Настоящее изобретение относится к новому соединению, которое обладает высокой активностью в качестве антагониста окситоцина и проявляет слабый антагонизм по отношению к вазопрессину.
Предпосылки к созданию изобретения
Преждевременные роды являются основной причиной пренатальной заболеваемости и смертности в Соединенных Штатах. Современные методы ингибирования преждевременных родов не всегда успешны и часто связаны со значительными побочными действиями. Поскольку матка является органом-мишенью для окситоцина и принимая во внимание, что окситоцин является фактором, значительно способствующим преждевременным родам, разработка сильного антагониста окситоцина должна привести к успешному ингибированию преждевременных родов с незначительными побочными действиями.
Структурно окситоцин (ОТ) и антидиуретический гормон (ADH), называемый также вазопрессином, сходны. Их сравнительные структуры приведены ниже.
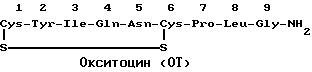
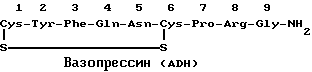
Различные исследователи в литературе описывают синтез антагонистов ADH, предназначенных для лечения гипертензии, и синтез антагонистов окситоцина. В 1960 г. Law, H. D. и V. Duvigneaud, J. Am., Chem. Soc., 82:4579, описали первый синтез антагониста окситоцина, представляющего собой 2-O-метилтирозин-OT. В 1967 г. Chan, Fear и Duvigneaud, Endoclinolody, 81:1267, описали 1-L-пеницилламинокситоцита и 1-дезаминопеницилламинокситоцина. Это было первое исследование, показывающее in vivo ингибирующее действие антагониста окситоцина на сокращение матки и ответную реакцию на окситоцин у анестезированных крыс.
В 1980 г. Sawyer, et al., Endocrinology, 160:81, описали синтез антагониста окситоцина, который объединял две характерные черты антагониста Law и Duvigneaud и антагониста Chan et al. Новый антагонист был (1-демаминопеницилламин-2-O-метилтирозин)окситоцином. Этот антагонист имел значение pA2 7, 8, определенное биоанализом на стимулирование родовой деятельности, pA2 является логарифмом отрицательным молярной концентрации антагониста, которая снижает восприимчивость к агонисту на 1/2. Это определение Schild, British J. Pharmacology, 2:189 (1947).
В 1983 г. Manning, et al., J. Med. Chem. 26:1607 - 161, описали синтез ряда антагонистов для ADH. Оказалось, что один из этих антагонистов обладает сильной активностью, противодействующей стимулированию родовой деятельности, и является (β,β-пентаметилен-β-меркаптопропионовая кислота1-D-Phe2-Ile4) аргининвазопрессином с pA2 8,2, или другими словами, он в 2,5 раза сильнее, чем антагонист, описанный Sawyer, et al., в 1980 (смотри стр. 1610, таблицу 1, соединение N 1). Этот антагонист окситоцина можно назвать (Pmp1-D-Phe2-Phe3- Ile4-Arg8)окситоцином. Родственный антагонист окситоцина, (Pmp1-D-Trp2-Phe3-Ile4- Arg8)окситоцин, описан Wilson and Gluoret, Abstract for Society for the Study of Reproduction Meeting July 14 - 17, 1986.
В 1981 г. Melin et al., Endocrinology, 88:173, получили антагонист окситоцина для ингибирования преждевременных родов. Они синтезировали 1-дезаминоэтилокситоцин, который имеет pA2 7,2. Они показали также, что это соединение ингибировало сокращение матки у крыс in vivo и у людей in vitro и in vivo (Fkerland, et al. , Obstet and Gynecol., 62:309, 1983). В 1985 г, Akerland, et al., Obstet and Gynecol., 64:499, описали синтез 1-дезамино-(D-Tyr(OC2H5)2-Thr4-Orn8)вазопрессина с pA2 8,3. Они испытывали это соединение in vitro на ткани матки человека и показали, что оно ингибирует сокращение матки.
В патенте 4597901 США описывается класс антагонистов вазопрессина, у которых цистеил-1 присутствует как в окситоцине, так и вазопрессине, и которые замещены β,β-циклопентаметилен-β-меркаптопропионовой кислотой.
Другие аминокислоты вазопрессина являются замещенными. Сообщается, что соединения полученного класса являются антагонистами вазопрессина, их биологическая активность проявляется в виде водного диуреза.
Сущность изобретения.
Настоящее изобретение относится к антагонисту окситоцина, который является аналогом окситоцина. В соединении настоящего изобретения цистеин-1 окситоцина замещен на β,β-(3-тиапентаметилен)-β-меркаптопропионовую кислоту. Кроме того, L-тирозин-2 замещен на D-триптофан, и пеницилламин замещен 1-цистеином в положении 6, а L-аргинин замещен в положении 8 L-лейцином. Полученное соединение, ((S)Pmp1-D-Trp2-Pen6-Arg8)окситоцин, как полагают, является новым соединением, как оказалось, обладает интересными свойствами. Оно высоко активно в качестве антагониста окситоцина. В то же время, хотя оно структурно близко вазопрессину и антагонистам вазопрессина, описанным в литературе, это новое соединение обладает минимальным антагонизмом к ADH. Если эти два антагонизма представить в виде соотношения, то соединение настоящего изобретения имеет очень высокое соотношение активностей анти-окситоцин/анти-ADH. Эта комбинация свойств очень благоприятна для терапевтического применения. Эффективное антиокситоциновое действие можно получить при минимальном побочном действии против ADH. Соединение настоящего изобретения, следовательно, пригодно для ингибирования сокращения мышц матки в ответ на окситоцин, его можно применять для подавления преждевременных родов.
Описание примеров осуществления изобретения.
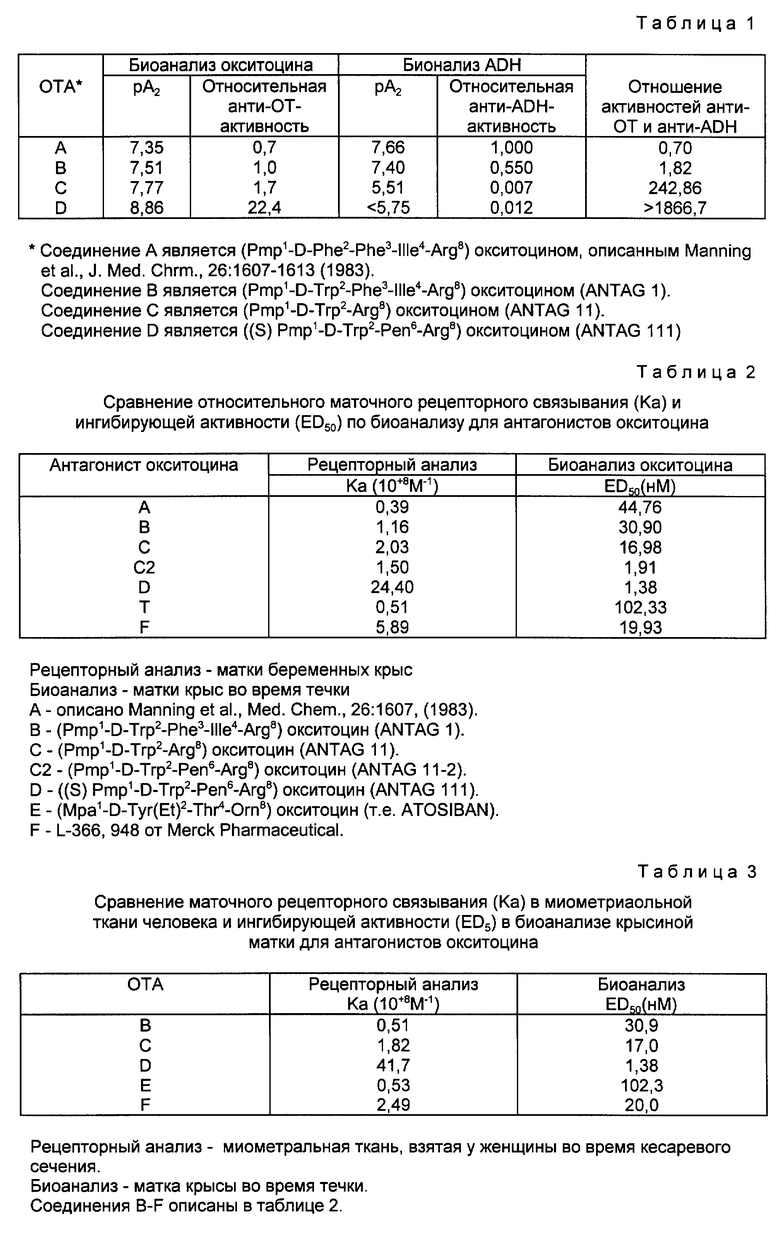
Антагонист окситоцина настоящего изобретения представлен формулой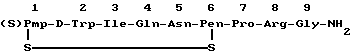
в которой
Pmp представляет собой β,β-(3-тиапентаметилен)-β-меркаптопропионовую кислоту, D-Trp представляет собой D-форму триптофана и Ile, Gln, Asn, Pen (Pen является пеницилламином), Pro, Arg представляют собой L-формы изолейцина, глутамина, аспарагина, пролина и аргинина соответственно.
Интересные свойства нового соединения настоящего изобретения представлены результатами биоисследований, описанных далее.
Биоисследование окситоцина.
Протокол, применяемый для методики биоислледования окситоцина, взят из методик, описанных в статье Sawyer et al., Endocrinology, 106:81 (1980), которые, в свою очередь, основаны на сообщениях Vunsick, Brit. J. Pharmacol., 3:328 (1960), и Holton, Brit. J. Pharmacol., 3:328 (1948). Подсчеты значений pA2 анализа описаны Schild, British J. Pharmacology, 2:189 (1947). Основное отличие настоящей методики от методик, описанных другими в данной области, в том, что площадь при сокращении интегрируют, тогда как большинство других методик подсчитывают амплитуду. Интегрирование обеспечивает значительно более совместимые и надежные результаты, хотя оценки pA2 являются приблизительно на порядок величин ниже, чем оценки, сообщенные с применением амплитуды сокращения в конечной точке.
Методика.
1. Животные - для исследования используют 1,5 см кусочки матки неоплодотворенной крысы (Hortzman) в период природной течки.
2. Буфер/ванна для анализа - применяют буфер Munsicks. Этот буфер содержит 0,5 мМ Mg++, который снижает величины pA2, но позволяет получать результаты, которые лучше коррелируют с данными in vivo (Sawer et al., 1980). Буфер при непрерывном контакте со смесью 95% кислорода и 5% диоксида углерода обеспечивает pH 7,4. Температура ванны для анализа 37oC. Применяют ванну на 10 мл, которая имеет водяную рубашку для поддержания температуры и входные и выходные отверстия для добавления и удаления буфера.
3. Полиграф/датчик - Кусочек применяемой для исследования ткани матки прикрепляли у одного конца и соединяли у другого конца с датчиком Statham Strain Gauge Force, который, в свою очередь, соединяли с Grass полиграфом Model 79 для мониторинга сокращений матки.
4. Схема исследований. (a) Ткань выдерживали в ванной для анализа в течение 1 часа при промывании новой порцией буфера каждые 15 мин. Все время ткань поддерживают при напряжении 1 г.
(b) Ткань стимулируют сначала окситоцином при 10 нМ для "акклиматизации" ткани и 4 нМ KCl для определения максимальной сократительной реакции.
(c) Затем строят кривую ответной реакции на кумулятивную дозу окситоцина и концентрацию окситоцина, эквивалентную приблизительно 80% от максимума, применяют для установления pA2 антагониста.
(d) Ткань подвергают воздействию окситоцином (Calbiochemical, San Diego, California) в течение 1 минуты и промывают. Соблюдается трехминутный интервал до добавления следующей дозы агониста или антагониста. Когда испытывают антагонист, его подают за пять минут до агониста. Агонист подают в течение одной минуты. Все реакции интегрируют при помощи интегратора 7P10 Grass. Это основное различие - между настоящей схемой и другими схемами в литературе, которые обычно измеряют амплитуду сокращений в качестве ответной реакции. Одну концентрацию окситоцина, соответствующую 80% максимальной ответной реакции, применяют для испытания антагониста. Применяют три различные концентрации антагонистов, две, которые будут снижать ответную реакцию на антагонист по меньшей мере на 50%, и одну, которая будет снижать ответную реакцию более чем на 50% (идеально 25%, 50% и 75%). Это повторяют три раза для каждой из трех доз антагониста.
(e) Подсчеты pA: Подсчитывают соотношение доза-ответная реакция (DR) для антагониста и диаграмму Schild строят нанесением Log (DR-1) в зависимости от Log концентрации антагониста. Нанесенную на диаграмму линию подсчитывают регрессивным анализом методом наименьших квадратов. pA2 представляет концентрацию антагониста в точке, где линия регресса пересекает 0 точку ординаты Log (DR-1). pA2 является отрицательным Log концентрации антагониста, которая снизит ответную реакцию на агонист на половину.
Как аналог окситоцина новое соединение настоящего изобретения можно определить как ((S)Pmp1-D-Trp2-Pen6-Arg8) окситоцин. Когда это соединение испытывали указанным выше анализом в качестве конкурирующего антагониста (антиметаболита) окситоцина, было найдено, что в среднем а 10 анализах величина pA2 была выше 8,86.
ADH-биоисследование.
Указанное выше соединение испытывали также на антагонизм к вазопрессину. Анти-ADH-активность можно определить измерением изменения в диурезе, обусловленном ADH, в присутствии и отсутствие антагониста. Пригодный ADH-анализ описан Sawyer et al., Endocrinology, 63:694 (1958). Когда испытание проводили по этой методике, то нашли, что соединение (( S)Pmp1-D-Trp2-Pen6-Arg8) окситоцин обладает очень слабой активностью в качестве антагониста вазопрессина. Соотношение антагонизма к окситоцину и антагонизма к ADH очень высокое, а именно превышает 1866, по сравнению с 200 для композиций, описанных в родовых заявках.
Благодаря активности как антагониста окситоцина при минимальном антагонизме для вазопрессина соединение настоящего изобретения будет пригодным для лечения симптомов, требующих антагонизма окситоцину, у людей и животных. Его можно применять для ингибирования сокращений матки и выделения молока, а также ингибирования преждевременных родов. Хотя структура этого соединения походит на структуру как окситоцина, так и вазопрессина, оно обладает не только повышенной антиокситоциновой активностью, но также очень пониженной анти-ADH-активностью. Это соединение можно также применять для ингибирования дисменореи или в качестве антидота для гиперстимулирования сокращения матки в процессе индуцирования родов окситоцином или для лечения гипертензии.
Соединение настоящего изобретения можно вводить женщинам различными известными способами. Для стационарных лечебных учреждений обычно следует выбирать внутривенное введение. Однако это соединение можно также вводить внутрибрюшинно, подкожно или внутримышечно. Возможно также пероральное введение. Если нужно, для перорального применения можно изготовлять таблетки или капсулы с покрытием, защищающим от деструкции в желудке и дающим возможность высвобождения его в кишечном тракте. На практике можно применять также подъязычное введение, обеспечивая пригодные дозы для этого соединения в таблетированных порошках, помещенных под язык. Это путь введения гормона окситоцина для индуцирования выделения молока из грудей продуцирующей молоко матери.
При этом лечении применяют эффективное, но нетоксичное количество соединения. Схему приема соединения настоящего изобретения для предотвращения или лечения симптомов выбирают в соответствии с различными факторами, включая тип, возраст, массу, пол, медицинское состояние женщины, тяжесть симптомов и способ введения соединения. Обычно практикующий врач может определить и прописать эффективное количество антагониста окситоцина на основе способа его введения для предотвращения или задержки прогресса состояния, которое нужно ингибировать. Например, эффективная доза может быть в пределах от 0,01 до 100 мг на 1 кг массы тела в день при введении внутривенным способом, например в стерильном обычном солевом растворе.
Соединение настоящего изобретения можно получить новым способом. В этом способе можно избежать замещения триптофана пептида, поскольку Trp-пептиды чувствительны к кислотам. Bodanszky et al., Med. Chem., 23:1258-1261 (1980) и Sawyer et al., Endocrinology, 106:81 (1980) получили (Trp8) окситоцин более трудными непрямыми способами, чтобы избежать кислотную обработку Trp-пептида. Эти способы описаны в номерах заявок США 07/289 780 и 07/433 644 и таким образом введены в изобретение ссылками.
Пример I. Синтез ((S)Pmp2-D-Trp2-Pen6-Arg8)окситоцина
Синтез производных β-меркаптопропионовой кислоты. Тетрагидротиопиран-4-он обрабатывают триэтилфосфоноацетатом методом Wadsworth and Emmons (Wadsworth, W. S., Ir. Emmons, W.D. (1973) в Organic Synthesis (Baumgarten, H. ed) Col. Vol. V, pp 547 - 549, John Wiley and Sons, NY), получая этил-4-тетрагидротиопиранилиден(TEP)ацетат. Присоединением 4-метилбензилмеркаптана (реакция Михаэля) по методу Yim and Huffman (Vim, N.C.F. and Huffman, W.F. (1983) Int. J. Pept. Prot. Res. 21, 568 - 570) и омылением получают тетрагидротиопиранил-4-(4-метилбензилтио)-4-уксусную кислоту или (S)Pmp(S-Med).
Аббревиатуру применяли в полном соответствии с рекомендациями комиссии по Биохимической номенклатуре IUPACIUB (J. Bio. Chem. 264, 688 - 673 (1989)). Если не указано особо, аминокислоты имеют L-конфигурацию. Другие применяемые аббревиатуры: OT, окситоцин; Pmp, β,β-пентаметилен-β-меркаптопропионовая кислота; (S)Pmp, β,β-(3-тиапентаметилен)-β-меркаптопропионовая кислота; Boc, трет-бутилоксикарбонил; Meb, 4-метилбензил; Tos, п-толуолсульфонил; ONp, 4-нитрофениловый эфир; DCM, дихлорметан; TFA, трифторуксусная кислота; EtOH, этанол; DIEA, диизопропилэтиламин; DMF, диметилформамид; DCC, дициклогексилкарбодиимид; HOBt, 1-гидроксибензотриазол; MeOH, метанол; CHL, хлороформ; Ac O, уксусный ангидрид; TEA, триэтиламин: MeCH, ацетонитрил; BuOH, н-бутанол; AcOH, уксусная кислота; Pyr, пиридин; Et2O, диэтиловый эфир; ЖХВР, жидкостная хроматография высокого разрешения; ТСХ, тонкослойная хроматография; PITC, фенилизотиоцианат, PTC, фенилтиокарбамоил; UV, ультрафиолет; OR, оптическое вращение.
Синтез пептида. Все защищенные пептидные предшественники антагонистов синтезировали вручную методом синтеза в твердой фазе (SP) (Merrifield, R.B. (1963) J. Am. Chem. Soc. 85, 2149-54). Стратегия синтеза Boc-аминокислот (Stewart, J. M. and Young, J.D. (1984) в Solid Phase Peptide Synthesis pp 1-176, Pierce Chemical Co., Rockford, IL) была следующей. Все аналоги (по положению 1) Pmp имели меркаптогруппу, защищенную 4-метилбензиловой группой. Окончание конденсации контролировали при помощи нингидриновой реакции (Kaiser, E. , Colescot, R.L., Bossinger, C.D. and Cook, P.I. (1970) Anal. Biochem. 34, 595-598). Защищенные пептиды удаляли из смолы аммонолизом (Manning, M. , (1968) J. Am. Chem. Soc., 90, 1348-1349). Защищенные пептиды освобождали от блокирующих групп на функциональных группах боковой цепи восстановлением системой Na/жидкий аммиак (du Vigneaud, V., Ressier, C., Swan, J. M. , Roberts, C.W., Katsoyannis, P.G. and Gordon, S. (1953) J. Am. Chem. Soc. , 75, 4879-4880) или жидкий HF-анизол (Sakakibara, S. and Shimonishi, Y. (1965) Bull. Chem. Sjc. Jpn., 38, 1412-1413) и дисульфгидрилпептиды циклизовали в очень разбавленном растворе (Manning, M., Lammek, B., and Kolodziejczyk, A. M. (1981) J. Med. Chem. 24, 701 - 706) в циклический дисульфид окислением феррицианидом калия (Hope, D.B., Murti, V.V.S. and Vigneaud, V. (1962) J. Biol. Chem. 237, 1563-1566). Свободные пептиды очищали от более низкомолекулярных побочных продуктов и солей гель-фильтрованием (Porath, J. and Flodin, P. (1959) Nature (London) 183, 1637-1659) на сефадексе G-15 (Manning, M., Wuu, T.C. and Baxter, J.W.M. (1968) J. Chromatogr, 38, 396-398) и препаративной жидкостной хроматографией высокого разрешения (ЖХВР) (Fluoret, G., Brieher, W., Mahan, K., and Wilson, L., Jr. (1991) J. Med. Chem. 34, 642 - 646). Чистоту пептида контролировали ТСХ, ЖХВР и аминокислотным анализом (Bidlingmeier, B.A., Cohen, S.A. and Tarvin, T.L. (1984) J. Chromatogr, 336, 93-104).
Пептидную последовательность каждого аналога собирали вручную SP-методом, применяя механический вибратор и специальный сосуд. Где возможно, некоторые пептиды освобождали от защитных групп жидким HF, применяя все оборудование из тефлона (Protein Research Foundation, Osaka, Japan). Boc-аминокислоты поставлял Bachem, синтетические или ионообменные смолы поставлял Biorad. Все другие реагенты получали от Aldrich Chemical Co., Pierce или Chemicfl Dynamics. Чистоту пептидов контролировали аналитической ЖХВР с применением ранее описанного устройства Millipore (Flouret, G., Brieher, W., Mahan, K. , and Wilson, L., Jr. (1991) J. Med. Chem. 34, 642-646) и аналитической колонки μ Bondapak C18 (30x0,39 см). Для препаративной ЖХВР мы применяли Gilson систему 71 для автопрепаративной ЖХВР, как описано ранее (FLuoret, G. , Brieher, W. , Mahan, K. , and Wilson, L., Jr. (1991) J. Med. Chem. 34, 642-646) и модуль препаративной колонки 21,4х25 см с предохранительным модулем, 5 см, оба модуля заполнены Dynamax-60A, 8 mkm, C18 (Rainin). Растворители, применяемые для хроматографии или синтеза, были сорта для ЖХВР (Fisher Scientific). Как для аналитической, так и препаративной ЖХВР, применяли системы растворителей;
(a) 0,05% TFA; (b) 60% MeCN-40% растворителя A. Чистоту пептидов контролировали также тонкослойной хроматографией (TCX) на силикагеле G, предварительно покрытом Uniplates (0.25 mm, Analtech). Применяли системы растворителей (соотношения даны в объемах):(A) н-BuOH-AcOH-H2O (4:1:1); (B) н-BuOH-AcOH-H2 (4: 1:5, верхняя фаза); (C) н-BuOH-AcOH-H2O (5:1:1); (D) н-BuOH-AcOH-H2 O-Pyr (5: 1: 1:1). Пептиды визуализировали реагентом Эрлиха или смесью хлор-толуидин (Stewart, J.M. and Young, J.D. (1984) in Solid Phase Peptide Synthesis, pp. 1-176; Pierce Chemical Co., Rockford, IL). Для аминокислотного анализа аналоги гидролизовали 6н. HCl в течение 24 час при 110oC и полученные аминокислотные компоненты превращали в производные реакцией с фенилизотиоцианатом и анализировали методом Waters Associates Picotag (применяя устройство Waters Picotag, как описано ранее (Flouret, G., Briher, W., Mahan, K., and Wilson, L., Jr. (1991) J. Med. Chem 34, 642-646). Оптическое вращение пептидов измеряли поляриметром Rudolph (точность ± 0,01o).
Твердофазный синтез защищенных пептидов. Вос-аминокислоты применяли для синтеза и для защиты функциональных групп боковых целей, Boc-Arg(Tos), Boc-Pen(Med)и (S)Pmp(Med). Мы применяли Boc-Gly-смолу (0,7 ммоль Boc-Gly/r), которую получали на хлорметилированной смоле 200-400 меш (BioRad), на 1% сшитой дивинилбензолом, этерификацией цезиевой солью соответствующей Boc-аминокислоты (Gisin, B.F., (1973) Hely. Chim. Acta 65, 1476-1482). Синтез с Boc-Gly-смолу (0,5-0,7 ммоль/г) проводили вручную путем требуемого числа циклов конденсации SP-методом синтеза, ранее модифицированным (Flouret, G., Brieher, W. , Nahan, K., and Wilson, L., Jr. (1991) J. Med. Chem. 34, 642-646). В каждом цикле Boc-группу удаляли 30%-ной трифторуксусной кислотой в DCM и после нейтрализации смолы 10% DIEA в DCM конденсацию проводили с трехкратным избытком Boc-аминокислоты и DCC. На соответствующих стадиях применяли шестимолярный Boc-Asn-ONp или Boc-Gln-ONp в DMF и избыток реагента удаляли осаждением водой. Завершение стадии конденсации контролировали при помощи нингидриновой реакции, которая обычно дает отрицательный ответ. Если реакция была положительной, стадию конденсации повторяли, но если реакция была только слабо положительной, то пептид блокировали ацетилированием смесью Ac2O-DIEA-DCM (1:1:8). Незащищенный Boc-D-Trp вводили в положение 2. Boc-группу затем удаляли 30%-ной TAF в DCM, содержащем 1% меркаптоэтанола и 10% анизола, и (S)(PmP(S-Meb) вводили реакцией трехмолярного избытка его в растворе ДМФА, активируемого DCC и HOBt. Конечный собранный пептид отделяли от смолы аммонолизом MeOH (25 мл), насыщенным аммиаком. Через 3 дня смолу отделяли фильтрованием и экстрагировали три раза горячим ДМФА. Фильтрат в метаноле и экстракты в ДМФА объединяли и выпаривали досуха. Остаток растворяли в ДМФА (2-3 мл) и защищенный амид пептида осаждали из объединенных экстрактов в ДМФА обработкой водой или смесью EtOH-Et2O, получая 400-600 мг защищенного пептида. Анализ защищенных пептидов, полученных после аммонолиза при помощи ТСХ, обычно показывал один основной компонент с небольшими примесями, поэтому их применяли сразу для удаления защитных групп и получения свободных аналогов.
Этил-4-тетрагидротиопиранилиденацетат: Этот эфир получали, как описано для получения этил-4-тетрагидропиранилиденацетата (Wadsworth, W. S., Jr. Emmons, W.D. (1973) in Organic Synthesis (Baumgarten, H. ed.) Coll. Vol. V, pp. 547-549, John Wiley and Sons, New York), и вакуумной перегонкой, в виде масла (выход 73%).
Тетрагидротиопиранил-4-(4-метилбензилтио)-4-уксусная кислота или (S)Pmp(4-S-Meb). Эту защищенную кислоту получали из предшествующего эфира методом Yim and Huffma, как описано для Pmp (Int. J. Prot. Res. 21, 568-570, 1983), т.пл. 113-115oC (выход 50-70%).
((S)Pmp1-D-Trp2-Pen6-Arg8)OT или Antag 111.
(S)Pmp(S-Meb)-D-Trp-Ile-Ghn-Asn-Pen(Meb)-Pro-Arg(Tos)-Gly-NH (600 мг), синтезированный SP-методом (начиная с 0,5 ммоля аминокислота-смола), как описано выше, растворяли в жидком аммиаке (200 мл), свежеперегнанном из натрия, и обрабатывали в безводных условиях куском натрия до образования бледно-голубого цвета в течение около 15-30 сек. После выпаривания аммиака в вакууме твердый остаток растворяли в 20 мл 50%-ной AcOH. Этот растворенный пептид добавляли в дезаэрированную воду (2 л) (этот большой объем можно значительно снизить модифицированной методикой), pH устанавливали 7,0 добавлением концентрированного раствора гидроксида аммония, и циклизацию в дисульфидное производное пептида проводили титрованием дисульфгидрилпептида 0,01н. феррицианидом калия до образования постоянного желтого окрашивания и затем добавляли 20%-ный избыток раствора феррицианида калия. Через 20 мин ферроцианид и феррицианидные соли удаляли перемешиванием 10 мин с AGIX-2(CI)-ионообменной смолой (15 г) и затем пропусканием суспензии через колонку, содержащую дополнительную ионообменную смолу (15 г), применяя дополнительно 0,2 н. AcOH (100 мл) для промывания. Объединенный фильтрат и промывные жидкости лиофилизовали. Анализ полученного твердого продукта, содержащего пептид, проводили на аналитической колонке Bondapak C18 (30 х 0,39 см) с контролем по полосе 220 нм, элюируя изократно 55% растворителем B (растворитель A, 0,05% TFA, растворитель B, 60: MeCN-40%, 0,05% TFA) со скоростью 1,8 мл/мин. В этих условиях достигалось хорошее отделение примесей. Остаток растворяли в самом малом возможном объеме 50%-ной уксусной кислоты и вводили в колонку (115 х 26 см) с сефадексом G-15 и элюировали тем же самым растворителем со скоростью около 50-60 мл/час (6). Элюат контролировали на УФ-спектрофотометре по полосе 254 нм. Фракции, соответствующие основному пику, контролировали аналитической ЖХВР на аналитической колонке (30 х 0,39) с μ Bondapak C18 при элюировании 57% растворителем B и детектировании пептидов по полосе 220 нм. Чистые фракции по критерию ЖХВР объединяли и лиофилизовали. Остаток растворяли в 0,2н. AcOH (20 мл) и вводили в препаративную колонку с Dynamax-60A, 8 мкм, C18 (Ranin) и предохранительным модулем 5 см. Градиент был от 0 до 45% в течение 45 мин, скорость элюирования была 5 мл/мин, контроль элюента проводили по полосе 280 нм. Центральные порции основного компонента были элюированы приблизительно через 3,5 часа. Более чистые фракции, определенные аналитической ЖХВР, объединяли и лиофилизовали, получая Antag 111 (240 мг, 42% от исходной смолы). Чистоту аналогов устанавливали тонкослойной хроматографией (ТСХ) в системах из 4 отдельных растворителей, аналитической ЖХВР и аминокислотным анализом. Аминокислотный анализ аналогов дал неожиданные соотношения ±10%. Присутствие D-триптофана в пептидах устанавливали УФ-спектроскопией у полосы 280 нм (13). Более низкое значение, найденное для триптофана, 0,96, заставляло предполагать, что пептидный лиофилизат мог иметь TFA и/или H2O.
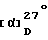 -39o (1N, AcOH)
-39o (1N, AcOH)
ТСХ: (A) н-BuOH-AcOH-H2O (4:1:1) - Rf 0,27
(B) н-BuOH-AcOH-H2O (4:1:5, верхняя фаза) - 0,42
(C) н-BuOH-AcOH-H2O (5:1:1) - 0,19
(D) н-BuOH-AcOH-H2O-Pyr (5:1:1) - 0,56
Пример II. Сравнительное испытание соединений
Для сравнения с ((S)Pmp1-D-Trp2-Pen-Arg8)) окситоцином (антагонист D в таблице 1) синтезировали три родственных соединения. Одно из них было соединением, описанным Manning et al., J. Med. Chem., 26:1607-1613 (1983). Это соединение можно назвать (Pmp1-D-Phe2-Phe3-Ile4-Arg8) окситоцином. В таблице 1 это соединение обозначено антагонистом A. Другое соединение было (Pmp1-D-Trp2-Phe3-Ile4-Arg8) окситоцином, описанным в заявке США N 07/239780, оно приведено в таблице 1 как антагонист B. Третье сравнительное соединение, (Pmp1-D-Trp2-Arg8)окситоцин, описано в заявке США N 07/433664, в таблице 1 оно приведено как антагонист C. Эти четыре соединения подвергали сравнительному изучению в биоанализах.
Биоисследование окситоцина.
Протокол, применяемый для методики биоисследования окситоцина, взят из методик, описанных в статье Sawyer et al., Endocrinology, 106:81 (1980), которые, в свою очередь, основаны на сообщениях Vunsick, Brit. J. Pharmacol., 3:328 (1960), и Holton, Brit. J. Pharmacol., 3:328 (1948). Подсчеты значений pA2 анализа описаны Schild, British J. Pharmacology, 2:189 (1947). Основным отличием настоящей методики от методик, описанных другими в данной области, является интегрирование площади при сокращении вместо простого подсчета амплитуды. Интегрирование обеспечивает значительно более совместимые и надежные результаты, хотя оценки pA2 приблизительно на порядок величин ниже, чем оценки, сообщенные с применением амплитуды сокращения в конечной точке.
Методика: Животные. Для анализа применяли 1,5 см кусочки матки неоплодотворенной крысы (Hortzman) в период природной течки.
Буфер/ванная для анализа. Применяют буфер Munsicks.
Этот буфер содержит 0,5 мМ Mg++, который снижает величины pA2, но позволяет получать результаты, которые лучше коррелируют с данными in vivo (Sawer et al. , 1980). Буфер при непрерывном контактировании со смесью 95% кислорода и 5% диоксида углерода обеспечивает pH 7,4. Температура ванных для анализа 37oC. Применяют ванну на 10 мл, которая имеет водяную рубашку для поддерживания температуры и входные и выходные отверстия для добавления и удаления буфера.
Полиграф/датчик. Кусочек применяемой для исследования ткани матки соединяют с датчиком Statham Strain Gauge Force, который, в свою очередь, соединяется с Grass полиграфом Model 79 для мониторинга сокращений.
Схема исследований. (a) Ткань выдерживают в ванной для анализа в течение 1 часа при промывании новой порцией буфера каждые 15 мин. Все время ткань поддерживают при напряжении 1 г.
(b) Ткань стимулируют сначала окситоцитом при 10 нМ для "акклиматизации" ткани и 4 мМ KCl для определения максимальной сократительной реакции.
(c) Затем строят кривую ответной реакции на кумулятивную дозу окситоцина и концентрацию окситоцина, эквивалентную приблизительно 80% от максимума, применяют для установления pA2 антагониста.
(d) Ткань подвергают воздействию окситоцитом (Calbiochemical, San Diego, California) в течение 1 минуты и промывают. Соблюдается трехминутный интервал до добавления следующей дозы агониста или антагониста. Когда испытывают антагонист, его подают за пять минут до агониста. Агонист подают в течение одной минуты. Все реакции интегрируют при помощи интегратора 7P10 Grass. Это основное различие между настоящей схемой и другими схемами в литературе, которые обычно измеряют амплитуду сокращений в качестве ответной реакции. Одну концентрацию окситоцина, соответствующую 80% максимальной ответной реакции, применяют для испытания антагониста. Применяют три различные концентрации антагонистов, две концентрации, которые будут снижать ответную реакцию на антагонист по меньшей мере на 50%, и одну, которая будет снижать ответную реакцию более чем на 50% (идеально 25%, 50% и 75%). Это повторяют три раза для каждой из трех доз антагониста.
Анти-ADH-активность измеряют путем изменения в моче антагониста под действием вазопрессина (ADH) в присутствии и отсутствие антагониста с целью определения специфичности антагониста. Анализ активности против ADH описан Sawyer et al., Endocrinology, 63:694 (1958).
Дополнительные изучения проводили для определения, отражают ли результаты биоанализа крыс связывающую аффинность к маточным рецепторам OT у крыс и человека. Сравнивали относительную связывающую аффинность 5 различных антагонистов окситоцина.
Исследование рецепторов окситоцина
Методика. Крысы. Ткань матки удаляли на 21 день беременности (роды в промежутке между 21,5 днем и 22,5 днем) у крыс Holtzman. Ткань освобождали от ее содержимого, промывали в охлажденном льдом буфере, нарезали на маленькие кусочки и хранили замороженными при -70oC до гомогенизации.
Люди. Нейромышечную ткань человека отбирали у пациентов во время кесаревого сечения с их согласия. Ткань промывали в холодном буфере, нарезали на маленькие кусочки и хранили замороженными при -70oC до гомогенизации.
Выделение рецепторов окситоцина. Рецепторы окситоцина (OTrs) находятся на клеточных мембранах и присутствуют в больших концентрациях в конце беременности в ткани матки. Замороженную ткань гомогенизируют в трис-буфере, гомогенизат фильтруют и фильтрат центрифугируют при 1000 г в течение 15 мин при 4oC. Супернатант центрифугируют при 40000 г 30 мин и осадок, содержащий клеточные мембраны, снова суспендируют в 10%-ной сахарозе. Затем проводят ультрацентрифугирование в градиенте плотностью, помещая суспензию в 10%-ной сахарозе сверху 35%-ной сахарозы и центрифугируя 30 мин в бакет-роторе при 105000 г. Мембраны с поверхности раздела 10%/35% сахарозы удаляют и снова суспендируют в содержащей трис-буфер EDTA в течение 30 мин. Эта процедура удаляет двухвалентные катионы и приводит к диссоциации любого OT, эндогенно связанного с рецептором. Эту смесь затем центрифугируют в течение 15 мин при 100000 г и осадок, содержащий мембранные OTrs, ресуспендируют в трис, PMSF, Mg++-буфере обработкой ультразвуком.
OT-рецепторный анализ. Для проведения анализа связывания применяют 0,1 мл меченого 20000 имп•мин-1 литием OT (New England Nuclear, 37,1 Ku/нмоль), 0,1 мл антагониста OT, добавляемого с повышаемой концентрацией, 0,25 мл буфера и 0,05 мл мембран (70-150 мкг белка). Пробирка на неспецифическое связывание имеет 100-кратное количество холодного OTA, добавленного в нее. Инкубирование проводят 30 мин при 30oC. Мембраны осаждают центрифугированием в ультраочищенных минипробирках (5 х 41 мм) в течение 30 мин при 105000 г. Полученный осадок, содержащий соединение 3H-OT, растворяют в 0,1 н. при 45oC в течение 30 мин и эту смесь затем помещают в сцинтилляционную жидкость и подсчет числа расп: мин-1 проводят в сцинтиляционном счетчике.
Данные анализируют методами подбора нелинейных кривых, применяя программу McPherson s EBDA (J. Pharmacol Methods 14:213-228, 1985) и Munson and Rodbard's LIGAND (Anal Biochim 107:220-239, 1980) для анализа методом насыщения и конкуренции с целью определения Kds и Kis.
Результаты сравнительного биоисследования и изучения рецепторов приведены в таблицах 1-4.
Как видно из данных таблицы 1, соединение D, представляющее собой новое соединение изобретения, проявляло более высокую антиокситоциновую активность, чем другие три соединения. Кроме того, оно имело значительно более низкую активность против ADH. Отношение активностей анти-OT/анти-ADH для соединения D выше, чем 1866, 70, тогда как такое отношение для соединения A было 0,7, для соединения B 1,8 и соединения C 242,9. Эти данные, следовательно, указывают, что соединение D, как можно предположить, будет продуцировать меньшие побочные действия (активность анти-ADH) при введении эффективной дозы как антагониста окситоцина, чем соединение A, B или C.
В таблице 2 приведено сравнение значений связывающей аффинности (Kas), полученных при анализе крысиных маточных рецепторов (Kas), с данными биоанализа. Корреляция Log10Ka с Log10ED50 была очень существенной (г = 0,92; p ниже 0,01). Сравнение относительной активности ANTAG 111 (соединение D) и ANTAG 1 (соединение B), определенных анализом крысиных маточных рецепторов и биоанализом, приведено в таблице 4. В обоих анализах ANTAG 111 приблизительно в 20 раз активнее, чем ANTAG 1.
В таблице 3 показано сравнение связывающей аффинности различных OTA к маточному OTr человека с данными биоанализа крыс. Корреляция Log10Ka и Log10ED50 была очень значимой (r = 0,92 p < 0 < 01). Сравнение относительной связывающей активности ANTAG 111 и ANTAG 1 для OTr человека (hOTr) показано в таблице 4. По этой оценке ANTAG 111 (соединение D) почти в 80 раз активнее, чем ANTAG 1. Следовательно, по-видимому при исследованиях на крысах можно недооценить относительную активность ANTAG 111 у людей. Эта возможность далее подтверждается изучениями in vivo, проведенными на беременных самках бабуинов и описанными ниже.
Пример III. Испытание соединений на беременных самках бабуинов.
Целью этого изучения было установление относительной in vivo активности четырех антагонистов окситоцина с применением в качестве модели ограниченных в действиях беременных самок бабуинов и сравнение этих результатов с предыдущими оценками активности, полученными анализами крыс и анализами человеческих OTr. Бабуин является отличной животной моделью благодаря его физиологической и анатомической близости человеку (смотри статьи нашей лаборатории: Am. J. Obstet Gynecol. 163:1815-1882, 1990; Am. J. Obstet Gynecol 165:456-560, 1991; Am J. Obstet Gynecol 165: 1487-1498, 1991; статьи других лабораторий: Endocrine Reviews 11: 124-150, 1990; 11:151-176, 1990). Беременных, ограниченных в действиях самок бабуинов изучали в период между 130 и 145 днями беременности (роды на 184 день). Антагонисты окситоцина вводили в виде одной ударной дозы (1 мг) внутриартериальной инъекцией, через 1 минуту инфузией вводили окситоцин. Окситоцин вводили инфузией непрерывно, начиная с 10 мЕ и удваивали дозу каждые 20 мин до мЕ/мин или до достижения значительной ответной реакции, т.е. сократительной способности (CF = (частота • среднюю амплитуду)/10 мин, т. е. CF более 50. Если не было значительной ответной реакции, через 24 часа испытание на окситоцин повторяли. Интервал ответной реакции на антагонисты (ARI) определяли умножением времени до первой значительной ответной реакции в минутах на соотношение сокращения и пульса (концентрация CF/OT). Результаты: Результаты приведены в таблице 4. Значения ARI очень хорошо коррелировали с оценками связывающей аффинности OTr крыс и человека (Ka) (r = 0,98; p ниже 0,01) у 4 из 5 антагонистов окситоцина. Один антагонист окситоцина (антагонист F в таблице 4) не проявил ингибирующую активность при испытываемых дозах, хотя он имел умеренно хорошую связывающую аффинность в анализах крысиных и человеческих OTr и биоанализе крыс. Этот антагонист окситоцина не получали в нашей лаборатории, и он не является аналогом окситоцина. Один мг самого лучшего испытанного антагониста окситоцина (соединение D, новое соединение настоящего изобретения) блокировал ответную реакцию на антагонист окситоцина не более чем в течение 24 час. Сравнение относительной активности различных ОТА и соединения B (ANTAG 1) показывает, что соединение D, ((S)Pmp1-D-Trp2-Pen6-Arg8)окситоцин (т.е. (ANTAG 111) почти в 130 активнее, чем соединение B. Говоря кратко, соотношения относительной активности (смотри таблицу 4) ANTAG 111 (соединения D) и ANTAG 1 (соединение B) по биоанализу крыс, анализу крысиных рецепторов, анализу человеческих рецепторов и биоанализу in vivo бабуинов были 22, 20, 82 и 133 соответственно. Эти данные указывают, что в анализах на крысах может недооцениваться эффективность ANTAG 111 в приматах.
Хотя настоящее изобретение описано в основном в связи с конкретными и предпочтительными примерами осуществления изобретения, понятно, что изобретение можно модифицировать, не выходя за пределы его объема. Следующая далее формула изобретения включает также все варианты использования или усовершенствования изобретения, вытекающие из общих принципов, и включает такие отклонения от настоящего описания изобретения, которые проявляются в пределах известной или обычной практики в области, к которой относится изобретение, или которые очевидны специалистам, работающим в данной области.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕПТАПЕПТИДНЫЕ АНАЛОГИ ОКСИТОЦИНА | 1997 |
|
RU2180668C2 |
| МОДУЛЯТОР NMDA-РЕЦЕПТОРА СО СТАБИЛИЗИРОВАННОЙ ВТОРИЧНОЙ СТРУКТУРОЙ И ЕГО ПРИМЕНЕНИЕ | 2011 |
|
RU2566821C2 |
| АНАЛОГИ ОКСИТОЦИНА | 2009 |
|
RU2496788C2 |
| ЗАЩИЩЕННЫЕ ПРОИЗВОДНЫЕ ВАЗОПРЕССИНА | 1997 |
|
RU2123498C1 |
| β-ЛАКТАМИЛЗАМЕЩЕННЫЕ АНАЛОГИ ФЕНИЛАЛАНИНА, ЦИСТЕИНА И СЕРИНА В КАЧЕСТВЕ АНТАГОНИСТОВ ВАЗОПРЕССИНА | 2006 |
|
RU2466991C2 |
| АНТАГОНИСТЫ LHRH, ИХ ПОЛУЧЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ПОЛУЧЕНИЕ, СПОСОБ ЛЕЧЕНИЯ ОПУХОЛЕЙ И БЕСПЛОДИЯ У МЛЕКОПИТАЮЩИХ | 2001 |
|
RU2248982C9 |
| ПРИМЕНЕНИЕ АНТАГОНИСТОВ ОКСИТОЦИНА ПРИ ВСПОМОГАТЕЛЬНОЙ РЕПРОДУКЦИИ | 2006 |
|
RU2385735C2 |
| ПРОИЗВОДНЫЕ ВАЗОТОЦИНА | 1991 |
|
RU2067586C1 |
| МОДУЛЯТОРЫ НМДА-РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2515615C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ ИНГИБИТОРА ЯКОРНОЙ ФУНКЦИИ БЕЛКА И СПОСОБ ОПРЕДЕЛЕНИЯ ПРИСУТСТВИЯ В КЛЕТКЕ КАЛЬЦИНЕЙРИНСВЯЗЫВАЮЩЕГО И ПКА СВЯЗЫВАЮЩЕГО ЯКОРНОГО БЕЛКА | 1995 |
|
RU2185441C2 |
Предложен антагонист окситоцина, представленный формулой (1), где (S) Pmp представляет собой β, β-(3-тиа-пентаметилен)-β-меркаптопропионовую кислоту, D-Trp представляет собой D-форму триптофана и Ilе, Gln, Acn, Pro и Arg представляют собой L-форму изолейцина, глутамина, аспарагина, пролина и аргинина соответственно, и Pen представляет собой остаток пенцилламина. Это соединение можно вводить беременным женщинам для задерживания преждевременных родов и при этом избежать нежелательных побочных действий из-за антагонизма антидиуретического гормона, вазопресина. 1 з.п. ф-лы, 4 табл.
 \
\
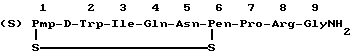
в которой (S)Pmp представляет собой β,β- (3-тиапентаметилен)- -β- меркаптопропионовую кислоту;
Pen - остаток пеницилламина,
обладающий свойствами антагониста окситоцина.
| Цистеинсодержащие пептиды для синтеза пептидных гормонов окситоцина и вазопрессина | 1974 |
|
SU523083A1 |
| Способ получения пептидов | 1976 |
|
SU639446A3 |
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| DE 4042008 A1, C 07 K 7/06, 1992 | |||
| Способ индикации температуры электрических индукционных аппаратов | 1975 |
|
SU649090A1 |
| US 4604378 A, C 07 K 7/16, 1986. | |||
