Лейкотриены составляют группу местнодействующих гормонов, продуцируемых живыми организмами из арахидоновой кислоты. Основными лейкотриенами являются лейкотриен B4 (обозначенный аббревиатурой ЛТB4), ЛТC4, ЛТD4 и ЛТE4. Биосинтезы этих лейкотриенов начинаются с действия фермента 5-липоксигеназы на арахидоновую кислоту для получения эпоксида, известного как лейкотриен A4 (ЛТA4), который преобразуется в другие лейкотриены путем последующих ферментативных стадий. Дальнейшие детали биосинтеза, а также метаболизм лейкотриенов описаны в книге Leukotrienes and Lipoxygenases, ed. J. Rokach, Elsevier, Amsterdam (1989). Действие лейкотриенов на живые организмы и их вклад в различные состояния заболеваний также обсуждаются в книге Rokach.
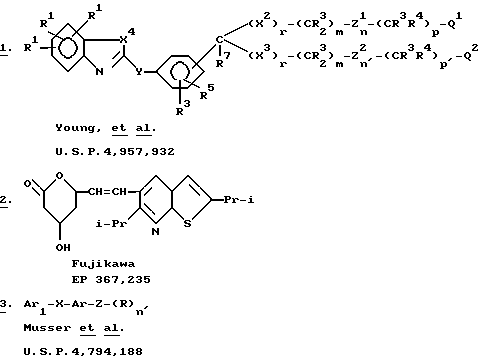
В патенте США N 4957932, Young et al., описываются соединения формулы I в качестве антагонистов лейкотриенов и ингибиторов биосинтеза лейкотриенов. Настоящие соединения отличаются от описанных ранее Young тем, что имеют другое гетероциклическое кольцо в левой части структуры. Fujikawa описывает тиено [2,3-b] пиридин 2 в EP N 367235, но место присоединения и природа основного заместителя отличаются от таковых у настоящих соединений. Musser et al. описывают соединение 3 в патенте US N 4794188, как являющееся ингибитором липоксигеназы и обладающее противовоспалительной и противоаллергической активностями. Однако соединение 3 отличается от настоящих соединений главным образом тем, что Ar1 отличается от нашей HETA группы. Таким образом, соединения по настоящему изобретению являются новыми.
Настоящее изобретение относится к 5,6-конденcированным гетероциклическим кислотам, обладающим активностью в качестве антагонистов лейкотриена, к способам их получения и к способам и фармацевтическим составам для применения этих соединений на млекопитающих (особенно на людях).
Благодаря их активности как антагонистов лейкотриена, соединения по настоящему изобретению являются полезными в качестве противоастматических, противоаллергических, противовоспалительных и цитозащитных агентов. Они являются полезными также при лечении ангины, спазмов головного мозга, нефрита почечных клубочков, гепатита, эндотоксикоза, увеита и отторжения аллотрансплантата.
Подробное описание изобретения
Соединения по изобретению наилучшим образом представлены формулой I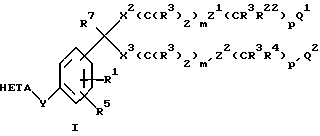
где R1 обозначает H или R2;
R2 обозначает низший алкил, низший алкенил, низший алкинил, -CF3, -CH2F, -CHF2, Ph(R26)2, CH2Ph(R26)2 или CH2CH2Ph(R26)2 или две R2-группы, присоединенные к одному и тому же атому, могут образовывать кольцо, содержащее до 8 атомов углерода и до 2 гетероатомов, выбранных из О, S и N;
R3 обозначает H или R2;
R4 обозначает R3, галоген, -NO2, -CN, -OR3, -SR2, N(R3)2, NR3COR7, S(O)R2 или S(O)2R2;
CR3R22 может быть остатком обычной аминокислоты;
R5 обозначает H, галоген, -NO2, -N3, -CN, -SR2, -S(O)R2, S(O)2R2, -N(R3)2, -COR3 или низший алкил;
R6 обозначает -(CH2)5-C(CR7)2-(CH2)5 -R8 или -CH2CON(R20)2;
R7 обозначает H или низший алкил;
R8 обозначает: A) моноциклический или бициклический гетероциклический радикал, содержащий от 3 до 12 углеродных атомов ядра и 1 или 2 гетероатома ядра, выбранных из N, S и O, и каждое кольцо в гетероциклическом радикале образовано из 5 или 6 атомов, или B) радикал W-R9;
R9 содержит до 21 атома углерода и является (1) углеводородным радикалом или (2) ацильным радикалом органической ациклической или моноциклической карбоновой кислоты, содержащей не более чем 1 гетероатом в кольце;
R10 обозначает -SR11, -OR15 или -N(R12)2;
R11 обозначает низший алкил, -COR14, Ph(R26)2, CH2Ph(R26)2 или CH2CH2Ph(R26)2;
R12 обозначает H, R11 или две группы R12, присоединенные к одному и тому же N, могут образовывать насыщенное кольцо из 5 или 6 членов, включающих атомы углерода и до двух гетероатомов, выбранных из О, S и N;
R13 обозначает низший алкил, низший алкенил, низший алкинил -CF3, Ph(R26)2, CH2Ph(Ph26)2 или CH2CH2Ph(R26)2;
R14 обозначает H или R13;
R15 обозначает H или R11;
R16 обозначает H, низший алкил или OH;
R17 обозначает низший алкил, низший алкенил, низший алкинил, Ph(R26)2, CH2Ph(R26)2 или CH2CH2Ph(R26)2;
R18 обозначает R13;
R19 обозначает H, низший алкил, низший алкенил, низший алкинил, -CF3, Ph, CH2Ph или CH2CH2Ph;
R20 обозначает H, низший алкил, Ph(R26)2, CH2Ph(R26)2 или CH2CH2Ph(R26)2, или две группы R20, присоединенные к одному и тому же N, могут образовывать насыщенное кольцо из 5 или 6 членов, включающих атомы углерода и до двух гетероатомов, выбранных из O, S и N;
R21 обозначает H или R17;
R22 обозначает R4, CHR7OR3 или CHR7SR2;
R23, R24 и R25 обозначают каждый независимо H, низший алкил, -CN, -CF3, COR3, CO2R7, CON(R20)2, OR3, SR2, S(O)R2, S(O)2R2, N(R12)2, галоген или электронную пару;
R26 обозначает H, низший алкил, -SR27, -OR28, -N(R28), -CO2R7, CON(R28)2, -COR7, -CN, CF3, NO2, SCF3 или галоген;
R27 обозначает низший алкил, фенил или бензил;
R28 обозначает R27, H или COR7, или две группы R28, присоединенные к одному и тому же N, могут образовывать насыщенное кольцо из 5 или 6 членов, включая атомы углерода и до двух гетероатомов, выбранных из О, S и N;
m и m' обозначают независимо 0-8;
p и p' обозначают независимо 0-8;
m + p равно 1-10, где X2 обозначает О, S, S(O) или S(O)2 и Z1 является связью;
m + p = 0-10, где Z1 является HET(R23R24R25);
m + p = 0-10, где X2 является CR3R16;
m' + p' = 1-10, где X3 обозначает О, S, S(O) или S(O)2, и Z2 является связью;
m' + p' = 0-10, где Z2 обозначает HET(R23R24R25);
m' + p'= 0-10, где X3 обозначает CR3R16;
S = 0-3;
Q1 обозначает тетразол-5-ил, -CO2R3, -CO2R6, -CONHS(O)2R13, -CN, -CON(R20)2, NR21S(O)2R13, -NR21CON(R20)2, -NR21COR14, OCON(R20)2, -COR19, -S(O)R18, -S(O)2R18, -S(O)2R18, -S(O)2N(R20)2,
-NO2, NR21CO2R17, -C(N(R12)2)=NR21, -C(R19)=NOH или C(R3)2OH; или, если Q1 обозначает CO2H и R22 обозначает -OH, -SH, CHR7OH или -NHR3, тогда Q1 и R22 и атомы углерода, к которым они присоединены, могут образовывать гетероциклическое кольцо путем отнятия воды;
Q2 обозначает H, OR15, низший алкил, галоген или Q1;
W обозначает O, S или NR3;
X1 обозначает О, S, -S(O)-, -S(O)2, =NR3, -C(R3)2- или связь;
X2 и X3 независимо обозначают O, S, S(O), S(O)2, CR3R16 или связь;
Y обозначает -CR3= CR3-, -C(R3)2-X1-, -X1-C(R3)2-, -C(R3)2-X1-C(R3)2-, -C≡C-, -CO-, -NR3CO-, -CONR3- O, S или NR3;
Z1 и Z2 являются независимо HET(R23R24R25) или связью;
HET обозначает дирадикал бензола, пиридина, фурана, тиофена или 1,2,5-тиадиазола;
HETA обозначает HE1 или HE2;
HE1 обозначает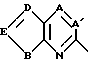
HE2 обозначает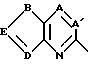
A и A1 обозначают N или CR5;
B обозначает O, S ила S(O);
D обозначает N или CR4;
E обозначает CR4, когда D обозначает CR4;
E обозначает CR3, когда D обозначает N;
или их фармацевтически приемлемыми солями.
Предпочтительными соединениями формулы I являются соединения формулы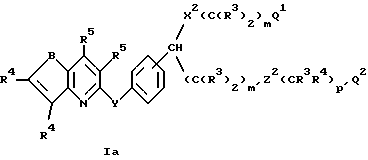
где B обозначает S или O;
R4 обозначает H, галоген, CN, CF3 или S(O)2R2;
R5 обозначает H или галоген;
m и m' каждый равен независимо 1-6;
p'= 0 или 1;
Q1 обозначает CO2R3, CO2R6, -CONHS(O)3R13, тетразол-5-ил или C(R3)2OH;
Q2 обозначает C(R3)2OR3, галоген или низший алкил;
X2 обозначает S или O;
Y обозначает -CH=CH-, -CH2-O-, -CH2-CH2-, -C≡C- или -CH(CH2)CH-;
Z2 обозначает HET(R23R24);
HET обозначает дирадикал бензола или тиофена;
и остальные заместители такие, как определены для формулы I.
Группа наиболее предпочтительных соединений формулы I описывается формулой Ib: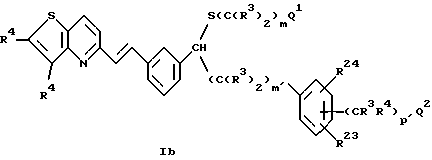
где R3 обозначает низший алкил или две группы R3, присоединенные к одному и тому же атому углерода, могут образовывать кольцо с от 3 до 6 членами, необязательно содержащее один атом кислорода или один атом серы;
R4обозначает H, галоген, -CN, CF3 или -S(О)2R2;
R23 и R24 обозначают независимо H, галоген или низший алкил;
m и m' равны независимо 1-5;
p' = 0 или 1;
Q1 обозначает -CO2R3, тетразол-5-ил или -CONHS(O)2R и
Q2 обозначает H, C(R3)2OH или OR15.
Определения
Следующие аббревиатуры имеют указанные далее значения:
Ac = ацетил
AIBN = 2,2'-азобисизобутиронитрил
Bn = бензил
DHP = 2,3-дигидро-4Н-пиран
DIBAL = диизобутилалюминийгидрид
DIPHOS = 1,2-бис(дифенилфосфино)этан
DMAP = 4-(диметиламино)пиридин
DMF = N,N-диметилформамид = ДМФ
DMSO = диметилсульфоксид
Et3N = триэтиламин
Fur = фурандиил
KHMDS = калий гексаметилдисилазан
LDA = литий диизопропиламид
MCPBA = мета-хлорнадбензойная кислота
MS = метансульфонил = мезил
MSO = метансульфонат = мезилат
NBS = N-бромсукцинимид
NCS = N-хлорсукцинимид
NSAID = нестероидный противовоспалительный препарат
PCC = пиридиний хлорхромат
PDC = пиридиний дихромат
Ph = фенил
Phe = бензолдиил
PPTS = пиридиний п-толуолсульфонат
pTSA = п-толуолсульфоновая кислота
Pye = пиридинил
r.t. = комнатная температура
rac. = рацемический
Tdz = 1,2,5-тиадиазол-3,4-диил
Tf = трифторметансульфонил = тритил
TfO = трифторметансульфонат = трифлат
Tn= 2- или 3-тиенил
THF = тетрагидрофуран = ТГФ
Thi = тиофендиил
THP = тетрагидропиран-2-ил
TLC = тонкослойная хроматография = ТСХ
Ts = п-толуолсульфонил = тозил
TsO = п-толуолосульфонат = тозилат
Tz = IH (или 2H)-тетразол-5-ил
C3H5 = аллил
Аббревиатура алкильных групп
Me = метил
Et = этил
n-Pr = нормальный пропил
i-Pr = изопропил
n-Bu = нормальный бутил
i-Bu = изобутил
s-Bu = вторичный бутил
t-Bu = третичный бутил
с - Pr = циклопропил
с - Bu = циклобутил
с - Pen = циклопентил
с-Hex = циклогексил
Термин алкил, алкенил и алкинил подразумевает линейную, разветвленную и циклическую структуры и их сочетания.
Термин "алкил" включает "циклоалкил" и "низший алкил" и охватывает углеродные фрагменты, имеющие до 20 атомов углерода. Примеры алкильных групп включают октил, нонил, ундецил, додецил, тридецил, тетрадецил, пентадецил, эйкозил, 3,7-диэтил-2,2-диметил -4-пропилнонил и тому подобное.
"Низший алкил" включает "низший циклоалкил" и подразумевает алкильные группы, состоящие из от 1 до 7 атомов углерода. Примеры низших алкильных групп включает метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил и тому подобное.
"Циклоалкил" включает "низший циклоалкил" и подразумевает углеводород, содержащий одно или более колец, состоящих из от 3 до 12 атомов углерода, при этом углеводород содержит всего до 20 атомов углерода. Примерами циклоалкильных групп являются циклопропил, циклопентил, циклогептил, адамантил, циклододецилметил, 2-этил-1-бицикло[4,4,0]децил и подобные.
"Низший циклоалкил" подразумевает углеводород, содержащий одно или более колец, состоящих из от 3 до 7 атомов углерода, при этом углеводород содержит всего до 7 атомов углерода. Примерами низших циклоалкильных групп являются циклопропил, циклопропилметил, циклобутил, 2-циклопентилэтил, циклогептил, бицикло 2,2,1-гепт-2-ил и подобные.
Термин "алкенил" включает "циклоалкенил" и "низший алкенил" и подразумевает алкенильные группы, состоящие из от 2 до 20 атомов углерода. Примеры алкенильных групп включают аллил, 5-децен-1-ил, 2-додецен-1-ил и подобные.
"Низший алкенил" включает "низший циклоалкенил" и подразумевает алкенильные группы, состоящие из от 2 до 7 атомов углерода. Примеры низших алкенильных групп включают винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и подобные.
"Циклоалкенил" включает "низший циклоалкенил" и подразумевает алкенильные группы, состоящие из от 3 до 20 атомов углерода, которые содержат кольцо, состоящее из от 3 до 12 атомов углерода, и в которых двойная связь может находиться где угодно в структуре. Примерами циклоалкенильных групп являются циклопропен-1-ил, циклогексен-3-ил, 2-виниладамант-1-ил, 5-метилендодец-1-ил и подобные.
"Низший циклоалкенил" подразумевает алкенильные группы, состоящие из от 3 до 7 атомов углерода, и которые содержат кольцо, содержащее от 3 до 7 атомов углерода, и в которых двойная связь может находиться где угодно в структуре. Примерами низших циклоалкенильных групп являются циклопропен-1-ил, циклогексен-3-ил, 2-циклопентен-1-ил и подобные.
Термин "алкинил" включает "циклоалкинил" и "низший алкинил" и подразумевает алкинильные группы, состоящие из от 2 до 20 атомов углерода. Примерами алкинильных групп являются этинил, 2-пентадецин-1-ил, 1-эйкозин-1-ил и подобные.
"Низший алкинил" включает "низший циклоалкинил" и подразумевает алкинильные группы, состоящие из от 2 до 7 атомов углерода. Примеры низших алкинильных групп включают этинил, пропаргил, 3-метил-1-пентинил, 2-гептинил и подобные.
"Циклоалкинил" включает "низший циклоалкинил" и подразумевает алкинильные группы, состоящие из от 5 до 20 атомов углерода, которые включают кольцо, состоящее из от 3 до 20 атомов углерода. Алкинильная тройная связь может находиться где угодно в группе при условии, что, если она находится в кольце, такое кольцо должно состоять из 10 членов или более. Примерами циклоалкинила являются циклододецин-3-ил, 3-циклогексил-1-пропин-1-ил и подобные.
"Низший циклоалкинил" подразумевает алкинильные группы, состоящие из от 5 до 7 атомов углерода, которые включают кольцо, состоящее из от 3 до 5 атомов углерода. Примерами низших циклоалкинильных групп являются циклопропилэтинил, 3-(циклобутил)-1-пропинил и подобные.
"Низший алкокси" подразумевает алкоксигруппы, состоящие из от 1 до 7 атомов углерода прямой, разветвленной или циклической конфигурации. Примеры низших алкоксигрупп включают метокси, этокси, пропокси, изопропокси, циклопропилокси, циклогексилокси и подобные.
"Низший алкилтио" подразумевает алкилтиогруппы, состоящие из от 1 до 7 атомов углерода прямой, разветвленной или циклической конфигурации. Примеры низших алкилтио групп включают метилтиопропилтио, изопропилтио, циклогептилтио и т.д. Для иллюстрации, пропилтиогруппа означает -SCH2CH2CH3.
"Низший алкилсульфонил" подразумевает алкилсульфонильные группы, состоящие из от 1 до 7 атомов углерода прямой, разветвленной или циклической конфигурации. Примеры низших алкилсульфонильных групп включает метилсульфонил, 2-бутилсульфонил, циклогексилметилсульфонил и т.д. Для иллюстрации, 2-бутилсульфонильная группа означает - S(O)2CH(CH3)CH2CH3.
Термин "алкилкарбонил" включает "низший алкилкарбонил" и подразумевает алкилкарбонильные группы, состоящие из от 1 до 20 атомов углерода прямой, разветвленной или циклической конфигурации. Примерами алкилкарбонильных групп являются формил, 2-метил-бутаноил, октадеканоил, 11-циклогексилундеканоил и подобные. Таким образом, 11-циклогексилдеканоильной группой является с-Нех-(CH2)10-CO-.
"Низший алкилкарбонил" подразумевает алкилкарбонильные группы, состоящие из от 1 до 8 атомов углерода, прямой, разветвленной или циклической конфигурации. Примерами низших алкилкарбонилгрупп являются формил, 2-метилбутаноил, циклогексилацетил и т.д. Для иллюстрации 2-метилбутаноильные группы означают -COCH(CH3)CH2CH3.
Термин Ph(R26)2 обозначает фенильную группу, замещенную двумя R26 заместителями.
Галоген включает F, Cl, Br и I.
Надо иметь в виду, что значение любого заместителя (например, R7, R12, R26 и т. д. ) в конкретной молекуле не зависит от его значения где-либо в другом месте молекулы. Так, -N(R12)2 представляет -NHH, -NHCH3, -NHC6H5 и т. д.
Кольца, образующиеся, когда соединяются две группы R2, включат циклопропан, циклобутан, циклопентан, циклогексан, циклогептан, циклооктан, оксетан, тетрагидрофуран, тетрагидропиран, тетрагидротиофен, тетрагидротиопиран, пирролидин, пиперидин, морфолин, тиаморфолин и пиперазин.
Гетероциклы, образованные, когда две группы R12, R20 или R27, соединяются через N, включают пирролидин, пиперидин, морфолин, тиаморфолин, пиперазин и N-метилпиперазин.
Когда Q1 и R22 и атомы углерода, к которому они присоединены, образуют кольцо, кольца, образованные таким образом, включают лактоны, лактамы и тиолактоны.
Пролекарственные эфиры по Q (то есть, когда Q=COOR6) предназначены для включения в эфиры, такие как описаны Saari et al., J. Med. Chem. 21, N 8, 746-753 (1978), Sakamoto et al., Chem. Pharm. Bull. 32, N 6, 2241-48 (1984) и Bundgaard et al., J. Med. Chem. 30, N 3, 451-454 (1987). Для значений R8 некоторыми представительными моноциклическими и бициклическими радикалами являются 2,5-диоксо-1-пирролидинил, (3-пиридинилкарбонил)амино, 1,3-дигидро-1,3-диоксо-2Н-изоиндол-2-ил, 1,3-дигидро-2Н-изoиндол-2-ил, 2,4-имидазолиндол-1-ил, 2,6-пиперидиндион-1-ил, 2-имидазолил, 2-оксо-1,3-диоксолен-4-ил, пиперидин-1-ил, морфолин-1-ил и пиперазин-1-ил.
Термин "обычная аминокислота" подразумевает следующие аминокислоты: аланин, аспарагин, аспарагиновая кислота, аргинин, цистеин, глутаминовая кислота, глутамин, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин и валин. (См. F.H.C . Crick, Simposium of the Society of Experimental Biology, 1958 (12), р. 140).
Оптические изомеры - Диастереомеры - Геометрические изомеры
Некоторые соединения, описанные здесь, имеют один или более асимметрических центров и могут, следовательно, образовывать диастереомеры и оптические изомеры. Настоящее изобретение охватывает такие возможные диастереомеры, а также их рацемические и разделенные, энантиомерно чистые формы и их фармацевтически приемлемые соли.
Некоторые из соединений, описанных здесь, содержат олефиновые двойные связи и, если не указано особо, охватывают как E, так и Z геометрические изомеры.
Соли
Фармацевтические композиции по настоящему изобретению содержат соединение формулы I в качестве активного ингредиента или его фармацевтически приемлемую соль и могут также содержать фармацевтически приемлемый носитель и необязательно другие терапевтические ингредиенты. Термин "фармацевтически приемлемая соль" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований, включающих неорганические основания и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и подобные. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в природе замещенные амины, циклические амины и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N, N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, гликозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и подобные.
Когда соединение по изобретению является основанием, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойнную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глютаминовую, бромисто-водородную, хлористо-водородную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памоевую, пантотеновую, фосфорную, янтарную, серную, виноградную, п-толуолсульфоновую и подобные. Особенно предпочтительными являются лимонная, бромистоводородная, хлористо-водородная, малеиновая, фосфорная, серная и виноградная кислоты.
Необходимо учесть, что при обсуждении способов лечения, которые представлены далее, отсылки к соединениям формулы I подразумевают также фармацевтически приемлемые соли.
Применение
Способность соединений формулы I подавлять действия лейкотриенов делает их полезными для профилактики или задержки симптомов, вызываемых лейкотриенами у человека. Эти подавления действий лейкотриена свидетельствует о том, что их соединения и фармацевтические композиции полезны при лечении, профилактике или улучшении состояний млекопитающих и особенно человека:
1) легочных заболеваний, включая болезни, такие как астма, хронический бронхит и относящиеся к закупоривающим дыхательные пути заболеваниям,
2) аллергии и аллергических реакций, таких как аллергический ринит, контактный дерматит, аллергический конъюнктивит и подобные,
3) воспалений, таких как артрит или кишечные воспалительные заболевания,
4) болей,
5) кожных заболеваний, таких как atopic экзема и тому подобное,
6) сердечно-сосудистых заболеваний, таких как ангина, ишемия миокарда, гипертония, агрегация тромбоцитов и тому подобное,
7) почечная недостаточность из-за ишемии, вызываемой иммунологической или химической (циклоспорин) этиологией,
8) мигрени или cluster головной боли,
9) глазных заболеваний, таких как увеит,
10) гепатита, полученного в результате химического, иммунологического или инфекционного раздражения,
11) травматических или шоковых состояний, таких как родовые травмы, эндотоксикоз и тому подобное,
12) отторжения аллотрансплантата,
13) защиты от побочных эффектов, связанных с терапевтическим назначением цитокинов, таких как интерлейкин II, и фактор некроза опухолей,
14) хронических заболеваний легких, таких как цистический фиброз, бронхит и другие заболевания мелких и крупных дыхательных путей, и
15) холецистит.
Таким образом, соединения по настоящему изобретению могут также использоваться для лечения или профилактики заболеваний млекопитающих (особенно человека), таких как эрозийный гастрит; эрозийное воспаление пищевода; понос; спазм сосудов головного мозга; преждевременные роды; самопроизвольный выкидыш; дисменоррея; ишемия; повреждения, вызванные вредными агентами, или омертвление тканей печени, поджелудочной железы, почек или миокарда; повреждения основной ткани печени, вызванные гепатоксичными агентами, такими как CCl4 и D-галактозамином; ишемическая почечная недостаточность; повреждения печени, вызванные болезнью; вызванные солями желчных кислот повреждения поджелудочной железы или желудка; клеточные нарушения, вызванные травмой или стрессом; и почечная недостаточность, вызванная глицерином.
Цитозащитная активность соединения может наблюдаться у животных и у человека путем фиксирования пониженной устойчивости слизистой желудка к вредному действию сильных раздражающих средств, например к вызывающему язву желудка действию аспирина или индометацина. В дополнение к ослаблению действия нестероидных противовоспалительных препаратов на желудочно-кишечный тракт, исследования на животных показывают, что цитозащитные соединения будут предохранять от повреждения желудка, вызываемого оральным введением сильных кислот, сильных оснований, этанола, гипертонических физиологических растворов и тому подобное.
Для измерения цитозащитной способности могут быть применены два испытания. Этими испытаниями являются: (A) исследование повреждения, вызванного этанолом, и (B) исследование язвы, вызванной индометацином, и они описаны в EP N 140684.
Уровни доз
Интенсивность профилактической или терапевтической дозы соединения формулы I будет, конечно, изменяться в зависимости от природы и серьезности состояния, которое подвергают лечению и от конкретного соединения формулы I и пути его введения. Она будет также изменяться в зависимости от возраста, веса и восприимчивости отдельного пациента. Обычно уровень дневной дозы колеблется для противоастматического, противоаллергического или противовоспалительного применения и обычно при использовании не для цитозащиты лежит в области от около 0,001 мг до около 100 мг на 1 кг веса тела млекопитающего, предпочтительно от 0,01 до 10 мг на 1 кг и более предпочтительно от 0,1 до 1 мг на 1 кг, в виде единичной или дробных доз. С другой стороны, в некоторых случаях могут быть необходимы дозы выше этих пределов.
Для применения, где используется композиция для внутривенного применения, подходящий уровень доз для противоастматического, противовоспалительного или противоаллергичесого применения составляет от около 0,001 мг до около 25 мг (предпочтительно от 0,01 мг до около 1 мг) соединения формулы I на 1 кг веса тела в день и для цитозащитного применения от около 0,1 мг до около 100 мг (предпочтительно от около 1 мг до около 100 мг и более предпочтительно от около 1 мг до около 10 мг) соединения формулы I на 1 кг веса тела в день.
В случае для применения, где используется композиция для орального применения, подходящий уровень доз для противоастматического, противовоспалительного или противоаллергического применения составляет, например, от около 0,01 мг до около 100 мг соединения формулы I на 1 кг веса тела в день, предпочтительно от около 0,1 мг до около 10 мг на 1 кг веса тела в день, и для цитозащитного применения от около 0,1 мг до около 100 мг (предпочтительно от около 1 мг до около 100 мг и более предпочтительно от около 10 мг до около 100 мг) соединения формулы I на 1 кг веса тела в день.
Для лечения заболеваний глаз могут быть использованы глазные препараты для глазного применения, содержащие 0,001-1% по весу растворов или суспензий соединений формулы I в подходящих глазных составах.
Точное количество соединения формулы I, предназначенное для использования в качестве цитозащитного агента, будет зависеть, кроме всего прочего, от того, где оно будет назначено: для лечения пораженных клеток или для предотвращения будущих поражений, от природы пораженных клеток (например, желудочно-кишечные язвы в сравнении с невротическим некрозом) и от природы вызывающего заболевание агента. Примером использования соединения формулы I для предотвращения дальнейших поранении могло бы быть назначение совместно с соединением формулы I и NSAID, что могло бы в противном случае вызвать такое поражение (например, индометацин). При таком применении соединение формулы I назначается от за 30 минут до и до 30 минут после введения NSAID. Предпочтительно его назначают до или одновременно с NSAlD (например, в комбинированной дозированной форме).
Фармацевтические композиции
Может быть использован любой подходящий путь введения для обеспечения млекопитающему, особенно человеку, эффективной дозировки соединения по настоящему изобретению. Например, можно использовать оральный, ректальный, наружный, парентеральный, глазной, легочный, носовой и тому подобное. Дозированные формы включают таблетки, лепешки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и тому подобное.
Фармацевтические композиции по настоящему изобретению содержат соединение формулы I в качестве активного ингредиента или его фармацевтически приемлемую соль и может также содержать фармацевтически приемлемый носитель и необязательно другие терапевтические ингредиенты. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксических оснований и кислот, включая неорганические основания или кислоты и органические основания или кислоты.
Композиции включают композиции, подходящие для орального, ректального, наружного, парентерального (включая подкожное, внутримышечное и внутривенное), глазного (офтальмологические), легочного (ингаляция через нос или рот) или носового введения, хотя наиболее подходящий путь в любом данном случае будет зависеть от природы и серьезности состояний, подвергаемых лечению, и от природы активного ингредиента. Удобным образом они могут быть представлены в единичной дозированной форме и получены любым из способов, хорошо известных специалистам.
Для введения путем ингаляции соединения по настоящему изобретению удобно использовать в виде аэрозольных брызг из баллончиков под давлением или распылителей. Соединения могут также приниматься в виде порошков, которые могут быть приготовлены в виде составов, и порошковая композиция может вдыхаться с помощью устройства для вдыхания порошка. Предпочтительной системой для ингаляции является аэрозоль для ингаляций с дозированной подачей (MDI - metered dose inhalation), который может состоять из суспензии или раствора соединения формулы I в подходящих газах-вытеснителях, таких как фторуглероды или углеводороды.
Подходящие наружные составы соединения формулы I включают трансдермальные устройства, аэрозоли, кремы, мази, лосьоны, антисептические порошки для присыпки ран и тому подобное.
При практическом применении соединения формулы I могут быть соединены в качестве активного ингредиента в однородной смеси с фармацевтическим носителем в соответствии с обычными фармацевтическими методами приготовления смесей. Носитель может принимать широкое разнообразие форм, в зависимости от желаемой для введения формы препарата, например оральной или парентеральной (включая внутривенную). При приготовлении композиций для оральных дозированных форм в случае жидких оральных препаратов, таких как, например, суспензии, эликсиры и растворы, можно использовать любую обычную фармацевтическую среду, такую как, например, вода, гликоли, масла, спирты, ароматизирующие агенты, консерванты, окрашивающие агенты и тому подобное; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие агенты, связующие, разрыхляющие агенты и тому подобное, в случае оральных твердых препаратов, таких как, например, порошки, капсулы и таблетки, с предпочтением, отдаваемым твердым оральным препаратам по сравнению с жидкими препаратами. Ввиду легкости введения таблетки и капсулы представляют наиболее удобную оральную форму дозированных единиц, в случае которой, очевидно, применяются твердые фармацевтические носители. Если желательно, таблетки могут быть покрыты с использованием стандартных водной или неводной технологий.
Кроме обычных дозированных форм, указанных выше, соединения формулы I могут также вводиться с помощью средств и/или выделяющих устройств с контролируемым высвобождением, таких как описаны в патентах США NN 3845770, 3916899, 3536809, 3598123, 3630200 и 4008719, описания которых включены здесь в качестве ссылок.
Фармацевтические композиции по настоящему изобретению, подходящие для орального введения, могут быть представлены в виде дробных единиц, таких как капсулы, облатки или таблетки, содержащие каждая заранее определенное количество активного ингредиента в виде порошка или гранул или в виде раствора или суспензии в водной среде, в неводной среде, в эмульсии типа масло-в-воде или в жидкой эмульсии типа вода-в-масле. Такие композиции могут быть получены любым из способов, применяемых в фармации, но все способы включают стадию связывания в ассоциацию активного ингредиента с носителем, который содержит один или более необходимых ингредиентов. Обычно композиции получают путем однородного и тщательного смешивания активного ингредиента с жидкими носителями или хорошо размельчающимися твердыми носителями или с обоими, и затем, если необходимо, придание продукту желаемого вида. Например, таблетка может быть получена прессованием или путем формовки необязательно с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть получены прессованием в соответствующей машине активного ингредиента в свободнотекущем виде, таком как порошок или гранулы, необязательно смешанного со связующим агентом, смазывающим агентом, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки могут быть получены формованием в подходящей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Желательно, чтобы каждая таблетка содержала от около 1 мг до около 500 мг активного ингредиента и каждая облатка или капсула содержала от около 1 до около 500 мг активного ингредиента.
Далее приведены примеры представителей фармацевтических дозированных форм соединений формулы I.
Суспензия для инъекции (внутримышечно), мг/мл:
Соединение формулы I - 10
Mетилцеллюлоза - 5,0
Твин 80 - 0,5
Бензиловый спирт - 9,0
Бензалконий хлорид - 1,0
Вода для инъекций - До общего объема 1 мл
Таблетка, мг/таблетка:
Соединение формулы I - 25
Микрокристаллическая целлюлоза - 415
Повидон - 14,0
Предварительно желатинированный крахмал - 43,5
Стеарат магния - 2,5 - 500
Капсула, мг/капсула:
Соединение формулы I - 25
Порошок лактозы - 573,5
Стеарат магния - 1,5 - 600
Аэрозоль, на баллончик:
Соединение формулы I, мг - 24
Лецитин, жидкий концентрат NF, мг - 1,2
Трихлорфторметан, NF, г - 4,025
Дихлордифторметан, NF, г - 12,15
Сочетание с другими лекарствами
Кроме соединений формулы I фармацевтические композиции по настоящему изобретению могут также содержать другие активные ингредиенты, такие как ингибиторы циклооксигеназы, нестероидные противовоспалительные лекарства (NSAID), периферические анальгетические агенты, такие как zomepirac diflunisal и тому подобное. Массовое отношение соединения формулы I ко второму активному ингредиенту может варьироваться и будет зависеть от эффективной дозы каждого ингредиента. Обычно будет использоваться эффективная доза каждого. Так например, когда соединение формулы I соединено с NSAID, массовое отношение соединения формулы I к NSAID будет обычно колебаться в области от около 1000:1 до около 1:1000, предпочтительно от около 200:1 до около 1:200. Сочетания соединения формулы I и других активных соединений будут обычно в вышеуказанной области, но в каждом случае будет использоваться эффективная доза каждого активного ингредиента.
NSAID может быть охарактеризован в виде пяти групп:
(1) производные пропионовой кислоты,
(2) производные уксусной кислоты,
(3) производные фенамовой кислоты,
(4) оксинамы, и
(5) производные бифенилкарбоновой кислоты,
или его фармацевтически приемлемая соль.
Производные пропионовой кислоты, которые могут быть использованы, включают: альминопрофен, беноксапрофен, буклоксовая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флубипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, прано-профен, супрофен, тиопрофеновую кислоту и тиоксапрофен. Близкие по структуре производные пропионовой кислоты, обладающие подобными анальгетическими и противовоспалительными свойствами, также предназначаются для включения в эту группу.
Таким образом, "производные пропионовой кислоты", как определено здесь, являются ненаркотическими анальгетическими/нестероидными противовоспалительными лекарственными препаратами, имеющими свободную группу -CH(CH3)COOH или -CH2CH2COOH (которая необязательно может быть в форме фармацевтически приемлемой солевой группы, например -CH(CH3)COO-Na+ или -CH2CH2COO-Na+), обычно присоединенной непосредственно или через карбонильную группу к кольцевой системе, предпочтительно к ароматической кольцевой системе.
Производные уксусной кислоты, которые могут использоваться, включают: индометацин, который является предпочтительным NSAID, ацеметацин, альклофенак, клиданак, диклофенак, фенклофенак, фенклозиновую кислоту, фентиазак, фурофенак, ибуфенак, изоксепак, сулиндак, тиопенак, толметин, зилометацин и зонепирак. Близкие по структуре производные уксусной кислоты, обладающие подобным анальгетическим и противовоспалительным свойствами, также предназначаются для включения в эту группу.
Таким образом, "производные уксусной кислоты", как определено здесь, являются ненаркотическими анальгетическими/нестероидными противовоспалительными лекарственными препаратами, имеющими свободную группу -CH2COOH (которая необязательно может быть в форме фармацевтически приемлемой солевой группы, например, -CH2COO-Na+), обычно присоединенную непосредственно к кольцевой системе, предпочтительно к ароматической или гетероциклической кольцевой системе.
Производные фенамовой кислоты, которые могут использоваться, включают: флуфенамовую кислоту, меклофенамовую кислоту, нифлумовую кислоту и толфенамовую кислоту. Близкие по структуре производные фенамовой кислоты, обладающие подобными анальгетическими и противовоспалительными свойствами, также предназначены для
включения в эту группу.
Таким образом, "производные фенамовой кислоты", как определено здесь, являются ненаркотическими анальгетическими/нестероидными противовоспалительными лекарственными препаратами, которые содержат основную структуру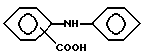
которая может содержать разнообразные заместители и в которых свободная группа -COOH может быть в форме фармацевтически приемлемой солевой группы, например -COO-Na+.
Производные бифенилкарбоновой кислоты, которые могут использоваться, включают: дифлунизал и флуфенизал. Близкие по структуре производные бифенилкарбоновой кислоты, обладающие подобными анальгетическими и противовоспалительными свойствами, также предназначаются для включения в эту группу.
Таким образом, "производные бифенилкарбоновой кислоты", как определено здесь, являются ненаркотическими анальгетическими/нестероидными противовоспалительными лекарственными препаратами, которые содержат основную структуру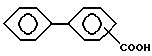
которая может содержать разнообразные заместители и в которых свободная группа -COOH может быть в форме фармацевтически приемлемой солевой группы, например -COO-Na+.
Оксикамы, которые могут использоваться по настоящему изобретению, включают: изоксикам, пироксикам, судоксикам и теноксикан. Близкие по структуре оксикамы, обладающие подобными анальгетическими и противовоспалительными свойствами, также предназначаются для включения в эту группу.
Таким образом, "оксикамы", как определено здесь, являются ненаркотическими анальгетическими/нестероидными противовоспалительными лекарственными препаратами, которые содержат основную структуру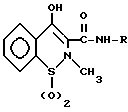
где R является арильной или гетероарильной кольцевой системой.
Могут использоваться также следующие NSAID: амфенак натрия, аминопрофен, анитразафен, антрафенин, ауранофин, бендазак, лизинат, бензиданин, бенпрозин, броперамол, буфезолак, цинметацин, ципроквазон, клоксимат, дазидамин, дебоксамет, делметацин, детомидин, дексиндопрофен, диацереин, ди-физаламин, дифенпирамид, эморфазон, энфенамовая кислота, эноликам, эпиризол, этерзалат, этодолак, этофенамат, фанетизол мезилат, фенклорок, фендозал, фенфлумизол, фепразон, флоктафенин, флуниксин, флуноксапрофен, флупроквазон, фопиртолин, форфозал, фурклопрофен, глюкаметацин, гаимезал, ибупроксам, изофезолак, изониксим, изопрофен, изоксикан лефетамин HCl, лефлуномид, лофемизол, лоназолак кальция, лотифазол, локсопрофен, лизин клониксинат, меклофенамат натрия, мезеклазон, набуметон, никтиндол, немезулид, орпаноксим, оксаметицин, оксападол, перизоксал цитрат, пимепрофен, пиметацин, пипроксен, пиразолак, перфенидон, проглюметацин малеат, проквазон, пиридокси профен, судоксикам, талметацин, талнифлумат, теноксикам, тиазолин бутазон, тиелавин B, тиарамид HCl, тифламизол, тимегадин, толпадол, триптамид и уфенамат.
Могут также использоваться следующие NSAID, обозначенные числовым кодом компании (см. , например, Pharmaprojects): 480156, AA861, AD1590, AFP802, AFP860, AI77B, AP504, AU800, BPPC, BW540C, CHIN01N127, CN100, EB382, EL508, F1044, GV3658, ITF182, KCNTEI6090, KME4, LA2851, MR714, MR897, MY309, ON03144, PR823, PV102, PV108, R830, RS2131, SCR152 SH440, SIR133, SPAS510, SQ27239, ST281, SY6001, TA60, TAI-901(4-бензоил-1-индaнкapбoнoвaя кислота), TVX2706, U60257, UR2301 и WY41770.
Наконец, NSAID, которые также могут использоваться, включают салицилаты, особенно ацетилсалициловую кислоту и фенилбутазоны и их фармацевтически приемлемые соли.
Кроме индометацина другими предпочтительными NSAID являются ацетилсалициловая кислота, диклофенак, фенбуфен, фенопрофен, флурбипрофен, ибупрофен, кетопрофен, напроксен, фенилбутазон, пироксикам, сулиндак и толметин.
Фармацевтические композиции, включающие соединения формулы I, могут также содержать ингибиторы биосинтеза, лейкотриенов, такие как описаны в Европейском патенте N 138481 (24 апреля 1985 г.), Европейском патенте N 115394 (8 августа 1984 г.), Европейском патенте N 136893 (10 апреля 1985 г.) и Европейском патенте N 140709 (8 мая 1985 г.), которые включены здесь в качестве ссылки.
Соединения формулы I могут также использоваться в сочетании с антагонистами лейкотриенов, такими как описаны в Европейском патенте N 106565 (24 апреля 1985 г.) и Европейском патенте N 104885 (4 апреля 1984 г.), которые включены здесь в качестве ссылки, и другими, известными в данной области, которые описаны в Европейских патентных заявках NN 56172 (21 июля 1982) и 61800 (10 июня 1982); и в описании патента Великобритании N 2058785 (15 апреля 1981), которые включены здесь в качестве ссылки.
Фармацевтические композиции, включающие соединения формулы I, могут также содержать второй активный ингредиент, антагонисты простагландина, такие, как описаны в Европейском патенте 11067 (28 мая 1980), или антагонисты тромбоксана, такие, как описаны в патенте США N 4237160. Они могут также содержать ингибиторы гистидин декарбоксилазы, такие как α-фторметилгистидин, описанный в патенте США N 4325961. Соединения формулы I могут также быть с успехом объединены с антагонистом H1- или H2-рецептора, такие как, например, ацетамазол, аминотиадиазолы, описанные в Европейском патенте N 40696 (2 декабря 1981), бенадрил, циметидин, фамотидин, фрамамин, гистадил, фенерган, рантидин, терфенадин и подобные соединения, такие, как описаны в патенте США NN 4283408, 4362736 и 4394508. Фармацевтические композиции могут также содержать ингибитор K+/H+ АТФазы, такой как омепразол, описанный в патенте США N 4255431, и тому подобное. Соединения формулы I могут быть также удобно объединены с большинством стабилизирующих клетки агентов, такими как 1,3-бис(2-карбоксихромон-5-илокси)-2-гидроксипропан и близкие по структуре соединения, раскрытые в описании патента Великобритании NN 1144905 и 1144906. Другая полезная фармацевтическая композиция, содержащая соединения формулы I в сочетании с антагонистами серотонина, такими как метилсергид, антагонистами серотонина, описанными в Nature, 316, 126-131 (1985), и тому подобными. Каждая из отсылок, приведенная в этом абзаце, включена здесь в качестве ссылки.
Другие успешные фармацевтические композиции содержат соединения формулы I в сочетании с антихолинергическими агентами, такими как ипратропий бромид, бронходиляторами, такими как бета агонист сальбутамол, метапротеренол, тербуталин, фенотерол и подобными, и антиастматическими лекарственными препаратами теофиллином, холин теофиллинатом и энпрофиллином, кальциевыми антагонистами нифедипином, дилтриаземом, нитрендипином, верапрамилом, нимодипином, фелодипином и т.д., и кортикостероидами гидрокортизоном, метилпреднизолоном, бетаметазоном, дексаметазоном, беклометазоном и тому подобными.
Способы синтеза
Соединения по настоящему изобретению могут быть получены согласно следующим способам (см. методы A -L, приведенные в конце описания). Температура указана в градусах Цельсия.
Способ A
Метиловый эфир II обрабатывают избытком восстанавливающего агента, такого как литийалюминийгидрид, в растворителе, таком как ТГФ, при 0oC с получением спирта, который окисляют с помощью реагента, такого как диоксид марганца, с получением альдегида III. Соединение III подвергают конденсации с ацетоном в основной среде с образованием тиенопиридина IV, который преобразуют в 2,3-дизамещенный тиенопиридин V в соответствии с методиками, описанными в способах B, C и D. Обработка тиенопиридина V галогенирующим агентом, таким как NBS, с последующим взаимодействием с трифенилфосфином дает фосфониевую соль VI. Взаимодействие VI с альдегидом VII в присутствии сильного основания, такого как трет-бутоксид калия, бис(триметилсилил)амид калия или бутиллития, с последующим гидролизом с помощью водного гидроксида натрия дает VIII. Примеры VII описаны в патенте США N 5104882 (способы D и I) и в Европейском патенте N 480717 (способ H), а также в настоящих примерах.
Способ B
Обработка тиенопиридина IV, полученного по способу A, хлорирующим агентом, таким как трихлоризоциануровая кислота или сульфурилхлорид, дает 2,3-дихлортиенопиридин Ve. Взаимодействие IV с хлором в концентрированной серной кислоте в присутствии сульфата серебра приводит к 3-хлортиенилпиридину Vf. Обработка IV сильным основанием, таким как алкиллитий или LDA, дает анион тиенопиридин-2-ила, который взаимодействует с различными электрофилами, давая различный заместитель по второму положению IV; например, анион 1) взаимодействует с NCS или хлором с получением 2-хлортиенопиридина Va; 2) взаимодействует с N-фтор-бис(бензолсульфонил)амидом (PhS(O)2)2NF или перхлоратом фтора (FClO4) с получением 2-фтортиенопиридина Vb; 3) взаимодействует с бpoмциaнoм (BrCN) с получением 2-цианотиенопиридина Vc; и 4) взаимодействует с ангидридом трифторметансульфоновой кислоты с получением 2-трифторметилсульфонилтиенопиридина Vd.
Способ C
2-Хлор- или 2-фтортиенопиридин (Va, b) преобразуют в различные 2,3-дизамещенные тиенопиридины путем следующей последовательности:
1) депротонирование 2-хлор- или 2-фтортиенопиридина (Va, b) с помощью сильного основания, такого как алкиллитий или LDA, дающее 2-хлор- или 2-фтортиенопиридин-3-ил анион;
2) взаимодействие аниона с различными электрофилами для образования 2,3-дизамещенных тиенопиридинов: например, взаимодействие с N-фтор-бис(бензолсульфонил)амидом или перхлоратом фтора с получением Vh; взаимодействие с ангидридом трифторметансульфоновой кислотой для получения Vi; взаимодействие с N-бромсукцинимидом или бромом для получения Vj; и взаимодействие с N-хлорсукцинимидом или хлором для получения Vk.
2-Хлор-3-фтортиенопиридин (Vh, X= Cl) преобразуют в 3-фтортиенопиридин (Vg) путем следующей последовательности:
1) взаимодействие с трет-бутиллитием в ТГФ;
2) протонирование с помощью воды.
Способ D
3-Хлор- или 3-фтортиенопиридин (Vf, g), полученный по способу B или способу C, депротонируют с помощью сильного основания, такого как алкиллития или LDA, для получения 3-хлор- или 3-фтортиенопиридино-2-ил аниона, который взаимодействует с различными электрофилами с получением 2,3-дизамещенных тиенопиридинов; например, взаимодействие с бромцианом дает VI; взаимодействие с ангидридом трифторметансульфоновой кислотой дает Vm; взаимодействие с хлорангидридом метансульфоновой кислоты дает Vn; взаимодействие с N-фтор-бис(бензолсульфонил)амидом или перхлоратом фтора дает Vo; и взаимодействие с N-хлорсукцинимидом или хлором дает Vp.
Способ E
Двойная связь в соединении VIII восстанавливается до одинарной связи бораном в ТГФ. Таким образом, обработка VIII избытком борана в ТГФ с последующим гидролизом метилового эфира дает кислоту IX.
Способ F
Йодпиридин XI взаимодействует с триметилсилилацетиленом (X) в присутствии комплекса Йодида меди (I) и хлорида трифенилфосфинпалладия (II) с получением фуранопиридина XII, который преобразуется в 2,3-дихлорфуранопиридин XIVa путем хлорирования с помощью трихлоризоциануровой кислоты или сульфурилхлорида или преобразуется в XIII путем снятия силильной группы с помощью фтористого водорода в присутствии пиридина. Оба XIVa и XIII преобразуются в различные 2,3-дизамещенные фуранопиридины XIV путем взаимодействий, описанных в способах B, C, D и J. В заключение XIV преобразуют в кислоту XV с использованием приемов, описанных в способе A.
Способ G
Альдегид III, полученный по способу A, подвергают конденсации с натриевой солью пировиноградной кислоты с последующей этерификацией метанолом в присутствии концентрированной соляной кислоты с получением метилового эфира XVI. Хлорирование XVI либо с помощью сульфурилхлорида, либо с помощью трихлоризоциануровой кислоты дает 2,3-дихлортиенопиридин XVII. XVII преобразуют в фосфониевую соль XVIII путем следующей последовательности:
1) восстановлением с помощью DIBAL в ТГФ;
2) замещением гидроксигруппы хлором взаимодействием с хлорирующим агентом, таким как тионилхлорид; и
3) взаимодействием с трифенилфосфином в органическом растворителе, таком как толуол или ацетонитрил. XVIII преобразуют в конечный продукт VIII методом, описанным в способе A.
Способ H
Соединение XIX обрабатывают хлорангидридом кислоты в присутствии основания с последующим взаимодействием с пентасульфидом фосфора в ТГФ в присутствии основания, такого как Na2CO3 с получением тиазолпиридина XX. Окисление XX с помощью MCPBA дает N-оксид, который взаимодействует с триметилсилицианидом и диалкилкарбамоилхлоридом с образованием нитрила XXI. Нитрил XXI преобразуют в фосфониевую соль путем следующей последовательности:
1) восстановление нитрила XXI с помощью DIBAL в ТГФ с получением альдегида;
2) восстановление альдегида с помощью NaBH4 в ТГФ-CH3OH;
3) получение мезилата спирта взаимодействием с мезилхлоридом в присутствии триэтиламина; и
4) взаимодействие мезилата с трифенилфосфином.
Фосфониевую соль преобразуют в конечную кислоту методами, описанными в способе A.
Способ I
Эфир тиофена XXIV, полученный согласно литературному методу (K.H.Weber and H. Daniel; Annalen (1979) 328; H.K.Gakhar, A.Khanna P.Baveja, Indian J. Chem. 16B (1928) 305), преобразуют в тиенопиридин XXV путем следующей последовательности:
1) восстановлением литийалюминийгидридом в ТГФ;
2) окислением оксидом марганца; и
3) конденсацией с ацетоном в присутствии основания, такого как гидроксид натрия.
XXV преобразуют в XXVI путем приемов, описанных в способе J. В заключение XXVI преобразуют в кислоту XXVII, используя приемы, описанные в способе A.
Способ J
Тиенопиридин XXV подвергают хлорированию либо сульфурилхлоридом, либо трихлорциануровой кислотой с получением 2,3-дихлортиенопиридина XXVIa.
Депротонирование XXV с помощью сильного основания, такого как алкиллитий или LDA, в ТГФ приводит к образованию тиенопиридин-2-ил аниона, который взаимодействует с N-хлорсукцинимидом или хлором с получением 2-хлортиенопиридина XXVIb; или он взаимодействует с N-фтор-бис(бензолсульфонил)амидом или перхлоратом фтора с получением 2-фтортиенопиридина XXVIc.
Депротонирование XXVIc либо с помощью алкиллития, либо с LDA с последующим взаимодействием с N-фтор-бис(бензолсульфонил)амидом или перхлоратом фтора дает дифтортиенопиридин XXVIi.
Депротонирование XXVIb либо с помощью алкиллития, либо с LDA с последующим взаимодействием с электрофильным агентом дает 2,3-дизамещенный тиенопиридин; например, взаимодействие с бромцианом дает XXVIe; взаимодействие с N-фтор-бис(бензолсульфонил)амидом или перхлоратом фтора дает XXXIf; взаимодействие с ангидридом трифторметансульфоновой кислоты дает XXVId.
Обработка XXVIa или XXVIf трет-бутиллитием с последующим гашением водным хлоридом аммония дает XXVIh или XXVIg соответственно.
Способ K
Кетон XXVIII преобразуют в хиральный аллильный спирт XXIX следующей последовательностью:
1) хиральное восстановление по методу Corey (комплекс BH3/оксазаборолидин) (J. Am. Chem. Soc., 1987, 109, 5551 и 7925);
2) взаимодействие α-бромметилакрилового эфира в присутствии основания;
3)восстановление с помощью DIBAL.
Обработка XXIX диазометаном/Pd (OAc)2, затем мезилхлоридом и триэтиловым амином с последующим замещением с цианидом натрия и затем гидролиз гидроксидом калия дает кислоту XXX. Кислоту XXX преобразуют в третичный спирт XXXI путем образования литиевого производного с помощью н-BuLi с последующим прибавлением ацетона. Оба, и XXX, и XXXI, преобразуют в альдегиды XXXII и XXXIII посредством следующих взаимодействий:
1) этерификацией диазометаном;
2) удалением ТНР-защитной группы с помощью PPTS, и
3) окислением с помощью оксида марганца.
Альдегиды XXXII и XXXIII преобразуют в конечную кислоту XXXIIIa путем приемов, описанных в способе A.
Способ L
3-Аминотиофен XXXIV преобразуют в аминокетон XXXV взаимодействием с бромкетоном XL (полученным из известного соединения α,α-дигидроксиацетона в две стадии:
1) монозащитой с помощью TBDMSCl;
2) бромированием с помощью CBr4 и DIPHOS в присутствии основания, такого как K2CO3.
XXXV преобразуют в тиенопиразин путем следующей последовательности:
1) бромированием по α-положению тиофенового кольца с помощью одного эквивалента брома;
2) обработкой бромпроизводного жидким аммиаком при -80oC; и
3) окислением кислородом.
XXXVI преобразуют во фтортиенопиразин XXXVII с помощью приемов, описанных в способе B.
Фосфониевую соль XXXVIII получают из XXXVII путем следующей последовательности:
1) удаление TBDMS эфира с помощью PPTS;
2) бромирование с помощью тетрабромида углерода и DIPHOS; и
3) взаимодействие с трифенилфосфином.
Конечный продукт XXXIX получают из фосфониевой соли XXXVIII с использованием приемов, описанных в способе A.
Представительные соединения
Табл. 1 и 2 иллюстрируют соединения, которые являются представительными соединениями по настоящему изобретению. В данных таблицах
Y1 обозначает -X2(C(R3)2)mZ1 (CR3R22)pQ1 и
W1 обозначает -X3(C(R3)2)m'Z2 (CR3R4)p'Q2 в формуле I.
Соединения в табл. 1 являются соединениями формулы Ic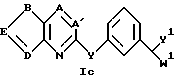
Соединения в табл. 2 являются соединениями формулы I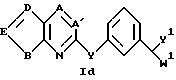
Исследования по определению биологической активности
Свойства лейкотриеновых антагонистов соединений по настоящему изобретению оценивают, используя следующие исследования:
1. Изучение связывания рецепторов [3H]ЛТD4 в ДМСО-дифференцированных клетках U 937 (человеческая моноцитарная клеточная линия).
2. Связывание рецепторов [3H]ЛТD4 на мембранах легких морских свинок.
3. Связывание рецепторов [3H]ЛТD4 на мембранах легких человека.
4. In vitro трахея морской свинки.
5. Исследования in vivo на анестезированных морских свинках.
Вышеуказанные испытания описаны T. R. Jcnes et al., Can. J. Physiol. Pharmacol. 1991, 69, 1847-1854.
Исследования на астматических крысах
Крысы получены из линии с врожденной астмой. Используются как самки (190-250 г), так и самцы (260-400 г).
Яичный альбумин (ЯА), степень чистоты V, кристаллизованный и лиофилизированный, получен от фирмы Sigma Chemical Cj., St. Louis. Гидроксид алюминия получен от фирмы Regis Chemical Company, Chicago. Бималеат метилсергида поставлен фирмой Sandoz Ltd., Basel.
Контрольное заражение и последующие регистрации дыхания проводят в чистой пластиковой коробке с внутренними размерами 10х6х4 дюймов. Верх коробки снимается; при использовании его крепко закрепляют четырьмя зажимами и герметичность поддерживают с помощью мягкой резиновой прокладки. Из центра каждого конца камеры вставлены через герметичные пломбы распылители De Vilbiss (N 40) и каждый конец коробки также имеет выходное отверстие. Пневмотахограф Fleisch N 0000 вставлен в один конец коробки и связан с датчиком измерения давления (РТ5-А), который затем присоединен к предварительному усилителю, Buxco Electronics (Buxco Electronics Inc., Sharon., Conn). Предварительный усилитель присоединен к динографу Beckman Tyre R Dynograph и к компьютеру Buxco, содержащему анализатор колебаний, Data Acquisition Logger, со специальным программным обеспечением. При аэрозолизации антигена выходные отверстия открыты и пневмотахограф изолирован от камеры. Отверстия закрыты и пневмотахограф и камера соединены при регистрации дыхательных моделей. Для контрольного заражения 2 мл 3% раствора антигена в физиологическом растворе помещают в каждый распылитель и аэрозоль поступает с воздухом из небольшого диафрагменного насоса Potter при 10 psi и скорости потока 8 литров/минуту.
Крыс активируют путем инъекции (подкожно) 1 мл суспензии, содержащей 1 мл ЯА и 200 мг гидроксида альбумина в физиологическом растворе. Они используются в период от 12 до 24 дней после активирования. С целью исключения серотонинового компонента из ответа крыс предварительно обрабатывали 3,0 мкг/кг метилсергида внутривенно за 5 минут до аэрозольного контрольного заражения. Крыс затем обрабатывают аэрозолем 3% ЯА в физиологическом растворе точно в течение 1 минуты, затем регистрируют их характеристики дыхания в течение дальнейших 30 минут. Продолжительность непрерывной одышки измеряют с помощью компьютера Buxco.
Соединения обычно вводят либо орально за 2-4 часа до контрольного заражения, либо внутривенно за 2 минуты до контрольного заражения. Их либо растворяют в физиологическом растворе или в 1% метоцела, либо суспендируют в 1% метоцела. Вводимый объем составляет 1 мл/кг (внутривенно) или 10 мл/кг (орально). Перед оральной обработкой крысы голодают в течение ночи. Активность соединений определяют по степени их способности уменьшать продолжительность одышки, вызванной антигеном, по сравнению с контрольной группой, обработанной индифферентным растворителем. Обычно соединение оценивают в серии доз и определяют ED50. Эта величина установлена как доза (мг/кг), которая могла бы ингибировать продолжительность симптомов на 50%.
Легочная механика у обученных чувствительных беличьих обезьян
Методика исследования включает усаживание обученных беличьих обезьян на стулья в камеру, обрабатываемую аэрозолем. В целях контроля регистрируют легочно-механические измерения параметров дыхания в течение периода около 30 минут для установления у каждой обезьяны нормального контрольного значения на этот день. При оральном введении соединения растворяют или суспендируют в 1% растворе метоцела (метилцеллюлоза, 65 HG, 400 сП) и дается в объеме 1 мл/кг веса тела. Для аэрозольного введения соединения используют ультразвуковой распылитель De Vilbiss. Период предварительной обработки варьируется от 5 минут до 4 часов перед тем, как обезьян подвергают контрольному заражению аэрозольными дозами либо лейкотриенов D4 (ЛТД4) либо антигена Ascaris suum; разбавление 1:25.
После контрольного заражения данные каждой минуты рассчитываются с помощью компьютера в виде процентного изменения относительно контрольных значений для каждого параметра дыхания, включая устойчивость дыхательных путей к воздействию (RL) и динамическую податливость (Cdyn). Результаты для каждого исследуемого соединения по существу являются полученными для минимального периода в 60 минут после контрольного заражения, которое затем сравнивают с ранее полученными естественными базовыми контрольными значениями для данной обезьяны. Кроме того, полные значения в течение 60 минут после контрольного заражения для каждой обезьяны (естественные базовые контрольные значения и данные исследований) отдельно усредняются и используются для расчета общего ингибирования в процентах исследуемым соединением реакции на ЛТД4 или антиген Ascaris. Для статистического анализа используют спаренный t-тест (ссылки: McFarlane, C. S. et al. Prostaglandins, 28, 173-182 (1984) и McFarlane, C.S. et al, Agents Actions, 22, 63-65 (1987)).
Предупреждение вызванного сужения просвета бронхов у овец с аллергией
A. Обоснование.
Некоторые аллергические овцы с известной чувствительностью к специфическому антигену (Ascaris suum) реагируют на ингаляционное контрольное заражение острыми и замедленными бронхиальными реакциями. Протекание во времени как острых, так и замедленных бронхиальных реакций соответствует протеканию во времени, наблюдаемому при астме, а фармакологическая модификация обеих реакций подобна обнаруженным у человека. Действие антигена на этих овцах в большой степени наблюдается на широких дыхательных путях и удобно регистрируется в виде изменений устойчивости легкого или специфической устойчивости легкого к оказываемому воздействию.
B. Методы.
Подготовка животных: используют взрослых овец с весом 35 кг (колебание от 18 до 50 кг). Все используемые животные удовлетворяют двум критериям: а) они имеют природную кожную реакцию на разбавления 1:1000 или 1:10000 экстракта Ascaris suum (Greer Diagnostics, Lenois, NC); и б) они предварительно отреагировали на ингаляцию контрольным заражением Ascaris suum как острым, так и замедленным сужением просвета бронхов (W.M. Abraham et al., Am. Rev. Resp. Dis., 28, 839-844 (1983)).
Измерения механики дыхательных путей: несдержанных овец удерживают в телеге в лежачем положении с фиксированными головами. После наружной анестезии носового прохода 2%-ным раствором лидокаина баллонный катетер продвигают через одну ноздрю в низкий пищевод. Животным затем через другую ноздрю вводят в трахею насадочную эндотрахеальную трубку, используя в качестве проводника жесткий бронхоскоп их оптического волокна. Плевральное давление оценивают с помощью баллонного катетера в пищеводе (наполненный одним мл воздуха), который расположен таким образом, чтобы вдох вызывал отклонение отрицательного давления с четко видимой сердечной осцилляцией. Боковое давление в трахее измеряют с помощью бокового катетера (внутренний диаметр 2,5 мм), введенного через и установленного наружно на кончике трубки, введенной в трахею через нос. Транслегочное давление, разница между трахеальным давлением и плевральным давлением, измеряют с помощью дифференциального датчика давления (DP45; Validyne Corp, Northridge, CA). Для изменения легочной устойчивости (RL) максимальный конец носотрахеальной трубки присоединяют к пневмотахографу (Fleusch, Dyna Sciects, Blue Bell, PA). Сигналы потока и транслегочное давление регистрируют на осциллографе (Model DR-12, Electronics for Medicine, White Plains NY), который присоединен к компьютеру PDP-II Digital (Digital Equipment Corp. , Maynard, MA) для линейной расстановки расчета RL по легочному давлению, объему дыхания, полученных ассимиляцией и потоком. Для определения RL используют анализ 10-15 вздохов. Объем грудного газа (Vtg) измеряют в организме плетизмографом для получения специфической легочной устойчивости (SRL = RLVtg).
Аэрозольные высвобождающие системы: аэрозоли экстракта Ascaris suum (1: 20) получены с использованием способных распылять медицинские препараты распылителей (Raindrop R, Puritan Bennett), которые дают аэрозоль с массовым средним аэродинамическим диаметром 6,2 мкМ (геометрическое стандартное отклонение 2,1), как определено анализатором электрического размера (Model. 3030; Thermal Systems, St. Paul, MN). Конец распылителя направляют в пластический t-фрагмент, один конец которого присоединен к носотрахеальной трубке, другой конец которой присоединен к дыхательной части респиратора Harvard. Аэрозоль высвобождается при вдыхаемом объеме 500 мл при скорости 20 раз в минуту. Таким образом, каждая овца получает эквивалентную дозу антигена как при плацебо, так и при испытаниях лекарств.
Экспериментальный протокол: перед контрольным заражением антигеном проводят основные измерения SRL, введение исследуемого соединения начинают за 1 час до контрольного заражения, измерение SRL повторяют и затем овец подвергают ингаляционному заражению антигеном Ascaris suum. Измерения SRL проводят сразу после контрольного заражения антигеном и через 1, 2, 3, 4, 5, 6, 6,5, 7, 7,5 и 8 часов после контрольного заражение антигеном. Плацебо и испытания лекарств разделяются по меньшей мере интервалом в 14 дней. При дальнейшем исследовании овцам дают дозу в виде пищевого комка исследуемого соединения с последующим введением исследуемого соединения за 0,5-1 часа до контрольного заражения Ascaris и спустя 8 часов после Ascaris, как описано выше.
Статистический анализ: для сравнения острой немедленной реакции на антиген и пика замедленной реакции контрольных и обработанных лекарством животных используют исследование Kruskal-Wallis one way ANOVA.
Изобретение далее иллюстрируется следующими неограничивающими примерами, в которых, если не указано другого:
(i) все операции проводят при комнатной или нормальной температуре, т.е. при температуре в области 18o-25oC;
(ii) упаривание растворителя проводят с помощью роторного испарителя при пониженном давлении (600-4000 паскаль: 4,5-30 мм рт.ст.) при температуре бани до 60oC;
(iii) протекание реакций контролируется тонкослойной хроматографией (ТСХ) и время реакции дано только для иллюстрации;
(iv) точки температур плавления являются неточными и "разл." указывает на разложение; представленные температуры плавления являются такими, которые определены для веществ, полученных как описано; полиморфизм может наблюдаться при одновременном выделении веществ с различными температурами плавления;
(v) структуру и чистоту всех конечных продуктов подтверждают по меньшей мере одним из следующих методов: ТСХ, масс-спектроскопия, спектроскопия ядерного магнитного резонанса (ЯМР), или микроаналитическими данными;
(vi) выходы представлены только для иллюстрации;
(vii) в том случае, когда они приведены, данные ЯМР представлены в виде значений (δ) для главных определяемых протонов, представленных в миллионных долях (м.д.) относительно триметилсилана (ТМС) в виде внутреннего стандарта, определенных при 300 МГц или 400 МГц, используя указанный растворитель; обычными аббревиатурами, используемыми для формы сигнала, являются: s - синглет; d - дублет; t - триплет; m - мультиплет; br - уширенный; и т.д.; кроме того, Ar - сигнал ароматики;
(viii) химические символы имеют их обычные значения; используются также следующие аббревиатуры: об. (объемное), масс (массовое), т.кип. (температура кипения), л (литр(ы)), мл (миллилитры), г (грамм(ы)), мг (миллиграмм(ы)), моль (молей), ммоль (миллимолей), экв. (эквивалент(ы)).
Пример 1. 1-(((1(R)-(3-(2-(3-Хлортиено[3,2-b] пиридин-5-ил) этенил)фенил-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)- -циклопропанацетат натрия.
Стадия 1: 3-Амино-2-формилтиофен.
К холодному (0oC) перемешиваемому раствору литийалюминийгидрида в ТГФ (380 мл, 1 M) прибавляют небольшими порциями метил-3-амино-2-тиофенкарбоксилат (30 г, 190 ммоль) в течение более 30 минут. Полученную смесь перемешивают 1 час при 0oC. Очень медленно, по каплям, прибавляют воду (15 мл) с последующим медленным прибавлением водного NaOH (15 мл, 3,5 М). Затем прибавляют еще воды (43 мл) и ТГФ (300 мл). Смесь хорошо перемешивают в течение 30 минут и затем фильтруют сквозь целит. Целит промывают дополнительным ТГФ. Фильтрат концентрируют до масла, которое растворяют в 2 л EtOAc. Раствор в EtOAc сушат над безводным MgSO4 и фильтруют. Полученный раствор сырого 3-амино-2-гидрокситиофена затем обрабатывают MnO2 (100 г). Смесь перемешивают при комнатной температуре в течение 20 часов и затем фильтруют сквозь целит. Фильтрат упаривают с получением 23,3 г (65%) указанного в заголовке соединения.
1H ЯМР (CDCl3) δ 6.10 (2H, br s), 6.54 (1H, d, J = 5 Гц), 7.48 (1H, d, J = 5 Гц), 9.57 (1H, s).
Стадия 2: Тиено[3,2-b]пиридин-5-карбоновая кислота.
К раствору 3-амино-2-формилтиофена (10 г, 78 ммоль) в EtOH (50 мл) прибавляют смесь NaOH (50 мл, 5%) и натриевой соли пировиноградной кислоты (17,16 г, 156 ммоль). Смесь нагревают до 60oC в течение 2 часов. Смесь охлаждают и промывают Et2O:EtOAc 1:1 и затем подкисляют 1 н. HCl до pH 3 при 0oC. Смесь фильтруют и твердый продукт сушат на воздухе с получением 10 г (71%) указанного в заголовке соединения.
1H ЯМР (CD3SOCD3) δ 7.68 (1H, d, J = 5.5 Гц), 8.00 (1H, d, J = 8.4 Гц), 8.28 (1H, d, J = 5.5 Гц), 8.65 (1H, d, J = 8.4 Гц).
Стадия 3: 3-Хлортиено[3,2-b]пиридин-5-карбоновая кислота.
К раствору Ag2SO4 (6,96 г, 22,3 ммоль) в конц. H2SO4 (60 мл) при 100oC прибавляют тиено[3,2-b]пиридин-5-карбоновую кислоту (4 г, 22,3 ммоль). Через быстро перемешиваемую смесь барботируют Cl2 в течение периода в 2 часа. Смесь охлаждают и затем выливают в лед (250 мл). AgCl выпадает в осадок и его фильтруют. Фильтрат разбавляют водой (500 мл) и дают кристаллизоваться при 0oC в течение ночи. Продукт фильтруют и сушат на воздухе с получением 3,04 г (64%) указанного в заголовке соединения.
1H ЯМР (CD3SOCD3) δ 8.10 (1H, d, J = 8.4 Гц), 8.39 (1H, s), 8.72 (H, d, J = 8.4 Гц).
Стадия 4: 3-Хлор-5-(хлорметил)тиено[3,2-b]пиридин.
Кислоту со стадии 3 этерифицируют избытком диазометана. К раствору соответствующего эфира (1,2 г, 5,6 ммоль) в ТГФ (10 мл) при -78oC прибавляют DIBAL (9,36 г, 1,5 М). Полученную смесь перемешивают при 0oC в течение 1 часа. Прибавляют метанол (0,5 мл) с последующим прибавлением HCl (10 мл, 0,5 М). Смесь экстрагируют EtOAc. Органический экстракт сушат над безводным MgSO4 и концентрируют в вакууме. Хроматография сырого продукта на силикагеле (элюирование с помощью 40% EtOAc в гексане) дает 900 мг (100%) соответствующего спирта. Затем спирт кипятят с обратным холодильником в S(O)Cl2 (5 мл) в течение 5 минут. Избыток реагента удаляют в вакууме. Затем прибавляют NaHCO3. Смесь экстрагируют с помощью EtOAc. Концентрация высушенного (безводным MgSO4) органического экстракта дает 1,2 г (98%) указанного в заголовке соединения.
Стадия 5: ((3-Хлортиено[3,2-b]пиридин-5-ил)трифенилфосфоний хлорида.
К раствору 3-хлор-5-(хлорметил)тиено 3,3-пиридина (1,2 г, 5,5 ммоль) в CH3CN (20 мл) прибавляют P(Ph)3 (2,88 г, 11 ммоль). Смесь кипятят с обратным холодильником в течение 20 часов и затем упаривают досуха. Прибавляют Et2O (8 мл). Смесь энергично перемешивают и кристаллическую соль фильтруют и промывают дополнительным Et2O с получением 2,1 г (81%) указанного в заголовке соединения.
1H ЯМР (CD3SOCD3) δ 5.75 (2H, d, J = 18.75 Гц), 7.48 (1H, d, J = 7.5 Гц), 7.65-8.00 (15H, m), 8.25 (1H, s), 8.55 (1H, d, J = 7.5 Гц).
Стадия 6: 1,1-Циклопропандиметанол циклический сульфит.
К раствору комплекса BH3 • ТГФ (1 M в ТГФ, 262 мл) прибавляют диэтил 1,1-циклопропандикарбоксилат (25 г, 134 ммоль) при 25oC в атмосфере N2. Раствор кипятят с обратным холодильником в течение 6 часов, охлаждают до комнатной температуры и осторожно прибавляют MeOH (300 мл). Раствор перемешивают 1 час и затем концентрируют до масла. Сырой диол растворяют в CH2Cl2 (234 мл) и по каплям прибавляют SOCl2 (15,9 г, 134 ммоль) в течение периода времени 15 минут при 25oC. После перемешивания еще в течение 15 минут смесь промывают водным NaHCO3. Органический экстракт сушат над Na2SO4, фильтруют и концентрируют с количественным получением указанного в заголовке соединения в виде белого твердого продукта.
Стадия 7: 1-(Гидроксиметил)циклопропанацетонитрил.
К раствору производного циклического сульфита со стадии 6 (14,7 г, 99 ммоль) в ДМФ (83 мл) прибавляют NaCN (9,74 г, 199 ммоль). Смесь нагревают при 90oC в течение 20 часов. После охлаждения прибавляют EtOAc (400 мл) и раствор промывают насыщенным раствором NaHCO3 (55 мл), насыщенным раствором NaCl и сушат над Na2SO4. Раствор концентрируют с получением 7,1 г (65%) указанного в заголовке соединения.
Стадия 8: 1-(Ацетилтиометил)циклопропанацетонитрил.
К раствору спирта со стадии 7 (42 г, 378 ммоль) в сухом CH2Cl2 (450 мл) при -30oC прибавляют Et3N (103,7 мл, 741 ммоль) с последующим прибавлением по каплям CH3S(O)2Cl (43,3 мл, 562 ммоль). Смесь нагревают до 25oC, промывают NaHCO3, сушат над Na2SO4 и концентрируют в вакууме с получением соответствующего мезилата. Мезилат затем растворяют в ДМФ (450 мл) и охлаждают до 0oC. Прибавляют тиоацетат калия (55,4 г, 485 ммоль) и смесь перемешивают при 25oC в течение 18 часов. Прибавляют EtOAc (1,5 л) и раствор промывают NaHCO3, сушат над Na2SO4 и концентрируют в вакууме с получением 45 г (70%) указанного в заголовке соединения.
Стадия 9: Метил 1-(меркаптометил)циклопропанацетат.
К раствору нитрила со стадии 8 (45 г, 266 ммоль) в MeOH (1,36 л) прибавляют H2O (84 мл) и конц. H2SO4 (168 мл). Смесь кипятят с обратным холодильником в течение 20 часов, охлаждают до 25oC, прибавляют H2O (1 л) и продукт экстрагируют CH2Cl2 (2х 1,5 л). Органический экстракт промывают H2O и сушат над Na2SO4. Концентрирование органического раствора дает 36 г (93%) указанного в заголовке соединения.
Стадия 10: 3-(((2-Тетрагидропиранил)окси)метил)бензальдегид.
Изопропилальдегид (150 г, 1,1 моль) растворяют в ТГФ (1 л) и EtOH (1 л) при 0oC. Порциями прибавляют NaBH4 (11,0 г, 291 моль) и смесь перемешивают 1 час при 0oC. Прибавление 25% водной NH4OAc и экстракция с помощью EtOAc (2х) с последующей очисткой путем флэш хроматографии (20% ---> 40% EtOAc в гексане) дает 60 г 3-(гидроксиметил)бензальдегида.
Спирт (0,44 ммоль) растворяют в CH2Cl2, (500 мл). Прибавляют DHP (50 г, 0,59 моль) и PTSA (1 г, 5 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. После концентрирования в вакууме остаток очищают с помощью флэш хроматографии (50 -> 15% EtOAc в гексане) с получением 85 г указанного в заголовке соединения.
Стадия 11: 1-(3-(((2-Тетрагидропиранил)окси)метил)фенил)-2- -пропен-1-ол.
К альдегиду со стадии 10 (85 г, 386 ммоль) в толуоле (1 л) при 0oC медленно прибавляют бромид магния в Et2O (450 мл, 1 М, 450 ммоль) в течение периода более 30 минут. После перемешивания в течение 1 часа при 0oC реакционную смесь гасят 25% водной NH4OAc и экстрагируют EtOAc (3х). Упаривание и очистка путем флэш хроматографии (15% -> 25% EtOAc в гексане) дает 82 г (86%) указанного в заголовке соединения.
Стадия 12: Этил 2-(3-(3-(((2-тетрагидропиранил)окси)метил)- фенил)-3-оксипропил)бензоат.
Аллиловый спирт со стадии 11 (24,8 г, 100 ммоль) и этил 2-бромбензоат (25,2 г, 110 ммоль) растворяют в ДМФ (200 мл). Прибавляют LiCl (4,2 г, 100 ммоль), LiOAc • 2H2O (25,5 г, 250 ммоль) и н-Bu4N+Cl- (55 г, 200 ммоль) и полученную смесь трижды дегазируют. Затем прибавляют Pd(OAc)2 (1 г) и смесь дегазируют еще три раза перед нагреванием ее при 100oC при перемешивании в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь выливают в H2О (600 мл), 10% водную NaHCO3 (200 мл) и Et2O. Сырой продукт экстрагируют Et2O (2х), промывают H2O и насыщенным раствором соли и сушат над Na2SO4 перед концентрированием в вакууме. Очистка на короткой колонке с силикагелем (20% EtOAc в гексане) дает 34 г (86%) указанного в заголовке соединения.
1H ЯМР (CD3COCD3): δ 8.02 (1H, bs), 7.92 (1H, d), 7.88 (1H, d), 7.65 (1H, d), 7.50 (3H, m), 7.32 (1H, bt), 4.8 (1H, d), 4.70 (1H, bs), 4.54 (1H, d), 4.3 (2H, q), 3.82 (1H, m), 3.50 (1H, m), 3.35 (2H, m), 1.9-1.45 (8H, m), 1.32 (3H, t).
Стадия 13: Этил 2-(3(S)-гидрокси-3-(3-(((2-(2-тетрагидропиранил) окси)метил)фенил)пропил)бензоат.
Кетоэфир со стадии 12 (24,8 г, 62,5 ммоль) растворяют в ТГФ (230 мл) и охлаждают до -45oC. По каплям прибавляют раствор в ТГФ (15 мл) аддукта тетрагидро-1-метил-3,3-дифенил-1H, 3H-пирроло- [1,2-c][1,3,2]оксазоборола • борана (J. Org. Chem. 56, 751 (1991), 4,55 г, 15,6 моль) и полученную смесь перемешивают 20 минут при -45oC. К этому раствору прибавляют по каплям 1,0 М боран в ТГФ (62,5 мл, 62,5 ммоль) в течение более 30 минут. Реакционную смесь перемешивают 1 час при -45oC и затем еще 2 часа при медленном нагревании до -20oC. После охлаждения раствора до -40oC его выливают в 25% водный NH4OAc (425 мл) и 1,0 М диэтаноламин (40 мл) при 0oC и энергично перемешивают в течение 20 минут. Указанное в заголовке соединение экстрагируют с помощью EtOAc (3х), сушат над MgSO4 и концентрируют при пониженном давлении. Сырое масло очищают путем флэш хроматографии (25% -> 50% EtOAc в гексане) с получением 22,6 г (91%) продукта в виде масла. [α]
Стадия 14: 1(S)-(3-(((2-Тетрагидропиранил)окси)метил)фенил)- -3-(2-(1-гидрокси-1-метилэтил)фенил)пропан-1-ол.
Безводный CeCl3 (17,25 г, 70 ммоль) кипятят в течение 2,5 часов в ТГФ (200 мл), используя насадку Дина-Старка, наполненную молекулярными ситами для удаления H2O. Суспензию охлаждают до -5oC и прибавляют по каплям MeMgCl (114 мл, 3 М в ТГФ, 340 ммоль) поддерживая внутреннюю температуру между -10oC и 0oC. Суспензию серого цвета перемешивают 2 часа перед тем, как медленно прибавив к ней гидроксиэфир со стадии 13 (27,1 г, 68 ммоль) в виде раствора в ТГФ (200 мл) через канюлю. Полученную смесь перемешивают 1,5 часа и при 0oC или ниже и затем медленно выливают в охлажденную льдом HOAc (1 л) и EtOAc (500 мл) и перемешивают в течение 30 минут. После установления pH, равным 6-7, сырое соединение экстрагируют EtOAc (2х) и объединенные органические фазы промывают насыщенным водным NaHCO3 и затем насыщенным раствором соли. Очистка на короткой колонке с силикагелем (от 30% до 50% EtOAc в гексане) дает 24,5 г (95%) указанного в заголовке соединения.
Стадия 15: Метил 1-(((1(R)-(3-(((2-тетрагидропиранил)окси)- метил)фенил)-3-(2-(1- гидрокси-1-метилэтил)фенил)пропил)тио)метил)- циклопропанацетат.
Диол со стадии 14 (17,9 г, 46,6 ммоль) растворяют в CH3CN (40 мл) и ДМФ (10 мл) и охлаждают до -42oC в атмосфере азота. Прибавляют диизопропил-этиламин (8,5 мл, 48,9 ммоль) и затем по каплям метансульфонилхлорид (3,6 мл, 46,6 моль). Раствор перемешивают 1,5 часа с помощью механической мешалки, поддерживая температуру между -42oC и -35oC; затем его охлаждают до -45oC. Прибавляют тиол со стадии 9 (7,84 г, 48,9 ммоль) и затем по каплям прибавляют ДМФ (15 мл). К реакционной смеси прибавляют трет-бутоксид калия в ТГФ (56 мл, 1,75 М, 97,9 ммоль) в течение более 20 минут с помощью шприца. Перемешивание продолжают в течение 5 часов при медленном нагревании от -35oC до -22oC, получая очень густой полупрозрачный гель. Реакцию гасят насыщенным водным NH4Cl (250 мл) и EtOAc (300 мл). Продукт экстрагируют EtOAc, промывают H2O и насыщенным раствором соли и сушат над MgSO4. Очистка путем флэш хроматографии (от 20 до 30% EtOAc в гексане) дает 16,8 г (68%) указанного в заголовке соединения.
Стадия 16: Метил 1-(((1(R)-(3-(гидроксиметил)фенил)-3-(2- -(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)цикпропанацетат.
К гидроксиэфиру со стадии 15 (9,02 г, 17,1 моль) в безводном MeOH (60 мл) в атмосфере азота прибавляют пиридин (50 мкл) и затем PPTS (1,1 г, 4,3 ммоль). Реакционную смесь перемешивают 3,5 часа при 55oC, затем при комнатной температуре в течение ночи перед тем как упаривать в вакууме. Остаток разбавляют EtOAc (500 мл) и промывают H2O, насыщенным водным NaHCO3, буфером NaH2PO4 (pH = 4.5) и насыщенным раствором соли. После высушивания над MgSO4 и упаривания растворителей остаток очищают путем флэш хроматографии (от 40% до 60% EtOAc в гексане) с получением 6,85 г (91%) указанного в заголовке соединения.
1H ЯМР (CD3COCD3): δ 7.41 (2H, m), 7.27 (3H, m), 7.09 (3H, m), 4.63 (2H, d), 4.19 (1H, t), 3.95 (1H, t), 3.88 (1H, s), 3.57 (3H, s), 3.1 (1H, ddd), 2.8 (1H, ddd), 2.5 (2H, s), 2.4 (2H, d), 2.17 (2H, m), 1.52 (6H, s), 0.52-0.35 (4H, m).
Стадия 17: Метил 1-(((1(R)-(3-(формилфенил)-3-(2-(1-гидрокси- -1-метилэтил)фенил)пропил)тио)метил)циклопропанацетат.
К дигидроксиэфиру со стадии 16 (6,8 г, 15,4 ммоль) в EtOAc (150 мл) при 50oC прибавляют MnO2 (6,7 г, 76,8 ммоль). После перемешивания в течение 30 минут при 50oC прибавляют еще MnO2 (6,7 г) и спустя 30 минут прибавляют третью порцию MnO2 (6,7 г). Спустя час теплую реакционную смесь фильтруют сквозь целит и лепешку промывают дополнительным количеством EtOAc. Упаривание растворителя дает желаемый альдегид 5,62 г (83%).
1H ЯМР (CD3COCD3): δ 10.4 (1H, s), 7.9 (1H, bs), 7.8 (2H, m), 7.58 (1H, t), 7.38 (1H, bd), 7.1 (3H, m), 4.1 (1H, t), 3.54 (3H, s), 3.13 (1H, ddd), 2.85 (1H, ddd), 2.51 (2H, s), 2.49 (2H, d), 2.2 (2H, m), 1.51 (6H, s), 0.52-0.32 (4H, m).
Стадия 18: Метил 1-(((1(R)-(3-(2-(3-хлортиено[3,2-b] пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)- метил)циклопропанацетат.
К суспензии фосфониевой соли со стадии 5 (409 мг, 0,85 ммоль) в сухом ТГФ (5 мл) при -78oC прибавляют раствор трет-бутоксида калия (0,716 мл, 1 M раствор в ТГФ). Смесь нагревают до комнатной температуры в течение 30 минут и затем охлаждают до -78oC перед прибавлением альдегида со стадии 17 (300 мг, 0,7 ммоль). Смесь перемешивают при -78oC в течение 30 минут, подогревают до 0oC в течение 15 минут. Прибавляют водный NH4OAc и смесь экстрагируют EtOAc. Органический экстракт промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют до масла. Хроматография сырого масла на силикагеле (элюирование 20% EtOAc в гексане) дает 420 мл (98%) указанного в заголовке соединения.
1H ЯМР (CD3COCD3): δ 0.35-0.55 (4H, m), 1.55 (6H, s), 2.1-2.3 (2H, m), 2.45 (2H, d, J = 7.5 Гц), 2.55 (2H, s), 2.8-2.95 (1H, m), 3.1-3.25 (1H, m), 3.55 (3H, s), 4.05 (1H, t, J = 7.5 Гц), 7.05-7.15 (4H, m), 7.4 (2H, d, J = 3.75 Гц), 7.5 (1H, d, J = 15 Гц), 7.6 (1H, m), 7.75 (1H, d, J = 7,5 Гц), 7.8 (1H, s), 7.85-7.95 (1H, d, J = 15 Гц), 8.05 (1H, s), 8.45 (1H, d, J = 8 Гц).
Стадия 19: Натрий 1-(((1(R)-(3-(2-(3-хлортиено[3,2-b] пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидроксиметилэтил)фенил)пропил)тио)- метил)циклопропанацетат
К раствору эфира со стадии 18 в ТГФ (1 мл) и MeOH (1 мл) прибавляют водный NaOH (1 н., 1,4 мл). Смесь перемешивают при 25oC в течение 20 часов. Прибавляют NH4Cl и смесь экстрагируют EtOAc. Органический экстракт промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют до масла. Хроматография сырого масла на силикагеле (элюирование 20% EtOAc/5% HOAc в гексане) дает 330 мг (79%) соответствующей кислоты. К этой кислоте в 3 мл EtOH прибавляют NaOH (1 н., 1,0 экв). Растворитель упаривают и продукт лиофилизируют с получением указанного в заголовке соединения.
Точная масса C33H33ClNO3S2Na (M+1): вычислено 614,1566
найдено 614,1566.
1H ЯМР (CD3COCD3): δ 0.2-0.43 (4H, m), 1.53 (6H, 2s), 2.26 (2H, m), 2.28 (2H, s), 2.6 (2H, s), 2.75-2.85 (1H, m), 2.95-3.3 (1H, m), 4.04 (1H, dd, J = 7.5 Гц, J' = 11.25 Гц), 7.01-7.08 (3H, m), 7.33-7.8 (3H, m), 7.42-7.47 (1H, d, J = 16.5 Гц), 7.53 (1H, d, J = 7 Гц), 7.65 (1H, s), 7.66 (1H, d, J = 8.5 Гц), 7.82-7.88 (1H, d, J = 17 Гц), 8.0 (1H, s), 8.34-8.37 (1H, d, J = 8.5 Гц).
Пример 2. 1-(((1(R-(3-(2-(Тиено[3,2-b] пиридин-5-ил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропил)тио) метил)циклопропанацетат натрия.
Стадия 1: ((Тиено[3,2-b]пиридин-5-ил)метил)трифенилофосфоний хлорид.
Применяя методы, описанные в стадиях 4-5 примера 1, тиено-[3,2-b]пиридин-5-карбоновую кислоту преобразуют в указанное в заголовке соединение.
1H ЯМР (CDCl3): δ 7.2 (2H, dd), 7.5-8.0 (17H, m), 8.2 (2H, m).
Стадия 2: 1-(((1(R)-(3-(2-(Тиено[3,2-b]пиридин-5-ил) этенил)-фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)-тио)метил) циклопропанацетат натрия.
Применяя методы, описанные в стадиях 18-19 примера 1, фосфониевую соль со стадии 1 преобразуют в указанное в заголовке соединение.
1H (CDCl3): δ 0.5 (4H, m), 1.6 (6H, 2s), 2.1 (2H, m), 2.4 (2H, m), 2.6 (2H, m), 2.9 (1H, m), 3.2 (1H, m), 4.0 (1H, t), 7.1 (3H, m), 7.2-7.5 (6H, m), 7.6 (2H, t), 7.8 (2H, t), 8.2 (1H, d, J = 8 Гц).
Анализ. Вычислено для C33H34NO3S2Na: C 68,37; H 5,91; N 2,42.
Найдено: C 68,54; H 5,96; N 2,46.
Пример 3. 1-(((1(R)-(3-(2-(3-Бромтиено[3,2-b]пиридин-5-ил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропил)тио) метил)циклопропанацетат натрия.
Стадия 1: Метил 3-бромтиено[3,2-b]пиридин-5-карбоксилат.
К раствору HCl (10%) в MeOH (10 мл) прибавляют тиено[3,2-b]пиридин-5-карбоновую кислоту (1,0 г, 5,6 ммоль, со стадии 2 примера 1) и смесь кипятят с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры растворитель удаляют упариванием и остаток распределяют между EtOAc и H2O. Твердый Na2CO3 прибавляют до тех пор, пока система не станет основной. Разделение, сушка и упаривание органического слоя приводят к получению 0,75 г (70%) метил тиено[3,2-b]пиридин-5-карбоксилата.
В раствор метил тиено[3,2-b]пиридин-5-карбоксилата (0,400 г, 2,07 ммоль) в 2 мл CHCl3 при 0oC барботируют HCl в течение 2 минут. Растворитель упаривают при пониженном давлении и твердый осадок в смеси брома (2 мл) и CHCl3 (2 мл) нагревают в запаянной трубке 12 часов при 70oC. После охлаждения прибавляют 10% раствор NaHCO3 и реакционную смесь экстрагируют CH2Cl2 (3x50 мл). Органические фазы промывают NaHCO3 и сушат над Na2SO4. Органические растворители упаривают и указанный в заголовке бромид очищают путем флэш хроматографии на силикагеле с EtOAc:гексаном 3:7 с получением 0,343 г (61%).
1H ЯМР (CDCl3): δ 4.06 (3H, m), 7.89 (1H, s), 8.19 (1H, d). 8.33 (1H, d).
MS, m/e 272 (m+ + 1).
Стадия 2: 3-Бромтиено[3,2-b]пиридин-5-метанол.
К раствору метилового эфира (0,388 г, 1,42 ммоль) со стадии 1 в 5 мл ТГФ при -78oC прибавляют DIBAL (3,55 ммоль) в течение более 5 минут. Реакционную смесь оставляют на 30 минут, после чего раствор охлаждают до 0oC и гасят MeOH. Прибавляют раствор тартрата калия и смесь экстрагируют EtOAc. Органическую фазу сушат над Na2SO4 и растворитель упаривают. Сырое масло очищают путем флэш хроматографии на силикагеле с EtOAc:гексаном 2:3 с получением 0,338 г (98%) указанного в заголовке спирта.
1H ЯМР (CDCl3): δ 3.97 (1H, t), 4.94 (2H, d), 7.29 (1H, d), 7.79 (1H, S), 8.16 (1H, d).
Стадия 3: 3-Бром-5-(хлорметил)тиено [3,2-b]пиридин.
Смесь тионилхлорида (5 мл) и спирта (0,331 г, 1,35 ммоль) со стадии 2 нагревают при 70oC в течение 30 минут, после чего растворитель упаривают. Остаток обрабатывают бикарбонатом и экстрагируют дихлорметаном. Органические фазы сушат над Na2SO4 и растворитель удаляют. Сырой продукт очищают путем флэш хроматографии на силикагеле с EtOAc:гексаном 5:95 с получением 0,170 г (48%) указанного в заголовке хлорида.
1H ЯМР (CD3COCD3): δ 4.90 (2H, s), 7.58 (1H, d), 8.20 (1H, s), 8.55 (1H, d).
Стадия 4: ((3-Бромтиено[3,2-b]пиридин-5-ил)метил)трифенилфосфоний хлорид.
Смесь хлорида (0,165 г, 0,62 ммоль) со стадии 3 и трифенилфосфина (0,325 г, 1,24 ммоль) в 4 мл ацетонитрила кипятят с обратным холодильником в течение 12 часов. После этого полученную суспензию охлаждают и растворитель упаривают. Сырой твердый продукт обрабатывают ацетоном: эфиром 1:1 с получением 0,296 г (91%) указанной в заголовке фосфониевой соли.
1H ЯМР (CDCl3): δ 6.04 (2H, d), 7.58-7.71 (10H, m), 7.94-7.99 (6H, m), 8.08 (1H, d), 8.26 (1H, d).
Стадия 5: Метил 1-(((1(R)-(3-(2-(3-бромтиено[3,2-b] пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)-фенил)пропил) тио)метил)циклопропанацетат.
К 1 молярному раствору трет-бутоксида калия (0,57 мл, 0,57 ммоль) прибавляют при -78oC суспензию фосфониевой соли (0,290 г, 0,55 ммоль) со стадии 4 в 3 мл ТГФ. Температуру доводят до 0oC в течение 20 минут, затем вновь понижают до -78oC с последующим прибавлением 0,5 молярного раствора метил 1-(((1(R)-(3-(формилфенил) -3-(2-(1-гидрокси-1-метилэтил)фенил) пропил)тио)метил) циклопропанацетата (1,47 мл, 0,44 моль) со стадии 17 примера 1. Баню выдерживают при 0oC в течение 1 часа и реакционную смесь гасят 25% водным NH4OAc. Органический растворитель упаривают и указанный в заголовке продукт очищают флэш хроматографией на силикагеле с помощью EtOAc:гексана 30:70 с получением 0,270 г (94%).
1H ЯМР (CD3COCD3): δ 0.38-0,51 (4H, m), 1.55 (6H, s), 2.22 (2H, m), 2.42 (2H, AB), 2.55 (2H, s), 2.89 (1H, m), 3.14 (1H, m), 3.57 (3H, s), 3.90 (1H, s), 4.0 (1H, t), 7.04-7.25 (3H, m), 7.40 (3H, m), 7.50 (1H, d), 7.5 (1H, m), 7.70 (1H, d), 7.76 (1H, s), 7.90 (1H, s), 8.13 (1H, s), 8.39 (1H, d).
Стадия 6: 1-(((1(R)-(3-(2-(3-Бромтиено[3,2-b]пиридин-5- -ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)- фенил)пропил)тио)метил)циклопропанацетат натрия.
2 н. раствор NaOH (0,41 мл, 0,82 ммоль) прибавляют к метиловому эфиру (0,279 г, 0,41 ммоль) со стадии 5 в 2 мл смеси метанол/ТГФ (0,5 мл/1,5 мл) и перемешивают 12 часов. Раствор выливают в 25% водный NH4Cl и экстрагируют EtOAc. Органический растворитель упаривают и сырое масло очищают флэш хроматографией на силикагеле с EtOAc/гексаном 40:60 с 2% уксусной кислотой с получением 0,224 г (86%) соответствующей карбоновой кислоты. Эту кислоту растворяют в этаноле и прибавляют 1 экв гидроксида натрия (1 н.). Растворитель удаляют и масло лиофилизуют с получением 0,231 г (99%) указанного в заголовке соединения.
Точная масса для C33H33BrNaNO3S2 + H+: вычислено 658,1060
найдено 658,1061.
Пример 4. 1-(((1(R)-(3-(2-(2,3-Дихлортиено[3,2-b]пирнидин-5-ил)этенил)фенил) -3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия.
Стадия 1: Метил 2,3-дихлортиено[3,2-b]пиридин-5-карбоксилат.
Смесь метил тиено[3,2-b] пиридин-5-карбоксилата (0,20 г, 1,03 ммоль) и трихлоризоциануровой кислоты (0,962 г, 4,14 ммоль) кипятят с обратным холодильником в CH3CN в течение 16 часов. Растворитель удаляют упариванием и сырой твердый продукт хроматографируют на силикагеле с 5% EtOAc в толуоле в качестве элюента с получением 0,189 г (70%) указанного в заголовке соединения.
1H ЯМР (C6D6): δ 3.55 (3H, s), 6.75 (1H, d, J=6.5 Гц), 7.75 (1H, d, J = 6.5 Гц).
Стадия 2: 2,3-Дихлор-5-(хлорметил)тиено[3,2-b]пиридин.
Используя методы, описанные в стадии 2 примера 3, метил 2,3-дихлортиено[3,2-b] пиридин-5-карбоксилат (0,100 г, 0,38 ммоль) преобразуют в 99% указанного в заголовке соединения.
1H ЯМР (CDCl3): δ 4.75 (2H, s), 7.50 (1H, d), 8.00 (1H, d).
Стадия 3: ((2,3-Дихлортиено [3,2-b]пиридин-5-ил)метил) трифенилфосфоний хлорид.
Используя методы, описанные в стадии 4 примера 3, 2,3-дихлор-5-(хлорметил)тиено[3,2-b]пиридин (0,078 г, 0,30 ммоль) преобразуют в 81% указанного в заголовке соединения.
1H ЯМР (CDCl3): δ 6.05 (1H, d), 7.50-8.00 (16H, m), 8.42 (1H, d).
Стадия 4: Метил 1-(((1(R)-(3-(2-(2,3-дихлортиено[3,2-b] пиридин-5-ил) этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил) тио)метил)циклопропанацетат.
Используя методы, описанные в стадии 18 примера 1, ((2,3-дихлортиено[3,2-b] пиридин-5-ил)метил)трифенилфосфоний хлорид (0,280 г, 0,54 ммоль) преобразуют в 77% указанного в заголовке соединения.
1H ЯМР (CD3COCD3): δ 0.45 (4H, m), 1.56 (6H, s), 2.20 (2H, m), 2.42 (2H, AB), 2.56 (2H, s), 2.88 (1H, m), 3.15 (1H, m), 3.58 (3H, s), 4.06 (1H, t), 7.13 (3H, m), 7.40-7.50 (4H, m), 7.59 (1H, m), 7.71 (1H, d), 7.76 (1H, s), 7.92 (1H, d), 8.31 (1H, d).
Стадия 5: 1-(((1(R)-(3-(2-(2,-Дихлортиено[3,2-b]пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)- фенил)пропил)тио)метил)циклопропанацетат натрия.
Используя методы, описанные в стадии 19 примера 1, вышеприведенный метиловый эфир (0,176 г, 0,27 ммоль) преобразуют в 86% указанного в заголовке соединения.
Анализ. Вычислено для C33H32Cl2NNaO3S2 • 1,5H2O:
C 58,66; H 5,22; N 2,07; Cl 10,49.
Найдено: C 58,78; H 5,15; N 2,27; Cl 11,06.
Пример 5. 1-(((1(R)-(3-(2-(3-Хлортиено[3,2-b]пиридин- -5-ил)этил)фенил)-3-(2-(1-гидpoкcи-1-метилэтил)фенил)пропил)тио) метил)циклопропанацетат натрия.
Стадия 1: Метил 1-(((1(R)-(3-(2-(3-хлортиено[3,2-b] пиридин- -5-ил)этил)фенил)-3-(2-(1-гидрокси-1-метилэтил)-фенил)пропил)тио)метил) циклопропанацетат.
К раствору олефина со стадии 18 примера 1 (270 г, 0,456 ммоль) в ТГФ при 0oC прибавляют BH3 в ТГФ (1 М) (1,36 мл, 1,37 ммоль). Смесь перемешивают в течение 5 часов при комнатной температуре. Прибавление 25% водной NH4OAc и экстракция EtOAc с последующей очисткой флэш хроматографией (15% EtOAc в толуоле) дает 110 мг (41%) насыщенного соединения.
Стадия 2: Натрий 1-(((1(R)-(3-(2-(3-хлортиено[3,2-b] пиридин- -5-ил)этил)фенил)-3-(2- (1-гидрокси-1-метилэтил)- фенил)пропил)тио) метил)циклопропанацетат.
Следуя методам, описанным для стадии 19 примера 1, эфир со стадии 1 гидролизуют до кислоты с выходом 90%.
1H ЯМР (300 МГц, CD3COCD3): δ 0.30-0.55 (4H, m), 1.50 (6H, 2s), 2.10-2.20 (2H, m), 2.40 (2H, m), 2.50 (2H, s), 2.80 (2H, m), 3.10 (1H, m), 3.15 (2H, m), 3.30 (2H, m), 3.45 (1H, m), 7.15-7.45 (8H, m), 8.00 (1H, s) 8.30 (1H, d).
Затем получают натриевую соль указанного в заголовке соединения.
Анализ. Вычислено для C33H35ClNS2O3Na • 3H2O:
C 59,19; H 6,18; N 2,09.
Найдено: C 59,16; H 5,92; N 2,08.
Пример 6. 1-(((1(R)-(3-(2-(2-Хлортиено[3,2-b]пиридин-5-ил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил) фенил)пропил)тио) метил)циклопропанацетат натрия.
Стадия 1: 2-Хлор-5-метилтиено[3,2-b]пиридин.
К раствору 5-метилтиено[3,2-b] пиридина (3,60 г, 24 ммоль) и N,N-диизопропиламина (100 мкл) в ТГФ (80 мл) при -78oC прибавляют по каплям 16 мл н-BuLi (1,6 М, 25,6 ммоль). Смесь перемешивают при -78oC в течение 20 минут и затем направляют через канюлю в раствор N-хлорсукцинимида (4,5 г, 34 ммоль) в ТГФ (300 мл) при -10oC. Смесь перемешивают при -10oC в течение 30 минут. Затем прибавляют насыщенный раствор NH4Cl и продукт экстрагируют EtOAc, сушат над MgSO4 и концентрируют до масла. Хроматография сырого масла на силикагеле (элюирование с помощью 15% EtOAc в гексане) дает 3,60 г (81%) указанного в заголовке соединения.
1H ЯМР (CDCl3): δ 2.65 (3H, s), 7.12 (1H, d, J = 7.5 Гц), 7.34 (1H, s). 7.90 (1H, d, J=7.5 Гц).
Стадия 2: 5-(Бромметил)-2-хлортиено[3,2-b]пиридин.
Смесь продукта со стадии 1 (0,371 г, 2,0 ммоль), N-бромсукцинимида (0,396 г, 2,2 ммоль) и бензоил пероксида в 10 мл тетрахлорида углерода кипятят при облучении солнечной лампой в течение 1 часа. После охлаждения до комнатной температуры растворитель удаляют и указанный в заголовке бромид очищают флэш хроматографией на силикагеле (элюирование 5% EtOAc в гексане) с получением 0,284 г (46%).
1H ЯМР (CDCl3): δ 4.63 (2Н, s), 7.38 (1H, s), 7.40 (1H, d), 8.04 (1H, d).
Стадия 3: ((2-Хлортиено [3,2-b]пиридин-5-ил)метил)трифенилфосфоний бромид.
Раствор бромида (0,304 г, 1,16 ммоль) со стадии 2 и трифенил фосфина (0,455 г, 1,73 ммоль) в 6 мл ацетонитрила перемешивают при комнатной температуре в течение 12 часов. Прибавляют эфир и твердый продукт промывают эфиром с получением 0,550 г (91%) указанной в заголовке фосфониевой соли.
1H ЯМР (CD3COCD3-CD3SOCD3): 5.68 2H, d), 7.37 (1H, s), 7.42 (1H, d), 7.75 (6H, m), 7.80-7.95 (9H, m), 8.38 (1H, d).
Стадия 4: 1-(((1(R)-(3-(2-(2-Хлортиено[3,2-b]пиридин-5- -ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)- фенил)пропил)тио)метил)циклопропанацетат натрия.
Используя методы, описанные в стадиях 18 и 19 примера 1, фосфоний бромид (0,484 г, 0,91 ммоль) со стадии 3 преобразуют в 86% указанного в заголовке соединения.
1H ЯМР (CDCl3) of the acid δ 0.38-0.61 (4H, m), 1.61 (3H, s), 1.64 (3H, s), 2.20 (2H, m), 2.31-2.45 (2H, m), 25 (1H, d, J = 14 Гц), 2.62 (1H, d, J = 13 Гц), 2.90 (1H, m), 3.19 (1H, m), 4.00 (1H, t), 7.08-7.19 (2H, m), 7.21-7.48 (8H, m), 7.57 (1H, d, J = 16 Гц), 7.69 (1H, s ), 7.96 (1H, d, J= 8.2 Гц).
Пример 7. 1-(((1(R)-(3-(2-(2-Фтортиено[3,2-b]пиридин- -5-ил)этенил)фенил)-3-(2-(1-гндрокси-1-метилэтил)фенил)пропил)тио) метил)циклопропанацетат натрия.
Стадия 1: 2-Фтор-5-метилтиено 3,2-b]пиридин.
К раствору 2,23 г (15 ммоль) 5-метилтиено[3,2-b]пиридина в 35 мл ТГФ прибавляют 80 мкл (0,6 ммоль) диизопропиламина и затем прибавляют 10,3 мл н-бутиллития (1,4 М в гексане) при -78oC. После перемешивания при -78oC в течение 15 минут прибавляют раствор 6,9 г (22 ммоль) N-фтор бис(бензолсульфонил)амида в 30 мл ТГФ. Реакционную смесь перемешивают при -78oC в течение 1 часа, нагревают до 0oC и перемешивают при 0oC в течение 2 часов. Водная обработка хлоридом аммония и этилацетатом с последующей хроматографической очисткой толуолом/этилацетатом = 6:1 дает 1,18 г (47%) указанного в заголовке соединения.
1H ЯМР (CDCl3): δ 7.87 (1H, d, J = 8 Гц), 7.11 (1H, d, J = 8 Гц), 6.88 (1H, d, J = 2,5 Гц), 2.64 (3H, s).
Другой продукт, идентифицированный как 2-(фенилсульфонил)-5-метилтиено [3,2-b]пиридин, также был выделен.
1H ЯМР (CDCl3): δ 2.69 (3H, s), 7.25 (1H, d), 7.27 (1H, s), 7.50-7.65 (3H, m), 8.05 (3H, m).
Стадия 2: 1-(((1(R)-(3-(2-(2-Фтортиено[3,2-b]пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио) метил)циклопропанацетат натрия.
Используя методы, описанные в стадиях 2-4 по примеру 6, получают указанное в заголовке соединение.
1H ЯМР δ 0.24-0.45 (4H, m), 1.5-1.53 (6H, 2s), 1.13-2.35 (2H, m), 2.35 (2H, s), 2.6 (2H, d, J = 5 Гц), 2.77-2.85 (1H, m), 3.1-3.25 (1H, m), 4.03 (1H, t, J = 7.5 Гц), 7.0-7.07 (4H, m), 7.3-7.37 (4H, m), 7.46 (1H, s), 7.49 (1H, d, J = 8 Гц), 7.68-7.71 (1H, d, J = 9 Гц), 7.76 (1H, s), 8.17 (1H, d, J = 8 Гц).
Пример 8. 1-(((1(R)-(3-(2-(2,3-Дифтортиено[3,2-b]пиридин-5-ил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил) циклопропанацетат натрия.
Стадия 1: 2,3-Дифтор-5-метилтиено[3,2-b]пиридан.
К раствору 2-фтор-5-метилтиено[3,2-b]пиридина (пример 7, стадия 1) (1,00 г, 6,00 ммоль) в 30 мл ТГФ при -78oC прибавляют н-бутиллитий (6,6 ммоль). Через 5 минут температуру поднимают до -20oC и в течение 0,5 минуты барботируют перхлорил фторид. Реакционную смесь нагревают до 0oC, выливают в 10% раствор NaHCO3 и экстрагируют EtOAc. Растворитель упаривают и указанное в заголовке соединение очищают флэш хроматографией на силикагеле с EtOAc/гексаном 3:1 с получением 0,376 г (34%).
1H ЯМР (CDCl3): δ 2.67 (3H, s), 7.81 (1H, d), 7.84 (1H, d).
Стадия 2: 5-Бромметил-2,3-дифтортиено[3,2-b]пиридин.
Продукт со стадии 1 (0,518 г, 2,8 ммоль), N-бромсукцинимид (0,548 г, 3,08 ммоль) и бензоил пероксид (0,034 г, 0,14 ммоль) в 12 мл тетрахлорида углерода кипятят при облучении солнечной лампой в течение 2 часов. После охлаждения до комнатной температуры растворитель удаляют и указанный в заголовке бромид очищают флэш хроматографией на силикагеле с толуолом с получением 0,341 г (46%).
1H ЯМР (CDCl3): δ 4.67 (2H, s), 7.49 (1H, d), 7.98 (1H, dd).
Стадия 3: ((2,3-Дифтортиено[3,2-b]пиридин-5-ил)метил) трифенилфосфоний бромид.
Раствор бромида (0,335 г, 1,27 ммоль) со стадии 2 и трифенилфосфина (0,366 г, 1,40 ммоль) в 4 мл ацетонитрила перемешивают при комнатной температуре в течение 20 часов. Растворитель удаляют и сырой твердый продукт обрабатывают ацетоном/эфиром 1:1 с получением 0,493 г (74%) указанной в заголовке фосфониевой соли.
1H ЯМР (CDCl3): δ 5.96 (2H, d), 7.63-8.04 (16H, m), 8.21 (1H, d).
Стадия 4: Метил 1-(((1(R)-(3-(2-(2,3-дифторфиено[3,2-b]пиридин-5-ил) этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил) тио)метил)циклопропанацетат.
Используя методы, описанные на стадии 18 примера 1, фосфоний бромид (0,484 г, 0,91 ммоль) со стадии 3 преобразуют в 86% указанного в заголовке соединения.
1H (CD3COCD3): δ 0.45 (4H, m), 1.55 (6H, s), 2.20 (2H, m), 2.40 (2H, AB system), 2.55 (2H, s), 2.89 (1H, m), 3.18 (1H, dt), 3.57 (3H, s), 3.90 (1H, s), 4.05 (1H, t), 7.10-7.25 (3H, m), 7.35-7.45 (4H, m), 7.56 (1H, m), 7.62 (1H, d), 7.75 (1H, s), 7.85 (1H, d), 8.23 (1Н, d).
Стадия 5: 1-(((1(R)-(3-(2-(2,3-Дифтортиено[3,2-b]пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил) тио)метил)циклопропанацетат натрия.
К раствору метилового эфира (0,200 г, 0,33 ммоль) со стадии 4 в смеси 1: 1 ТГФ: H2O (1,6 мл) прибавляют твердый LiOH (0,016 г 0,66 ммоль). После 2 дней перемешивания раствор выливают в 10% раствора NH4OAc и экстрагируют EtOAc. Органические растворители упаривают и сырое масло очищают флэш хроматографией на силикагеле с EtOAc:гексаном 4:6 с 2% уксусной кислотой с получением 0,183 г (94%) соответствующей карбоновой кислоты. Эту кислоту растворяют в этаноле и прибавляют 1 эквивалент гидроксида натрия. Растворители удаляют и полученное масло обрабатывают водой и лиофилизуют с получением 0,183 г (96%) указанного в заголовке соединения.
Анализ. Вычислено для C33H32F2NNaO3S2 • 2H2O:
C 60,80; H 5,58; N 2,5.
Найдено C 60,85; H 5,11; N 2,14.
Пример 9. 1-(((1(R)-(3-(2-(2-Хлор-3-фтортиено[3,2-b] пиридин- 5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил) циклопропанацетат натрия.
Стадия 1: 2-Хлор-3-фтор-5-метилтиено[3,2-b]пиридин.
К раствору 550 мг (3 ммоль) 2-хлор-5-метилтиено[3,2-b]пиридина (пример 6, стадия 1) и 14 мкл (0,1 ммоль) диизопропиламина в 12 мл ТГФ при -78oC прибавляют 2,3 мл н-бутиллития (1,4 М в гексане). После перемешивания в течение 10 минут при -78oC FClO4 (газ) барботируют в реакционную смесь в течение 15 секунд. Темно-красный цвет немедленно превращается в желтый. Реакционную смесь перемешивают при -78oC в течение 15 минут, подогревают до 0oC и перемешивают в течение 15 минут. Прибавляют водный хлорид аммония и продукт экстрагируют этилацетатом. Хроматографическая очистка на силикагеле с помощью толуола/этилацетата = 10:1 дает 340 мг (57%) указанного в заголовке продукта.
1H ЯМР (CDCl3): δ 7.88 (1H, dd, J = 8 Гц, J' = 0.5 Гц), 7.20 (1H, d, J = 8 Гц), 2.70 (3H, s).
Стадия 2: 5-(Бромметил)-2-хлор-3-фтортиено[3,2-b]пиридин.
Используя методы, описанные на стадии 2 примера 8, получают указанное в заголовке соединение.
1H ЯМР (CDCl3): δ 8.0 (1H, dd, J = 8 Гц, J'= 0,5 Гц), 7.52 (1H, d, J = 8 Гц), 4.69 (2H, s).
Стадия 3: ((2-Хлор-3-фтортиено[3,2-b]пиридин-5-ил)метил)- трифенилфосфоний бромид.
Используя методы, описанные на стадии 3 примера 8, получают указанное в заголовке соединение.
1H ЯМР (DMSO-d6): δ 8.46 (1H, d, J=8 Гц), 7.88-7.68 (15H, m), 7.42 (1H, d, J=8 Гц), 5.67 (2H, d, J = 14 Гц).
Стадия 4: Метил 1-(((1(R)-(3-(2-(2-хлор-3-фтортиено[3,2-b]пиридин-5-ил)этенил)фенил) -3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил) циклопропанацетат.
Используя методы, описанные на стадии 18 примера 1, из фосфониевой соли со стадии 3 получают указанное в заголовке соединение.
1H ЯМР (CDCl3): δ 7.95 (d, J = 8 Гц, 1H), 7.70 (d, J = 14 Гц, 1H), 7.60 (s, 1H), 7.49 (m, 2H), 7.38-7.08 (m, 7H), 3.92 (t, J = 7 Гц, 1H), 3.60 (s, 3H), 3.12 (m, 1H), 2.85 (m, 1H), 2.49 (s, 2H), 2.38 (s, 2H), 2.20 (m, 2H), 1.60 (s, 3H), 1.58 (s, 3H), 0.50 (m, 4H).
Стадия 5: 1-(((1(R)-(3-(2-(2-Хлор-3-фтортиено[3,2-b] пиридин-5-ил)этенил)фенил)- 3-(2-(1-гидрокси-1-метилэтил)-фенил)пропил)тио)метил)циклопропанацетат натрия.
Используя методы, описанные на стадии 19 примера 1, метиловый эфир со стадии 4 преобразуют в указанное в заголовке соединение.
1H (CDCl3): δ 7.90 (d, J=8 Гц, 1H), 7.66 (d, J = 5, 1H), 7.62 (d, J = 9 Гц, 1H), 7.50 (d, J = 9 Гц, 1H), 7.42 (m, 1H), 7.35-7.05 (m, 7H), 3.97 (t, J = 7 Гц, 1H), 3.16 (m, 1H), 2.88 (m, 1H), 2.58-2.34 (m, 4H) 2.18 (m, 2H), 1.60 (s, 3H), 1.59 (s, 3H), 0,47 (m, 4H).
Пример 10. 1-(((1(R)-(3-(2-(3-Хлор-2-фтортиено[3,2-b] -пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил) пропил)тио)метил)циклопропанацетат натрия.
Стадия 1: 3-Хлор-2-фтор-5-метилтиено[3,2-b]пиридин.
К раствору 461 мг (2,74 ммоль) 2-фтор-5-метилтиено[3,2-b] пиридина (пример 7, стадия 1) и 14 мкл (0,1 ммоль) диизопропиламина в 12 мл ТГФ при -78oC прибавляют 2,15 мл н-бутиллития (1,4 М гексане). После перемешивания в течение 10 минут при -78oC прибавляют раствор 585 мг (4,4 ммоль) N-хлорсукцинамида в 10 мл ТГФ. Смесь перемешивают при -78oC в течение 20 минут, нагревают до 0oC, перемешивают при 0oC в течение 30 минут и затем распределяют между водным хлоридом аммония и этилацетатом. Хроматографическая очистка на силикагеле с помощью гексана/этилацетата = 8:1 дает 350 мг (63%) указанного в заголовке продукта.
1H ЯМР (CDCl3): δ 7.88 (d, J = 8 Гц, 1H), 7.18 (d, J = 8 Гц, 1H), 2.70 (s, 3H).
Стадия 2: 1-(((1(R)-(3-(2-(3-Хлор-2-фтортиено[3,2-b] пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)- фенил)пропил)тио)метил)циклопропанацетат натрия.
Используя методы, описанные на стадиях 2-5 примера 8, получают указанное в заголовке соединение из продукта со стадии 1.
1H ЯМР (CD3COCD3): δ 0.2-0.55 (4H, m), 1.5 (3H, s), 1.55 (3H, s), 2.1 (2H, m), 2.25 (2H, s), 2.65 (2H, s), 2.70-2.85 (1H, m), 3.15-3.25 (1H, m), 4.05 (1H, t, J = 7.5 Гц), 6.95-7.1 (3H, m), 7.3-7.4 (4H, m), 7.5 (1H, d, J = 7.5 Гц), 7.65 (1H, d, J = 7.5 Гц), 7.7 (1H, s), 7.85 (1H, d, J = 15 Гц), 8.28 (1H, d, J = 7.5 Гц).
Пример 11. 1-(((1(R)-(3-(2-(2-(Фенилсульфонил)тиено[3,2-b]пиридин-5-ил) этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)- метил)циклопропанацетат натрия.
Используя методы, описанные на стадиях 2-4 примера 6, получают указанное в заголовке соединение из 2-(фенилсульфонил)-5- -метилтиено[3,2-b]пиридина, выделенного на стадии 1 примера 7.
1H ЯМР of the acid (CDCl3): δ 0.36-0.53 (3H, m), 0.58 (1H, m), 1.61 (3H, s), 1.63 (3H, s), 2.05 (1H, s), 2.19 (2H, m), 2.34-2.52 (3H, m), 2.60 (1H, d), 2.90 (1H, m), 3,19 (1H, m), 4.0 (1H, t), 7.07-7.20 (3H, m), 7.23-7.38 (4H, m), 7.43 (1H, m), 7.50-7.70 (6H, m), 8.03-8.13 (4H, m).
Анализ. Вычислено для C39H38NNaO5S3 • 3,6H2O:
C 59,69; H 5,81; N 1,78.
Найдено: C 59,68; H 5,70; N 1,52.
Пример 12. 1-(((1(R)-(3-(2-(2,3-Дихлорфуро[3,2-b]пиридин-5-ил)этенил)фенил)-3-(2- (1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия.
Стадия 1: 2-(Триметилсилил)-6-метилфуро[3,2-b]пиридин.
Смесь 2-йод-5-метилпиридин-3-ола (20 г, 85 ммоль), CuI (2,1 г, 11 ммоль), триметилсилилацетилена (23,4 г, 238 ммоль) и бис(трифенилфосфин)палладий (II) хлорида (5,37 г, 7,65 ммоль) в Et3N (380 мл) кипятят с обратным холодильником в течение 20 часов. Смесь охлаждают и разбавляют эфиром к фильтруют сквозь целит, фильтрат концентрируют в вакууме и остаток хроматографируют на силикагеле (элюирование с 10% EtOAc в гексане) с получением 15 г (86%) указанного в заголовке соединения.
1H ЯМР (CD3COCD3): δ 0.40 (9H, s), 2.54 (3H, s), 7.12 (1H, d, J = 8 Гц), 7.14 (1H, s), 7.75 (1H, d, J = 8 Гц).
Стадия 2: 2,3-Диxлop-5-метилфуро[3,2-b]пиридин.
К раствору 2-триметилсилил-5-метилфурано[3,2-b] пиридина (1,05 г, 5,15 ммоль) в CH2Cl2 (16 мл) при 0oC прибавляют трихлоризоциануровую кислоту (1,2 г, 5,15 ммоль). Смесь перемешивают при 0oC в течение 30 минут и затем при комнатной температуре в течение 20 часов. Прибавляют 30% раствор EtOAc в гексане. Полученную смесь фильтруют сквозь короткий слой силикагеля и элюируют с дополнительным 30% EtOAc в гексане. Упаривание фильтрата дает 1 г (96%) указанного в заголовке соединения.
1H ЯМР (CD3COCD3): δ 2.61 (3H, s), 7.33 (1H, d, J = 8 Гц), 7.84 (1H, d, J = 8 Гц).
Стадия 3: ((2,3-Дихлорфуро[3,2-b]пиридин-5-ил)трифенилфосфоний бромид.
К раствору 2,3-дихлор-5-метилфурано[3,2-b]пиридина (0,5 г, 2,47 ммоль) в CCl4 (15 мл) прибавляют N-бромсукцинимид (0,44 г, 2,47 ммоль) и бензоил пероксид (2 мг). Смесь перемешивают и подвергают фотолизу при облучении солнечной лампой в течение 1 часа.
Полученную смесь охлаждают и фильтруют сквозь целит. Упаривание фильтрата дает масло, которое растворяют в CH3CN (10 мл). К смеси прибавляют трифенилфосфин (1,29 г, 4,94 ммоль) и смесь перемешивают при комнатной температуре в течение 20 часов. Растворитель удаляют и остаток растирают в эфире с получением после фильтрации 1 г (77%) указанного в заголовке соединения.
1H ЯМР (CDCl3): δ 5.9 (2H, d, J = 15 Гц), 7.55-8.0 (16H, m), 8.2 (1H, d, J = 9 Гц).
Стадия 4: Метил 1-(((1(R-(3-(2-(2,3-дихлорфуро[3,2-b] пиридин-5- ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил пропил) тио)метил)циклопропанацетат.
Используя методы, описанные на стадии 18 примера 1, фосфониевую соль со стадии 3 преобразуют в указанное в заголовке соединение с 92% выходом.
1H ЯМР (CD3COCD3): δ 0.40-0.50 (4H, m), 1.53 (6H, s ), 2.20 (2H, m), 2.40 (2H, AB system), 2.57 (2H, s), 2.90 (1H, m), 3.15 (1H, s), 3.59 (3H, s), 3.9 (1H, s), 4.05 (1H, t), 7.10 (3H, m), 7.40 (1H, m), 7.55 (1H, m), 7.62 (1H, d), 7.74-7.80 (2H, m), 7.91 (1H, d).
Стадия 5: 1-(((1(R)-(3-(2-(2,3-Дихлорфуро[3,2-b] пиридин-5-ил) этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)-фенил)пропил) тио)метил)циклопропанацетет натрия.
Используя методы, описанные на стадии 19 примера 1, метиловый эфир со стадии 4 (0,176 г, 0,27 ммоль) преобразуют с 86% выходом в указанное в заголовке соединение.
Анализ. Вычислено для C33H32Cl2NNaO4S • 1,5H2O:
C 60,08; H 5,36; N 2,12; Cl 10,75.
Найдено: C 60,04; H 5,01; N 2,06; Cl 11,07.
Пример 13. 1-(((1(R)-(3-(2-(3-Хлорфуро[3,2-b]пиридин- -5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио) метил)циклопропанацетат натрия.
Стадия 1: 3-Хлор-5-метилфуро[3,2-b]пиридин.
К раствору 2,3-дихлор-5-метилфуро[3,2-b]пиридина (0,661 г, 3,27 ммоль) (пример 12, стадия 2) в 16 мл ТГФ прибавляют 1,7 М раствор трет-бутиллития (4,04 мл, 6,87 ммоль). Через 30 минут раствор гасят при -78oC метанолом и раствором NH4Cl. Реакционную смесь доводят до комнатной температуры и экстрагируют EtOAc. Растворитель упаривают и указанное в заголовке соединение очищают флэш хроматографией на силикагеле с EtOAc:гексаном 1:4 c получением 0,444 г (81%).
1H ЯМР (CDCl3): δ 2.71 (3H, s), 7.16 (1H, d), 7.65 (1H, d), 7.84 (1H, s).
Стадия 2: 5-Бромметил-3-хлорфуро[3,2-b]пиридин.
Используя методы, описанные на стадии 2 примера 8, 3-хлор-5-метилфуро[3,2-b]пиридин (0,245 г, 1,46 ммоль) преобразуют с 46% выходом в указанное в заголовке соединение.
1H ЯМР (CDCl3): δ 4.70 (2H, s), 7.50 (1H, d), 7.78 (1H, d), 7.90 (1H, s).
Стадия 3: ((3-Хлорфуро[3,2-b]пиридин-5-ил)метил)трифенилфосфоний бромид.
Используя методы, описанные на стадии 3 примера 8, 5-бромметил-3-хлорфурано[3,2-b] пиридин (0,162 г, 0,65 ммоль) преобразуют с 86% выходом в указанное в заголовке соединение.
1H ЯМР (CDCl3): δ 5.90 (2H, m), 7.55-8.00 (17H, m), 8.25 (1H, d).
Стадия 4: Метил 1-(((1(R)-(3-(2-(3-хлорфуро[3,2-b] пиридин- -5-ил)этенил)фенил)-3-(2- (1-гидрокси-1-метилэтил)- фенил)пропил)тио)метил) циклопропанацетат.
Используя методы, описанные на стадии 18 примера 1, ((3-хлорфуро[3,2-b] пиридин-5-ил)метил)трифенил фосфоний бромид (0,273 г, 0,53 ммоль) преобразуют с 75% выходом в указанное в заголовке соединение.
1H ЯМР (CD3COCD3): δ 0.40-0.55 (4H, m), 1.55 (6H, s), 2.23 (2H, m), 2.40 (2H, AB system), 2.58 (2H, s), 2.90 (1H, m), 3.20 (1H, m), 3.60 (3H, s), 3.90 (1H, s), 4.05 (1H, t), 7.13 (2H, m), 7.40-7.60 (6H, m), 7.65 (1H, d), 7.70-7.85 (2H, m), 7.95 (1H, d), 8.30 (1H, s).
Стадия 5: 1-(((1(R)-(3-(2-(3-Хлорфуро[3,2-b]пиридин-5-ил)этенил)фенил-3- (2-(1-гидрокси-1-метилэтил)-фенил)пропил)тио)метил)циклопропанацетат натрия.
Используя методы, описанные на стадии 5 примера 8, метиловый эфир со стадии 4 (0,160 г, 0,28 ммоль) преобразуют с выходом 83% в указанное в заголовке соединение.
Анализ. Вычислено для C33H33ClNNaOS4 • 2H2O:
C 62,49; H 5,89; N 2,21; C1 5,59.
Найдено: C 62,23; H 5,33; N 2,20; Cl 5,34.
Пример 14. (R) 1-((3-(2-Бромфенил)-1-(3-(2-(2,3- дихлортиено[3,2-b]пиридин-5-ил)этенил)-фенил)пропокси)метил) циклопропанацетат натрия.
Стадия 1: 3-(2-Бромфенил)-1-(3-(((2-тетрагидропиранил)окси)- метил)фенил)-1-пропанон.
Смесь аллилового спирта со стадии 11 примера 1 (30,14 г, 121 ммоль), 1,2-дибромбензола (16 мл), Pd(OAc)2 (830 мг), LiCl (5,38 г), LiOAc • 2H2O (31,6 г) и н-Bu4NCl (67,96 г) в 240 мл дегазируют и нагревают при 85oC в атмосфере N2 в течение 30 минут и при 90oC в течение 45 минут. Смесь добавляют ко льду и 25% NH4OAc (2 л). Указанный в заголовке кетон экстрагируют EtOAc, сушат над Na2SO4 и очищают флэш хроматографией на силикагеле с EtOAc:гексаном 10:90; выход 29,53 г (60%).
1H ЯМР (CDCl3): δ 7.97 (1H, s), 7.90 (1H, d), 7.57 (2H, t), 7.45 (1H, dd), 7.32 (1H, dd), 7.24 (1H, dd), 7.09 (1H, m), 4.83 (1H, d), 4.47 (1H, t), 4.55 (1H, d), 3.92 (1H, m), 3.58 (1H, m), 3,32 (2H, m), 3.20 (2H, m), 1,95-1.45 (6H, m).
Стадия 2: 3-(2-Бромфенил)-1-(3-(((2-тетрагидропиранил)окси)- метил)фенил)-1-пропанол.
Раствор кетона со стадии 1 (29,00 г, 72 ммоль) в 260 мл безводного ТГФ при -55oC (температура реакционной смеси) по каплям прибавляют к раствору (S)-тетрагидро-1-метил-3,3-дифенил-1H, 3H-пирроло[1,2-c][1,3,2] оксазоборола (4,07 г, 0,2 экв.; J.Org. Chem. 56, 751 (1991)) в 70 мл ТГФ и затем прибавляют 1,0 М борана в ТГФ (75 мл). Смеси дают нагреться до -20oC в течение более 3 часов. Затем ее охлаждают до -45oC, гасят 10% водным диэтаноламином и нагревают до комнатной температуры. Затем прибавляют 25% водный NH4OAc и хиральный спирт экстрагируют с помощью EtOAc, сушат над Na2SO4 и фильтруют сквозь силикагель с помощью EtOAc:толуола от 5:95 до 10:90; выход: 27,52 г, 94%.
1H ЯМР (CDCl3): δ 7.53 (1H, d), 7.40-7.16 (6H, m), 7.05 (1H, m), 4.80 (1H, d), 4.72 (2H, m), 4.50 (1H, d), 3.93 (1H, m), 3.55 (1H, m), 2.90 (1H, m), 2.80 (1H, m), 2.0 (2H, m), 1.95 (1H, d, OH), 1.90-1.48 (6H, m).
Стадия 3: Метил 2-((3-(2-бромфенил) -1(R)-(3-(((2-тетрагидропиранил) окси)метил)фенил) пропокси)метил)пропеноат.
При 0oC 95% NaH (2,4 г, 100 ммоль) порциями прибавляют к перемешиваемому раствору спирта со стадии 2 (29,5 г, 73 ммоль) в 400 мл ДМФ и смесь перемешивают при 0oC в течение 1 часа. Затем прибавляют метил 2-(бромметил)акрилат (10 мл, 88 ммоль) и смесь перемешивают при 0oC в течение 8 часов и при комнатной температуре в течение ночи. Гасят насыщенным водным NH4Cl и продукт экстрагируют эфиром, промывают насыщенным раствором соли, сушат над Na2SO4 и очищают флэш хроматографией с EtOAc:гексаном 1:5; выход 20,00 г (82%).
1H ЯМР (CDCl3): δ 7.50 (1H, d, J = 7.5 Гц), 7.37-7.17 (6H, m), 7.04 (1H, m), 6.33 (1H, br, s), 5.97 (1H, br, s), 4.79 (1H, d, J = 11 Гц), 4.70 (1H, m), 4.50 (1H, d, J = 11 Гц), 4.35 (1H, dd), 4.12 (1H, d), 4.02 (1H, d), 3.92 (1H, m), 3.73 (3H, s), 3.55 (1H, m), 2.90 (1H, m), 2.78 (1H, m), 2.12 (1H, m), 2.00 (1H, m), 1.90-1.50 (6H, m).
Стадия 4: 2-((3-(2-Бромфенил)-1(R)-(3-(((2-тетрагидропиранил)- окси)метил)фенил) пропокси)метил)-2-пропен-1-ол.
К раствору эфира со стадии 3 (29,69 г, 59 ммоль) в 300 мл CH2Cl2 при -78oC медленно прибавляют 1,5 М раствор диизобутилалюминийгидрида в толуоле (99 мл, 149 ммоль) и смесь перемешивают при -78oC в течение 30 минут. Затем прибавляют 2 М виноградной кислоты и раствор нейтрализуют 10 н. NaOH. Продукт экстрагируют EtOAc, сушат над Na2SO4 и концентрируют с получением 26,90 г, 96%, указанного в заголовке спирта.
1H ЯМР (CDCl3): δ 7.52 (1H, d), 7.38-7.13 (6H, m), 7.03 (1H, m), 5.14 (2H, AB system), 4.80 (1H, d), 4.74 (1H, t), 4.53 (1H, d), 4.33 (1H, dd), 4.30 (2H, AB system), 3.97 (1H, d), 3.88 (1H, m), 3.55 (1H, m), 2.90 (1H, m), 2.75 (1H, m), 2.19-1.50 (9H, m).
Стадия 5: 1-((3-(2-Бромфеннл) -1(R)-(3-(((2-тетрагидропиранил)- окси)метил)фенил)пропокси) метил)циклопропанметанол.
При 0oC Pd(OAc)2 (500 мг) и ≈ 0,4 М CH2N2 в эфире (1,84) одновременно порциями прибавляют к раствору аллилового спирта со стадии 4 (20,45 г, 43,0 ммоль) в 80 мл ТГФ. Когда реакция завершается, смесь фильтруют через небольшую лепешку силикагеля и концентрируют. Остаток очищают флэш хроматографией на силикагеле с EtOAc:толуолом 15:85 с получением 12,40 г (59%) указанного в заголовке продукта.
1H ЯМР (CDCl3): δ 7.51 (1H, d), 7.38-7.14 (6H, m), 7.05 (1H, m), 4.79 (1H, 2d), 4.72 (1H, br s), 4.50 (1H, 2d), 4.25 (1H, dd), 3.92 (1H, m), 3.65 (1H, m), 3.54 (2H, m), 3.28 (2H, AB system), 2.90 (1H, m), 2.78 (1H, m), 2.65 (1H, m), 2.18-1.50 (8H, m), 0.55 (2H, m), 0.43 (2H, m).
Стадия 6: 1-((3-(2-Бромфенил) -1(R)-(3-(((2-тетрагиропиранил)- окси)метил)фенил)пропокси) метил)циклопропанацетонитрил.
К раствору спирта со стадии 5 (15,30 г, 31,3 ммоль) в 200 мл CH2Cl2 при -40oC прибавляют метансульфонилхлорид (2,90 г, 37,5 ммоль) и триэтиламин (6,50 г, 46,6 ммоль) и раствор перемешивают при -40oC в течение 30 минут и при 0oC в течение 1 часа. Затем прибавляют водный насыщенный NaHCO3 и мезилат экстрагируют CH2Cl2, сушат над Na2SO4 и дважды промывают толуолом. К раствору этого мезилата в 240 мл безводного диметилсульфоксида прибавляют NaCN (7,69 г, 157 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Затем прибавляют воду (1 л), 250 мл насыщенного NaHCO3 и продукт экстрагируют эфиром, промывают насыщенным раствором соли, сушат над Na2SO4 и очищают флэш хроматографией с EtOAc:толуолом 5:95; выход: 13,43 г (86%).
1H ЯМР (CDCl3): δ 7.54 (1H, d), 7.38-7.15 (6H, m), 7.06 (1H, m) 4.80 (1H, d), 4.72 (1H, m), 4.50 (1H, d), 4.26 (1H, dd), 3.94 (1H, m), 3.57 (1H, m), 3.32 (1H, d), 3.09 (1H, d), 2.93 (1H, m), 2.81 (1H, m), 2.75 (1H, d), 2.45 (1H, d), 2.18-1.50 (8H, m), 0,68-0.49 (4H, m).
Стадия 7: 1-((3-(2-Бромфенил) -1(R)-(3-(((2-тетрагидропиранил)- окси)метил)фенил)пропокси) метил)циклопропануксусная кислота.
Смесь нитрила со стадии 6 (13,22 г, 26,5 ммоль), 8 н. KOH (330 мл) и EtOH (130 мл) нагревают с обратным холодильником в течение 17 часов. Затем при комнатной температуре прибавляют 25% водный NH4OAc (500 мл) и АсОН (190 мл) (давая pH ≈ 6) и продукт экстрагируют EtOAc и сушат над Na3SO4. Флэш хроматография остатка с EtOAc:толуолом:AcOH 10:90:1 дает 10,34 г (75%) указанного в заголовке соединения.
1H ЯМР (CDCl3): δ 7.52 (1H, d), 7.35-7.10 (6H, m), 7.06 (1H, m), 4.79 (1H, d), 4.74 (1H, m), 4.54 (1H, d), 4.38 (1H, m), 3.94 (1H, m), 3.60 (1H, m), 3.39 (1/2H, d), 3.28 (1/2H, d), 3.20 (1/2H, d), 3.03 (1/2H, d), 2.89 (1H, m), 2.78 (1H, m), 2.78 (1/2H, d), 2.63 (1/2H, d), 2.45 (1/2H, d), 2.28 (1/2H, d), 2.18 (1H, m), 2.05 (1H, m), 1.95-1.50 (6H, m), 0.62-0.43 (4H, m).
Стадия 8: Метил 1-((3-(2-бромфенил) -1(R)-(3-гидроксиметил)- фенил)пропокси) метил)циклопропанацетат.
Кислоту со стадии 7 (1,816 г, 3,51 ммоль) этерифицируют CH2N2 при 0oC в эфире: ТГФ. Избыток CH2N2 гасят AcOH и продукт концентрируют и промывают дважды толуолом. Этот эфир растворяют в 20 мл MeOH и затем прибавляют пиридин (7 мкл) и п-толуолсульфонат пиридина (220 г, 0,88 ммоль). Через 6 дней перемешивания растворитель упаривают, затем прибавляют 25% водный NH4OAc и продукт экстрагируют EtOAc, сушат над Na2SO4 и очищают с помощью флэш хроматографии на силикагеле EtOAc:гексаном 30:70; выход 1,53 г, (97%).
1H ЯМР (CDCl3): δ 7.50 (1H, d), 7.35-7.18 (6H, m), 7.04 (1H, m), 4.70 (2H, d), 4.21 (1H, dd), 3.63 (3H, s), 3.18 (1H, AB system), 2.93 (1H, m), 2.78 (1H, m), 2.45 (2H, s), 2.08 (1H, m), 1.98 (1H, m), 1.95 (1H, t, OH), 0.58-0.40 (4H, m).
Стадия 9: (R)1-((3-(2-Бромфенил)-1-(3-(2-(2,3-дихлортиено- [3,2-b]пиридин-5-ил)этенил)фенил)пропокси)метил)циклопропанацетат натрия
Используя методы по примеру 1, стадии 17-19, но применяя на стадии 18 ((2,3-дихлортиено[3,2-b] пиридин-5-ил)метил)трифенилфосфоний бромид (пример 4, стадия 3), из эфира стадии 8 получают указанный в заголовке продукт.
1H ЯМР (free acid CDCl3): δ 8.02 (1H, d), 7.68 (1H, d), 7.60-7.48 (4H, m), 7.43-7.34 (2H, m), 7.28-7.19 (3H, m), 7.07 (1H, m), 4.34 (1H, dd), 3.38 (1H, d), 3.19 (1H, d), 2.93 (1H, m), 2.80 (1H, m), 2.70 (1H, d), 2.49 (1H, d), 2.18 (1H, m), 2.08 (1H, m), 0.64-0.47 (4H, m).
Пример 15. 1-((1(R)-(3-(2-(2,3-Дихлортиено[3,2-b] пиридин-5-ил)- этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил) пропокси)метил)- циклопропанацетат натрия.
Стадия 1: Метил 1-((3-(2-(1-гидрокси -1-метилэтил)фенил)- 1(R)-(3-(((2-тетрагидропиранил) окси)метил)фенил) пропокси)метил) циклопропанацетат.
К охлажденному раствору по примеру 14, стадия 7 (2,216 г, 4,28 ммоль) в 30 мл ТГФ при -100oC прибавляют 1,6 М BuLi в гексане (5,9 мл) и смесь перемешивают при -78oC в течение 30 минут. Затем прибавляют ацетон (630 мкл, 8,6 ммоль) и смесь перемешивают при -78oC в течение 1 часа и затем дают нагреться до -20oC. Затем прибавляют насыщенный водный NH4Cl и продукты экстрагировали EtOAc. При 0oC прибавляют диазометан ≈ 0,5 М. Когда этерификация заканчивается, избыток CH2N2 гасят AcOH. Раствор сушат над Na2SO4 концентрируют и промывают толуолом. Флэш хроматография остатка с EtOAc:гексаном от 15: 85 до 35: 65 дает, во-первых, оставшееся исходное вещество (дебромированное), во-вторых, продукт α-присоединения ацетона и, в-третьих, указанный в заголовке продукт.
1H ЯМР (CDCl3): δ 7.40 (1H, d), 7.34-7.08 (7H, m), 4.80 (1H, d), 4.72 (1H, m), 4.50 (1H, d), 4.33 (1H, dd), 3.93 (1H, m), 3.64 (3H, s), 3,57 (IH, m), 3.30 (1H, d), 3.20 (1H, m), 3.14 (1H, d), 2.96 (1H, m), 2.58 (1H, d), 2.33 (1H, d), 2.17-1.48 (8H, m), 1.65 (6H, 2s), 1.27 (1H, s, OH), 0.51 (4H, m).
Стадия 2: 1-((1(R)-(3-(2-(2,3- Дихлортиено[3,2-b]пиридин- -5-ил)этенил)фенил)-3- (2-(1-гидрокси-1-метилэтил)фенил)пропокси) метил)циклопропанацетат натрия.
Используя методы по примеру 1, стадии 16-19, но применяя на стадии 18 ((2,3-дихлортиено[3,2-b] пиридин-5-ил)метил) трифенилфосфоний бромид (пример 4, стадия 3), из эфира со стадии 1 получают указанную в заголовке натриевую соль.
11H ЯМР (free acid CDCl3): δ 8.00 (1H, d), 7.70 (1H, d), 7.60-7.50 (3H, m), 7.42-7.30 (3H, m), 7.26 (2H, m), 7.20-7.08 (2H, m), 4.45 (1H, dd), 3.30 (1H, m), 3.31 (1H, d), 3.20 (1H, d), 2.95 (1H, m), 2.58 (1H, d), 2.38 (1H, d), 2.18 (1H, m), 2.07 (1H, m), 1.70 (6H, 2s), 0.64-0.47 (4H, m).
Пример 16. 1-(((3-(4-Циклопропилфенил)-1(R)-(3-(2- (2,3-дихлортиено[3,2-b] пиридин-5-ил) этенил)фенил)пропил)тио) метил)циклопропанацетат натрия.
Стадия 1: 3-(1-Гидрокси-2 -пропен-1-ил)бензонитрил.
К 3-цианобензальдегиду (25 г, 0,190 ммоль) в ТГФ (576 мл) по каплям прибавляют при -10oC винилмагнийбромид в ТГФ (202 мл, 0,201 ммоль). Через 15 минут реакционную смесь выливают в холодный раствор 25% водного ацетата аммония и экстрагируют EtOac. Полученную смесь очищают с помощью флэш хроматографии с получением 17,5 г (60%) указанного в заголовке соединения.
Стадия 2: 3-(1-Гидрокси-2-пропен-1-ил)бензальдегид.
К нитрилу (стадия 1) (17,0 г, 0,107 ммоль) в ТГФ (465 мл) при -78oC прибавляют по каплям раствор DIBAL (157 мл, 0,235 ммоль). Полученную смесь медленно охлаждают до 0oC. После завершения реакции реакционную смесь выливают в 10% раствор виноградной кислоты (1 л). После перемешивания в течение 1 часа указанный в заголовке продукт экстрагируют EtOAc и очищают путем флэш хроматографии (от 40% до 50% EtOAc в гексане) с получением 15 г (88%) альдегида.
Стадия 3: 1-(3-(2-(2,3-Дихлортиено [3,2-b]пиридин-5-ил)- этенил)фенил)-2-пропен-1-ол.
К суспензии фосфониевой соли по примеру 4, стадия 3 (10 г, 19,4 ммоль) в ТГФ (110 мл) при -78oC прибавляют 1 M трет-бутоксида калия в ТГФ (17,8 мл, 17,8 ммоль). Через 10 минут при 0oC смесь желтого цвета доводят до комнатной температуры в течение периода 10 минут и затем охлаждают до -78oC. Затем прибавляют альдегид со стадии 2 (2,63 г, 16,23 ммоль) в ТГФ (40 мл) и реакционную смесь перемешивают 1 час при 0oC и 1 час при комнатной температуре. Реакционную смесь нейтрализуют путем прибавления раствора 25% водного ацетата аммония, экстрагируют EtOAc и очищают с помощью флэш хроматографии с получением 4,0 г (70%) олефинового продукта.
Стадия 4: 3-(4-Циклопропилфенил)-1-(3- (2-(2,3-дихлортиено [3,2-b]пиридин-5-ил) этенил)фенил)пропан-1-он.
Через смесь аллилового спирта со стадии 3 (1,0 г, 2,77 ммоль 4-(йодфенил)циклопропана (1,35 г, 5,50 ммоль), хлорида лития (135 мг), ацетата лития (749 мг) и ацетата палладия (50 мг) в ДМФ (6,98 мл) барботируют азот. Смесь нагревают в атмосфере азота при 70oC в течение 10 минут. После обработки 25% водным раствором ацетата аммония и EtOAc органическую фазу упаривают досуха. Полученный твердый продукт обрабатывают ацетоном с получением 650 мг кетона в виде твердого белого продукта. Фильтрат очищают на силикагеле с получением более 200 мг кетона.
Стадия 5: 3-(4-Циклопропилфенил)-1(S)(3-(2- (2,3-дихлортиено [3,2-b]пиридин-5-ил) этенил)фенил)пропан-1-ол.
К раствору в CH2Cl2 (4,0 мл) (1)-B-хлордиизопинокамфилборана (904 мг, 2,82 ммоль) при -30oC прибавляют раствор кетона со стадии 4 (45 мг, 0,934 ммоль) в CH2Cl2 (4,6 мл). Температуру медленно доводят до 0oC в течение более 3 часов. Затем прибавляют насыщенным раствор NH4Cl и смесь перемешивают в течение ночи при комнатной температуре. После нейтрализации 25% водным раствором NH4OAc продукт экстрагируют EtOAc. После упаривания к остатку прибавляют эфир и затем 1 н. раствор HCl. Гидрохлоридную соль фильтруют и три раза промывают эфиром. К суспензии соли в воде и EtOAc прибавляют 1 н. раствор NaOH и диэтаноламина (10%). После упаривания получают 270 мг (60%) желаемого хирального спирта.
Стадия 6: 5-(2-(3-(3- (4-Циклопропилфенил)-1(S)-(метансульфонилокси) пропил)фенил)этенил) -2,3-дихлортиено[3,2-b]пиридин.
К раствору спирта (стадия 5) (230 мг, 0,47 ммоль) в CH2Cl (2,5 мл) при -40oC прибавляют Et3N (100 мкл, 0,717 ммоль) и MsCl (45,0 мкл, 0,547 ммоль). Полученную смесь затем нагревают до 0oC. После периода в 10 минут прибавляют насыщенный раствор NaHCO3. Мезилат экстрагируют CH2Cl2, сушат над Na2SO4 упаривают и дважды перегоняют с толуолом и в таком виде используют на следующей стадии.
Стадия 7: 1-(((3-(4-Циклопрофилфенил)-1(R)-(3- (2-(2,3-дихлортиено [3,2-b] пиридин-5-ил)этенил)фенил)пропил)тио)метил)- циклопропанацетат натрия.
В раствор тиоловой кислоты, полученной гидролизом эфира со стадии 9 примера 1 (63,0 мг, 0,431 ммоль), в ТГФ (1,7 мл) барботируют N2. Затем в течение более 15 минут при -15oC по каплям прибавляют н-бутиллитий. После периода 15 минут при -15oC температуру медленно доводят до -5oC. К полученной суспензии при -20oC прибавляют раствор мезилата (стадия 6) (230 мг, 0,411 ммоль) в ТГФ (1,7 мл). Температуру медленно поднимают до -5oC, затем до 0oC и до комнатной температуры. Через 2 часа прозрачный раствор гасят прибавлением 25% водного раствора NH4OAc, экстрагируют EtOAc и сушат над Na2SO4. Указанный в заголовке продукт очищают флэш хроматографией 50% EtOAc в гексане и далее 50% EtOAc в гексане с 1% HOAc с получением 160 мг (75%) продукта.
1H ЯМР (300 МГц, CD3COCD3: δ 0,30-0.50 (4H, m), 0.60-0.85 (4H, m), 1.85 (1H, m), 2.15 (2H, m), 2.48 (2H, s), 2.55 (2H, AB system), 2.60 (2H, m), 3.95 (1H, t), 7.00 (4H, AA BB system), 7.30-7.45 (3H, m), 7.60 (1H, m), 7.68 (1H, s), 7.75 (1H, d), 7.89 (1H, d), 8.49 (1H, d).
Пример 123. (R)1-(((3-(2-(1-Гидрокси-1-метилэтил)фенил)-1- -(3-(2-(2-метилтиазоло[5,4-b]пиридин-5-ил)этенил)фенил)пропил)- тио)метил)циклопропанацетат натрия.
Стадия 1: N-Ацетил-2-хлор-3-пиридинамин.
К раствору 2-хлор-3-пиридинамина (14,9 г, 116 ммоль) в 300 мл ТГФ прибавляют K2CO3 (32 г, 232 ммоль) и ацетилхлорид (12 мл, 169 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Прибавляют насыщенный NH4Cl и продукт экстрагируют EtOAc, сушат над Na2SO4 и фильтруют сквозь силикагель с получением 20,81 г указанного в заголовке продукта.
1H ЯМР (CDCl3): δ 8.73 (1H, d), 8.13 (1H, d), 7.65 (1H, br s, NH), 7.28 (1H, dd), 2.27 (3H, s).
Стадия 2: 2-Метилтиазоло[5,4-b]пиридин.
Пентасульфид фосфора (56,5 г), Na2CO3 (13,7 г) перемешивают вместе в 400 мл ТГФ в течение ≈ 30 минут. К этому раствору прибавляют раствор продукта со стадии 1 (17,32 г) в 100 мл ТГФ и смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 2 М NaOH (500 мл) и смесь перемешивают при комнатной температуре в течение 2 часов. Продукт экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над Na2SO4 и очищают флэш хроматографией на силикагеле с EtOAc:толуолом 20:80; выход: 12,07 г (83%).
1H ЯМР (CDCl3): δ 8.54 (1H, d), 8.18 (1H, d), 7.40 (1H, dd), 2.88 (3H, s).
Стадия 3: 2-Метилтиазоло[5,4-b]пиридин N-оксид.
К раствору продукта со стадии 2 (8,00 г) в 400 мл CH2Cl2 прибавляют м-хлорнадбензойную кислоту (26,0 г) и смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 0,5 М NaOH и очищают флэш хроматографией на силикагеле с ацетоном:толуолом:метанолом 40:40:20.
1H ЯМР (CDCl3): δ 8.29 (1H, d), 7.86 (1H, d), 7.38 (1H, dd), 2.88 (3H, s).
Стадия 4: 5-Циано-2-метилтиазоло[5,4-b]пиридин.
К раствору продукта со стадии 3 (4,706 г, 28,3 ммоль) в 60 мл CH2Cl2 прибавляют триметилсилилцианид (7,6 мл, 57 ммоль) и смесь перемешивают при комнатной температуре в течение 30 минут. Затем прибавляют диметилкарбомоилхлорид (5,2 мл, 56 ммоль) и смесь кипятят с обратным холодильником в течение ночи. При 0oC прибавляют 2 н. NaOH (60 мл) и смесь перемешивают при этой температуре один час. Продукт экстрагируют EtOAc, сушат над Na2SO4 и очищают флэш хроматографией на силикагеле с EtOAc:толуолом 10:90; выход: 4,50 г (91%).
1H ЯМР (CDCl3): δ 8.26 (1H, d), 7.80 (1H, d), 2.93 (2H, s).
Стадия 5: 5-Формил-2-метилтиазоло[5,4-b]пиридин.
К суспензии продукта со стадии 4 (4,42 г, 25 ммоль) в 100 мл безводного ТГФ при -78oC прибавляют по каплям 1,5 М раствор диизобутилалюминийгидрида в толуоле (40 мл) и смесь перемешивают при -78oC в течение 2 часов. Затем прибавляют 10% раствор виноградной кислоты и смесь перемешивают при комнатной температуре в течение 2 часов, нейтрализуют 10 н. NaOH и экстрагируют EtOAc. Указанный в заголовке продукт сушат над Na2SO4 и очищают флэш хроматографией на силикагеле с EtOAc:гексаном 20:80 с получением 3,733 г (83%) в виде белого твердого продукта.
1H ЯМР (CDCl3): δ 10.13 (1H, s), 8.33 (1H, d), 8.12 (1H, d), 2.95 (3H, s).
Стадия 6: 5-(Гидроксиметил)-2-метилтиазоло[5,4-b]пиридин.
К суспензии продукта со стадии 5 (3,733 г, 21 ммоль) в 200 мл EtOH при 0oC прибавляют NaBH4 (800 мг, 21 ммоль) и смесь перемешивают при 0oC в течение 5 минут. Затем медленно прибавляют насыщенный водный NH4Cl и смесь экстрагируют EtOAc: ТГФ 1:1, сушат над Na2SO4 и очищают флэш хроматографией на силикагеле с ацетоном:толуолом 30:70; выход: 3,49 г (92%).
1H ЯМР (CDCl3): δ 8.15 (1H, d), 7.37 (1H, d), 4.89 (2H, d), 3.45 (1H, t, OH), 2.87 (3H, s).
Стадия 7: 5-(((Метансульфонил)окси)метил)-2-метилтиазоло- [5,4-b]пиридин.
К раствору спирта со стадии 6 (303 мг, 1,68 ммоль) в 17 мл CH2Cl2 при -40oC прибавляют триэтиламин (350 мкл, 2,5 ммоль) и метансульфонилхлорид (170 мкл, 2,2 ммоль) и раствор перемешивают при -40oC в течение 30 минут и при 0oC в течение 2 часов. Прибавляют насыщенный водный NaHCO3 и продукт экстрагируют CH2Cl2, сушат над Na2SO4, концентрируют и удаленную воду дважды промывают толуолом.
1H ЯМР (CDCl3): δ 8.23 (1H, d), 7.48 (1H, d), 5.43 (2H, s), 3.10 (3H, s), 2.88 (3H, s).
Стадия 8: ((2-Метилтиазоло[5,4-b] пиридин-5-ил)метил- трифенилфосфоний метансульфонат.
Раствор мезилата со стадии 7 (1,68 ммоль) и трифенилфосфина (660 мг, 2,52 ммоль) в 8 мл безводного CH3CN нагревают с обратным холодильником в течение 2 часов. Растворитель упаривают и масло дважды обрабатывают эфиром и эфир дважды декантируют. Опять обрабатывают 25 мл эфира через 2,5 дня. Растворитель декантируют с получением очень гигроскопичного твердого продукта, который сушат в вакууме; выход: 732 мг (84%).
1H ЯМР (DMSO): δ 8.20 (1H, d), 7.65-7.90 (15H, m), 7.41 (1H, d), 5.58 (2H, d), 2.77 (3H, s), 2.29 (3H, s).
Стадия 9: (R) 1-(((3-(2-(1-Гидрокси-1-метилэтил)-1-(3-(2-(2- метилтиазоло[5,4-b] пиридин-5-ил)-этенил)фенил)пропил)тио)метил)- циклопропанацетат натрия.
Используя методы, описанные в стадиях 18-19 примера 1, из фосфониевой соли со стадии 8 получают указанное в заголовке соединение.
Анализ. Вычислено для C33H35N2O3S2Na • 3,6H2O:
C 60,09; H 6,45; N 4,25.
Найдено: C 60,04; H 6,41; N 4,28.

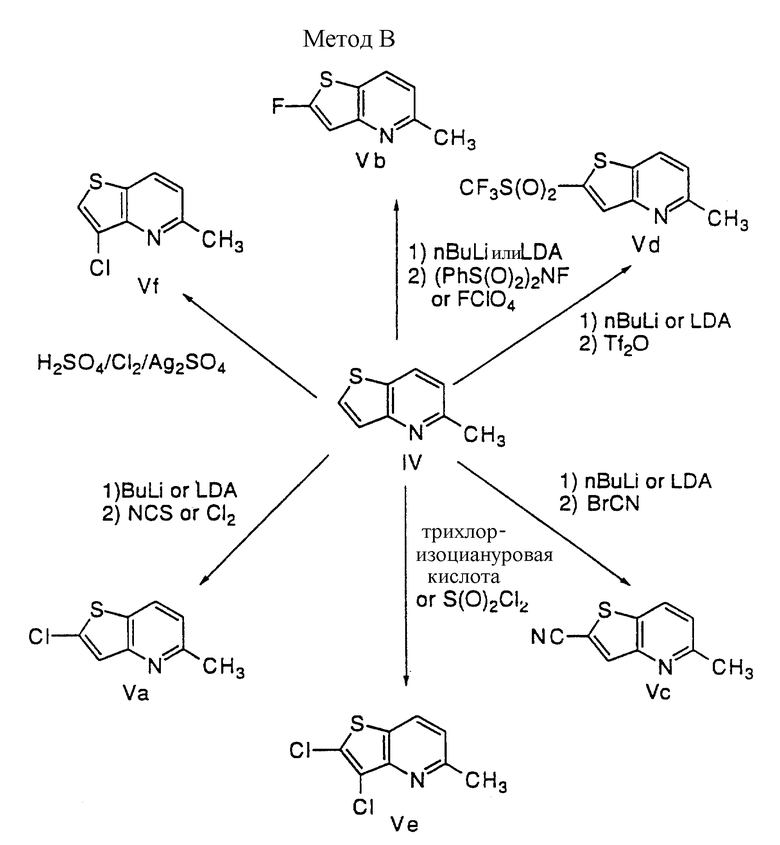
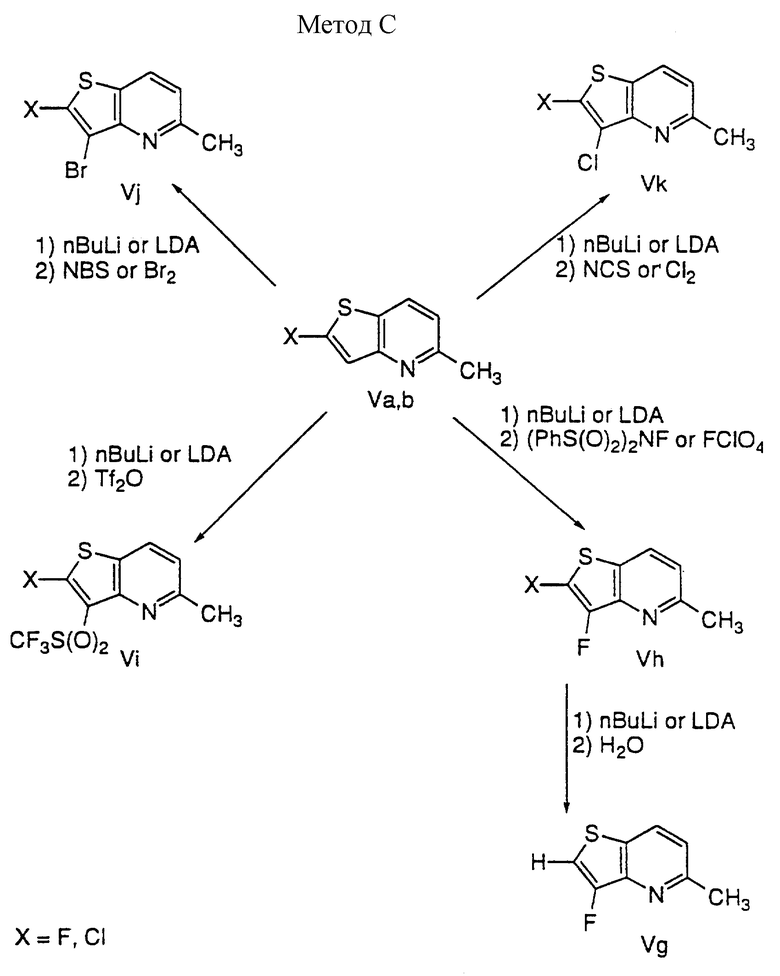

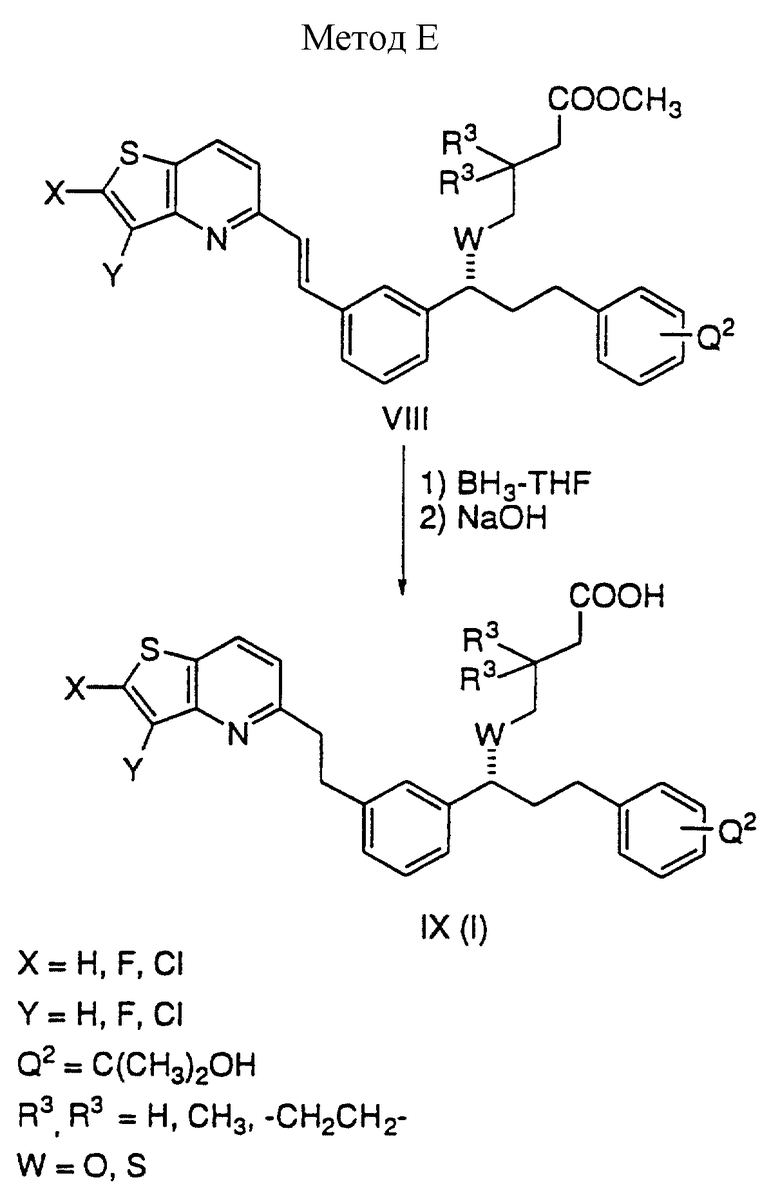
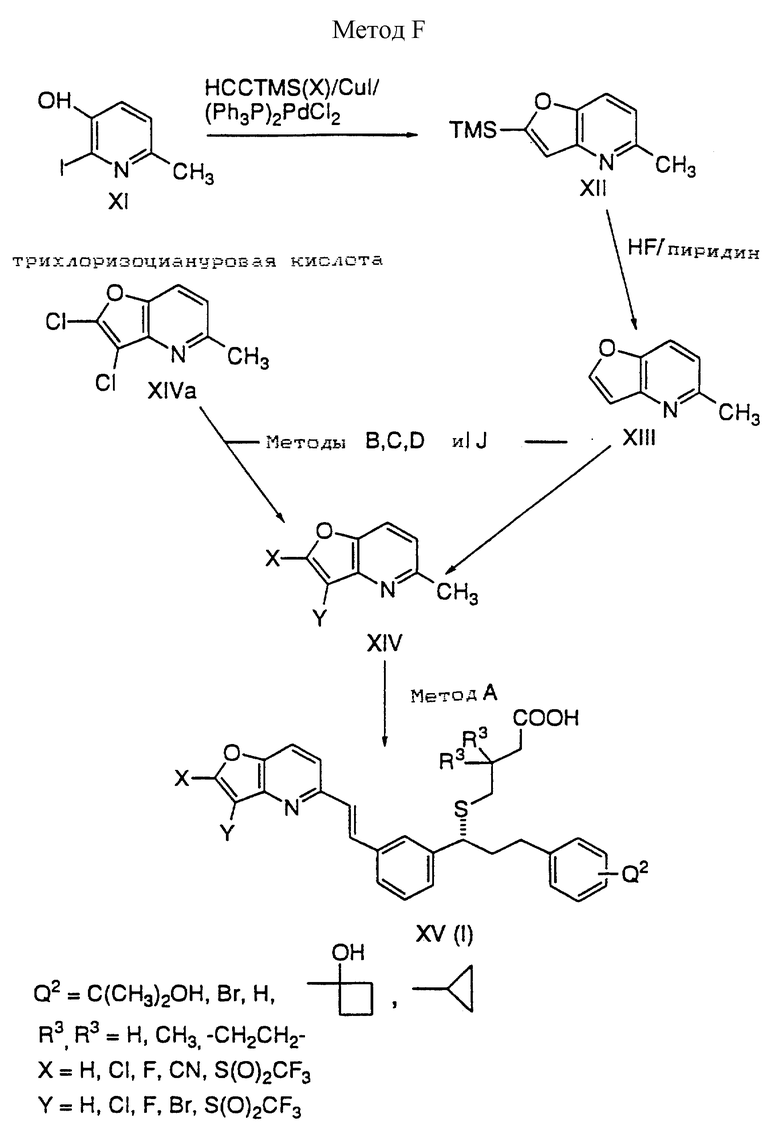

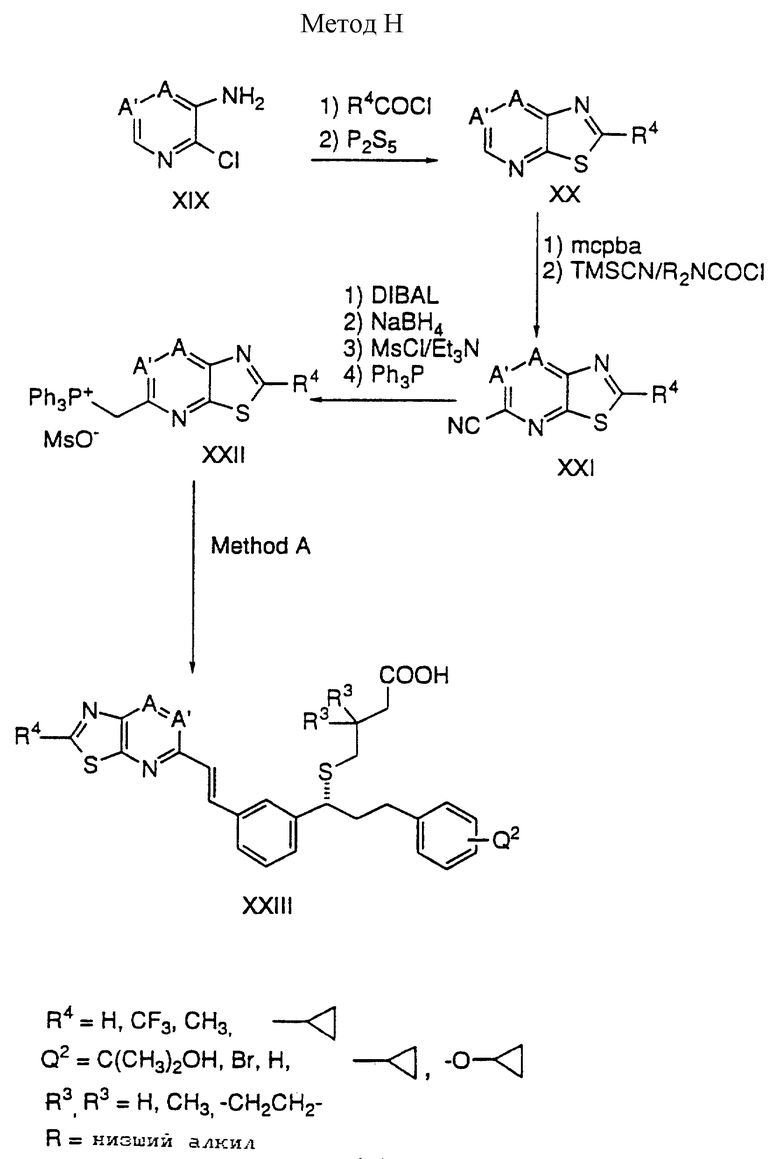
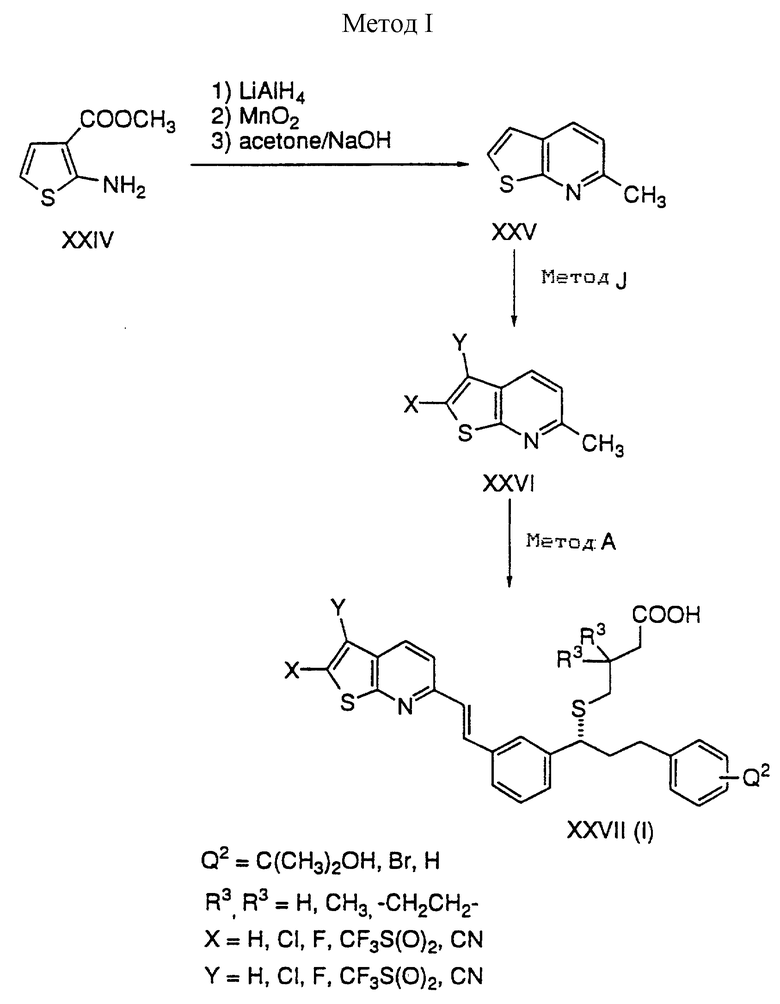
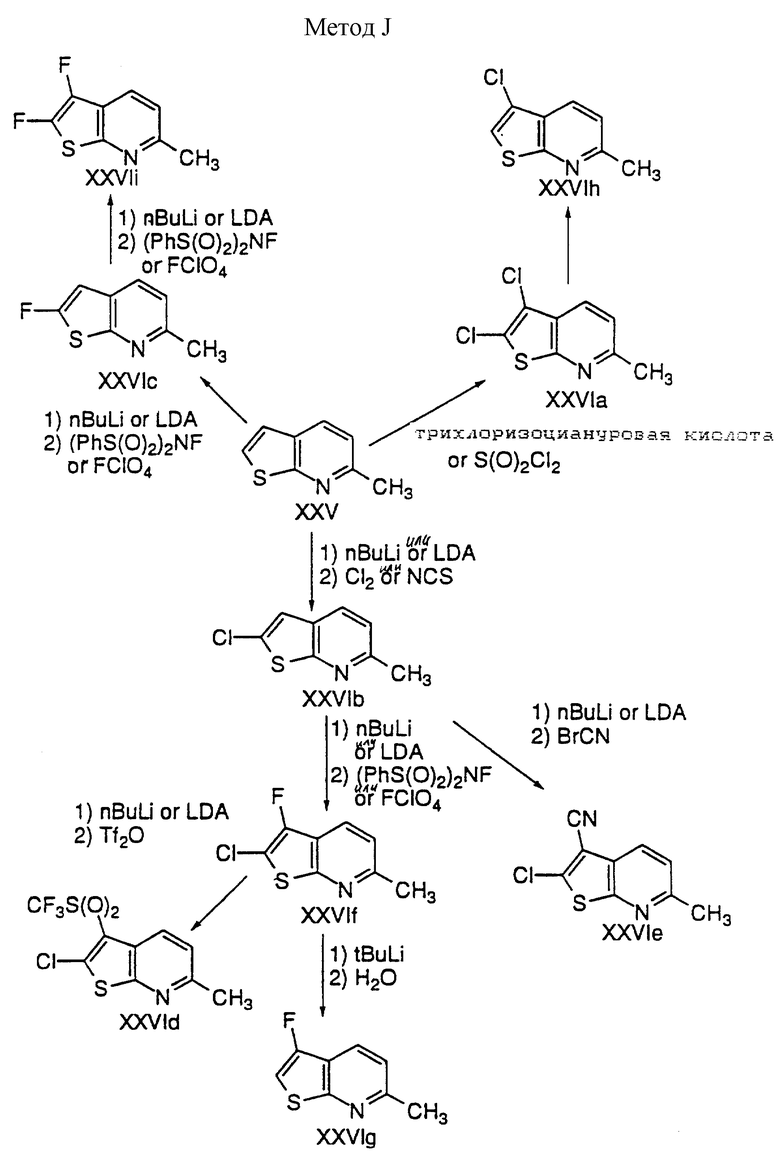
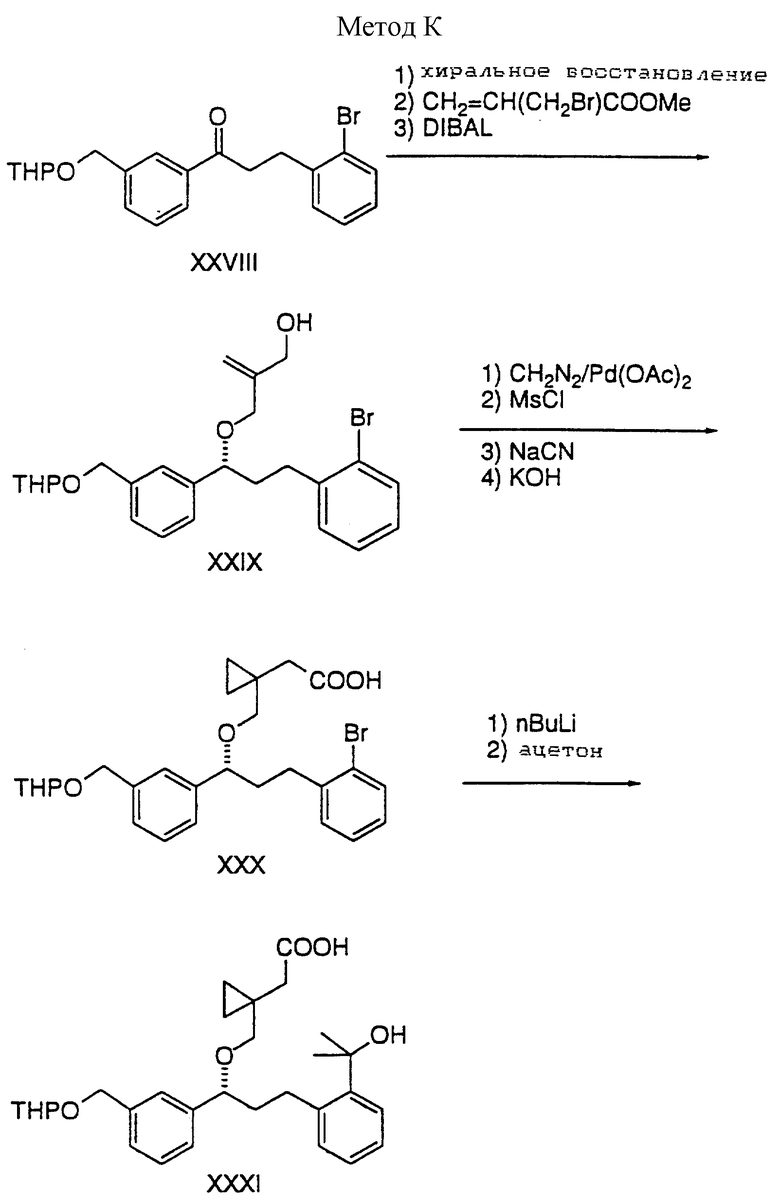
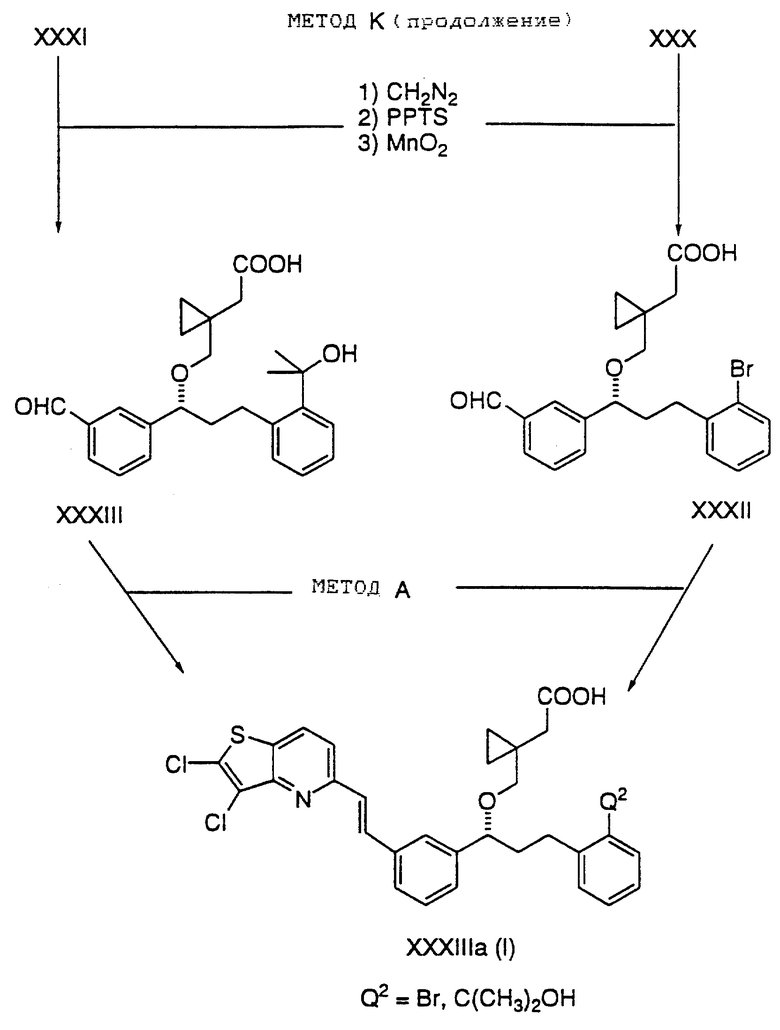
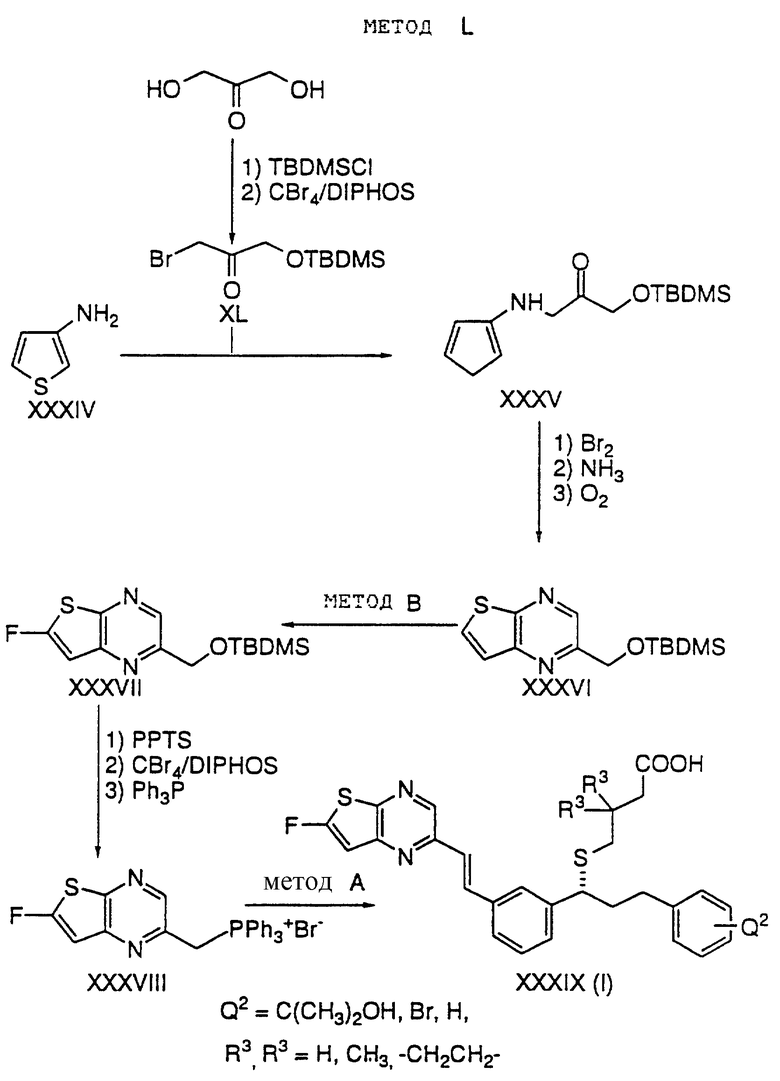
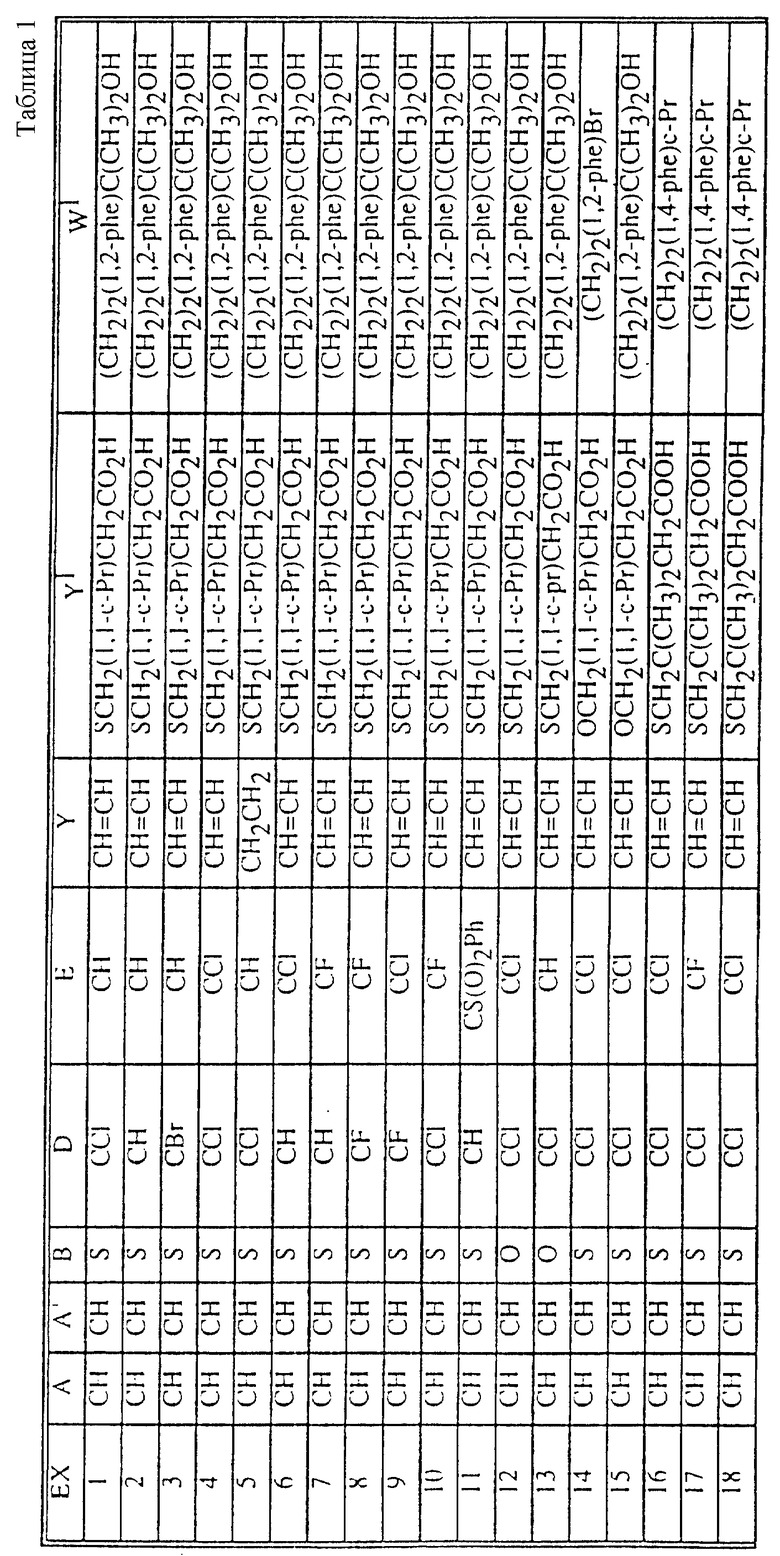
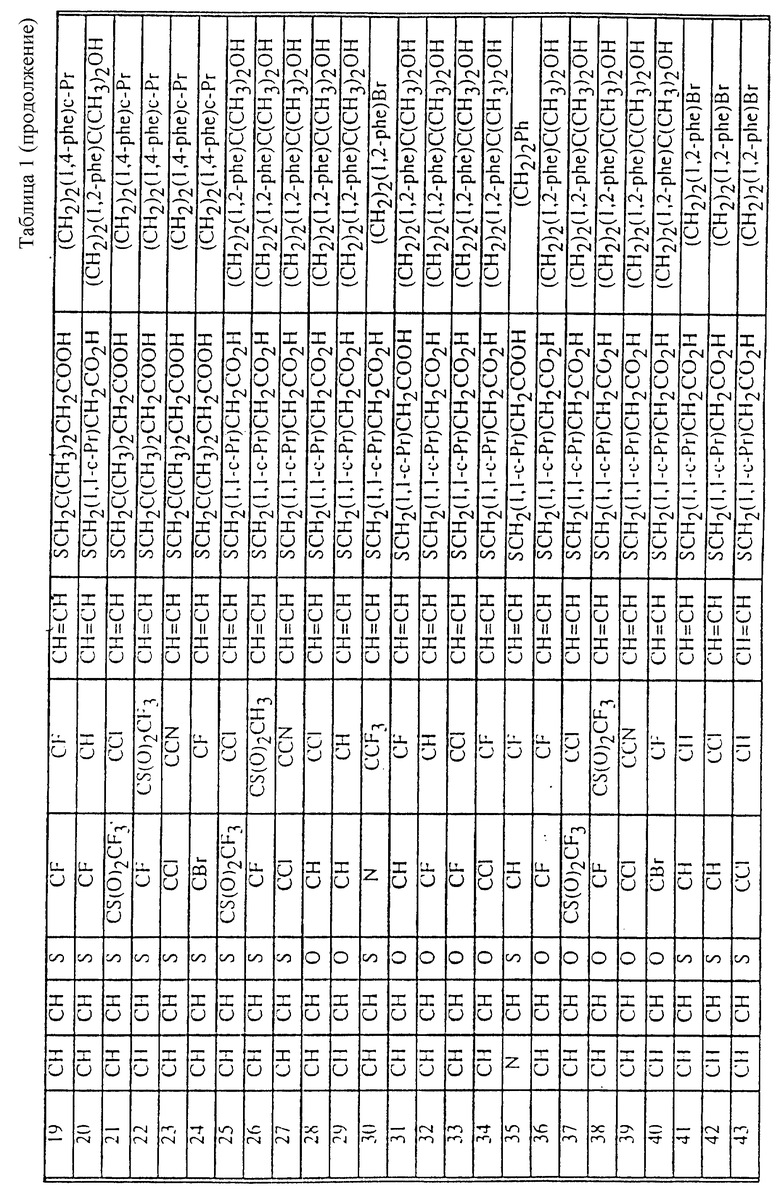
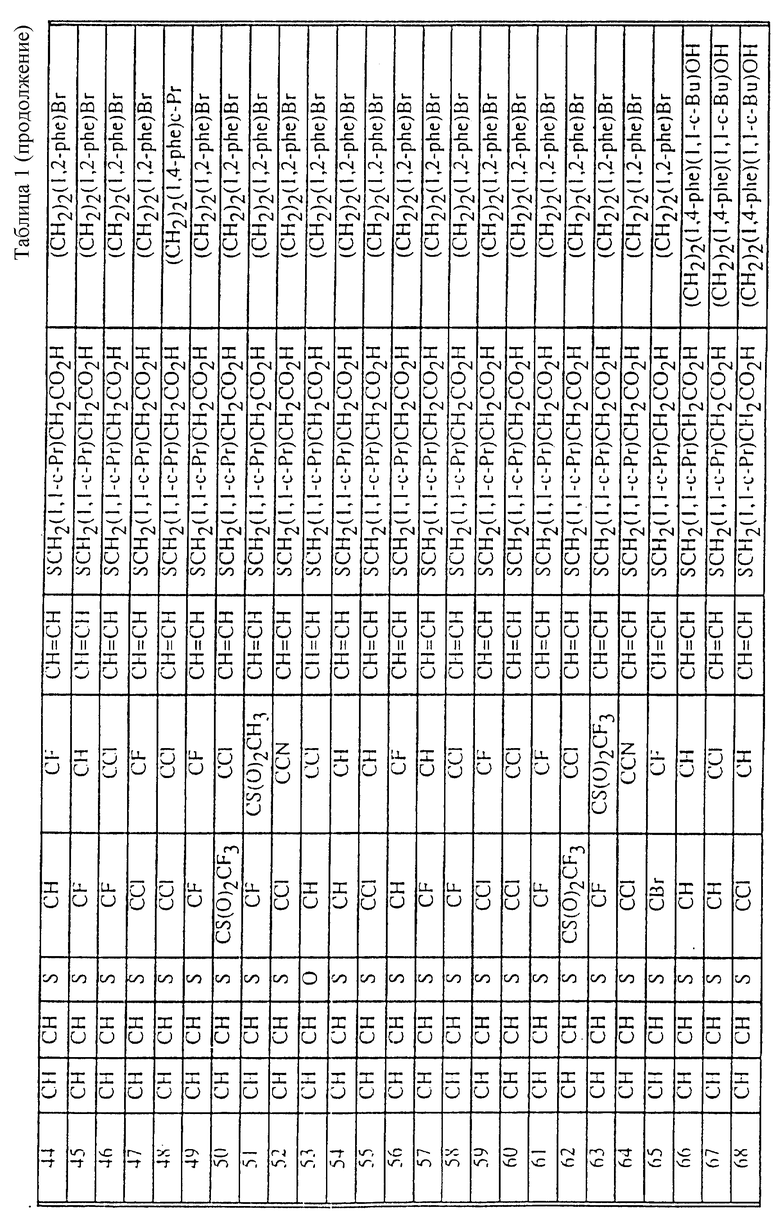
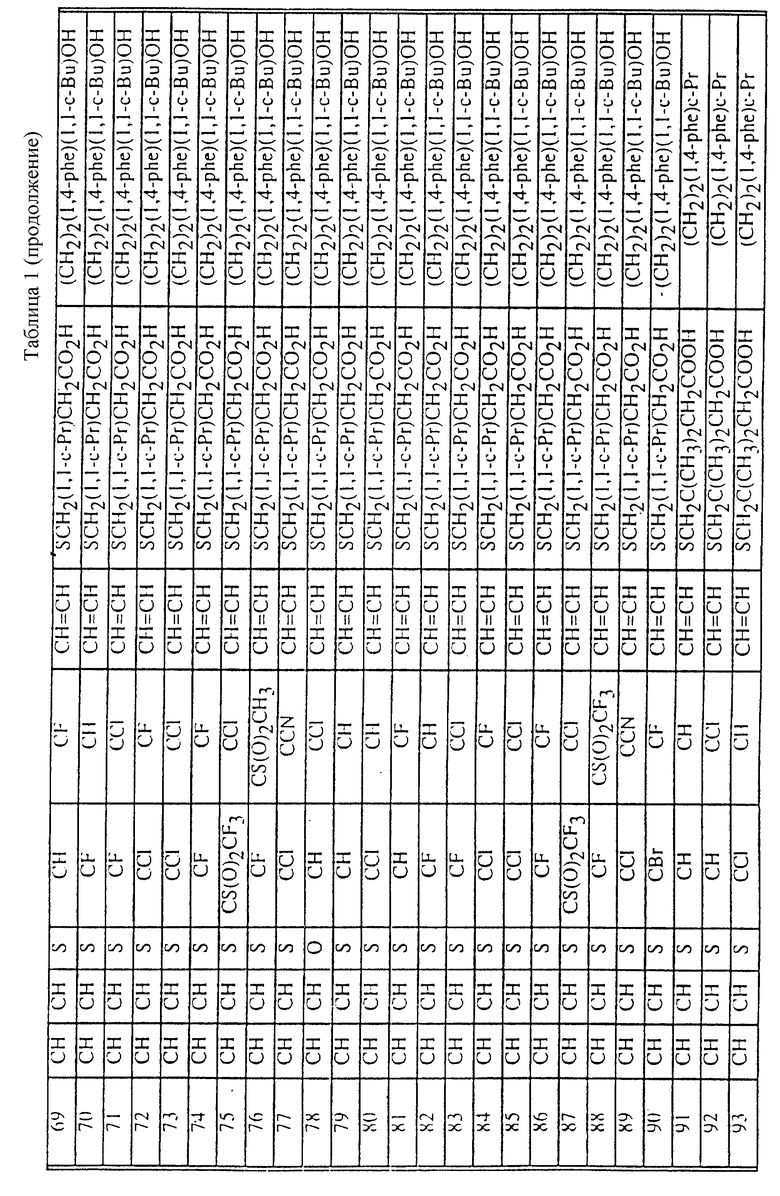
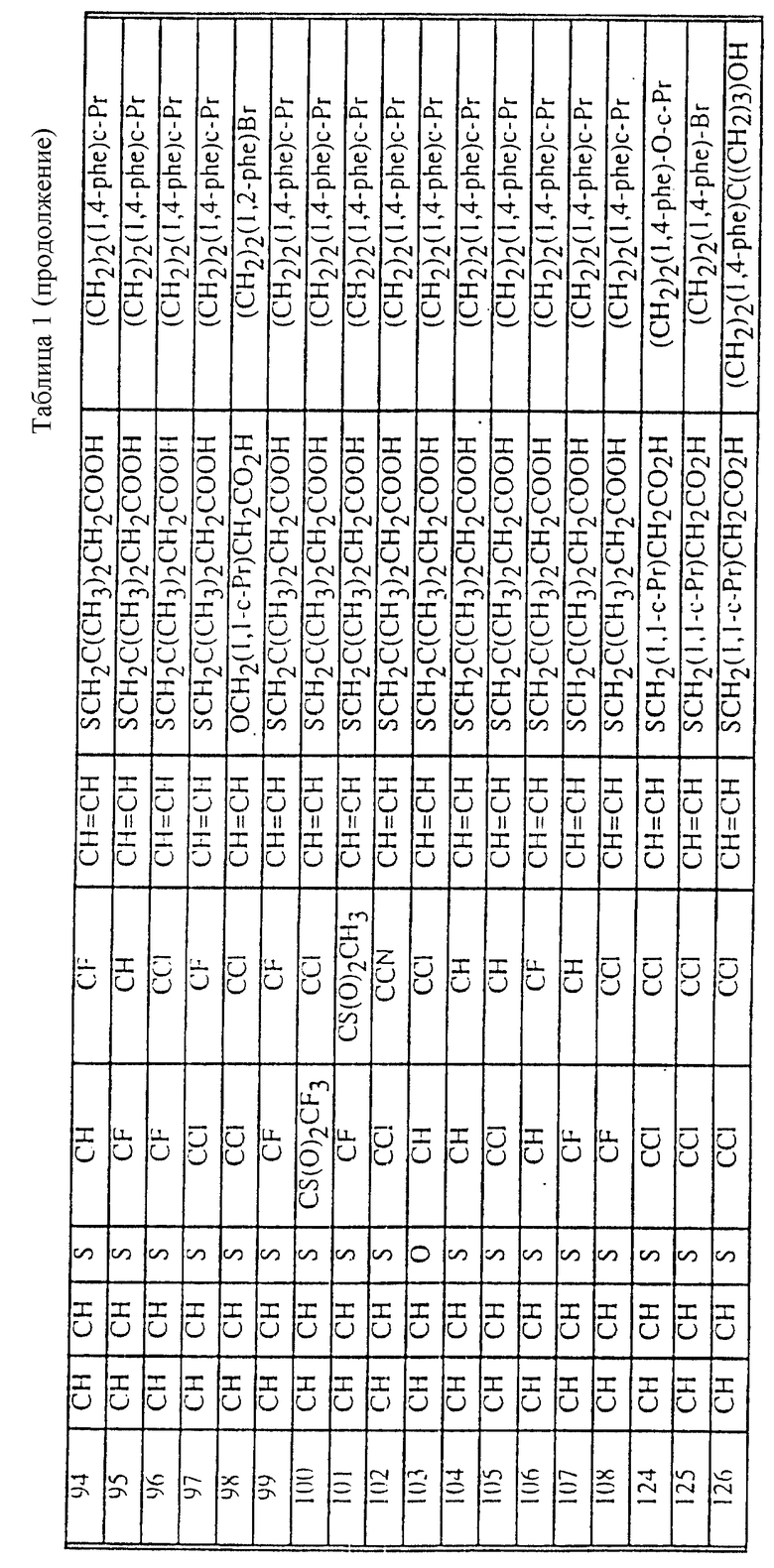
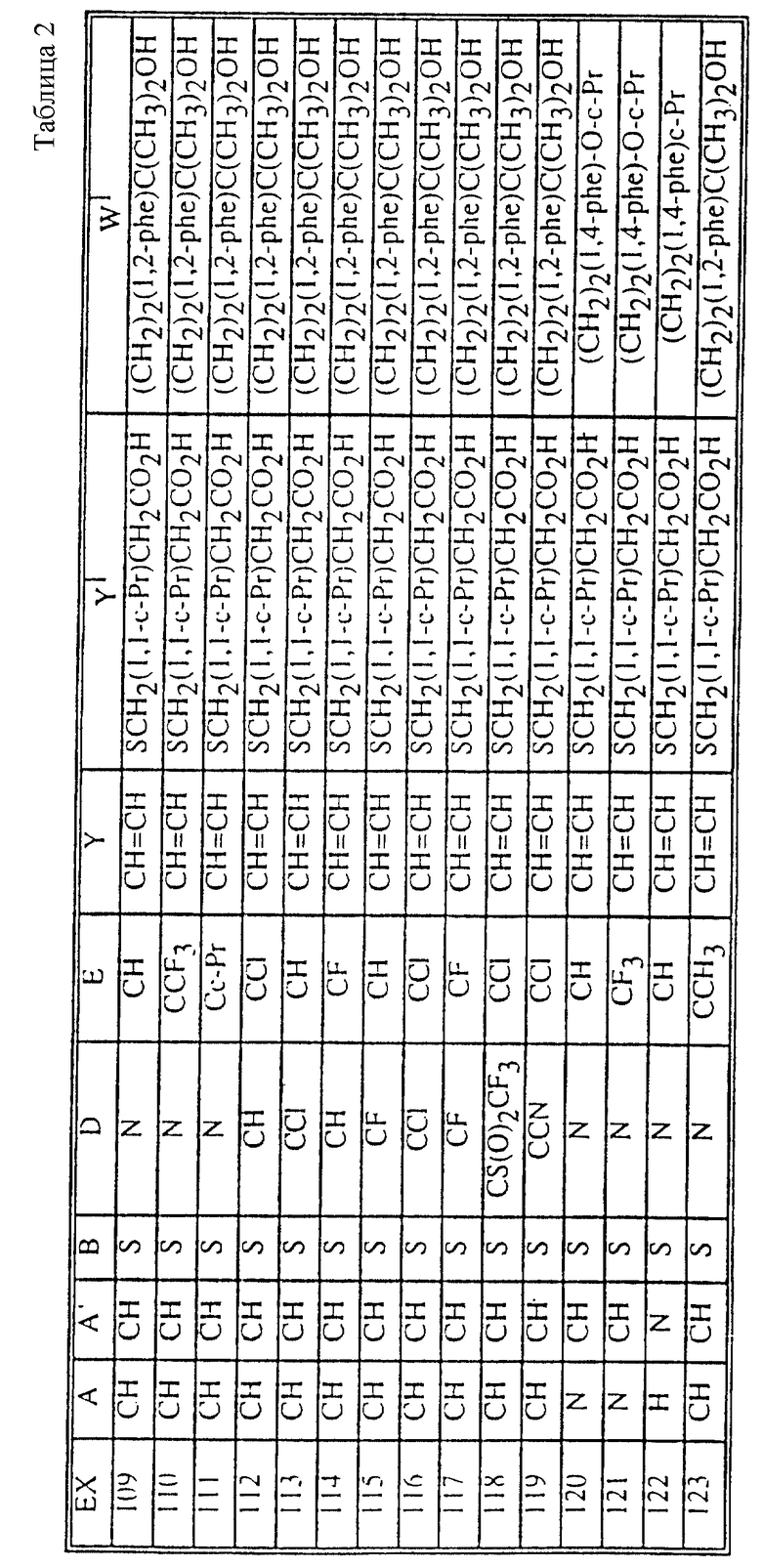
Описываются новые диарил-5,6-конденсированные гетероциклические кислоты общей формулы (I), где значения НЕТА, Y, R1; R3-R7, X2, X3, Z1, Z2, R22, p, m, Q1, Q2 указаны в п.1 формулы изобретения, которые являются антагонистами действия лейкотриенов. Соединения полезны в качестве противоастмастических, противоаллергических, противовоспалительных и цитозащитных агентов. Они также полезны при лечении ангины, спазмов головного мозга, нефрита почечных клубочков, гепатита, эндотоксикоза, увеита и отторжения аллотрансплантата. Описывается также фармацевтическая композиция и способ защиты от воздействия лейкотриенов. 3 с. и 8 з.п. ф-лы, 2 табл.
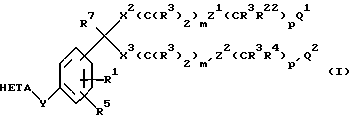
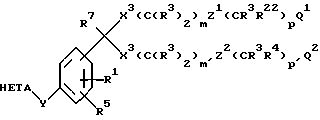
где НЕТА обозначает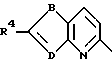
В - S или O;
D - N или CR4;
R1, R5 и R7 каждый, Н;
R3 - Н, низший алкил или две R3-группы, присоединенные к одному и тому же атому углерода, могут образовывать 3-6-членное углеводородное кольцо;
R4 - Н, низший алкил, галоген или S(О)2R2, где R2 - фенил;
m и m1 каждый независимо равен 1 - 6;
р и р1 каждый независимо равен 0 или 1;
Q1 - СО2R3;
Q2 - водород, С(R3)2ОR3, ОR15 или циклоалкил С3-C7;
Х2 - S или О;
Х3 - связь;
Y - -СН=СН-;
Z1 - связь;
Z2 - НЕТ(R23R24), где НЕТ обозначает дирадикал бензола;
R15, R22, R23 и R24 каждый - водород,
или их фармацевтически приемлемые соли.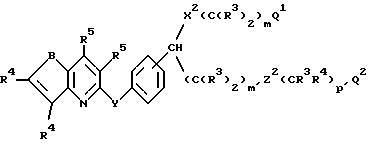
где В - S или О;
R3 - Н, низший алкил или две R3-группы, присоединенные к одному и тому же атому углерода, образуют 3-6-членное углеводородное кольцо;
R4 - Н, галоген или S(О)2R2, где R2 - фенил;
R5 - Н;
m и m1 каждый независимо равен 1 - 6;
р1 = 0 или 1;
Q1 - СО2R3;
Q2 - С(R3)2ОR3;
Х2 - S или О;
Y - -СН=СН-;
Z2 - НЕТ(R23R24), где НЕТ - дирадикал бензола;
R23 и R24 - водород.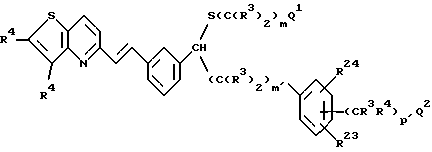
где R3 - Н, низший алкил или две R3-группы, присоединенные к одному и тому же атому углерода, образуют 3-6-членное углеводородное кольцо;
R4 - Н, галоген или S(О)2R2, где R2 - фенил;
R23 и R24 - Н;
m и m' каждый независимо равен 1 - 5;
р1 = 0 или 1;
Q1 - СО2R3;
Q2 - Н, С(R3)2ОН или ОR15, где R15 - водород.
1-(((1(R)-(3-(2-(3-хлортиено[3,2-b] пиридин-5-ил-)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(тиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(3-бромтиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(2,3-дихлортиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(3-хлортиено[3,2-b] пиридин-5-ил)этил)фенил)-3-(2-(1-гидрокси-1-метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(2-хлортиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(2-фтортиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(2,3-дифтортиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(2-хлор-3-фтортиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(3-хлор-2-фтортиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(2-фенилсульфонил)тиено[3,2-b]пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(2,3-дихлорфуро[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
1-(((1(R)-(3-(2-(3-хлорфуро[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропил)тио)метил)циклопропанацетат натрия;
(R) 1-((3-(2-бромфенил)-1-(3-(2-(2,3-дихлортиено[3,2-b] пиридин-5-ил)этенил)фенил)пропокси)метил)циклопропанацетат натрия;
1-((1(R)-(3-(2-(2,3- дихлортиено[3,2-b] пиридин-5-ил)этенил)фенил)-3-(2-(1-гидрокси-1- метилэтил)фенил)пропокси)метил)циклопропанацетат натрия;
1-(((3-(4-циклопропилфенил)-1(R)-(3-(2-(2,3-дихлортиено[3,2-b] пиридин-5-ил)этенил)фенил пропил)тио)метил)циклопропанацетат натрия;
(R) 1-(((3-(2-(1-гидрокси-1-метилэтил)фенил)-1-(3-(2-(2-метилтиазоло[5,4-b] пиридин-5-ил)этенил)фенил) пропил)тио)метил)циклопропанацетат натрия.
| 0 |
|
SU318084A1 | |
| Способ получения конденсированных производных пирролинона или их гидрохлоридов | 1983 |
|
SU1382400A3 |
| US 4957932 А, 18.09.1990. | |||

