г).
ТСХ Rf = 0,15 (CH2Cl2-CH3OH-AcOH 9:1:1).
Метиловый эфир 3-s-(бутилсульфониламино)-4-/4-N-трет- бутилоксикарбонилпиперидин-4-ил)бутилоксифенил/масляной кислоты (5-3).
К соединению 5-2 (0,36 г, 0,8 ммоля), триэтиламину (170 мкл, 1,2 ммоля), 4-диметиламинопиридину (12 мг, 0,1 ммоля) и ТГФ (5 мл) при 0oC и перемешивании прибавляют н-бутилсульфонилхлорид (130 мкл, 1 ммоль). Охлаждающую баню удаляют и перемешивание продолжают 6 ч. Реакционную смесь разбавляют 10 мл EtOAc, промывают водой (2 • 5 мл) и рассолом, сушат (MgSO4) и концентрируют. Очисткой сырого продукта реакции вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (4:1) получают в виде масла соединение 5-3 (180 мг).
1H-ЯМР (300 МГц, CDCl3) 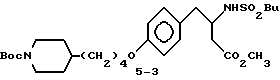 : 1,12 (2H, м), 1,25-1,83 (13H, м), 1,29 (3H, т), 1,47 (9H, с), 2,68 (6H, м), 2,87 (2H, д), 3,73 (3H, с), 3,93 (2H, т), 4,08 (1H, м), 4,72 (1H, д), 6,87 (2H, д), 7,12 (2H, д).
: 1,12 (2H, м), 1,25-1,83 (13H, м), 1,29 (3H, т), 1,47 (9H, с), 2,68 (6H, м), 2,87 (2H, д), 3,73 (3H, с), 3,93 (2H, т), 4,08 (1H, м), 4,72 (1H, д), 6,87 (2H, д), 7,12 (2H, д).
δ
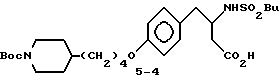
3-s-(бутилсульфониламино)-4-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил)бутилоксифенил/масляная кислота (5-4).
К соединению 5-3 (175 мг, 0,33 ммоля) в CH3OH (4 мл) добавляют 1 н. NaOH (1 мл, 1 ммоль) с последующим непрерывным перемешивании 20 ч при комнатной температуре. Реакционную смесь разбавляют 15 мл EtOAc, промывают 10 мл 5% KHSO4 и рассолом, сушат (MgSO4) и концентрированием получают в виде масла соединение 5-4 (160 мг).
ТСХ Rf = 0,31 (CH2Cl2 - CH3OH - AcOH 9:0,5:0,5).
3-s-(бутилсульфониламино)-4-/4-(пиперидин-4-ил)бутилоксифенил/масляная кислота (5-5).
К перемешиваемому раствору соединения 5-4 (160 мг, 0,3 ммоля), CH2Cl2 (2 мл) и анизола (100 мкл) прибавляют при 0oC CF3CO2H (1 мл). После выдерживания 1,5 ч при 0oC растворители испаряют и очисткой сырого продукта реакции вытеснительной хроматографией с элюированием смесью этанол-H2O-конц. NH4OH (10:0,8:0,8) получают соединение 5-5 (42 мг) в виде твердого вещества.
1H-ЯМР (300 МГц, D2O-CF3CO2D) 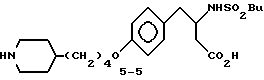 : 0,82 (3H, т), 1,1-1,7 (11H, м), 1,8 (2H, м), 1,98 (2H, м), 2,48 (2H, т), 2,72 (3H, м), 3 (3H, м), 3,43 (2H, м), 3,96 (1H, м), 4,1 (2H, т), 7,01 (2H, д), 7,32 (2H, д).
: 0,82 (3H, т), 1,1-1,7 (11H, м), 1,8 (2H, м), 1,98 (2H, м), 2,48 (2H, т), 2,72 (3H, м), 3 (3H, м), 3,43 (2H, м), 3,96 (1H, м), 4,1 (2H, т), 7,01 (2H, д), 7,32 (2H, д).
Схема 6 приведена в конце описания.
Метиловый эфир 2-s-(N-бензилоксикарбониламино)-3-/4-(3- хлорпропилокси)фенил/пропионовой кислоты (6-1).
Обработкой раствора соединения 1-1 (0,95 г, 2,9 ммоля) и 3-хлор-1-тозилоксипропана (0,84 г, 3,19 ммоля) в ДМФА карбонатом цезия (0,47 г, 1,45 ммоля) получают раствор, который перемешивают в течение ночи при комнатной температуре. Затем реакционную смесь разбавляют H2O и экстрагируют эфиром. Эфирный экстракт промывают рассолом, сушат (Na2SO4) и удалением растворителя получают маслянистый остаток. Очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (95:5) получают соединение 6-1 в виде прозрачного масла.
Rf = 0,5 (силикагель, элюирование смесью гексан-EtOAc 70:30).
Метиловый эфир 2-s-(бензилоксикарбноиламино)-3-/4-(3-йод-пропилокси)фенил/пропионовой кислоты (6-2).
К раствору соединения 6-1 (0,6 г, 1,5 ммоля) в ацетоне добавляют йодистый натрий (1,1 г, 7,5 ммоля) и полученный раствор кипятят 16 ч. Затем реакционную смесь разбавляют эфиром, промывают водой, рассолом и сушат (Na2SO4). Удалением растворителя получают масло, очисткой которого вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (90:1) получают в виде прозрачного масла соединение 6-2.
1H-ЯМР (300 МГц, CDCl3) δ : 1,85-2,08 (4H, м), 3,04 (2H, м), 3,26 (2H, т), 3,71 (3H, с), 3,95 (2H, т), 4,6 (1H, м), 5-5,21 (3H, м), 6,78 (2H, д), 6,99 (2H, д), 7,33 (5H, м).
Метиловый эфир 2-s-(N-бензилоксикарбониламино)-3-/4-(2,6-диметилпиперизин-4-ил)пропилоксифенил/пропионовой кислоты (6-3).
Раствор соединения 6-2 (0,1 г, 0,2 ммоля) и 2,6-диметилпиперазина (0,068 г, 0,6 ммоля) в 1 мл ТГФ перемешивают 20 ч при комнатной температуре. Отгонкой растворителя при низком давлении получают соединение 6-3 в виде прозрачного масла.
1H-ЯМР (300 МГц, CDCl3) δ : 1,45 (4H, д), 1,82 (3H, м), 2,65 (2H, м), 2,79 (2H, м), 3,05 (1H, м), 3,18 (2H, ш.д), 3,6 (1H, м), 3,72 (3H, с), 3,96 (2H, м), 4,62 (1H, м), 5,1 (2H, с), 5,21 (1H, м), 6,79 (2H, д), 7 (2H, д), 7,35 (5H, ш.с).
2-(N-бензилоксикарбониламино)-3-/4-(2,6-диметилпиперазин-4-ил)пропилоксифенил/пропионовая кислота (6-4).
К раствору соединения 6-3 (0,09 г, 0,2 ммоля) в метаноле добавляют 1 н. NaOH (0,7 мл) и смесь выдерживают 16 ч при комнатной температуре. Удалением растворителя получают сырую кислоту, которую очищают вытеснительной хроматографией на силикагеле с элюированием смесью изопропанол-NH4OH-H2O (10:1:1) с получением чистого соединения 6-4.
Rf = 0,25.
1H-ЯМР (300 МГц, CD3OD) δ : 1,65-1,85 (4H, м), 2,6-2,7 (2H, м), 2,8-2,95 (6H, м), 3,11 (8H, м), 3,52 (2H, м), 3,65-3,75 (2H, м), 3,82 (2H, т), 4,17 (1H, м), 4,7 (2H, с), 4,85 (2H, м), 6,63 (2H, д), 6,92 (2H, д), 7,1 (5H, ш. с).
Схема 7 приведена в конце описания.
Метиловый эфир 2-s-(N-бензилоксикарбониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил) пропилоксифенил/пропионовой кислоты (7-1).
К раствору соединения 1-1 (4 г, 2,6 ммоля) и 3-(N-Boc-пиперидин-4-ил)пропилйодида (1,1 г, 3,3 ммоля) в 40 мл ДМФА добавляют карбонат цезия (0,4 г, 1,35 ммоля) и полученный раствор перемешивают 20 ч при комнатной температуре. Растворитель удаляют, остаток переносят в EtOAc, промывают водой, рассолом и сушат (Na2SO4). Удалением растворителя получают остаток, очисткой которого вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (4: 1) получают в виде прозрачного масла чистое соединение 7-1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,1 (2H, м), 1,37-1,45 (11H, м), 1,65-1,82 (4H, м), 2,68 (2H, м), 3,03 (2H, м), 3,71 (3H, с), 3,9 (2H, т), 4,08 (2H, ш. д. ), 4,61 (1H, м), 5,1 (1H, с), 5,18 (1H, м), 6,79 (2H, д), 7 (2H, д), 7,35 (5H, ш.с.).
2-(s)-(N-бензилоксикарбониламино)-3-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил)пропилоксифенил/пропионовая кислота (7-2).
К соединению 7-1 (0,5 г, 0,9 ммоля) в метаноле (12 мл) добавляют 1 н. NaOH (3 мл) и смесь выдерживают 16 ч при комнатной температуре. Затем растворитель удаляют и остаток подкисляют 5%-ным раствором KHSO4, несколько раз экстрагируют EtOAc, объединенные органические экстракты промывают рассолом и сушат (Na2SO4). Удалением растворителя получают прозрачное масло.
1H-ЯМР (300 МГц, CDCl3) δ: 1,1 (2H, м), 1,37-1,52 (12H, м), 1,62-1,85 (5H, м), 2,66 (2H, т), 3,1 (2H, м), 4,89 (2H, т), 4,1 (4H, м), 4,62 (1H, м), 5,09 (1H, с), 5,19 (1H, м), 6,79 (2H, д), 7,03 (2Н, д), 7,34 (5H, ш.с.).
Метиловый эфир 3-s-(N-бензилоксикарбониламино)-4-/4-(N-трет-бутилоксикарбонилпиперидин-4-ил)пропилоксифенил)бутановой кислоты (7-3).
К перемешиваемому раствору соединения 7-2 (1,6 г, 2,9 ммоля) в EtOAc добавляют при -15oC изобутилхлорформат (2,9 ммоля) и N-метилморфолин (2,9 ммоля) и полученный раствор перемешивают 0,5 ч при -15oC. Затем добавляют диазометан (5 ммолей в Et2O) и реакционную смесь перемешивают 20 мин при 0oC. Реакционную смесь продувают аргоном, разбавляют EtOAc и промывают водой. Органическую фазу сушат (MgSO4) и удалением растворителя получают целевой диазокетон.
1H-ЯМР (300 МГц, CDCl3) δ: 1,1 (2H, м), 1,35-1,5 (12H, м), 1,55-1,85 (6H, м), 2,68 (2H, ш.т.), 2,95 (2H, д) 3,9 (2H, т), 4,09 (3H, м), 4,42 (1H, м), 5,06 (1H, м), 5,2 (1H, м) 5,35 (1H, м), 6,8 (2H, д), 7,06 (2H, д), 7,35 (5H, ш.с.).
Полученный диазокетон (1,63 г, 2,9 ммоля) растворяют в CH3OH (20 мл) и при комнатной температуре обрабатывают раствором бензоата серебра (0,22 мг, 0,96 ммоля) и триэтиламина (1,25 мл) в CH3OH (5 мл). Спустя несколько минут, реакционная смесь становится черной, происходит выделение газа. Через 0,5 ч растворитель удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью гексан-EtOAc (4:1) получают в виде масла соединение 7-3.
1H-ЯМР (300 МГц, CDCl3) δ: 1,12 (2H, м), 1,37-1,47 (12H, м), 1,6 (2H, с), 1,65-1,83 (4H, м), 2,49 (2H, м), 2,62-2,91(4H, м), 3,67 (3H, с), 3,9 (2H, т), 4,03-4,2 (4H, м), 5,08 (2H, c), 5,24 (1H, м), 6,79 (2H, д), 7,05 (2H, д), 7,32 (5H, ш.с.).
3-s-(N-бензилоксикарбониламино)-4-/4(пиперидин-4-ил)пропилоксифенил/бутановая кислота (7-4).
К раствору соединения 7-3 (0,3 г, 0,53 ммоля) добавляют 1 н. NaOH (1,7 мл) и полученную смесь перемешивают 16 ч при комнатной температуре. Растворитель удаляют, остаток подкисляют 5%-ным водным раствором KHSO4 и экстрагируют несколько раз EtOAс. Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и удалением растворителя получают целевую кислоту.
1H-ЯМР (300 МГц, CD3OD) δ: 1,1 (2H, м), 1,4-1,52 (12H, м), 1,65-1,84 (6H, м), 2,54-2,93 (8H, м), 3,92 (2H, т), 4,05-4,12 (3H, м), 5,1 (2H, с), 6,71 (2H, д), 7,08 (2H, д), 7,35 (5H, м).
Полученную кислоту растворяют в CH2Cl2 (4 мл), к раствору добавляют анизол (0,41 ммоля) и затем при 0oC прибавляют трифторуксусную кислоту (2 мл). После перемешивания 2,5 ч при 0oC растворители удаляют и очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью EtOH-NH4OH-H2O (10:1:1) получают чистое соединение 7-4 в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD) δ: 1,3-1,5 (4H, м), 1,6 (1H, м), 1,75-1,85 (2H, м), 1,95 (2H, д), 2,54 (2H, м), 2,72 (2H, м), 2,93 (2H, т), 3,32 (6H, м), 3,92 (2H, т), 4,11 (1H, м), 4,95 (2H, м), 6,75 (2H, д), 7,05 (2H, д), 7,25 (5H, м).
Схема 8 приведена в конце описания.
δ
Метиловый эфир 2-s-(гексаноиламино)-3-(4-йодфенил)пропионовой кислоты (8-2).
Суспензию соединения 8-1 (1,01 г, 2,96 ммоля) в 20 мл CH2C12 охлаждают до 0oC, добавляют пиридин (1,43 мл, 17,7 ммоля) с последующим прибавлением гексаноилхлорида (1,25 мл, 8,88 ммоля). Спустя 20 мин, наблюдается полное исчезновение соединения 8-1. Осторожно добавляют воду (25 мл) и полученную смесь экстрагируют EtOAc (150 мл). Отделенную органическую фазу промывают 10% KHSO4, рассолом, сушат (Na2SO4) и удалением растворителя получают белое твердое вещество. Очисткой полученного продукта вытеснительной хроматографией на силикагеле с элюированием 5% Et2O в CHCl3 получают чистое соединение 8-2 (1,07 г) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) 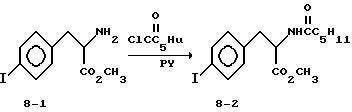 : 0,88 (3H, т), 1,27 (4H, м), 1,6 (2H, м), 2,09 (2H, т), 3,05 (2H, м), 3,75 (3H, с), 4,88 (1H, м), 5,93 (1H, м), 6,83 (2H, д), 7,6 (2H, д).
: 0,88 (3H, т), 1,27 (4H, м), 1,6 (2H, м), 2,09 (2H, т), 3,05 (2H, м), 3,75 (3H, с), 4,88 (1H, м), 5,93 (1H, м), 6,83 (2H, д), 7,6 (2H, д).
δ
5-(N-трет-бутилоксикарбонилпиперидин-4-ил)-1-триметил-1-силилпент-3-ен-1-ин (8-4).
Суспензию 3-триметилсилил-(2-пропинил)трифенилфосфоний-бромида (3 г, 6,62 ммоля) (Олдрич) в 50 мл ТГФ охлаждают до -78oC и обрабатывают прибавлением по каплям н-BuLi (6,62 ммоля). Полученный раствор оставляют нагреваться до 40oC и после перемешивания 0,5 ч получают темно-красный раствор. После охлаждения до -78oC к реакционной смеси добавляют соединение 8-3 (1,07 г, 4,73 ммоля) в 15 мл ТГФ и оставляют нагреваться при перемешивании в течение 1 ч до 0oC. Реакционную смесь нейтрализуют 50 мл H2O и экстрагируют EtOAc (200 мл). Органическую фазу отделяют, сушат (Na2SO4) и испарением получают остаток, очискной которого вытеснительной хроматографией на силикагеле с элюированием 10% EtOAc в гексане получают чистое соединение 8-4 (2,02 г).
Rf = 0,3.
1Н-ЯМР (300 МГц, CDCl3) 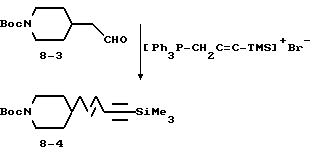 : 0,1 (9H, с), 0,7-1,1 (4H, м), 1,1-1,4 (13H, м), 1,4-1,6 (3H, м), 1,83 (8H, м), 2,4-2,6 (3H, м), 3,85 (3H, м), 5,35 (1H, т), 6 (1H, м).
: 0,1 (9H, с), 0,7-1,1 (4H, м), 1,1-1,4 (13H, м), 1,4-1,6 (3H, м), 1,83 (8H, м), 2,4-2,6 (3H, м), 3,85 (3H, м), 5,35 (1H, т), 6 (1H, м).
δ
5-(N-трет-бутилоксикарбонилпиперидин-4-ил)пент-3-ен-1-ин (8-5).
Раствор соединения 8-4 (0,815 г, 2,54 ммоля) в 60 мл ТГФ обрабатывают 12 мл H2O и гидратом гидроксида лития (0,96 г, 2,28 ммоля). Реакционную смесь перемешивают 6 ч при комнатной температуре, и за это время окраска становится темно-оранжевой. Затем реакционную смесь разбавляют Et2O (75 мл), водную фазу отделяют и промывают Et2O (3•75 мл). Объединенные органические экстракты промывают рассолом, сушат и испаряют. Очисткой полученного остатка вытеснительной хроматографией на силикагеле с элюированием 10% EtOAc в гексане получают 0,63 г чистого соединения 8-5.
1Н-ЯМР (300 МГц, CDCl3) 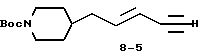 : 1,1-1,25 (3H, м), 1,25-1,5 (11H, м), 1,6-1,75 (3H, м), 2,06 (2H, т), 2,3 (1H, т), 2,6-2,78 (2H, м), 4,07 (2H, м), 5,51 (1H, м), 6,22 (1H, м).
: 1,1-1,25 (3H, м), 1,25-1,5 (11H, м), 1,6-1,75 (3H, м), 2,06 (2H, т), 2,3 (1H, т), 2,6-2,78 (2H, м), 4,07 (2H, м), 5,51 (1H, м), 6,22 (1H, м).
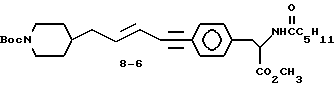
Метиловый эфир 2-s-(гексаноиламино)-3-/4-(5-N-трет-бутил-оксикарбонилпиперидин-4-ил)-пент-3-ен-1-инфенил/пропионовой кислоты (8-6).
Раствор соединения 8-5 (0,3 г, 1,2 ммоля) и соединения (0,58 г, 1,4 ммоля) в диэтиламине (6 мл) продувают N2, добавляют бистрифенилфосфинпалладийхлорид (0,049 г, 0,07 ммоля), затем йодид меди (1) (7 мг, 0,035 ммоля), после чего снова продувают N2. Спустя несколько минут, реакционная смесь становится гомогенной и раствор перемешивают 16 ч при комнатной температуре.
Растворитель удаляют в высоком вакууме, остаток растворяют в буфере с pH 7 и экстрагируют Et2O. Органический экстракт промывают 10% KHSO4, рассолом, сушат (Na2SO4) и испаряют. Очисткой остатка вытеснительной хроматографией на силикагеле с элюированием 20% EtOAc в гексане получают 0,28 г чистого соединения 8-6.
Rf = 0,3 (20% EtOAc в гексане).
1Н-ЯМР (300 МГц, CDCl3) δ: 0,9 (3H, м) 1,05-1,4 (9H, м), 1,52 (6H, с), 1,58-1,75 (4H, м), 2,07 (2H, м), 1,7 (2H, м), 3,14 (2H, м), 3,75 (2H, м), 4,1 (2H, м), 4,89 (1H, м), 5,7 (1H, м), 5,94 (1H, м), 6,18 (1H, м), 7,03 (2H, м), 7,38 (2H, м).

2-s-(гексаноиламино)-3-[4-(5-пиперидин-4-ил)пептилфенил] -пропионовая кислота (8-7).
Соединение 8-6 (0,275 г, 0,52 ммоля) растворяют в EtOH и 2 мл H2O с 5 каплями HOAc. Добавляют Pd-C (100 мг) и полученную суспензию гидрируют в трясучке Паара 4 ч под давлением 50 фунт/кв.дюйм (3,5 ат). Реакционную смесь фильтруют через Солка-Флок и растворитель удаляют. Очисткой полученного остатка вытеснительной хроматографией с элюированием 35% EtOAc в гексане получают 0,22 г метилового эфира 2-s-гексаноиламино-3-[4-(5-N-трет-бутилоксикарбонилпиперидин-4-ил)пептилфенилпропионовой кислоты.
1Н-ЯМР (300 МГц, CDCl3) δ: 0,85 (3H, т), 1-1,35 (12H, м), 1,45 (9H, с), 1,5-1,65 (6H, м), 2,15 (2H, т), 2,5-2,65 (4H, м), 3,05 (2H, м), 3,71 (3H, с), 4,04 (2H, м), 4,83 (1H, м), 5,96 (1H, м), 6,98 (2H, д), 7,04 (2H, д).
Полученный эфир (0,17 г, 0,32 ммоля) суспендируют в 10 мл смеси ТГФ-H2O (1:1) и CH3OH (2 мл), добавляют гидрат гидроксида лития (0,067 г, 1,6 ммоля) и реакционную смесь перемешивают 2 ч при комнатной температуре. Затем растворитель удаляют и остаток переносят в H2O. Затем добавлением 10% KHSO4 устанавливают pH 2-3 и экстрагируют EtOAc. Органические экстракты промывают рассолом, сушат (Na2SO4) и испарением получают 0,5 г целевой кислоты.
1Н-ЯМР (300 МГц, CDCl3) δ: 0,85 (3H, м), 0,95-1,42 (15H, м), 1,47 (9H, с), 1,5-1,7 (7H, м), 2,18 (2H, м), 2,48-2,72 (5H, м), 5,02-5,3 (2H, м), 4,03 (2H, м), 4,84 (1H, м), 6,05 (1H, м), 7,06 (4H, с).
Полученную кислоту (0,15 г, 0,29 ммоля) растворяют в EtOAc (25 мл), охлаждают до -70oC и обрабатывают 10 мин газообразным HCl. Температуру оставляют повышаться в течение 0,5 ч до -20oC. Реакционную смесь продувают N2 и растворитель удаляют. Очисткой остатка вытеснительной хроматографией на силикагеле с элюированием смесью EtOH-H2O-NH4OH (9:1:1) получают чистое соединение 8-7 (0,04 г) в виде твердого вещества.
1Н-ЯМР (300 МГц, CD3OD) δ: 0,79 (3H, т), 1,05-1,3 (9H, м), 1,32-1,56 (4H, м), 1,74 (2H, д), 2,03 (2H, м), 2,42 (2H, м), 2,7-2,85 (3H, м), 3,04 (1H, дд), 3,21 (2H, м), 4,38 (1H, м), 6,92 (2H, д), 7 (2H, д).
Приемлемые альтернативные защитные группы, которые могут быть использованы в препаративных способах изобретения, включают бензиловый эфир, циклогексиловый эфир, 4-нитробензиловый эфир, трет-бутиловый эфир, 4-пиридилметиловый эфир, бензилоксикарбонил, изоникотиноилоксикарбонил, о-хлорбензилоксикарбонил, п-нитробензилоксикарбонил, п-метоксибензилоксикарбонил, трет-амилоксикарбонил, изоборнилоксикарбонил, адамантилоксикарбонил, 2-(4-бифенил)-2-пропилоксикарбонил и 9-флуоренилметоксикарбонил.
Помимо соединений, охарактеризованных выше в примерах, дополнительные соединения изобретения приведены в табл. 1 (примеры 18-57). Эти соединения синтезированы применением путей синтеза и способов, иллюстрируемых вышеприведенными схемами реакций и примерами и их вариациями, хорошо известными специалистам и не требующими ненужных экспериментов. Все указанные в табл. 1 переменные даются со ссылкой на следующую общую формулу: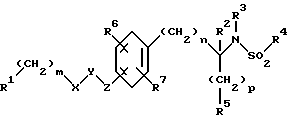
Пример 58. У здоровых людей добровольцев венопункцией отбирают кровь в 0,1 объема смеси кислота-цитрат-декстроза (85 мМ цитрата натрия, 64 мМ лимонной кислоты, 110 мМ декстрозы). Обогащенную тромбоцитами плазму получают центрифугированием 12 мин при 40 • g. Добавляют PGE1 (5 мг/мл) и тромбоциты собирают центрифугированием 12 мин при 800 • g. Осадок тромбоцитов вновь суспендируют в буфере для человеческих тромбоцитов (140 мМ NaCl, 7,9 мМ KCl, 3,3 мМ Na2HPO4, 6 мМ HEPES, 2% альбумина бычьей сыворотки, 0,1% декстрозы, pH 7,2) и фильтруют через Сефарозу 2B, предварительно уравновешенную буфером для человеческих тромбоцитов. Тромбоциты подсчитывают и их концентрацию устанавливают в 2 • 108/мл в буфере для человеческих тромбоцитов. Добавляют человеческий фибриноген (10 - 100 мг/мл) и CaCl2 (1 мМ) и агрегацию инициируют добавлением 10 мМ АДФ. Агрегацию регулируют повышением начальной степени светоизлучения.
Хотя изобретение описано и проиллюстрировано ссылками на его определенные рекомендуемые воплощения, для специалиста очевидно, что в него могут быть внесены различные изменения, модификации и замены без отхода от духа и объема изобретения. К примеру, эффективные дозировки, отличные от вышеуказанных рекомендуемых, могут применяться как следствие вариаций в реакции млекопитающего, излечиваемого от тяжких нарушений свертывания крови и эмболии, или при иных показаниях для соединений изобретения, указанных выше.
Аналогично, характерные наблюдаемые фармакологические реакции могут меняться в соответствии и в зависимости от конкретного выбранного активного компонента и присутствия или отсутствия фармацевтического носителя, а также от типа препарата и используемого пути введения, и такие ожидаемые вариации или отличия в результате могут считаться соответствующими целям и практике изобретения. Таким образом, имеется в виду, что изобретение ограничивается только объемом нижеследующей формулы изобретения и что эта формула изобретения будет интерпретироваться настолько широко, насколько это приемлемо в разумных пределах.
Результаты исследований in vitro показывают, что соединения согласно изобретению являются сильными ингибиторами связывания фибриногена с рецепторными сайтами тромбоцитов крови. Ингибирование стимулируемых АДФ тромбоцитов показано в табл. 2, которая дает сравнение концентрации (дозировки) соединения, необходимого для ингибирования агрегации на 50%, относительно контроля, в котором соединение отсутствует.
Описание исследования.
Цели исследования.
Целью данного двойного слепого исследования с произвольным варьированием дозировок, проводимого на фоне контроля с использованием плацебо, были проверка безопасности и толерантности препарата тирофибан (МК-383), нового непептидного соединения, антагониста рецептора тромбоцитов 11b/111a, на фоне терапии с применением внутривенно гепарина и аспарина; изучение фармакодинамики и фармакокинетики тирофибана; оценка случаев нежелательных проявлений в области сердечной деятельности (срочная повторная реваскуляризация, инфаркт миокарда и летальный исход) с применением тирофибана на фоне плацебо в очень сложной группе пациентов, перенесших коронарную ангиопластическую операцию.
Предпосылки.
Внезапная закупорка сосудов осложняет проведение от 4 до 8% ангиопластических процедур. Последние клинические данные дали основание предполагать, что активные вещества, ведущие себя как антагонисты рецепторов тромбоцитарного гликопротеина 11b/111a, могут снизить случаемость нежелательных ишемических осложнений после проведения коронарной ангиопластической процедуры.
Методика.
73 пациента, поделенные на три группы, получали тирофибан в трех последовательно увеличивающихся дозировках, а 20 пациентов получали плацебо. Внутри каждой группы пациенты получали произвольно либо тирофибан, либо плацебо по произвольно выбранной схеме 3:1. Пилюли в дозировке 5 • 10 и 10 мг/кг и непрерывные внутривенные вливания (16 - 24 ч) в дозировке 0,05; 0,10 и 0,15 мг/кг/мин назначались пациентам соответственно I, II и III групп. Больные получали сопутствующие гепарин и аспирин при ангиопластических процедурах. Данные по пациентам, получавшим плацебо (только гепарин и аспирин), были собраны по всем группам для сравнения. Были проведены измерения фармакодинамического эффекта тирофибана на агрегацию тромбоцитов ex vivo при стимулировании до 5 ммоль/л аденозин дифосфатом (АДФ), а также замеры продолжительности кровотечений. По всем пациентам была проведена оценка клинических показаний, хотя была только ограниченная возможность установить клинически ощутимые различия (снижение на одну треть числа клинических явлений) между группами (всего 5%).
Результаты.
Действие тирофибана ассоциировалось с зависимым от дозы ингибированием ex vivo агрегации тромбоцитов при посредстве АДФ, которое сохранялось в течение внутривенного вливания и быстро исчезает, как только лекарство прекращают вводить. Нежелательные случаи кровотечения, связанные в значительной степени с кровоизлиянием в участке подхода сосудов, слегка возрастали при введении самой высокой дозы. Нежелательные технические проявления были нечасты у всех больных, и если случались у небольшого числа пациентов внутри каждой группы, то особо не различались по клинической картине.
Выводы.
Данное исследование дало возможность установить рациональный и в основном хорошо переносимый режим дозировки для назначения тирофибана в качестве дополнительной терапии для сложных пациентов после ангиопластики.
Результаты.
Пациенты, принимающие участие в исследованиях.
Демографические характеристики 73 пациентов, принимающих тирофибан, и 20 пациентов, принимавших плацебо, представлены в табл. 1. Приблизительно 48% пациентов были включены в список только на основании морфологии комплексных коронарных патологических изменений.
Фармакодинамические реакции.
Среднепроцентное ингибирование агрегации тромбоцитов ex vivo в ответ на 5 моль/л АДФ и средняя продолжительность кровотечения при 2-часовом вливании лекарства показаны в табл. 2. Ингибирование агрегации тромбоцитов наступало быстро, как только начинался прием лекарства, и через 5 мин от начала приема пилюль тирофибана достигало от 93 до 96% в группах II и III соответственно. Ингибирование агрегации тромбоцитов сохранялось при непрерывном внутривенном вливании тирофибана и было наибольше заметным при наиболее высоких дозах в группах II и III. Увеличение ингибирования с увеличением дозы тирофибана через 5 мин, через 2 ч и в конце вливания (p < 0,001 по методу jonckheere) было весьма значительным. Не было замечено ингибирования агрегации тромбоцитов у пациентов, получавших только гепарин (плацебо), разница между дозами тирофибана и плацебо во все моменты приема лекарства была также весьма значительной (p < 0,001). Хотя не было видно никакого влияния гепарина на агрегацию тромбоцитов ex vivo, наблюдалось 2,1-кратное (до 11 мин) увеличение средней продолжительности кровотечения, отмеченного после 2-часового лечения в группе, принимавшей плацебо. Среднее повышение в группе I было в 2,9 раза (до 19,5 мин) (p = 0,118 [NS] против плацебо). Средняя длительность кровотечения превысила 30 мин для групп II и III (p = 0,003 и 0,001 соответственно против плацебо группы, показавшей увеличение длительности кровотечения). Ингибирование агрегации тромбоцитов прекращалось сразу же после прекращения вливания тирофибана. Когда удалялись фасции, закрывающие доступ к кровеносному сосуду через 4 ч после прекращения лекарственного вливания по программе исследований, процент ингибирования агрегации составлял менее 50% для всех групп.
Нежелательные клинические явления.
При проведении данных испытаний не было летальных исходов, инфарктов миокарда или непредвиденной необходимости операций по коронарному шунтированию. Случаи срочной повторной рекатетеризации при повторной коронарной ангиопластики или того и другого отражены в табл. 3. Необходимость реваскуляризации была крайне низкой как при вливании лекарства, так и перед выпиской из больницы. Хотя явно было, что случаи повторной реваскуляризации во время изучения вливания лекарства были реже (2,7% для пациентов, которых лечили тирофибаном, против 5% для тех групп, которые принимали плацебо), все эти случаи были так редки (менее 1% в каждой группе), что никакого формального статического сравнения не проводилось.
Кровотечение.
При проведении данных испытаний не было никаких нейтрализующих сосудистых операций или внутривенных или забрюшинных кровотечений. Одному пациенту из группы III, получавшей тирофибан, сделали переливание эритроцитарного препарата дозой в 1 ед. и одному пациенту из группы, получившей плацебо, сделали переливание 2 ед. эритроцитарного препарата. Общее количество случаев кровотечения составляло 4,8; 3,3; 13,6; 5 соответственно в лечебных группах I, II и III, а также в группе плацебо. Излучение лекарственного вливания было прервано у одного пациента из группы III из-за открывшегося кровотечения из десен, анализ показал значительное снижение гематокрита, оказавшееся, как было позднее установлено, ложным. У одного пациента проявилась другая тромбоцитопения с показателем 70000 мм3 во время вливаний лекарства по режиму опыта, но, как только ему прекратили вводить внутривенный гепарин, количество тромбоцитов в крови нормализовалось до 93000 мм3, несмотря на то, что ему еще продолжали в течение 12 ч делать вливания тирофибана.
Рекатетеризация и повторная раваскуляризация во время вливаний лекарства по режиму исследований и при выписке из больницы приведены в табл. 4.
МК-383 (L-тирозин, N-(n-бутилсульфонил)-О-[4-бутил-(4-пиперидинил)]моногидрохлорид моногидрат) представляет собой сильнодействующий и специфический антагонист рецептора тромбоцитарного фибриногена, что может быть использовано для предупреждения процессов, ведущих к окклюзивному образованию тромбов в полости кровеносных сосудов. Были проведены 2 серии испытаний стадии I (контрольная группа получала плацебо) с участием 56 здоровых добровольцев на безопасность, толерантность, фармакокинетику и фармакодинамику препарата МК-383 в виде вливаний продолжительностью в 1 и 4 ч на фоне действия аспирина и при прекращении приема аспирина. Когда здоровым мужчинам делали непрерывные вливания в количестве до 0,4 мкг/кг/мин в течение 1 ч или до 0,2 мкг/кг/мин в течение 4 ч, это воспринималось как хорошо переносимое реверсивное средство торможения функции тромбоцитов. При скорости вливания 0,25 и 0,15 мкг/кг/мин в течение 1 и 4 ч соответственно препарат МК-383 увеличивал базовую продолжительность кровотечения от 2 до 2,5 раза и блокировал вызванную аденозин дифосфатом (АДФ) агрегацию тромбоцитов не менее чем на 80%. К фармакокинетическим показателям МК-383 можно добавить плазменный клиренс в среднем 329 мл/мин, устойчивый объем распределения 76 и продолжительность периода полувыведения, составляющую 1,6 ч. Процент экскретированной с мочой дозы составлял 37%. Коррекции между плазменной концентрацией МК-383 (C) и блокированием агрегации тромбоцитов были изучены на максимально эффективной модели сигмовидной кишки. Плазменная концентрация, дающая 50% блокирования (C-50) для МК-383, у здоровых добровольцев составляет примерно 13 нг/мл с коэффициентов Хилла больше 5. Экспоненциальная эмпирическая модель, основанная на простом анализе динамического распределения, лучше всего описывает отношение (C) МК-383 с моделью расширения продолжительности кровотечения (ВТЕ). Модель указывает, что концентрация МК-383 в плазме, необходимая для удвоения ВТЕ, приблизительно равна 30 нг/мл (т.е. в 2,5 раза больше, чем C-50 для блокирования вызванной АДФ агрегации тромбоцитов). На фармакокинетику МК-383 не оказал влияния предварительный прием 325 мг аспирина за 1 день и 1 ч перед его вливанием. Наоборот, предварительный причем аспирина снизил C-50 и увеличил продолжительность кровотечения, что заставляет предполагать, что аспирин может оказать дополнительный эффект в отношении блокирования функции тромбоцитов.
Испытуемые соединения приведены в табл. 5.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИЗОКСАЗОЛИНЫ И ИЗОКСАЗОЛЫ, СПОСОБ ПОДАВЛЕНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОДАВЛЯЮЩАЯ АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1994 |
|
RU2149871C1 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| ПРОИЗВОДНЫЕ ДИАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ АРИТМИИ | 1994 |
|
RU2155587C2 |
| ДИАРИЛ-5,6-КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЗАЩИТЫ ОТ ВОЗДЕЙСТВИЯ ЛЕЙКОТРИЕНОВ | 1993 |
|
RU2154065C2 |
| ЗАМЕЩЕННЫЕ МОРФОЛИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПРОТИВОДЕЙСТВИЯ ВЕЩЕСТВУ Р ИЛИ БЛОКИРОВАНИЯ РЕЦЕПТОРОВ НЕЙРОКИНИНА-1 | 1995 |
|
RU2170233C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ СНИЖЕНИЯ КОЛИЧЕСТВА ТАХИКИНИНОВ ПРИ ЛЕЧЕНИИ ИЛИ ПРОФИЛАКТИКЕ ФИЗИОЛОГИЧЕСКИХ СОСТОЯНИЙ, АССОЦИИРУЕМЫХ С ИЗБЫТКОМ ТАХИКИНИНОВ | 1994 |
|
RU2131426C1 |
| ПРОИЗВОДНЫЕ ФЕНОКСИФЕНИЛУКСУСНОЙ КИСЛОТЫ | 1994 |
|
RU2139273C1 |
| ТЕТРАГИДРОПИРАНИЛЦИКЛОПЕНТИЛТЕТРАГИДРОПИРИДОПИРИДИНОВЫЕ МОДУЛЯТОРЫ АКТИВНОСТИ РЕЦЕПТОРА ХЕМОКИНА | 2003 |
|
RU2285004C2 |
| ИНГИБИТОРЫ HIV ПРОТЕАЗЫ | 1994 |
|
RU2137768C1 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609210C2 |
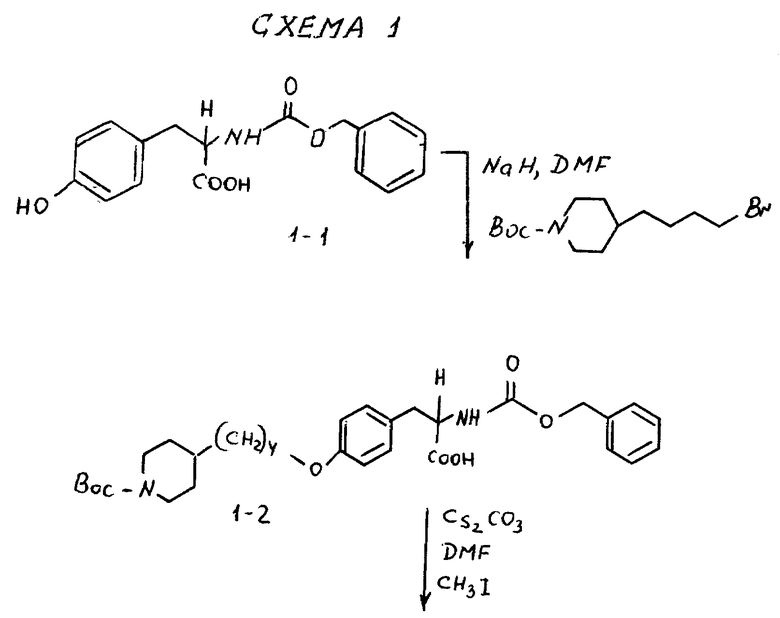
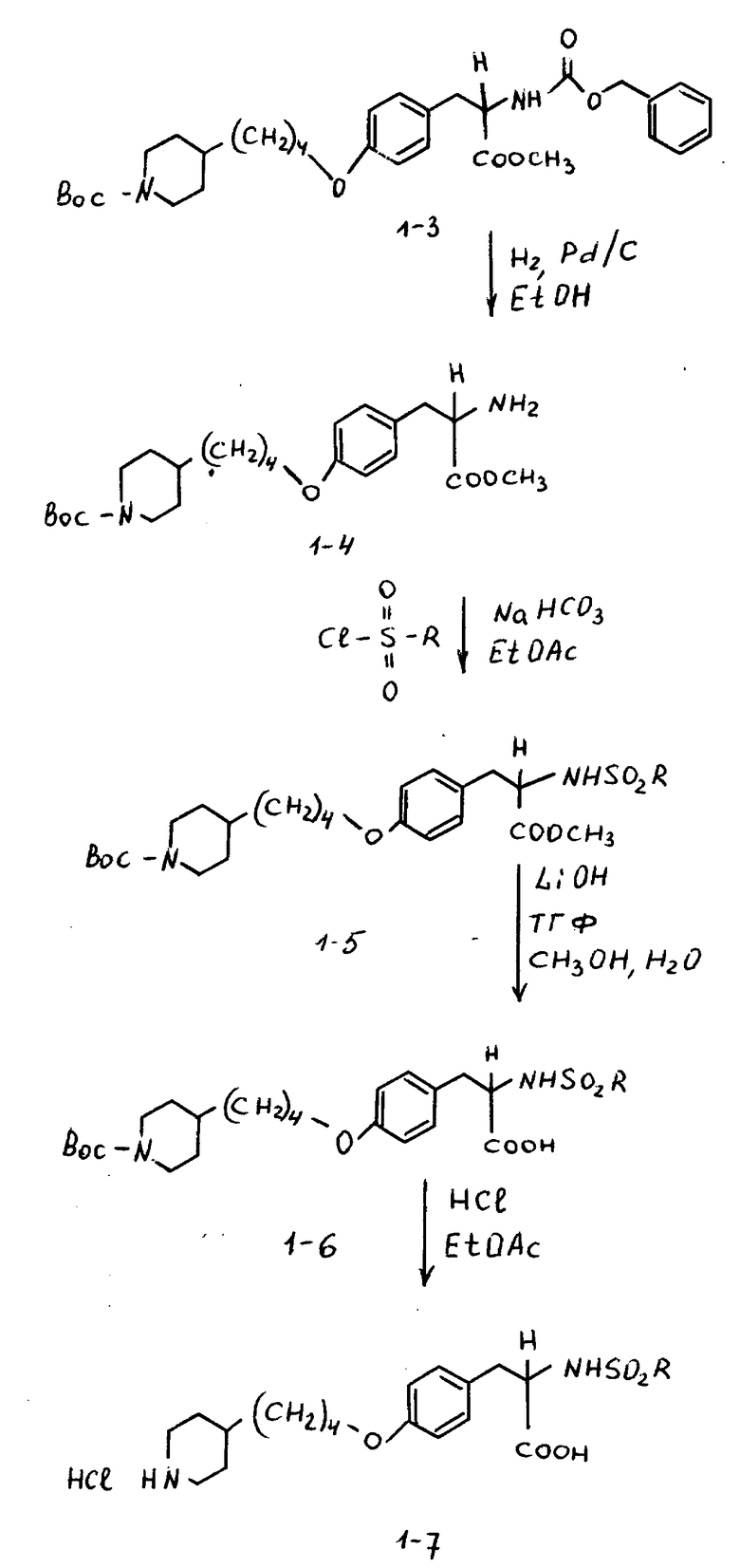
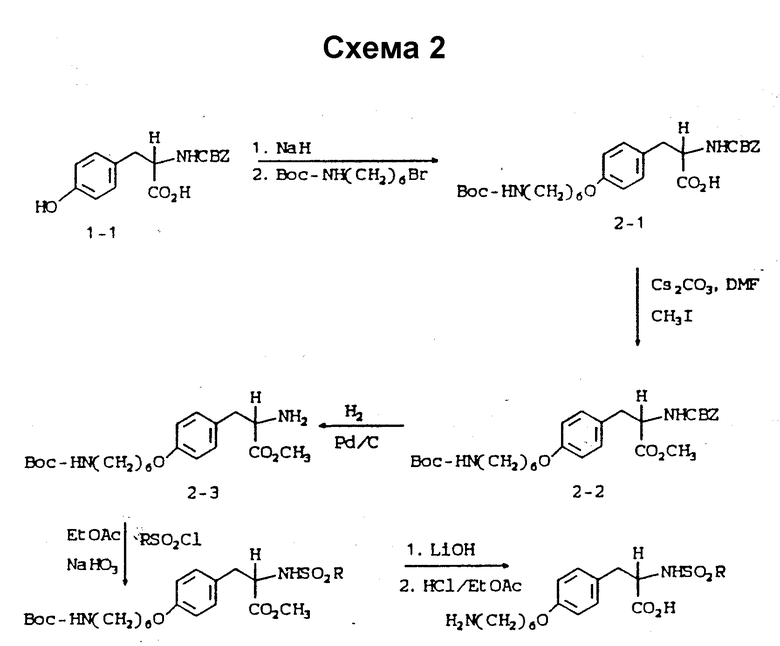

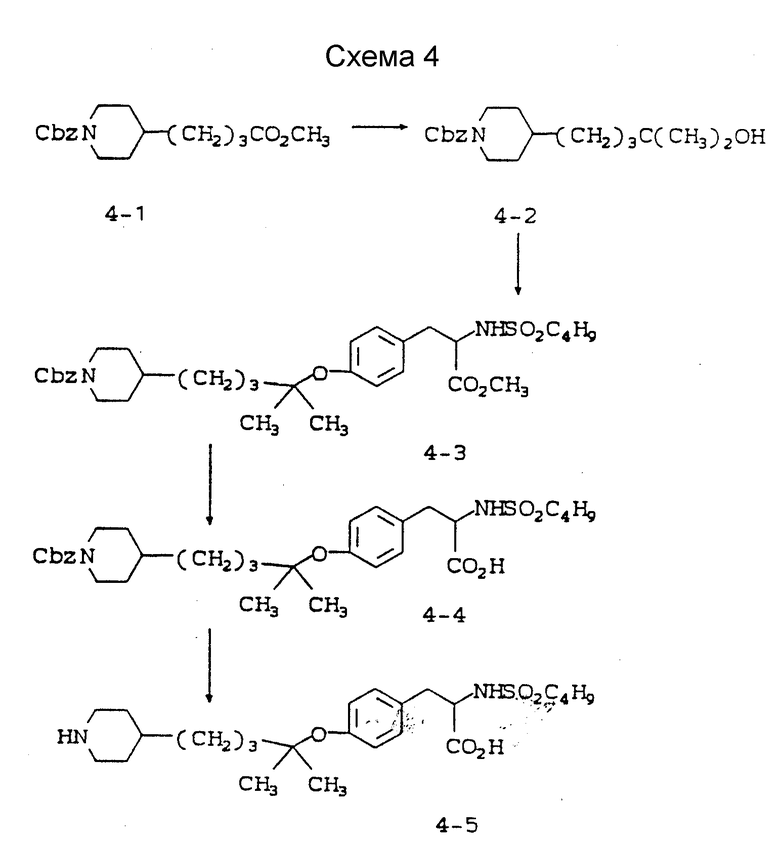
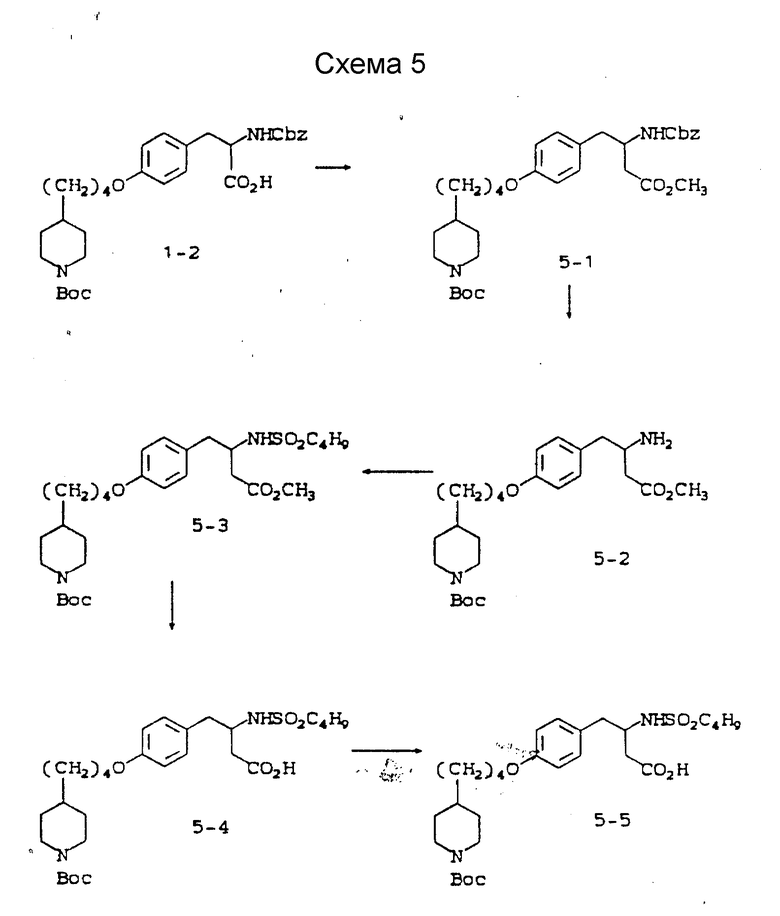
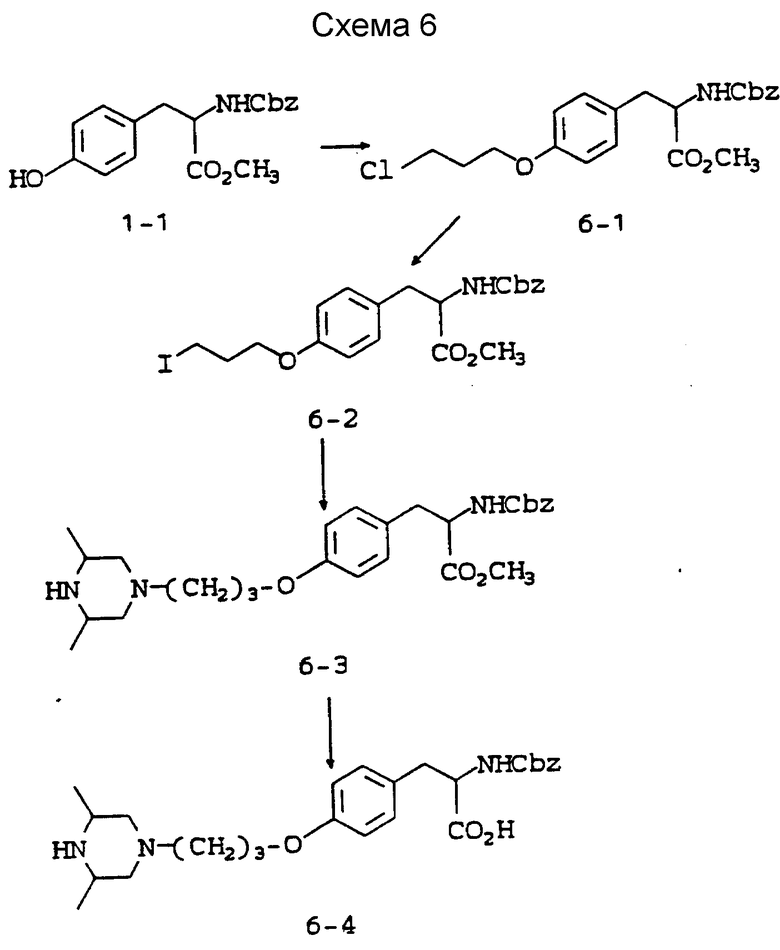
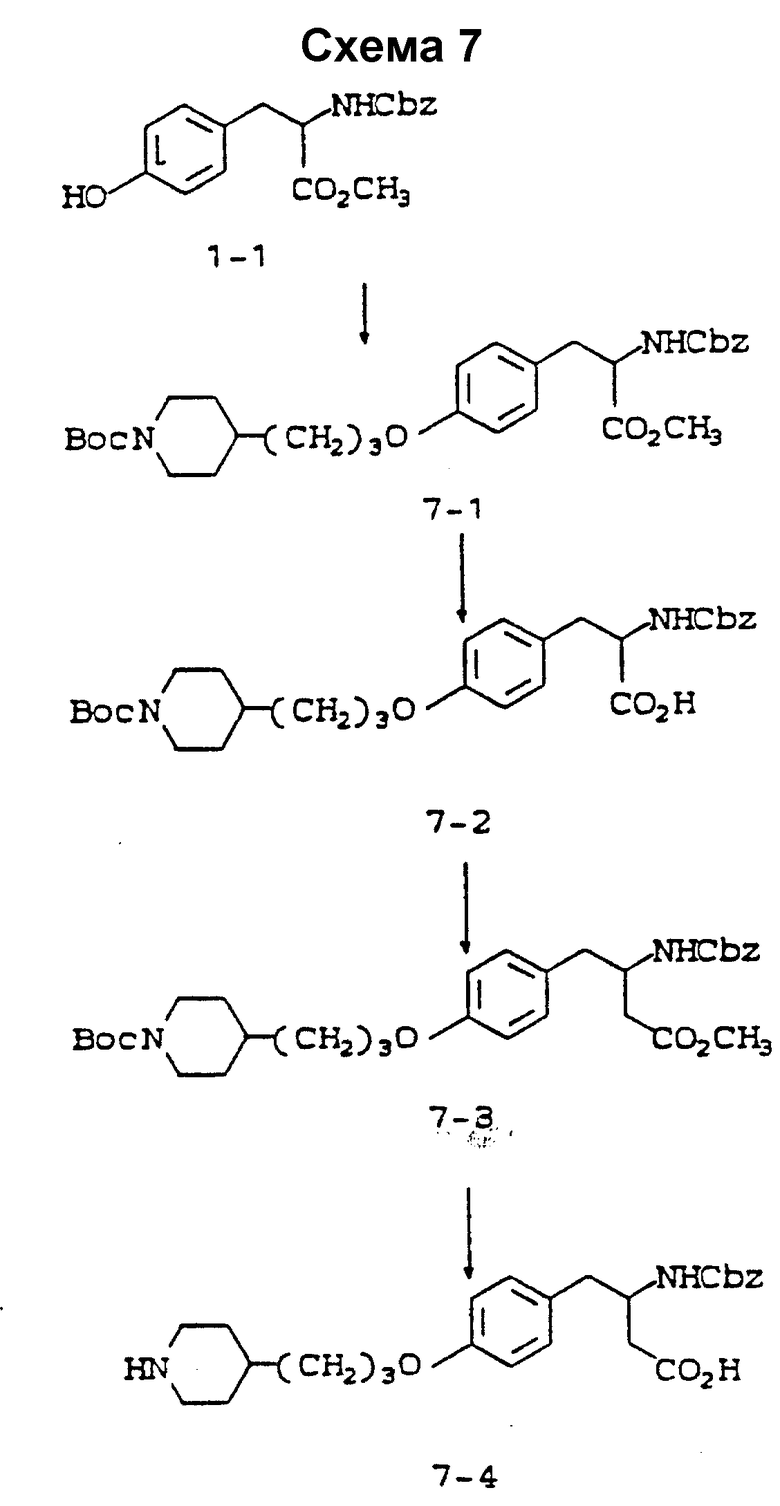
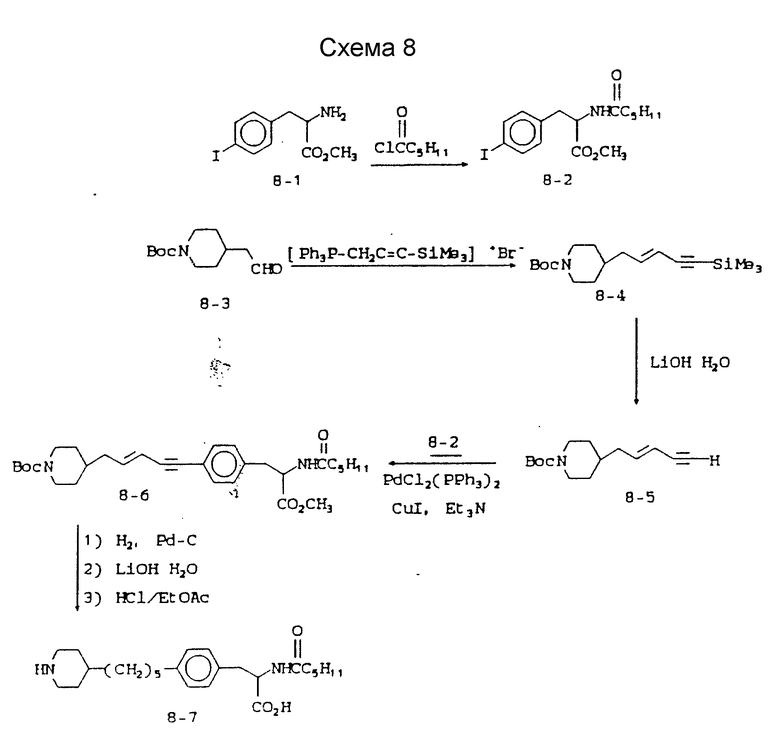
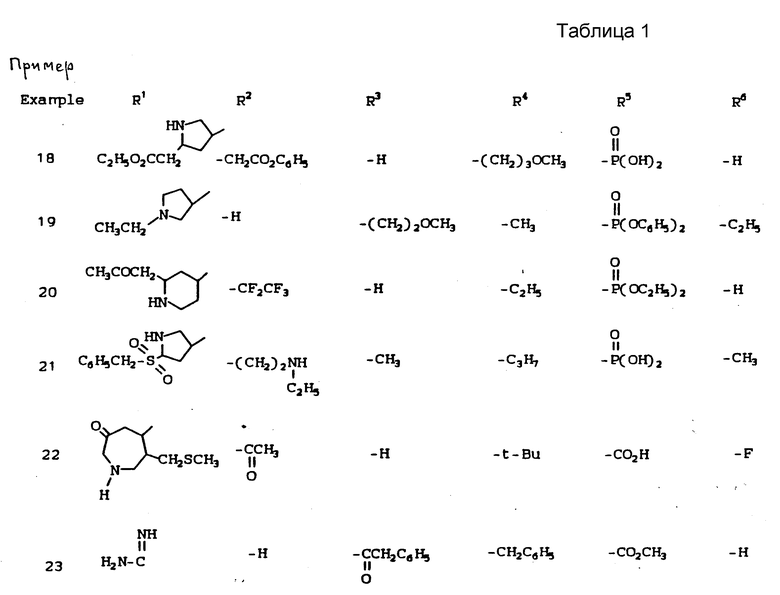
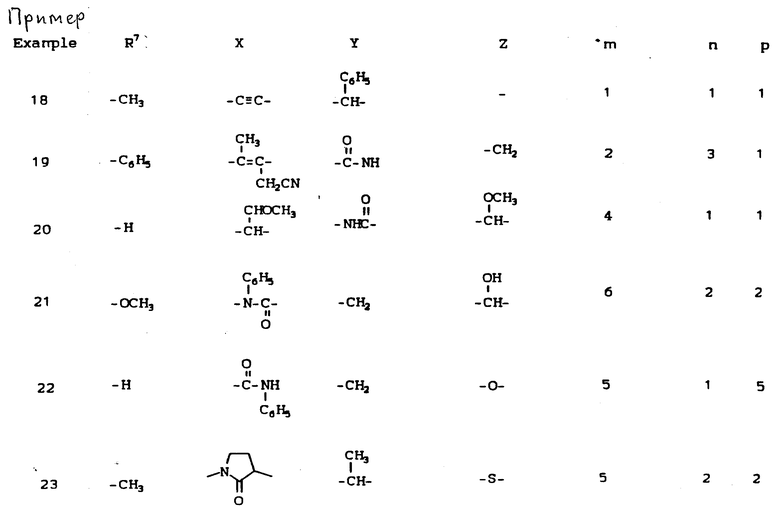
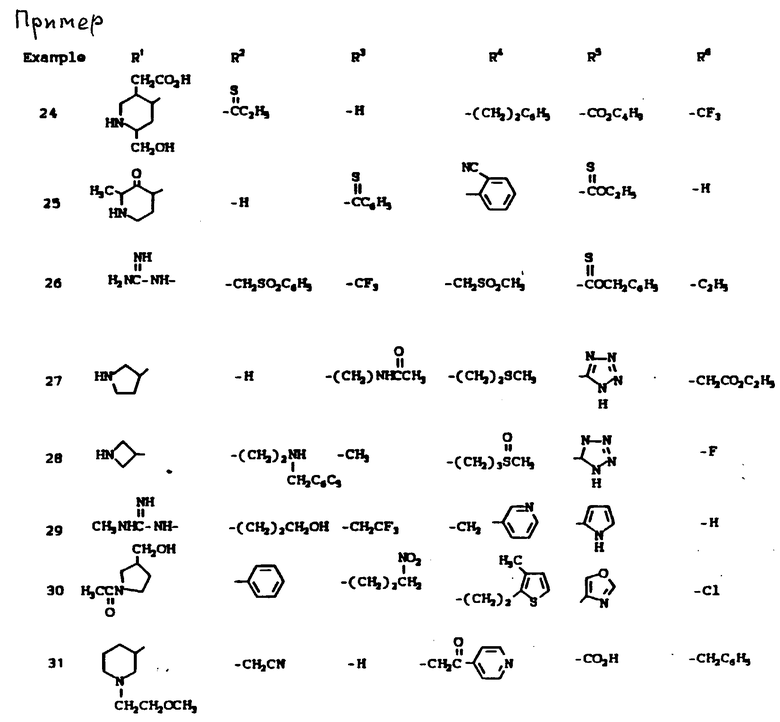
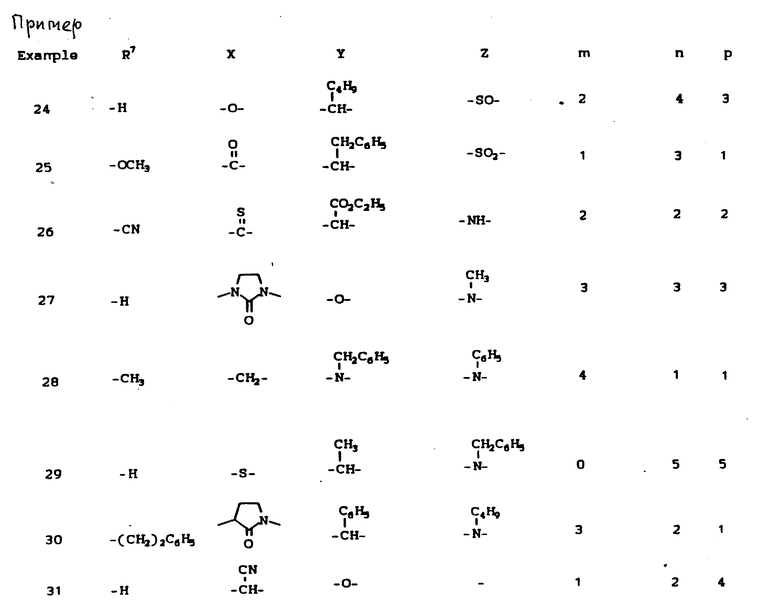

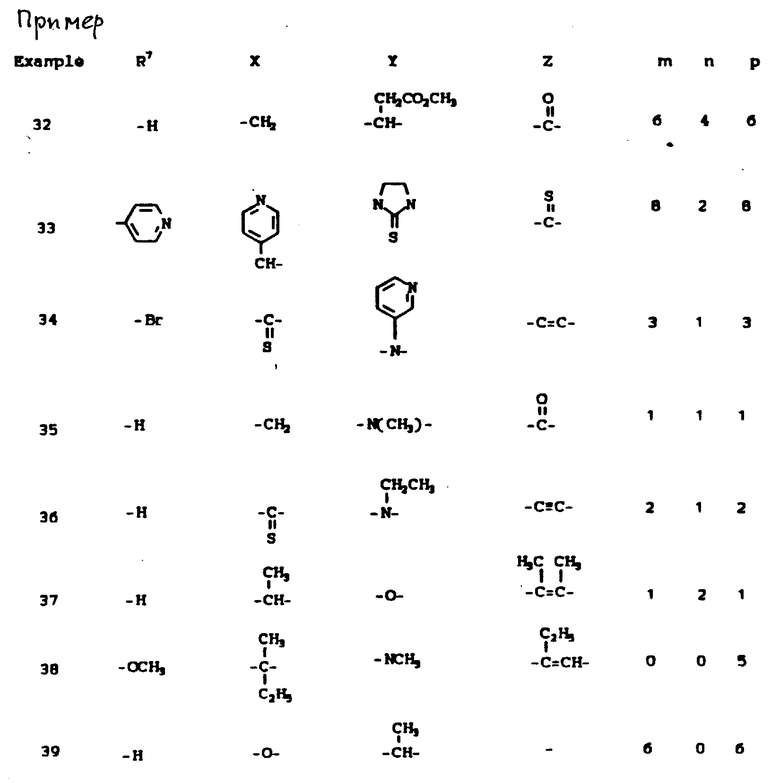
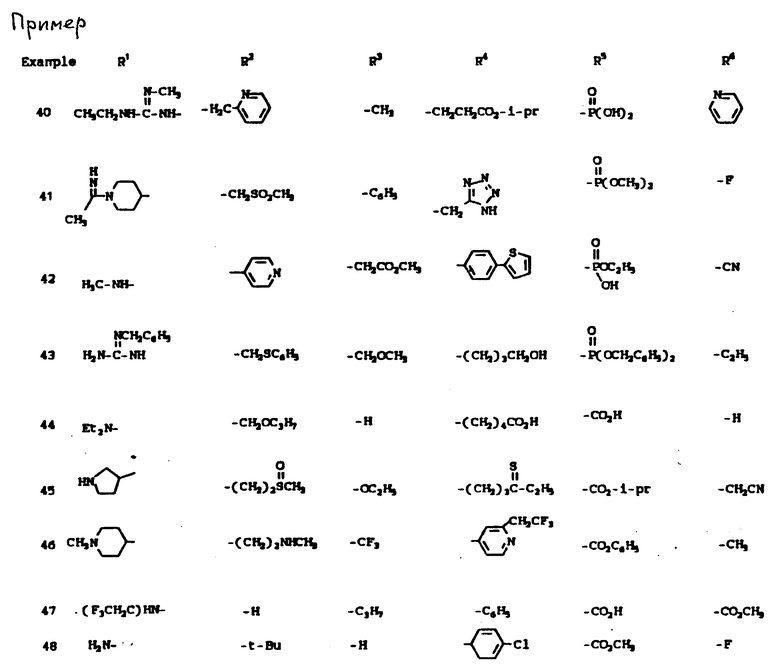

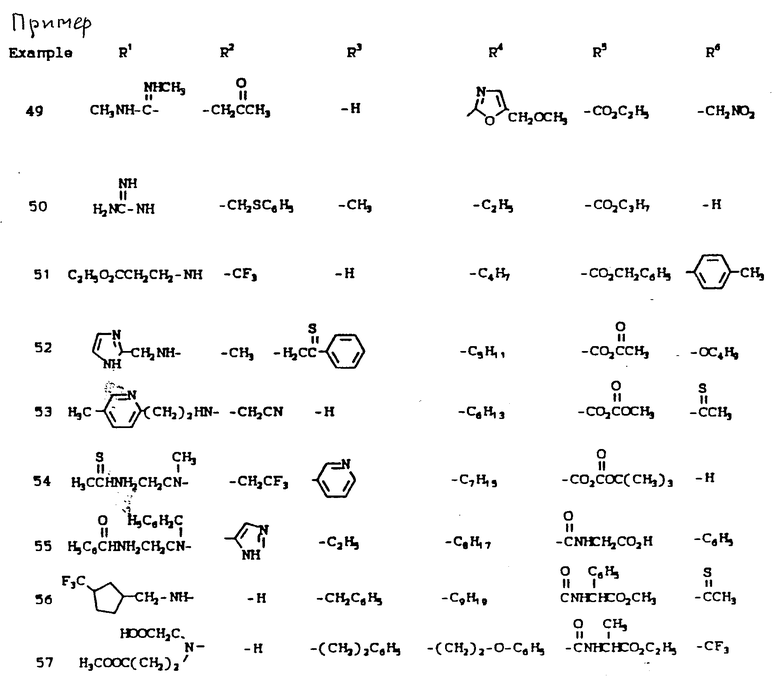
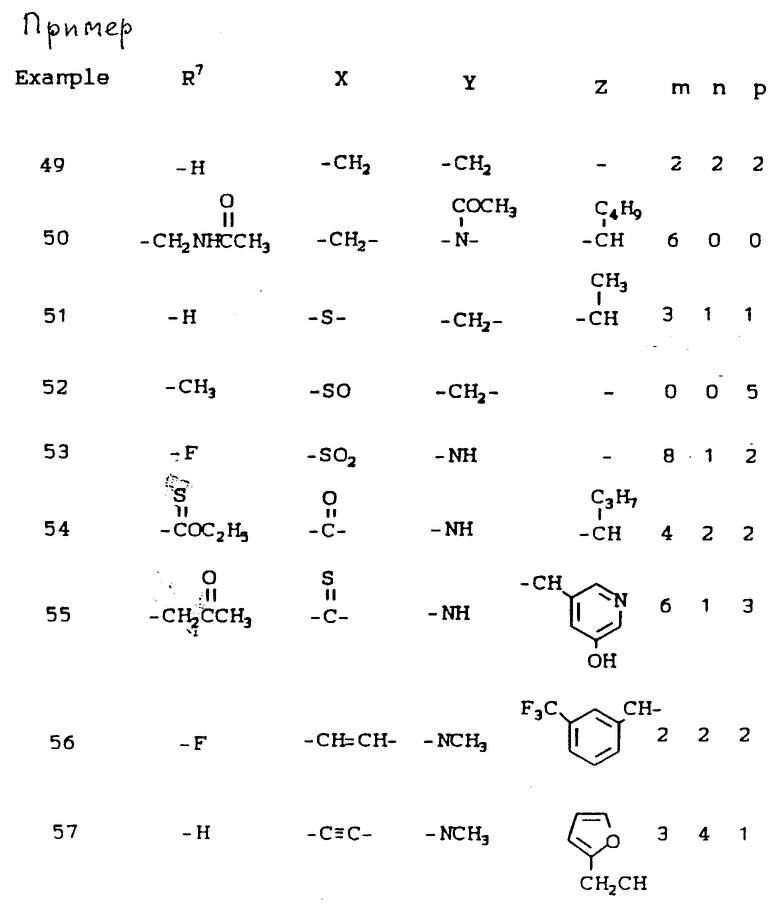
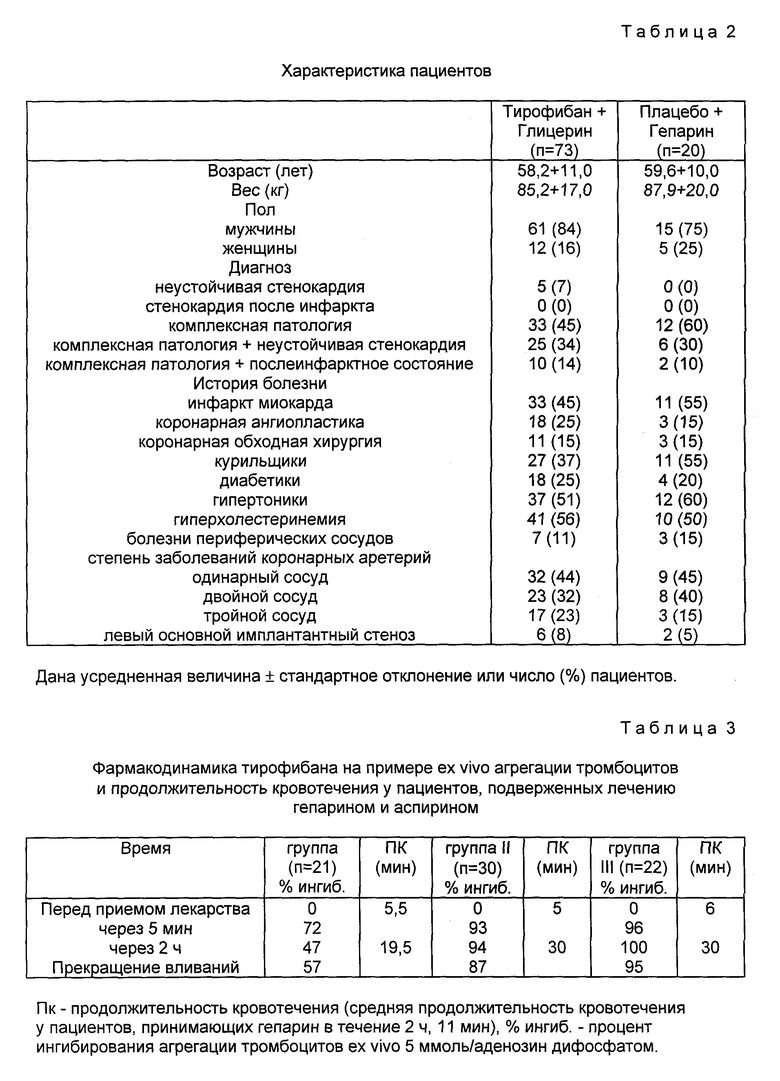
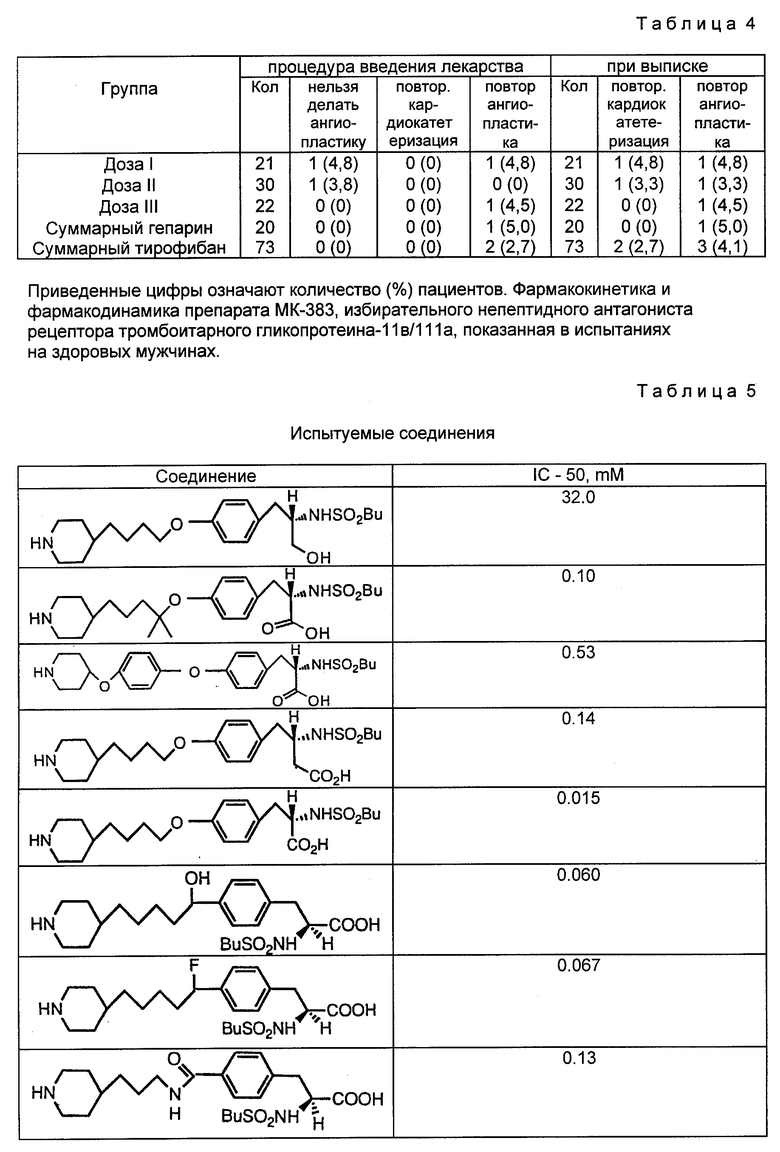
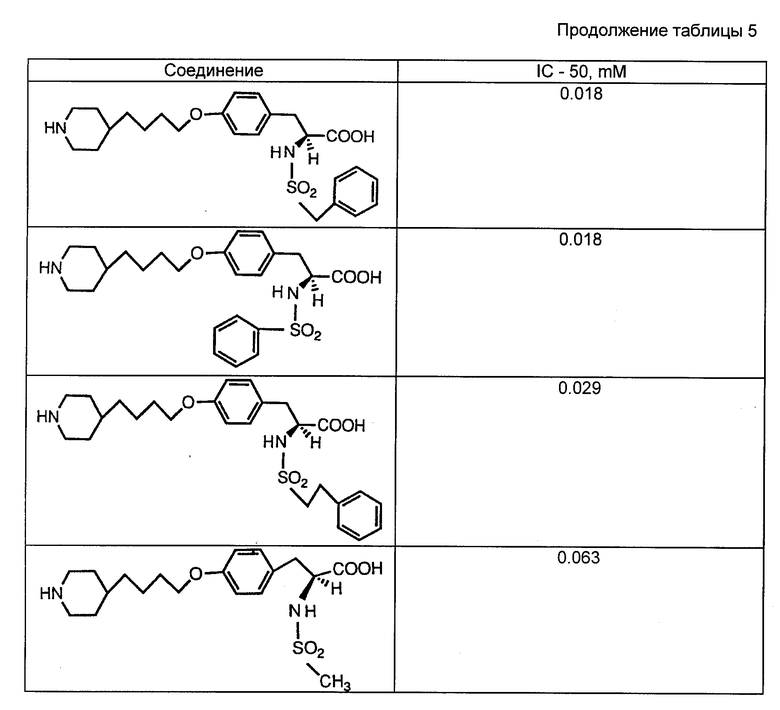
Производные сульфонамида формулы I, где R - шестичленный гетероцикл с атомом азота или NR6R7, R - водород, R7 - водород или группа формулы Ph-  = NH, H2N-
= NH, H2N- 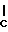 - NH, CH3 -
- NH, CH3 -  = NH; R2 = R3 = H; m = 4-6, n = 1; P=0 или 1, R4 - C1-10-алкил, бензил, стирил, фенил, тиенил или замещенный нафтил; R5-COOR11, R11 - H или метил, которые представляют собой антагонисты 11b/111a рецептора фибриногена, препятствуют агрегации тромбоцитов и полезны для профилактики и лечения заболеваний, вызываемых образованием тромба. 4 с. и 9 з.п. ф-лы, 5 табл., 8 схем.
= NH; R2 = R3 = H; m = 4-6, n = 1; P=0 или 1, R4 - C1-10-алкил, бензил, стирил, фенил, тиенил или замещенный нафтил; R5-COOR11, R11 - H или метил, которые представляют собой антагонисты 11b/111a рецептора фибриногена, препятствуют агрегации тромбоцитов и полезны для профилактики и лечения заболеваний, вызываемых образованием тромба. 4 с. и 9 з.п. ф-лы, 5 табл., 8 схем.
Соединение I: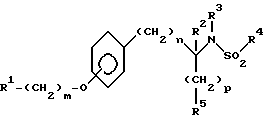
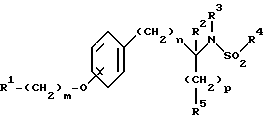
в которой R1 - шестичленное гетероциклическое кольцо, содержащее гетероатом азота, или группа NR6R7, в которой R6 - водород, R7 - водород или группа формулы
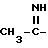
R2 и R3 - водород;
m = 4 - 6;
n = 1;
p = 0 или 1;
R4 - C1 - C10-алкил, бензил, стирил, фенил, тиенил или замещенный нафтил;
R5 - группа COOR11, в которой R11 - водород или метил,
или их фармацевтически приемлемые соли.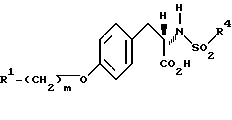
в которой R1 - шестичленное гетероциклическое кольцо, содержащее гетероатом азота или группа NR6R7, в которой R6 и R7 - водород;
R4 - C1 - C10-алкил, бензил, стирил, фенил или замещенный нафтил;
m = 0 - 6,
или их соли.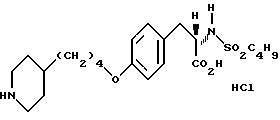
5. Соединение по п.3, имеющее формулу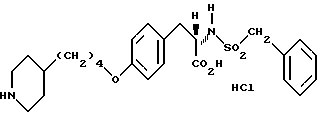
6. Соединение по п.3, имеющее формулу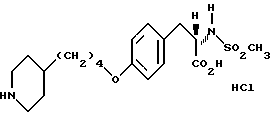
7. Соединение по п.3, имеющее формулу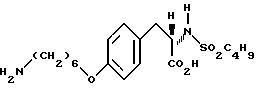
8. Соединение по п.1, имеющее формулу
9. Фармацевтическая композиция для ингибирования агрегации тромбоцитов, содержащая производное сульфонамида в качестве активного агента и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве производного сульфонамида она содержит соединение общей формулы I по п.1 в эффективном количестве.
| US 4122255 (Lilly Industries Limited) 24.10.78, A 61 K 31/445. |
