Изобретение касается клонированных ДНК, кодирующих человеческий эритропоэтин, экспрессии указанных ДНК и получения in vitro активного эритропоэтина человека.
Эритропоэтин (далее ЭПО) представляет собой циркулирующий гликопротеид, который стимулирует образование эритроцита в высших организмах (см. Carnot и др. , Compt. Rend, 143:384 (1906)). ЭПО иногда упоминается еще и как фактор, стимулирующий эритропоэз.
Продолжительность жизни человеческих эритроцитов составляет около 120 дней. Таким образом, около 1/120 общего числа эритроцитов разрушается ежедневно в ретикуло-эндотелиальной системе. Однако относительно постоянное число эритроцитов продуцируется ежедневно для поддержания постоянного уровня эритроцитов (Guyton, Textbook of Medikal Physiology, стр. 56-60, W.B. Saunders Co., Филадельфия (1976)).
Эритроциты продуцируются путем созревания и дифференцировки эритробластов в костном мозге, и ЭПО является фактором, который воздействует на менее дифференцированные клетки и индуцирует их дифференцировку в эритроциты (Guyton, выше).
ЭПО представляет собой многообещающее терапевтическое средство для клинического лечения анемии, и в частности ренальной анемии. К сожалению, использование ЭПО еще не является общепринятым в практической терапии вследствие его малой доступности.
При использовании ЭПО в качестве терапевтического средства особое значение приобретает вопрос антигенности. Предпочтительно, чтобы ЭПО был получен из сырья человеческого происхождения, например, из крови или мочи пациентов, страдающих апластической анемией или подобными болезнями, при которых выделяются большие количества ЭПО. Эти исходные материалы, однако, существуют в ограниченном количестве (См., например, White и др., Rec, Progr. Horm. Res., 16-219 (1960); Espada и др., Biochem. Med. 3:475 (1970); Fischer, Phurm. Rev., 24:459 (1972) и Gordon, Vitam, Horm. (N.Y.) 31:105 (1973).
Получение ЭПО, как правило, осуществляется путем концентрирования и очистки мочи пациентов с названными заболеваниями (см., например, патенты США NN 439780, 4303650 и 3865801, раскрытие которых введено сюда в качестве ссылки). Ограниченный запас такой мочи служит препятствием практическому использованию ЭПО. Трудность же в использовании мочи от здоровых людей заключается в низком содержании в ней ЭПО по сравнению с мочой пациентов, страдающих анемией. Кроме того, моча здоровых людей содержит определенные ингибирующие факторы, поэтому удовлетворительный терапевтический эффект может быть получен только при условии последующей значительной очистки получаемого таким образом ЭПО.
ЭПО также может быть выделен из плазмы крови овцы, причем выделение ЭПО из этого источника обеспечивает создание достаточно сильнодействующих и устойчивых водорастворимых препаратов (см. Coldwasser, Control Cellular Dif. Develop. , Часть A, стр. 487-494, Alan R. Liss, Inc., Нью-Йорк (1981), которая введена сюда в качестве ссылки). ЭПО овцы, однако, может обладать антигенным эффектом.
Таким образом, традиционные методики выделения и очистки, используемые при получении ЭПО из естественных источников снабжения, являются малопригодными для его массового производства.
Sugimoto и др. в патенте США N 4377513 описывают один способ массового производства ЭПО, предусматривающий размножение in vivo лимфобластоидных клеток человека, таких как Namalva, BAL L - 1, TAL L - 1, TAL L - 1 и JBL.
В специальной литературе появилось также описание получения ЭПО с использованием методов генетической инженерии. Однако еще не опубликовано ни пригодного раскрытия указанных способов, ни химической природы продукта. В противоположность этому настоящая заявка предлагает пригодный для массового производства способ получения белков, проявляющих биологические свойства человеческого ЭПО. С помощью этих методик можно также получить белки, которые могут химически отличаться от аутентичного человеческого ЭПО и все же обнаруживать аналогичные (а в некоторых случаях и улучшенные) свойства. Для удобства все такие белки, проявляющие биологические свойства человеческого ЭПО, упоминаются далее как ЭПО.
Настоящее изобретение касается клонирования последовательности ДНК, которая обеспечивает высокие уровни экспрессии человеческого ЭПО. Также описываются пригодные векторы экспрессии и клеточные системы для получения ЭПО, схемы очистки рекомбинантного продукта, приводятся его характеристики.
Как описывается более подробно ниже, ЭПО был выделен в частично очищенной форме, подвергнут дальнейшей очистке до гомогенности и переварен трипсином для получения специфических фрагментов. Эти фрагменты были очищены и подвергнуты определению последовательностей. На основе этих последовательностей сконструированы и синтезированы олигонуклеотиды, которые затем были использованы для скрининга человеческой геномной библиотеки, ген ЭПО был выделен с целью выделения гена ЭПО.
На базе мРНК из печени плода человека (в возрасте 20 недель) была получена и подвергнута скринингу кДНК-библиотека. Выделены три клона кДНК ЭПО (после скрининга > 750000 рекомбинантов). Два из них были определены как имеющие полную длину. Образец E. coli, трансформированной полноразмерной последовательностью, кодирующей ЭПО, был депонирован в АТСС, Rockville, Maryland под номером 40153. Эти кДНК были экспрессированы как в трансформированных вирусом SV-40 клетках обезьяны (клеточная линия COS-1; Gluzman, Cell 23: 175-182 (1981)), так и в клетках яичника китайского хомячка (клеточная линия CHO; Urlaub, G. и Chasin, L.A. Proc. Natl. Acad. Sci США 77: 4216-4280 (1980)). ЭПО, полученные в клетках млекопитающих, являются биологическими активными in vitro и in vivo.
Таблица 1 представляет собой последовательность экзона из 87 пар оснований гена ЭПО человека.
Фиг. 1 иллюстрирует обнаружение мРНК ЭПО в мРНК печени плода человека.
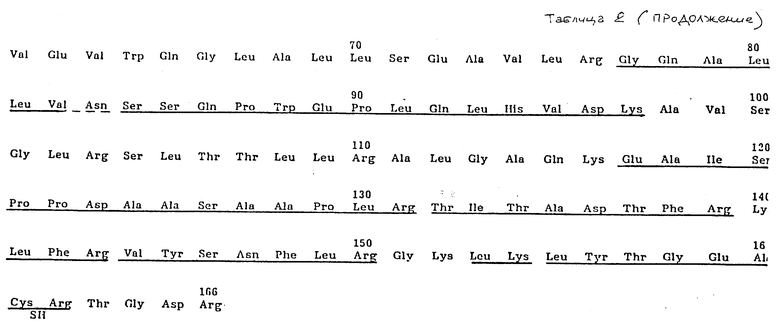
Таблица 2 представляет аминокислотную последовательность ЭПО, выведенную из нуклеотидной последовательности клона λ-HEPOF L 13.
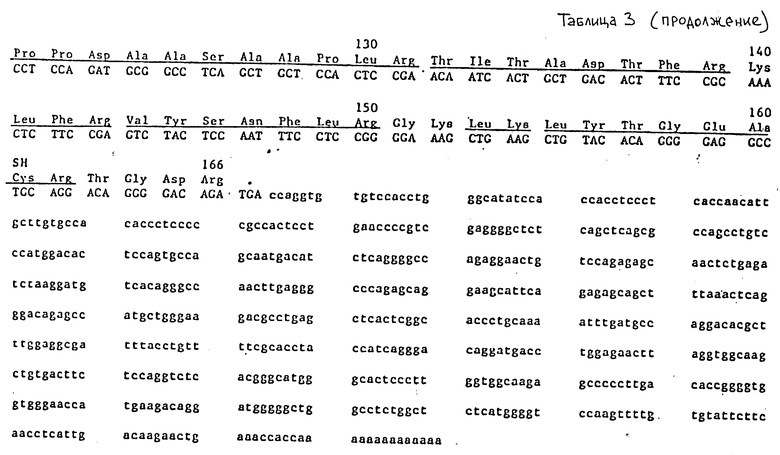
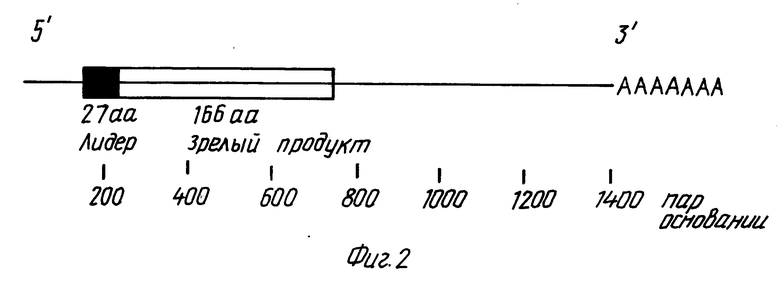
Таблица 3 представляет нуклеотидную последовательность кДНК ЭПО в λ-HEPOF L 13 (показана схематически на фиг. 2) и аминокислотную последовательность, выведенную из нее.
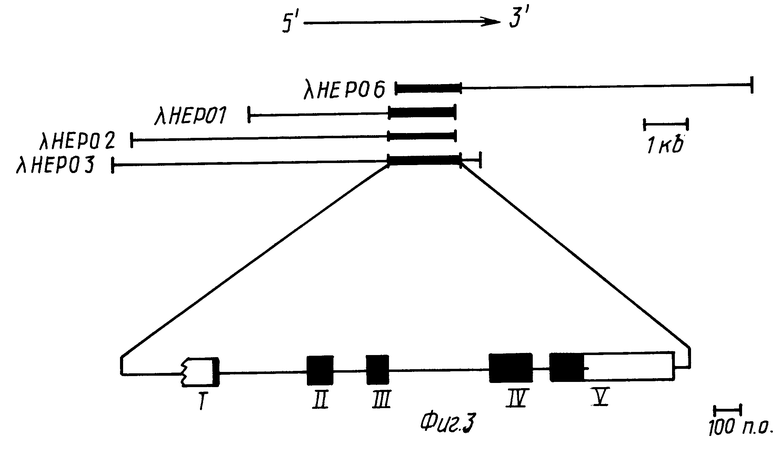
Фиг. 3 иллюстрирует относительные положения вставок ДНК четырех независимых геномных клонов человеческого ЭПО.
Фиг. 4 представляет собой карту интронной и экзонной структуры гена ЭПО человека.
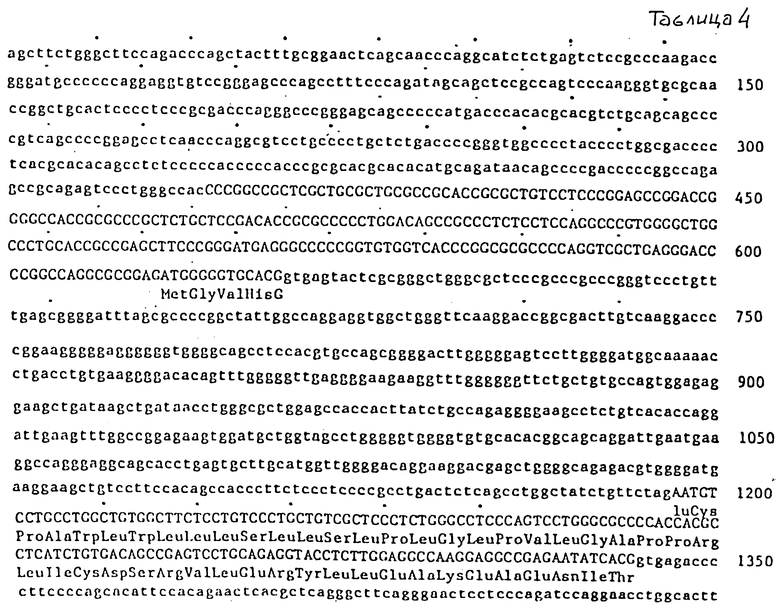
Таблица 4 иллюстрирует ДНК-последовательность гена ЭПО, схематически показанного на фиг. 4.
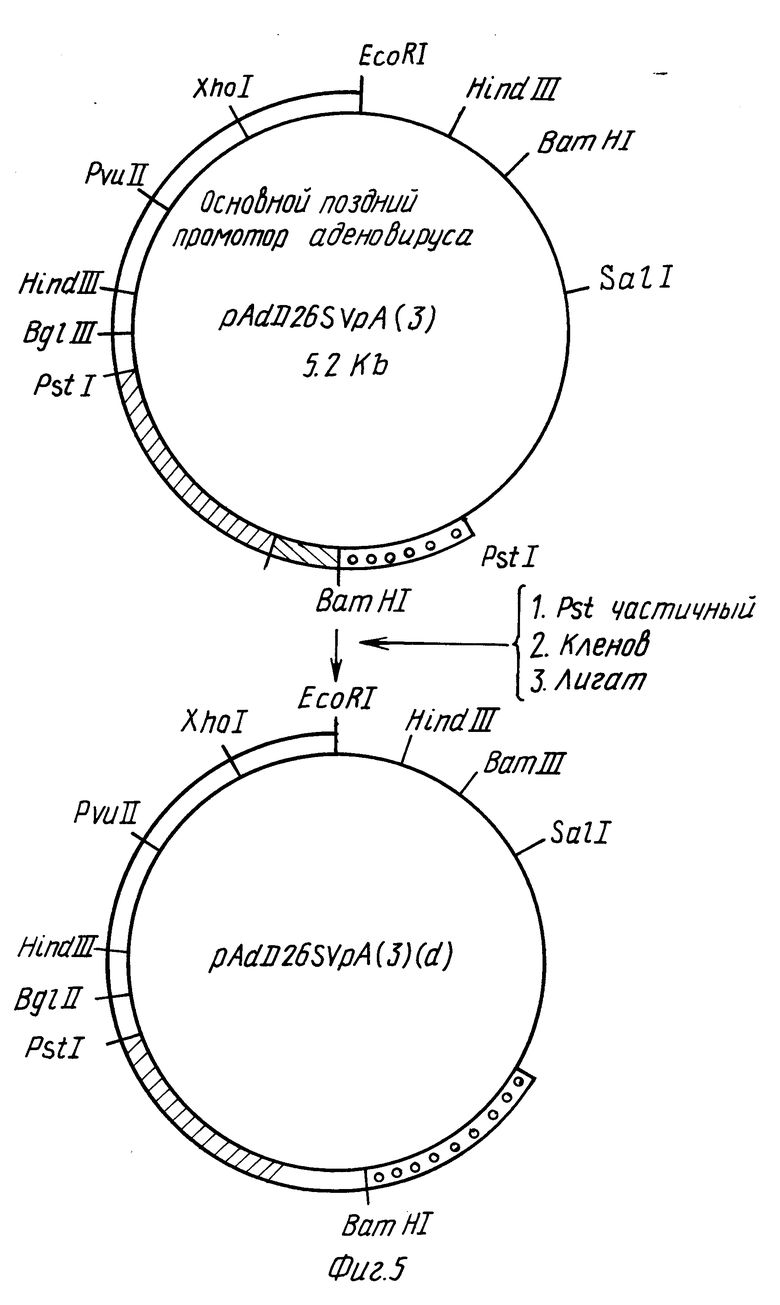
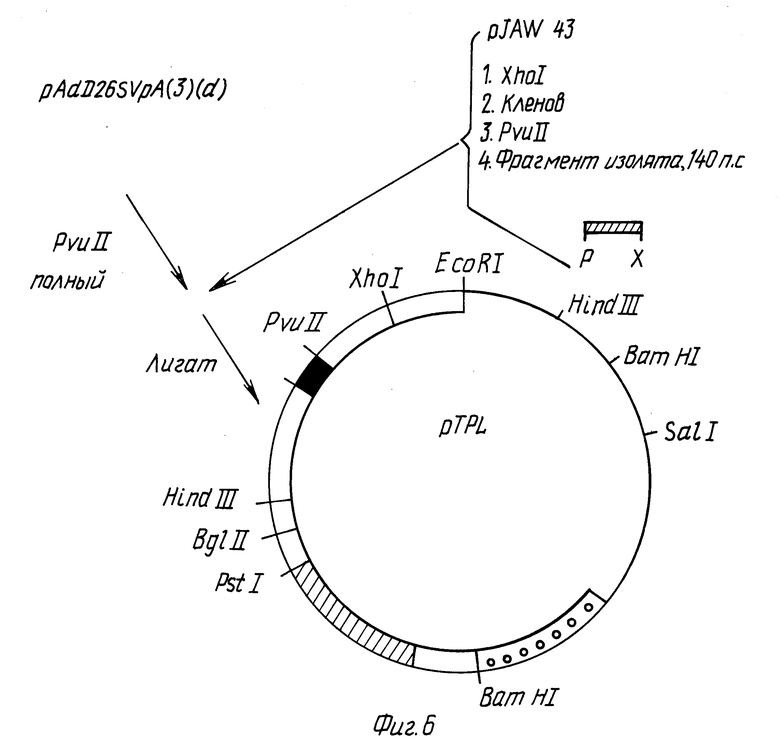
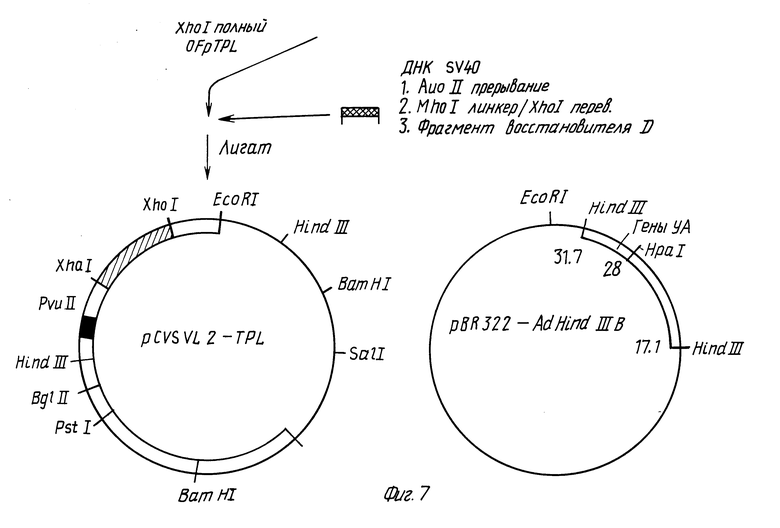
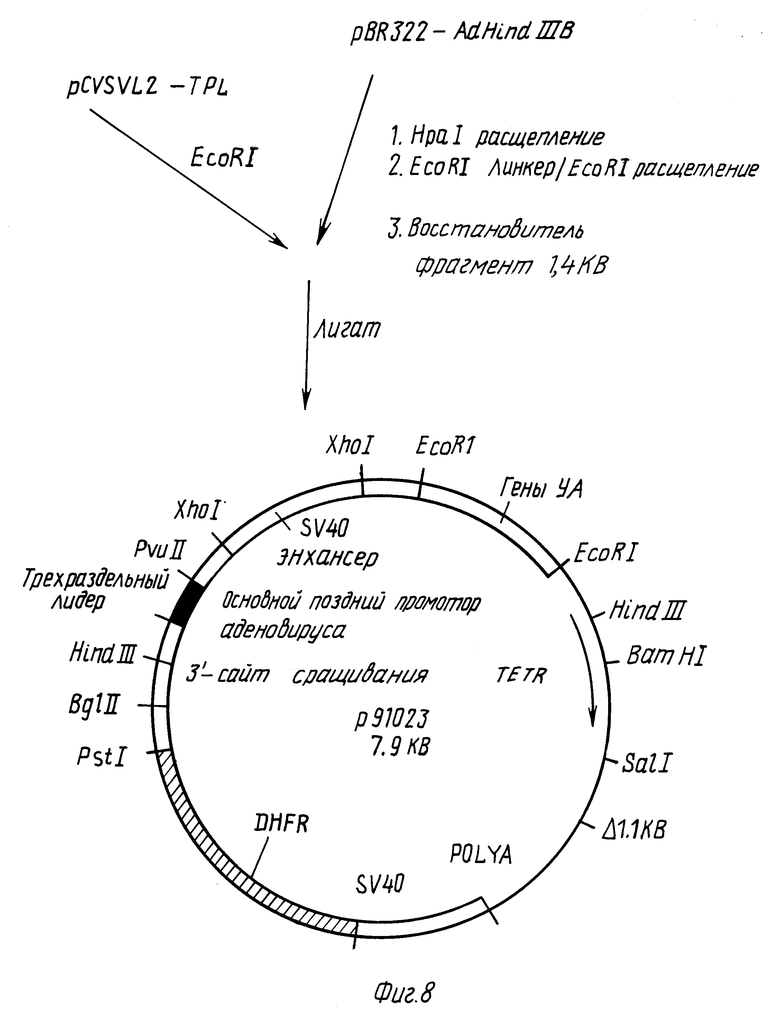
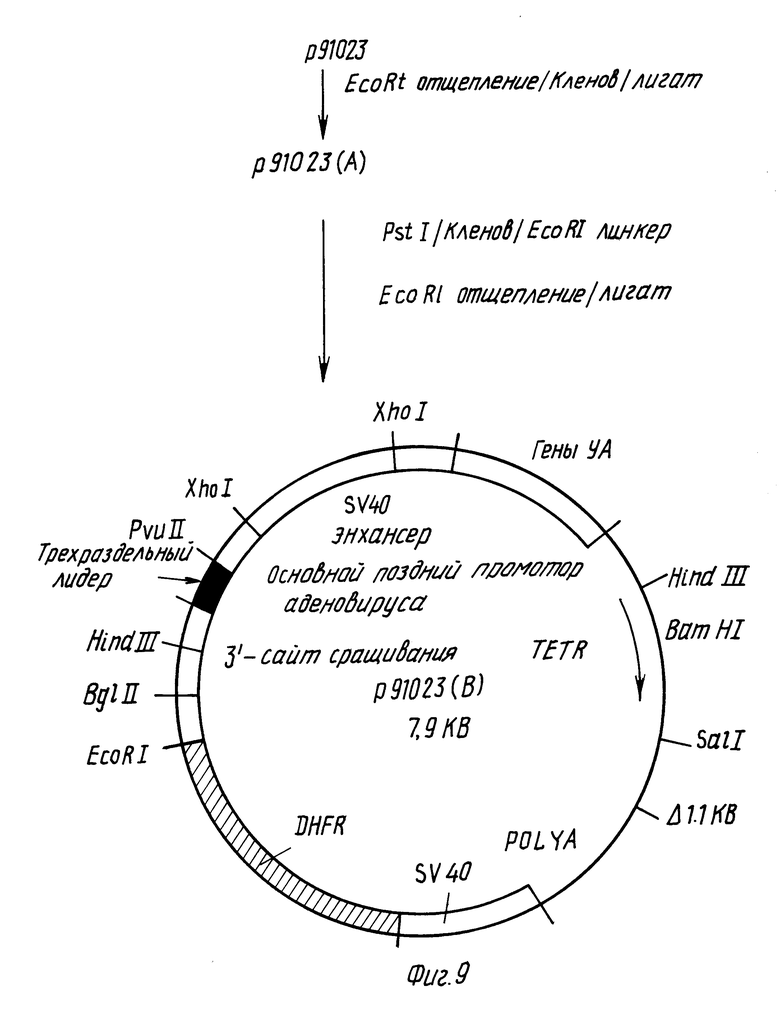
Фиг. 5 - 9 иллюстрируют конструирование вектора p91023(B).
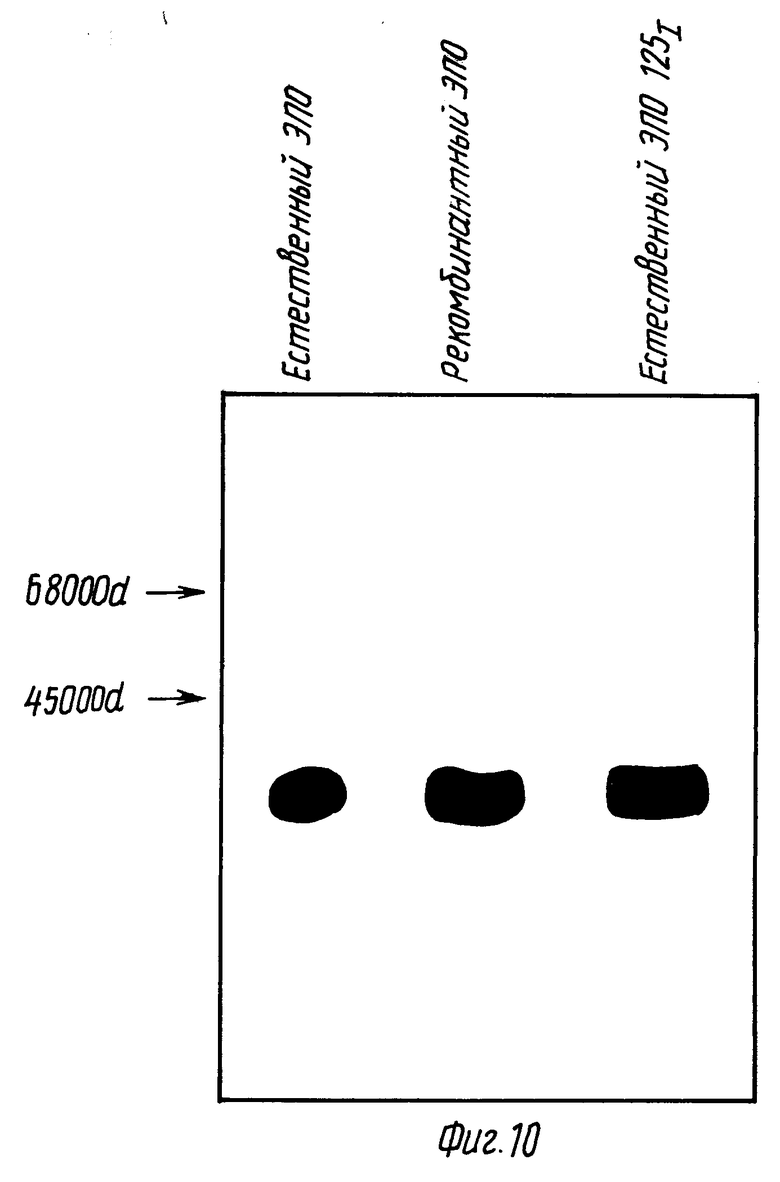
Фиг. 10 изображает результаты сравнительного анализа в SDS-полиакриламидном геле ЭПО, полученного в клетках COS-1, и нативного ЭПО.
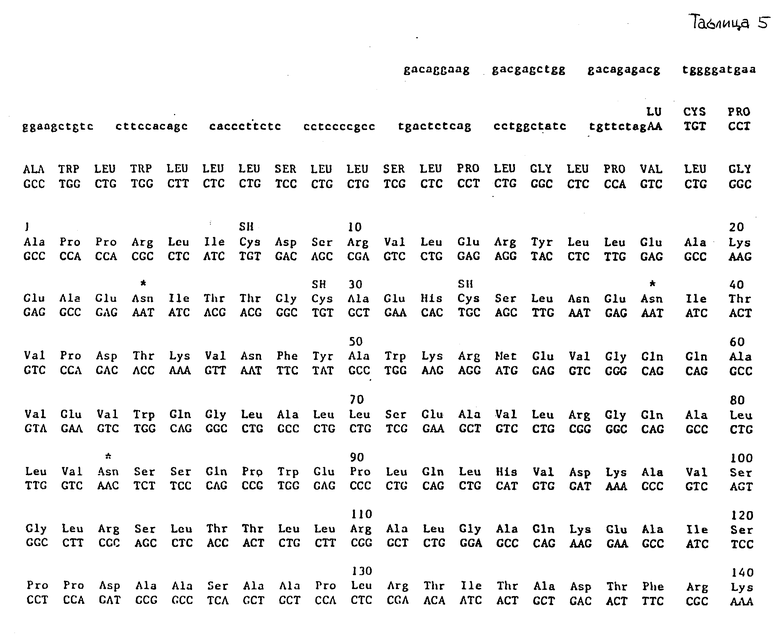
Таблица 5 иллюстрирует нуклеотидную и аминокислотную последовательности вставки клона λ-HEPOF L 6.
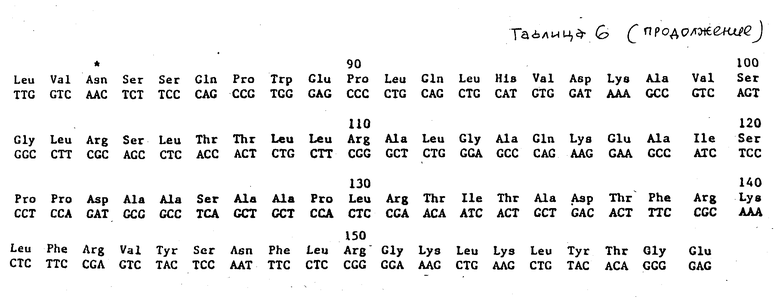
Таблица 6 иллюстрирует нуклеотидную и аминокислотную последовательности вставки клона λ-HEPOF L 8.
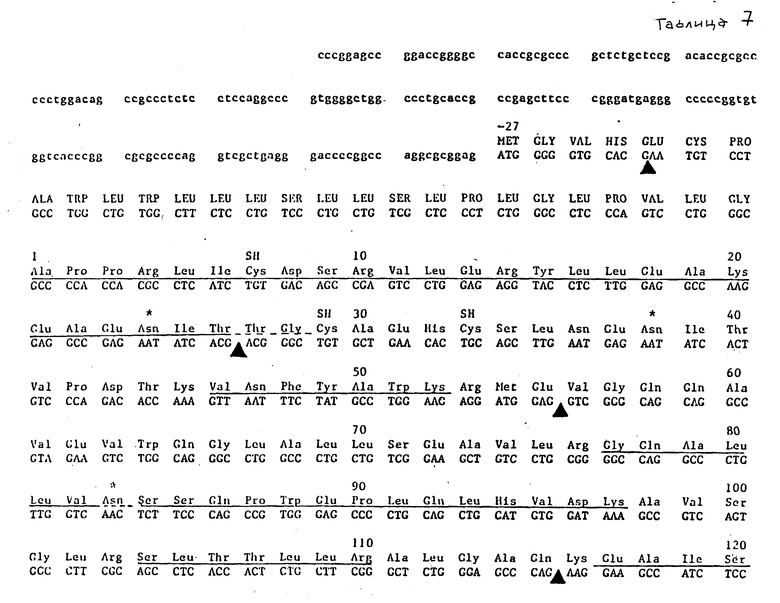
Таблица 7 иллюстрирует нуклеотидную и аминокислотную последовательности вставки клона λ-HEPOF L 13.
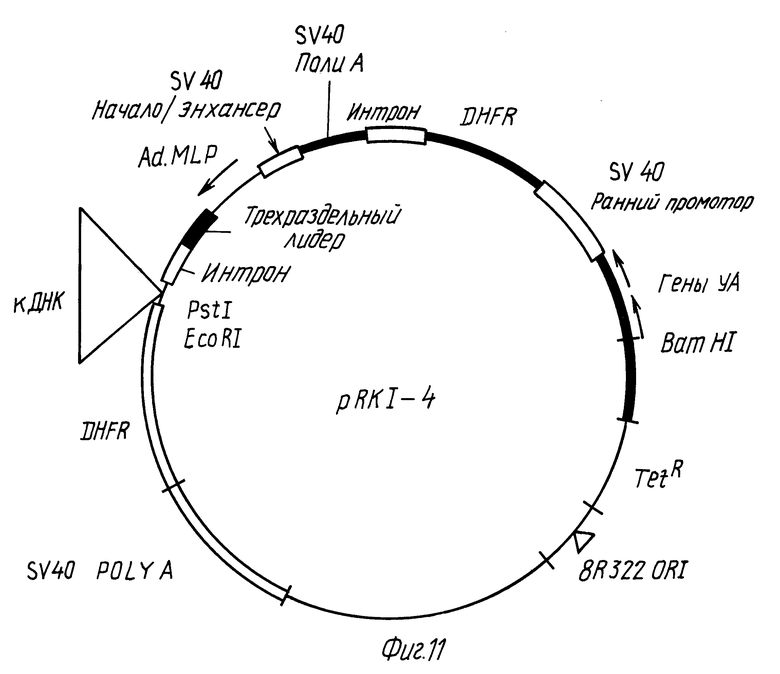
Фиг. 11 представляет схематическое изображение плазмиды pRK 1-4.
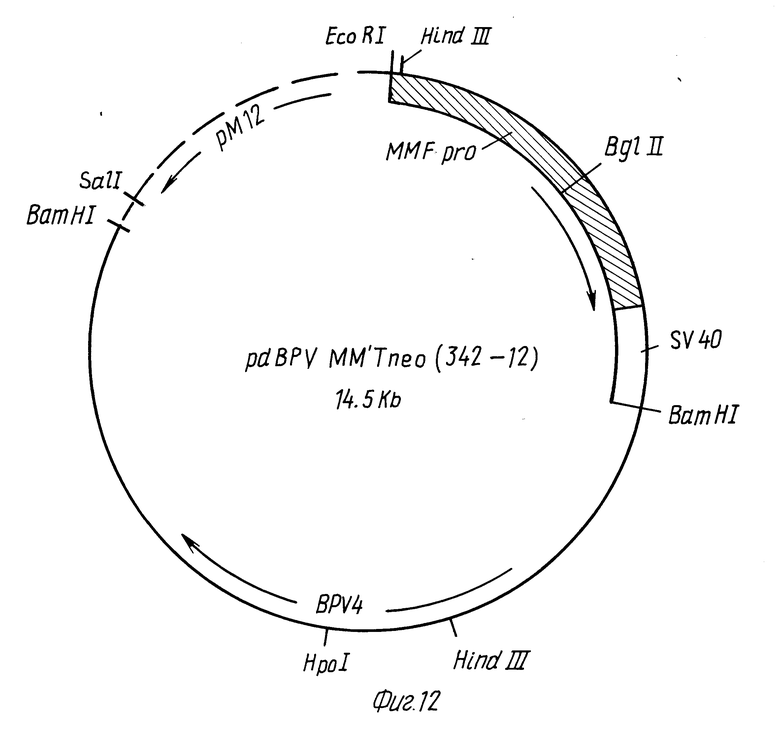
Фиг. 12 представляет собой схематическое изображение плазмиды pdBPY-MMTneo (342-12).
Настоящее изобретение касается клонирования последовательностей ДНК, кодирующих ЭПО, полученного путем экспрессии этих последовательностей рекомбинантного продукта.
A. Выделение геномного клона человеческого ЭПО
Человеческий ЭПО был очищен до гомогенности из мочи пациентов, страдающих апластической анемией, как описано ниже. Полное переваривание этого очищенного ЭПО трипсином дало фрагменты, которые были разделены высокоэффективной жидкостной хроматографией с обращенной фазой, восстановлены из градиентных фракций и подвергнуты микросеквенированию. Последовательности триптических фрагментов подчеркнуты в таблицах 2 и 3 и обсуждены более подробно ниже. Две из аминокислотных последовательностей (Val-Asn-Phe-Tyr-Ala-Trp-Lys и Val-Tyr-Ser-Asn-Phe-Leu-Arg) были выбраны для конструирования олигонуклеотидных зондов (олигонуклеотидный пул 17-членников с 32-кратным вырождением и олигонуклеотидный пул 18-членников с 128-кратным вырождением - для первого триптического фрагмента, а также два пула 14-членников, каждый с 48-кратным вырождением, - для второго триптического фрагмента). 32-кратный дегенеративный 17-членник был использован для скрининга человеческой геномной ДНК-библиотеки в векторе Ch4 A (22) с использованием модификации метода амплификации in situ Woo и O'Malley [4] для получения фильтров для скрининга.
Арабские числа в скобках [1 - 59] используются в настоящем описании для ссылок на публикации, которые приводятся в числовом порядке в конце описания настоящего изобретения.
Фаг, гибридизирующийся с 17-членником, был упакован, распределен на малые группы и опробован с 14-членными и 18-членными пулами. Фаг, гибридизирующийся с 17-членниками, 18-членниками и 14-членниками, был очищен, а фрагменты были субклонированы в векторы М13 для секвенирования путем дидезокси-метода обрыва цепи, описанного Sanger и Conlson [23].
Последовательность области одного из клонов, гибридизирующаяся с 32-кратным дегенеративным 17-членником, показана в табл. 1. Эта последовательность ДНК содержит внутри открытой рамки считывания нуклеотидную последовательность, кодирующую триптический фрагмент, использованный для построения 17-членного пула олигонуклеотидов. Кроме того, анализ последовательности ДНК показывает, что гибридизирующаяся с 17-членником область содержится внутри 87 вр-экзона.
Подтверждение того, что эти два клона (обозначены здесь λ-HEPO1 и λ-HEPO2) являются геномными клонами ЭПО, получено путем секвенирования дополнительных экзонов, содержащих области кодирования и другого триптического фрагмента.
B. Выделение клонов кДНК ЭПО.
Northern-(56) анализ человеческой фетальной (возраст 20 недель) печеночной мРНК осуществляли с использованием 95-членного олигонуклеотидного однонитевого зонда, полученного из клона М13, содержащего участок 87 вр-экзона, описанного в табл. 1. Как показано на фиг. 1, в мРНК фетальной печени обнаруживается сильный сигнал. Точная идентификация этой полосы как мРНК ЭПО была достигнута с использованием того же зонда для скрининга λ-кДНК библиотеке мРНК фетальной печени [25]. Было получено несколько гибридизирующихся клонов (с частотой приблизительно 1 положительный на 250000 подвергнутых скринингу рекомбинантов). Полная нуклеотидная и выведенная аминокислотная последовательности для двух клонов ( λ-HEPOF L 13 и λ-HEPOF L 8) представлены в табл. 5 и 6. ЭПО-кодирующая информация содержится внутри 594 нуклеотидов в 5'-половине кДНК, включающих кодирование сильно гидрофобного 27-аминокислотного лидера и 166-аминокислотного зрелого белка.
Идентификация N-конца зрелого белка была основана на N-концевой последовательности белка, выделенного из мочи людей с апластической анемией (табл. 1 и Goldwasser [26], Sue и Sytkowski [27] и Yangawa [21]). В настоящее время неизвестно, представляет ли этот N-конец (Ala-Pro-Pro-Arg---) фактический N-конец для ЭПО в системе кровообращения или происходит некоторое расщепление его в почке или моче.
Аминокислотные последовательности, которые подчеркнуты в табл. 2 и 3, обозначают триптические фрагменты и участок N-конца, для которых была получена информация с белковой последовательности. Выведенная аминокислотная последовательность точно согласовывается с последовательностями триптических фрагментов, которые были секвенированы, подтверждая, что выделенный ген кодирует человеческий ЭПО.
C. Структура и последовательность гена ЭПО человека
Относительные положения вставок ДНК четырех независимых геномных клонов ЭПО человека показаны на фиг. 3. Гибридизационный анализ этих клонированных ДНК при помощи олигонуклеотидных зондов определил положение гена ЭПО внутри области размером приблизительно 3,3 кв, показанной затемненной линией на фиг. 3. Полное секвенирование этой области (см. пример 4) и сравнение с клонами кДНК привели к получению карты интронной и экзонной структуры гена ЭПО, показанной на фиг. 4. Ген ЭПО разделяется на 5 экзонов. Часть экзона 1, полные экзоны II, III и IV, а также часть экзона V содержат белок-кодирующую информацию. Оставшиеся части экзонов I и V представляют 5'- и 3'-нетранслируемые последовательности, соответственно.
6. Экспрессия ЭПО в COS-клетках
Для того чтобы наглядно показать, что биологически активный ЭПО может экспрессироваться в системе культуры клеток in vitro, осуществляли исследования экспрессии в клетках COS [57]. Вектор, используемый для переходных исследований, а именно p91023 (B), описан в примере 5. Этот вектор содержит основной поздний промотор аденовируса, последовательность полиаденилирования, сайт начала репликации и энхансер SV40, а также ген VA аденовируса. Вставка кДНК в λ-HEPOF L 13 (см. таблицу 6) была встроена в вектор p91023 (B) ниже основного позднего промотора аденовируса. Этот новый вектор идентифицирован как pPTF-L 13.
Через двадцать четыре часа после трансфекции этой конструкции в штамм М6 клеток COS-1 (Horowitz и др., T.Mol. Appl. Genet 2:147-149 (1983) (клетки промыли и перевели в свободные от сыворотки среды и через 48 час клетки собрали). Затем с использованием количественного радиоиммунного анализа для ЭПО (57) исследовали уровень высвобождения ЭПО в культуральную надосадочную жидкость. В отдельном эксперименте вектор, содержащий кДНК ЭПО из λ-HEPOF L 13 был введен в клетки COS-1; среды далее собирали, как описано выше. ЭПО в средах затем определялся количественно любым из двух биологических анализов, проводимых in vitro, а именно с 3H-тимидином и CFV-E [12, 29], и любым из двух анализов in vivo, а именно на гипоксической мыши и голодающей крысе [29, 30] (см. таблицу 9 и пример 17). Эти результаты показывают, что в клетках COS-1 продуцируется биологически активный ЭПО. Western-блоттинг с использованием поликлонального антитела анти-ЭПО показал, что эритропоэтин, продуцируемый клетками COS, имеет подвижность в SDS-полиакриламидных гелях, идентичную подвижности нативного ЭПО, полученного из человеческой мочи (пример 8). Таким образом, гликозилирование продуцированного в COS-1 ЭПО может быть аналогичным тому, что происходит в случае нативного ЭПО.
Различные векторы, содержащие другие промоторы, также могут быть использованы в COS-клетках или в клетках других млекопитающих. Примеры этих иных промоторов, пригодных в практике настоящего изобретения, включают ранние и поздние промоторы SV-40, генный промотор металлотионеина мыши, промотор, обнаруживаемый в длинных концевых повторах ретровирусов птиц или млекопитающих. Примерами других типов клеток, пригодных в практике настоящего изобретения, являются такие клетки млекопитающих, как CHO (яичник китайского хомячка), С127 (эпителий обезьяны), ЗТ3 (мышиный фибробласт), CU-1 (почка африканской зеленой мартышки).
Примеры экспрессии ЭПО в СНО, С127 и ЗТ3 представлены в примерах 10 и 11 (СНО) и 13 (С127 и ЗТ3).
Рекомбинантный ЭПО, полученный в клетках СНО, как в примере 11, был очищен традиционными методами колоночной хроматографии. Относительные количества сахаров, присутствующих в гликопротеиде, анализировали двумя самостоятельными методами (1) Reinhold, Methods in Enzymol, 50:244-249 (Метанолиз) и (II) Takemoto, H. и др. Anal. Biochem. 145:245 (1985) (пирил-аминирование, совместно с самостоятельным определением сиаловой кислоты). Результаты, полученные каждым из этих способов, полностью согласовывались друг с другом. Таким образом было проделано несколько определений, что дало средние значения, где N-ацетилглюкозамин для сравнительных целей дан как значение 1:
Сахар - Относительный молярный уровень
N-Ацетилглюкозамин - 1
Гексозы: - 1,4
Галактоза - 0,9
Манноза - 0,5
N-Ацетилнейроминовая кислота - 1
Фукоза - 0,2
N-Ацетилгалактозамин - 0,1
Примечательно, что значительные уровни содержания фукозы N-ацетилгалактозамина воспроизводимо наблюдались при использовании обоих самостоятельных методов анализа на сахар. Присутствие N-ацетилгалактозамина указывает на наличие O-сцепленного гликозилирования белка. Наличие O-сцепленного гликозилирования далее подтверждалось анализом в SDS-PAGE. В частности, вслед за ферментативным отщеплением всего N-сцепленного углевода на гликопротеидах с использованием эндо-F N-гликозид-пептидазы молекулярная масса белка еще более снижалась после последовательного переваривания нейраминидазой.
Биологическая активность in vitro очищенного рекомбинантного ЭПО была определена методом C.Krystal, Exp. Hematol., 11:649 (1983) (биопроба на пролиферацию клеток селезенки). После многократных определений удельная активность in vitro очищенного рекомбинантного ЭПО была найдена составляющей более, чем 200000 единиц/мг белка. Среднее значение находилось в интервале 275000-300000 единиц/мг белка. Кроме того, также наблюдались значения выше чем 300000. Отношения активности in vivo (анализ полицитемических мышей, Kazal и Erslev, hm. Clinical Lab. Sci., Том B, стр. 91 (1975)) к активности in vitro, полученные для рекомбинантного продукта, находились в интервале 0,7-1,3.
Интересно сравнивать представленную выше характеристику гликопротеида с характеристикой рекомбинантного CHO-продуцируемого ЭПО, ранее изложенной в Международной заявке на патент N WO 85/02810 (опубликована 20 июня 1985 года). Аналогичный сравнительный анализ на сахар, представленный на стр. 65 данной заявки, показывает, что значение для фукозы и N-ацетилгалактозамина равны 0, тогда как отношение гексоза: N-ацетилгалактозамин равно 15,09: 1. Отсутствие N-ацетилгалактозамина указывает на отсутствие O-сцепленного гликозилирования в полученном гликопротеиде. В противоположность этому охарактеризованный выше рекомбинантный CHO-продуцируемый ЭПО настоящего изобретения содержит значительные и воспроизводимо обнаруживаемые количества как фукозы, так и N-ацетилгалактозамина, менее 1/10 относительного количества гексоз и отличается наличием O-сцепленного гликозилирования. Высокая удельная активность вышеописанного рекомбинантного ЭПО может быть непосредственно связана с особенностями его гликозилирования.
Биологически активный ЭПО, полученный экспрессией клонированных последовательностей ДНК для ЭПО настоящего изобретения, может быть использован для лечения. Количество активного компонента, безусловно, будет зависеть от жесткости режима, выбранного пути введения и удельной активности активного ЭПО, и в конечном счете будет определяться лечащим врачом или ветеринаром. Это количество активного ЭПО, определенное в каждом случае лечащим врачом, упоминается здесь как "эффективное для лечения количество ЭПО". Например, при лечении индуцированной гипопролиферативной анемии, ассоциированной с хронической почечной недостаточностью, у овцы эффективное суточное количество ЭПО найдено равным 10 единицам/кг в течение 15-40 дней. См. Eschbach и др., J. Clin. Invest., 73:434 (1984).
Активный рекомбинантный ЭПО может вводиться любым путем, подходящим для используемых условий. Предпочтительна инъекция ЭПО в кровоток млекопитающего. Специалисту, однако, ясно, что предпочтительный путь введения изменяется в зависимости от выбранного режима.
Активный ЭПО можно вводить как чистое (или в основном чистое) соединение, но предпочтительно иметь его в виде фармацевтической композиции или препарата.
Композиции настоящего изобретения как для ветеринарного, так и для человеческого применения, содержат активный рекомбинантный ЭПО, как описано выше, вместе с одним или более фармацевтически приемлемыми носителями и, при необходимости, другими терапевтическими средствами. Носитель (носители) может быть "приемлемым" в смысле его совместимости с другими компонентами композиции и невредным для его реципиента. Желательно, чтобы композиция не включала окислитель и другие вещества. Композиции могут быть получены любым из способов, хорошо известных в области фармацевтики. Все способы включают стадию связывания активного компонента с носителем, который составляет один или более вспомогательных компонентов. В основном, рецептуры приготавливают путем однородного и тщательного смешивания активного компонента с жидкими носителями, или тонкоизмельченными твердыми носителями, или с обоими сразу, после чего, если необходимо, осуществляют формование продукта.
Композиции, пригодные для парэнтерального введения, включают стерильные водные растворы активного компонента с растворами, предпочтительно изотоническими. Такие композиции можно приготавливать путем растворения твердого активного компонента в воде для получения водного раствора. Придание стерильности указанному раствору может достигаться в одноразовых или многодозовых контейнерах, например, запаянных ампулах или пробирках.
Для лучшего понимания настоящего изобретения, действительный объем которого изложен в прилагаемой формуле изобретения, приводятся нижеследующие примеры. Понятно, что в предлагаемых методах могут быть выполнены модификации в пределах сущности настоящего изобретения.
Примеры
Пример 1: Выделение геномного клона ЭПО.
ЭПО очищают от мочи пациентов, страдающих апластической анемией, в основном, как описано ранее (Miyake и др., J. Biol. Chem., 252:5558 (1977)), за исключением того, что фенольную обработку заменяют термообработкой при температуре 80oC в течение 5 минут для инактивации нейраминидазы. Конечной стадией очистки является фракционирование на колонке C-4 Vydac высокоэффективной жидкостной хроматографии (Группа Сепараций) с использованием градиента ацетонитрила от 0 до 95% с 0,1% трифторуксусной кислотой (ТФК) в течение 100 минут. Положение ЭПО в градиенте определяют электрофорезом в геле и анализом N-концевой последовательности [21, 26, 27] основных пиков. ЭПО элюируют при 53% ацетонитрила. Фракции, содержащие ЭПО, выпаривают до 100 мкл, доводят до pH 7,0 бикарбонатом аммония, переваривают полностью 2%-ным ТРСК-обработанным трипсином (Worthington) в течение 18 ч при температуре 37oC. Триптический перевар затем подвергают ВЭЖХ с обращенной фазой, как описано выше. Определяют оптическую плотность при 280 и 214 нм. Хорошо отделенные пики выпаривают почти насухо и подвергают непосредственно анализу N-концевой аминокислотной последовательности [59] , используя газофазный секвенатор Модели 480Ф Applied Biocyctems. Полученные последовательности приведены в табл. 2 и 3. Как описано выше, два из этих триптических фрагментов отбирают для синтеза олигонуклеотидных зондов. Из последовательности Val-Asn-Phe-Tyr-Ala-Trp-Lys (аминокислоты 46 - 52 в табл. 2 и 3) получают 17-членник с 32-кратной вырожденностью.
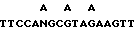
и 18-членник с 128-кратной вырожденностью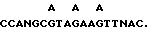
Из последовательности Val-Tyr-Ser-Asn-Phe-Leu-Arg (аминокислоты 144 - 150 в табл. 2 и 3) получают два пула 14-меров, каждый - с 32-кратной вырожденностью.
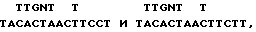
которые отличаются в первом положении лейцинового кодона. Олигонуклеотиды помечают на 5'-конце 32P, используя полинуклеотидкиназу (New England Biolabs) и гамма 32P-ATP (New England Nuclear). Удельная активность олигонуклеотидов варьирует между 1000 и 3000 дюйм3/ммол олигонуклеотида. Геномную ДНК-библиотеку человека в бактериофаге лямбда (Lawn и др., [22]) подвергают скринингу с использованием модификации методики амплификации in situ, первоначально описанной Woo и др. [47]. Приблизительно 3,5 • 105 фагов высевают с плотностью 6000 фагов на 150 мм чашку Петри (среды NZCYM) и инкубируют при температуре 37oC до тех пор, пока пятна не станут видимыми, однако маленькими (приблизительно 0,5 мм). После охлаждения при температуре 4oC в течение 1 ч дупликатные реплики картин пятен переносят на найлоновые мембраны (New England Nuclear) и инкубируют в течение ночи при температуре 37oC на чашках со свежими средами NZCYM. Затем денатурируют и нейтрализуют флотированием каждого в течение 10 мин на тонкой пленке 0,5H NaOH - 1 M NaCl и 0,5 M Tris (pH 8) - 1M NaCl, соответственно. Вслед за вакуумной сушкой при температуре 80oC в течение 2 часов фильтраты промывают в 5 x SSC, 0,5% SDS в течение 1 часа, и клеточные остатки на поверхности фильтров удаляют путем осторожного соскоба сухой тканью. Это соскабливание снижает фоновое связывание зонда с фильтрами. Фильтры затем промывают водой и предварительно гибридизируют от 4 до 8 ч при температуре 48oC в 3М растворе тетраметиламмонийхлорида, 10 мМ NaPO4 (pH 6,8), 5 x D-Enhardt's, 0,5% SDS и 10 мМ ЭДТА. 17-членник, меченый 32P, затем прибавляют при концентрации 0,1 пмол/мл и осуществляют гибридизацию при температуре 48oC в течение 72 ч. Затем фильтры последовательно промывают в 2 • SSC (0,3 M NaCl - 0,03 M цитрат натрия, pH 7) при комнатной температуре, в течение 1 ч в 3 M TMAC1 - 10 мМ NaPO4 (pH 6,8) при комнатной температуре и от 5 до 15 мин - при температуре гибридизации. Приблизительно 120 сильных дупликатных сигналов обнаруживают после 2-дневной авторадиографии. Положительные сигналы отбирают, группируют в пулы по 8, пересеивают и вновь подвергают скринингу в трипликате с использованием 1/2 пула 14-членников на каждом из двух фильтров и 17-мера - на третьем фильтре. Условия и 17-членник для высевания и гибридизации такие же, как описано выше, за исключением того, что гибридизацию для 14-членника осуществляют при температуре 37oC. Вслед за авторадиографией 17-членный зонд удаляют с фильтра в 50%-ном формамиде в течение 20 минут при комнатной температуре, а фильтр повторно гибридизируют при температуре 52oC с 18-членным зондом. Два независимых фага гибридизируются со всеми тремя зондами. ДНК одного из этих фагов (обозначен здесь λ-HEPO1) переваривают полностью с помощью San 3A и субклонируют в М13 для анализа ДНК-последовательности с использованием дидезокси-метода обрыва цепи, предложенного Sanger и Conlson, [23]. Нуклеотидная последовательность и выведенная аминокислотная последовательность открытой рамки считывания, кодирующей триптический фрагмент ЭПО (подчеркнутая область), описаны здесь. Интронные последовательности даны в строчных буквах, экзонные последовательности (87 нуклеотидов) даны в прописных буквах. Последовательности, которые согласуются с сайтами акцептора сращивания (a) и донора (d), подчеркнуты (см. табл. 4).
Пример 2: Northern - анализ мРНК печени плода человека.
5 мкг мРНК человеческой фетальной печени (20-недельной) и мРНК печени взрослого подвергают электрофорезу в 0,8%-ном агарозном формальдегидном геле и переносят на нитроцеллюлозу, используя метод Derman и др., Cell, 23:731 (1981). Из М13, содержащего вставку, показанную в таблице 1, затем получают однонитевой зонд. Праймером является 20-членник, происходящий из того же триптического фрагмента, что и первоначальный 17-членный зонд. Получение зонда проводят, как описано Anderson и др., PNAS, [49], за исключением того, что вслед за отщеплением Smal (которое продуцирует желательный зонд длиной 95 нуклеотидов, содержащий 74 нуклеотидов кодирующей последовательности) малый фрагмент очищают из М13 хроматографией на колонке с Сефарозой C14B и 0,1 N NaOH - 0,2 M NaCl. Фильтр гибридизируют с зондом приблизительно до 5 • 106 отсчетов в минуту в течение 12 часов при температуре 68oC, промывают в 2 x SSC при температуре 68oC и подвергают воздействию в течение 6 дней усиливающего экрана. Однонитевая маркерная мРНК из 1200 нуклеотидов (указана стрелкой) движется по соседней "дорожке" (фиг. 1).
Пример 3: кДНК из фетальной печени.
Зонд, идентичный описанному в примере 2, получают и используют для скрининга библиотеки кДНК фетальной печени, полученной в векторе λ-Ch21A (Toole и др., Nature, [25]) (1984), используя стандартные методики скрининга пятен (Berten, Davis, Science, [53] (1978)). Три самостоятельных положительных клона (обозначены здесь λ-HEPOF L6, 1350 пар оснований; λ-HEPOF L8, 700 п. о. и λ-HEPOF L13, 1400 п.о.) выделяют после скрининга 1 • 106 пятен. Полные вставки клонов λ-HEPOF L13 и λ-HEPOF L6 секвенировали после субклонирования в M13 полностью (табл. 7 и 5, соответственно). λ-HEPOF L8 секвенировали частично, оставшиеся участки подразумеваются идентичными другим двум клонам (табл. 6). 5'- и 3'-нетранслируемые последовательности представлены строчными буквами. Кодирующая область представлена прописными буквами.
В отношении табл. 2 и 3 следует упомянуть, что выведенная аминокислотная последовательность, показанная ниже нуклеотидной последовательности, пронумерована, начиная с цифры 1, для первой аминокислоты зрелого белка. Лидерный пептид указан прописными буквами. Остатки цистеина в зрелом белке дополнительно указаны SH, и потенциальные N-сцепленные сайты гликозилирования обозначены звездочкой. Аминокислоты, которые подчеркнуты, отмечают те остатки, которые идентифицированы непосредственно секвенированием N-конца или секвенированием триптических фрагментов ЭПО, которые описаны в примере 1. Частичное подчеркивание указывает остатки в аминокислотной последовательности, которые не удалось определить однозначно. кДНК-клоны λ-HEPOF L6, λ-HEPOF L8 и λ-HEPOF L13 депонированы и доступны из American Type Culture Collection, Rockville, Maryland, под входящими номерами АТСС 40156, АТСС 40152 и АТСС 40153, соответственно.
Пример 4: Структура гена ЭПО.
Относительные размеры и положения четырех самостоятельных геномных клонов (λ-HEPO1,2,3 и 6) из библиотеки Hae-111/AluI проиллюстрированы перекрывающимися линиями на фиг. 3. Утолщенная линия указывает положение гена ЭПО. Масштаб (в Kb) и положения известных сайтов расщепления эндонуклеазами рестрикции приводятся. Область, содержащая ген ЭПО, полностью секвенирована для обоих тяжей с использованием направленных экзонуклеаза 111-генерированных рядов делеций через эту область. Схематическое изображение пяти экзонов, кодирующих мРНК ЭПО, показано на фиг. 4. Точная 5'-граница экзона 1 в настоящее время неизвестна. Белок-кодирующие участки экзонов закрашены. Полная нуклеотидная последовательность экзонов показана в таблице 4. Известные пределы каждого экзона очерчены сплошными вертикальными линиями. Геномные клоны λ-HEPO1, λ-HEPO2, λ-HEPO3 и λ-HEPO6 депонированы и доступны из American Type Culture Collection, Rockville, Maryland, под входящими номерами АТС 40154, АТСС 40155, АТСС 40150 и АТСС 40151, соответственно.
Пример 5: Конструирование вектора p91023 (B).
Вектором для трансформации является pAdD26sVpA (3), описанный Kaufman и др. , Mol. Cell Biol., 2:1304 (1982). Конструкция этого вектора показана на фиг. 5. Эта плазмида содержит кДНК-ген дигидрофолатредуктазы мыши (DFHP), который находится под транскрипционным контролем основного позднего промотора аденовируса 2 (Ad2). 5'-сайт сращивания указан в аденовирусной ДНК, а 3'-сайт сращивания, происходящий из гена иммуноглобулина, присутствует между основным поздним промотором аденовируса 2 и DFHP-кодирующей последовательностью. Ранний сайт полиаденилирования SV40 присутствует по направлению ниже DFHP-кодирующей последовательности. Прокариотический участок pAdD26-SVpA (3) происходит из psVod/Mellon и др., Cell, 27:279 (1981) и не содержит последовательностей pBR322, известных тем, что они ингибируют репликацию в клетках млекопитающих (Lusky и др., Nature, 293:79 (1981).
Как показано на фиг. 5, pAdD26sVpA (3) переводят сначала в плазмиду pAdD26sVpA (3) (d) делецией одного из двух сайтов Pst 1. Это осуществляют путем частичного переваривания Pst 1, используя недостаточность фермента таким образом, что получают субпопуляцию линеаризованных плазмид, в которой только один сайт Pst 1 расщеплен, и последующей обработкой фрагментов Кленова, сшиванием для рециркуляции и скринингом на деленцию сайта Pst 1, расположенного в 3'-положении по отношению к последовательности полиаденилирования SV40.
В плазмиду pAdD26SVpA (3) (d) вставляют трехраздельный лидер аденовируса и гены, ассоциированные с вирусом (VA гены). pAdD26SVpA (3) (d) расщепляют Pvull для создания линейной молекулы. Затем pJAW 43/zatn и др., Cell, 16:851 (1979)) переваривают Xhol, обрабатывают фрагментом Кленова, переваривают PvuIl, и фрагмент из 140 п.о. выделяют электрофорезом в акриламидном геле [6] в трис-боратном буфере (Maniatis и др., выше). Фрагмент 140 п.о. затем сшивают с PvuII-переваренной плазмидой pAdD26SVpA(3) (d) (фиг. 6). Продукт сшивания используют для трансформации E.coli с приобретением устойчивости к тетрациклину; колонии подвергают скринингу с использованием методики Grunstein - Hoghess. Применяя меченый 32P зонд, гибридизирующийся с фрагментом из 140 п.о., получают положительные колонии, из которых выделяют ДНК для испытания того, является ли реконструированный сайт PvuII 5' или 3'-концом вставленной ДНК из 140 п.о. Правильная ориентация сайта PvuII - на 5'-стороне вставки 140 п.о. Эта плазмида обозначена pTPL на фиг. 6.
Фрагмент ДНК вируса SV40, содержащий последовательность энхансера SV40, получают путем переваривания ДНК SV40 Ava 11, затупления концов фрагментом Кленова, сшивания Xho 1-линкеров с фрагментами от переваривания Xho 1 и выделения наибольшего фрагмента (D) посредством электрофореза в геле. Этот фрагмент затем сшивают с pTPL, расщепленной Xho 1, получая плазмиду pCYSVL 2-TPL. Ориентация фрагмента D SV40 в pCYSVL 2-TPL такова, что поздний промотор вируса SV40 находится в такой же ориентации, что и основной поздний промотор аденовируса.
Для введения генов, связанных с аденовирусом (генов VA), в pCYSVL 2-TPL, вначале конструируют плазмиду pBR322, которая содержит фрагмент в Hind 111 аденовируса типа 2. ДНК аденовируса типа 2 переваривают Hind 111 и фрагмент B выделяют электрофорезом в геле. Этот фрагмент вставляют в плазмиду pBR322, которая предварительно была переварена Hind 111. После трансформации E.coli устойчивые к ампициллину рекомбинанты подвергают скринингу на наличие вставки фрагмента в Hind 111; ориентацию вставки определяют путем переваривания рестрикционным ферментом. pBR322 - Ad Hind 111 B содержит фрагмент B Hind 111 аденовируса типа 2 в ориентации, изображенной на фиг. 7.
Как показано на фиг. 8, гены VA получают из плазмиды pBR322 - Ad Hind 111 B путем переваривания Hpa 1, добавления линкеров EcoRI и переваривания EcoRI с последующим восстановлением фрагмента размером 1,4 кб. Фрагмент, имеющий липкие концы EcoRI, затем вшивают в EcoRI-сайт pPTL, ранее переваренной EcoRI. После трансформирования HB101 E.coli и отбора трансформантов по устойчивости к тетрациклину колонии подвергают скринингу посредством фильтр-гибридизации с ДНК, специфичной для генов VA. Из положительно гибридизирующихся клонов получают ДНК и охарактеризовывают перевариванием рестрикционными эндонуклеазами. Полученная плазмида обозначена p91023.
Как показано на фиг. 9, два сайта EcoR1 в плазмиде p91023 отщепляют путем полного расщепления p91023 при помощи EcoRI и восстановления двух ДНК-фрагментов (около 7 кб и около 1,3 кб). Последний фрагмент содержит гены VA. Концы обоих фрагментов заполняют с использованием фрагмента Кленова, и два фрагмента затем сшивают один к другому. Плазмиду p91023(A), содержащую гены VA и являющуюся аналогичной плазмиде p91023, однако потерявшую интерстициальный фрагмент для двух сайтов EcoRI, идентифицируют посредством скрининга Gruns tein - Hogness фрагментов гена VA, а также традиционного рестрикционного анализа.
Один Pst 1-сайт в плазмиде p91023 (A) отщепляют и заменяют на EcoRI-сайт. p91023(A) полностью расщепляют при помощи Pst 1 и обрабатывают фрагментом Кленова. EcoRI-линкеры сшивают с тупоконечным сайтом Psr1 плазмиды p91023(A). Линейную плазмиду p91023(A) с EcoRI-линкерами, присоединенными в тупоконечном сайте Pst1, отделяют от несшитых линкеров и полностью переваривают EcoRI, после чего повторно сшивают. Плазмиду p91023(B), как изображено на фиг. 9, восстанавливают и идентифицируют как имеющую структуру, аналогичную плазмиде p91023(A), но вместо прежнего сайта Pst1 содержащую сайт EcoRI. Плазмида p91023(B) депонирована и доступна из American Type Culture Collection, Rockville, Maryland, под входящим номером АТСС 39754.
Пример 6: Экспрессия кДНК ЭПО в CO5-клетках.
Вставки кДНК из λ-EPOF L6 и λ-EPOF L13 встраивают в плазмиду p91023(B), образуя pRTF L6 и pRTF L13, соответственно. 8 мкг каждой из очищенных ДНК затем используют для трансфекции 5 • 106 клеток COS с помощью DEAE-декстранового метода (ниже). Через 12 ч клетки промывают и обрабатывают Хлорохином (0,1 мМ) в течение 2 ч, промывают вновь и выдерживают в течение 24 ч в средах (10 мл), содержащих 10%-ную фетальную телячью сыворотку. Среды меняют на новые (по 4 мл), свободные от сыворотки, и собирают через 48 часов.
ЭПО определяют количественно радиоиммунным анализом, как описано Sherwood и Goldwasser [54]. Антитело поставлено Доктором Judith Sherwood. Йодированный изотопный индикатор получают из гомогеннного ЭПО, как описано в примере 1. Чувствительность пробы составляет приблизительно 1 нг/мл. Результаты приведены ниже в таблице 8.
pPTF L13 депонирована и доступна из American Type Culture Collection, Rockville, Maryland, под входящим номером АТСС 39990.
Пример 7: Экспрессия кДНК ЭПО в COS-клетках.
кДНК из λ-HEPOF L13 переносят в плазмиду p91023(B), трансформируют в клетки COS-1 и собирают, как описано выше (пример 6), за исключением того, что хлорохинную обработку опускают.
Биологическую активность ЭПО измеряют in vitro с использованием либо колониеобразующей пробы с клетками фетальной печени мыши в качестве источника CFV-E, либо метода поглощения 3H-тимидина, используя клетки селезенки от мышей, инъекцированных фенилгидразином. Чувствительности этих анализов составляют приблизительно 25 мЕдиниц/мл. Биологическую активность in vivo измеряют с использованием либо метода гипоксической мыши, либо метода голодающей крысы. Чувствительность этих анализов составляет приблизительно 100 мЕдиниц/мл. С имитированной кондиционированной средой активность ни в одном случае не обнаружена. Результаты исследования ЭПО, экспрессированного клоном EPOF L13, показаны ниже в табл. 9. Приведенные активности выражены в единицах/мл, и использованием в качестве стандарта торгового, количественно определенного ЭПО (Toyobo, 1 пс.).
Пример 8: Анализ ЭПО из COS-клеток в SDS ПAA-геле.
180 нг ЭПО, полученного из среды клеток COS, трансфецированных кДНК (из λ-HEPOF L13) в составе вектора 91023(B) (выше), подвергают электрофорезу в 10%-ном ПАА-геле в присутствии SDS (по Laewlli) и электропередают на нитроцеллюлозную бумагу (Towbin и др. , Proc. Natl. Acad. Sci. США, 76:4350 (1979)). Фильтр зондируют антителом анти-ЭПО, как описано в табл. 8, промывают и повторно зондируют белком A125J. Фильтр авторадиографируют в течение двух дней. Нативный гомогенный ЭПО, описанный в примере 1, также подвергают электрофорезу или до (дорожка B), или после йодирования (дорожка C) (см. фиг. 10). Используемые маркеры включают метионин, меченый 35S, сывороточный альбумин (68000 д) и яичный альбумин (45000 д).
Пример 9: Конструирование pRKI-4.
Из плазмиды PSV2DHFR/Subramani и др., Mol Cell. Biol. 1:854-864 (1981)) выделяют фрагмент BamHI-Pvule, содержащий ранний промотор SV40, смежный с геном дигидрофолятредуктазы мыши (DHFR), энхансер SV40, малый антигенный интрон t и последовательность полиаденилирования SV40 (фрагмент A). Другие фрагменты получают из вектора p91023(A) (выше) следующим образом: p91023(A) переваривают Pst 1 в единственном сайте Pst1 около промотора аденовируса для линеаризации плазмиды и либо сшивают с синтетическими конвертерами Pst1 - EcoRI и циклизуют (создавая сайты Pst 1 - EcoR1 - Pst 1 в первоначальном сайте Pst1 p91023(B')), либо обрабатывают большим фрагментом ДНК-полимеразы 1 для разрушения сайтов Pst1 и сшивают с синтетическим EcoRI - линкером и рециклизуют (создавая EcoRI-сайт в первоначальном сайте Pst1 p91023(B). Каждую из двух полученных плазмид p91023(B) и p91023(B') переваривают Xba и EcoRI для получения двух фрагментов (F и G). Путем соединения фрагмента G из плазмиды p91023(B) и фрагмента F из плазмиды p91023(B'), а также фрагмента G из плазмиды p91023(B) и фрагмента F из плазмиды p91023(B') создают две новые плазмиды, которые содержат либо сайт EcoRI - Pst1, либо сайт Pst1 - EcoRI. Вектор, где Pst1-сайт близок к основному позднему промотору аденовируса назван p91023(C).
Вектор p91023(C) полностью переваривают Xho1, и полученную линеаризованную ДНК с липкими концами затупляют путем реакции заполнения концов с помощью большого фрагмента ДНК-полимеразы 1 E.coli. К этой ДНК пришивают фрагмент Hind 111 - EcoRI из 340 п.о., содержащий энхансер SV40, полученный, как описано ниже.
Фрагмент Hind 111 - Pvull из вируса SV 40, который содержит начало репликации, а также энхансер вставляют в плазмиду с lac/Little и др., Mol.Biol. Med., 1:473-488 (1983)), которую предварительно переваривают BamHI, заполняют липкие концы с помощью большого фрагмента ДНК-полимеразы 1 и переваривают ДНК Hind 111. В полученной плазмиде (c SVHPlac) BamH1-сайт регенерирует при сшивании с тупым концом Pvull. Из плазмиды с SVHPlac получают фрагмент EcoRI-Hind 111 и лигируют его с фрагментом EcoRI - Hind 111 pSV Od/Mellon и др., выше), который содержит плазмидную основу репликации. Полученную плазмиду обозначают pSVHPOd. Затем получают фрагмент EcoRI - Hind 111 из 340 п.о. плазмиды pSVHPOd, содержащий ori SV40/энхансер, затупляют с обоих концов при помощи большого фрагмента ДНК-полимеразы 1 и сшивают с Xhol- переваренным, тупоконечным вектором p91023(C), описанным выше. Полученную плазмиду (p91023 (C)/Xho/ плюс EcoRI/Hind 111), в которой ориентация фрагмента Hind 111 - EcoRI такова, что BamH1-сайт внутри фрагмента является близлежащим к гену VA, называют pES105. Плазмиду pES105 переваривают BamH1 и Pvull, а также отдельно Pvull, и выделяют фрагмент BamH1 - Pvull, содержащий основной поздний промотор аденовируса (фрагмент B), и фрагмент Pvull, содержащий плазмидный ген (устойчивости/устойчивости к тетрациклину), а также другие последовательности (фрагмент C). Фрагменты A, B и C сшивают и полученную плазмиду, показанную на фиг. 11, выделяют и обозначают pRKI-4. Плазмида pRKI-4 депонирована American Type Culture Collection Rockville, Maryland, где она доступна под входящим номером АТСС 39940.
Пример 10: Экспрессия кДНК ЭПО в CHO- клетках - Метод 1.
ДНК (20 мкг) плазмиды pRTF L 13, описанной выше (пример 6), расщепляют рестрикционной эндонуклеазой Cla 1 и сшивают с Cla 1-расщепленной ДНК плазмиды pAdD265SVp(A) 1 (2 мкг), которая содержит интактный ген дигидрофолятредуктазы (DHFR), приводимый в действие основным поздним промотором аденовируса (Kaufman и Sharp, Mol.and Cell Biol., 2:1304-1319 (1982)). Эту сшитую ДНК используют для трансфекции DHFR-отрицательных клеток CHO (DUKX-B11, Chasin L. A. и UrlaubG. (1980) PNAS 77:4216-4220). После двухдневного выращивания клетки, которые включили по крайней мере один ген DHFR, отбирают в альфа-средах, не содержащих нуклеотидов, и пополняют 10%-ной диализированной фетальной бычьей сывороткой. После культивирования в избирательных средах, в течение двух недель колонии извлекают, объединяют в группы из 10-100 колоний, повторно высевают и выращивают до слияния в альфа-средах, не содержащих нуклеотидов. Среды от пулов, выращенных в условиях метотрексатной селекции, испытывают на содержащие ЭПО посредством радиоиммуноанализа (RIA). Пулы, которые показывают наличие ЭПО, выращивают в присутствии метотрексата (0,02 мкМ). Субклонированный из положительного пула штамм Cla 4,4,02 ЭПО высвобождает 460 нг-мл ЭПО в среду, содержащую 0,02 мкМ МТХ (табл. 10). Cla 44,02-7 ЭПО представляет собой отобранную клеточную линию для получения ЭПО, которая депонирована American Type Culture Colection под входящим номером АТСС CRL 8695. Для пулов, которые являются отрицательными при RIA, метотрексат-устойчивые колонии, полученные их эквивалентных культур, растущих в присутствии метоткрексата (0,02 мкМ), вновь подвергают проверке в пулах на наличие ЭПО путем RIA. Те культуры, которые не являются положительными, субклонируют и подвергают культированию при еще более высоких концентрациях метотрексата.
Ступенчатая селекция с метотрексатом (MTX) достигается повторными циклами культивирования клеток в присутствии возрастающих концентраций метотрексата и селекции для выживших микроорганизмов. При каждом цикле ЭПО выявляют в надосадочном слое культуры с помощью RIA и анализа биологической активности in vitro. Уровни используемого метотрексата в каждой ступенчатой амплификации составляют 0,02 мкМ, 0,1 мкМ и 0,5 мкМ. как показано в табл. 10, после первого цикла селекция при 0,02 мкМ MTX в культуральные среды высвобождаются значительные уровни ЭПО.
Пример 11: Экспрессия кДНК ЭПО в CHO-клетках -Метод II.
ДНК клона λ-HEPOF L 13 переваривают EcoRI, и малый фрагмент RI. содержащий ген ЭПО, субклонируют в EcoRI-сайт плазмиды pRKI-4 (см. пример 10). Эту рекомбинантную ДНК (pRKF L 13) затем используют для трансфекции DHFR-отрицательных клеток CHO прямо (без переваривания), а селекцию и амплификацию осуществляют, как описано в примере 10 выше.
ДНК RKF L 13 также вставляют в клетки CHO путем слияния протопластов и микроинъекции. Плазмида pRKF L 13 депонирована и доступна из American Type Culture Collection Rockvill, Maryland, под входящим номером АТСС 39989.
Предпочтительный клон единичной колонии депонирован и доступен из American Type Culture Collection Rockville, Maryland, под входящим номером ТСС CRL 8695.
Пример 12: Экспрессия геномного клона ЭПО в клетках COS-1.
Вектором, используемым для экспрессии геномного клона ЭПО, является pSVOd/Mellon и др. (выше). ДНК из pSVOd переваривают полностью при помощи Hind 111 и затупляют с использованием фрагмента ДНК-полимеразы 1. Геномный клон λ-HERO3 полностью переваривают EcoRI и Hind 111, и 4,0 кб фрагмент, содержащий ген ЭПО, выделяют и затупляют, как описано выше. Нуклеотидная последовательность этого фрагмента от Hind 111-сайта до области, находящейся выше сигнала полиаденилирования, показана на фиг. 4 и в табл. 4. Фрагмент гена ЭПО вставляют в плазмидный фрагмент psvod, и правильно построенные рекомбинанты в обеих ориентациях выделяют и проверяют. Плазмида pCZ2-1 имеет ген ЭПО в ориентации "a" (т.е. с 5'-концом гена ЭПО, приближенным к началу SV 40), а плазмида pCZ1-3 содержит ген в противоположной ориентации (ориентация "b").
Плазмиды pCZ1-3 и pCZ2-1 трансфекцируют в клетки COS-1, как описано в примере 7, среды собирают и анализируют на наличие иммунологически реактивного ЭПО. Приблизительно 31 нг/мл ЭПО обнаружено в отстоявшихся культурах после введения pCZ2-1 и 16-31 нг/мл после введения pCZ1-3.
Кодирующие фрагменты геномных клонов HEPO1, HEPO2 и HEPO6 могут быть вставлены в клетки COS для экспрессии аналогичным образом.
Пример 13: Экспрессия в клетках C127 и ЗТЗ.
a. Конструирование pBPYEPO
Плазмиду, содержащую кДНК-последовательность ЭПО под транскрипционным контролем металлотионеинового промотора мыши и связанную с полной ДНК вируса бычьей папилломы, получают следующим образом.
pEPO49f
Плазмида pSP6/5 приобретена y Promeda Biotec. Эту плазмиду полностью переваривают с EcoRI. EcoRI-фрагмент размером 1340 п.о. из λ-HEPOF L 13 вставляют в нее при помощи ДНК-лигазы. Полученную плазмиду, в которой 5'-конец гена ЭПО является ближайшим к промотору SP6 (как определено перевариванием BglI и Hind 111), обозначают pEPO49f. В этой ориентации сайт BamH1 в полилинкере pSP6/5 непосредственно примыкает к 5'-концу гена ЭПО.
pMMTneo BRY
Плазмиду pdBPV-MMTneo (342-12) (Law и др. , Mol. and Cell Biol., 3: 2110-2115 (1983), показанную на фиг.12, переваривают до готовности BamH1 для получения двух фрагментов: большого фрагмента размером ≈ 8 кб, содержащего геном ВРУ, и меньшего фрагмента размером ≈ 6,5 кб, содержащего pML2 начало репликации и устойчивости к ампициллину, металлотионеиновый промотор, устойчивости к неомицину и сигнал полиаденилирования SV40. Переваренную ДНК рециклизуют ДНК-лигазой, и плазмиды, которые содержат только фрагмент размеров 6,8 кб, идентифицируют перевариваниями рестрикционными эндонуклеазами EcoRI и BamH1. Одна такая плазмида обозначена pMMTneo BPY.
pEPO15a
pMMTneo BPY полностью переваривают BglII. pEPO49f переваривают до готовности BamH1 и BglII и получают в геле 700 п.о. фрагмент, содержащий целую ЭПО-кодирующую область. BglII-переваренную pMMTneo BPY и фрагмент BamH1/BglII 700 п. о. сшивают, и полученные плазмиды идентифицируют гибридизацией в колониях при помощи олигонуклеотидного d/GGTCATCTGTCCCCTGTCC) зонда, являющегося специфичным к гену ЭПО. Из плазмид, которые являются положительными данными гибридизации, одна (pEPO15a), которая имеет кДНК ЭПО в ориентации, так, что 5'-конец кДНК ЭПО является близлежащим к металлотионеиновому промотору, идентифицируется путем переваривания EcoR1 и Kpn1.
pBPY-EPO
Плазмиду pEPO15a подвергают расщеплению BamH1 для линеаризации. Плазмиду pdBPY-MMTneo (342-12) также полностью переваривают BamH1 с получением двух фрагментов размером 6,5 и 8 кб. Фрагмент, который содержит целый геном вируса бычьей папилломы, выделяют в геле. Расщепленную pEPO15a и фрагмент длиной 8 кб сшивают друг с другом в плазмиду pBPY-EPO, которую идентифицируют гибридизацией в колониях с использованием олигонуклеотидного зонда d/P-CCACACCCGGTACACA-OH), являющегося специфичным к геному BPY. Переваривание ДНК pBPY-EPO с помощью Hind 111 указывает на то, что направление транскрипции генома BPY является таким же, как и направление транскрипции металлотионеинового промотора (аналогично pdBPY-MMTneo /342-12/). Плазмида pdBPY-MMTneo/342-12/ доступна из American Tyre Culture Collection, Rockville, Maryland, под входящим номером АТСС 37224.
b. Экспрессия
Метод 1
Получают ДНК pBPY-EPO и приблизительно 25 мкг ее используют для трансфекции 1•106 клеток C127/Lowy и др., J.of Virol., 26:291-298 (1978), используя стандартные методики осаждения фосфата кальция (Crahm и др., Virology, 52:456-467 (1973)). Через пять часов после трансфекции среды удаляют, клетки подвергают шоку глицерином, промывают и прибавляют свежую α-среду, содержащую 10%-ную фетальную бычью сыворотку. Через сорок восемь часов клетки трипсинизируют, разбавляют в отношении 1: 10 DME средой, содержащей 500 мкг/мл G418 (Sonthern и др., Mol. Appl. Genet., 1:327-341 (1982)), и выращивают в течение двух-трех недель. G418-устойчивые колонии затем переносят индивидуально в лунки для микротитрования и выращивают в присутствии G418. Клетки затем промывают, прибавляют свежие среды, содержащие 10%-ную фетальную бычью сыворотку, которые собирают через 24 часа. Кондиционированные среды испытывают и выявляют присутствие ЭПО радиоиммунным анализом и биологической пробой in vitro.
Метод II
Клетки C127 или ЗТ3 котрансфецируют 25 мкг pBPY-EPO и 2 мг psvrueo (Souther и др., выше), как описано в Методе I. Это составляет приблизительно 10-кратный молярный избыток pBPY-EPO. Вслед за трансфекцией применяют методику, аналогичную описанной в методе I.
Метод III
Клетки C127 трансфецируют 30 мкг pBPY-EPO, как описано в методе I. Вслед за трансфекцией и разведение (1:10) свежие среды меняют каждые три дня. Приблизительно через 2 недели появляются очаги BPY-транформированных клеток. Отдельные очаги собирают в 1 см гнезда микротитровальной чашки, выращивают до субсливающегося монослоя и анализируют на присутствие ЭПО с помощью иммунного анализа и исследования биологической активности.
C. Определение in vitro активности рекомбинантного ЕРО, экспрессированного в клетках C127 и 3Т3
ЭПО, экспрессированный в клетках C127 и 3Т3 методом II тестировали на наличие биологической активности. Биологическая активность очищенного рекомбинантного ЭПО in vitro анализировалась по методу G. Krystal, Exp. Hematol. 11:649 (1983) (биоанализ пролиферации клеток селезенки) с определением белка на основе аминокислотных данных. Согласно многочисленным определениям специфическая активность in vitro рекомбинантного ЭПО, очищенного из клеток C127, составляла 267000 ед/мг белка. Специфическая активность in vitro рекомбинантного ЭПО, очищенного из клеток 3Т3, была 244000 ед/мг белка.
В обоих случаях значительные уровни N-ацетилгалактозамина были определены двумя независимыми способами (Reinhold, Methods in Enzymol. 50:244-249 (Methanolysis) и Take moto H. et al., Anal. Biochem. 145:245 - (1985)), что свидетельствует об O-связанном гликозилировании белка.
Пример 14: Очистка ЭПО.
Кондиционированные среды после культивирования COS-клеток, трансформированных рекомбинантным вектором, содержащие примерно 200 мкг/л ЭПО, концентрируют с использованием ультрафильтрационных мембран для молекулярного веса 10000, например, таких как Millipoze Pellican с мембраной 0,465 кв.м., а затем диафильтруют против 10 мМ буферного раствора фосфата натрия при pH 7. Концентрированные и диафильтрованные кондиционированные среды содержат примерно 2,5 мг ЭПО в 380 мг общего белка. Раствор ЭПО дополнительно концентрируют до 186 мл, и осажденные белки извлекают центрифугированием при 110000 x g в течение 30 мин.
pH надосадочного слоя, который содержит ЭПО, доводят до 5,5 50%-ной уксусной кислотой, оставляют при температуре 4oC в течение 30 мин, а образовавшийся осадок отделяют центрифугированием при 13000 x g в течение 30 мин.
Хроматография на карбоксиметил-сефарозе
Супернатант от центрифугирования, содержащий примерно 200 мкг ЭПО (24 мг общего белка), наносят на колонку, упакованную СМ-Сефарозой (20 мл), уравновешенной 10 мМ ацетата натрия, pH 5,5, промывают 40 мл того же буферного раствора. ЭПО, связанный с СМ-Сефарозой, элюируют 100 мл градиента NaCl (0 - 1М) в 10 мМ растворе фосфата натрия, pH 5,5. Фракции, содержащие ЭПО (общее количество 50 мкг), объединяют и концентрируют до 2 мл с использованием ультрафильтрационной мембраны Americon YM10.
ВЭЖХ с обращенной фазой
Концентрированные фракции с СМ-Сефарозы, содержащие ЭПО, подвергают дальнейшей очистке ВЭЖХ с обращенной фазой, используя колонку Vydac C-4. ЭПО подают на колонку, уравновешенную 10%-ным растворителем B (растворитель A представляет собой 0,1% CF3CO2H в воде; растворитель B представляет собой 0,1% CP3CO2H в CF3CN), с низкой скоростью подачи 1 мл/мин. Колонку промывают 10% B в течение 10 мин, и ЭПО элюируют линейным градиентом B (10 - 70% в течение 60 минут). Фракции, содержащие ЭПО, объединяют. Получают ≈ 40 мкг ЭПО на 120 мкг общего белка; раствор лиофилизируют. Лиофилизированный ЭПО повторно растворяют в 0,1М Tris-HCl при pH 7,5, содержащем 0,15М NaCl, и повторно подвергают ВЭЖХ с обращенной фазой. Фракции, содержащие ЭПО, объединяют и анализируют электрофорезом в SDS-полиакриламидном (10%) геле (Lamelli U.K., Nature). Объединенные фракции ЭПО содержат 15,5 мкг ЭПО в 25 мкг общего протеина. Аналогичным образом проводят очистку ЭПО, получаемого с помощью других видов клеток млекопитающих согласно настоящему изобретению.
Пример 15: Приготовление фармацевтический композиции.
В стерильном, снабженном мешалкой 100-литровом котле с двойными стенками V2A растворяют вспомогательные вещества:
Мочевина - 700,0 г
Хлорид натрия - 70,0 г
Твин 20 (Tween 20) - 7,0 г
Однозамещенный фосфат натрия • 1 H2O - 38,4 г
Двузамещенный фосфат натрия • 2 H2O - 350,0 г
Хлорид кальция • 2 H2O - 8,4 г
Глицин - 105,0 г
L-Лейцин - 140,0 г
L-Изолейцин - 140,0 г
L-Треонин - 35,0 г
L-Глутаминовая кислота - 35,0 г
L-Фенилаланин - 70,0 г
Вода для инъекционных целей - 70,0 г
К 30 л этого раствора вспомогательных веществ прибавляют порцию в 214,3 мл эритропоэтинового сырья с титром 140000 единиц/мл и доводят до окончательного объема 35 л и основательно перемешивают. Остатком раствора вспомогательных веществ промывают фильтрационную систему. Раствор исходной смеси стерильно фильтруют на мембранном фильтре с размером пор 0,2 мкм. Стерильно профильтрованный раствор разливают по 1-миллилитровым склянкам в асептических условиях и сушат вымораживанием в лиофилизационной установке с соблюдением следующих критериев:
Время замораживания: 12 - 14 часов при -40oC.
Основная сушка: температура раствора +10oC, давление 10-1 мбар, время 48 - 60 часов.
Дополнительная сушка: температура раствора +20oC, давление 10-3 мбар, время 4 - 6 часов.
Таким образом получают объемистое, с открытыми порами, сухое вещество для инъекций, сохраняющееся не менее 2 лет в холодильнике и 1 год при комнатной температуре и растворяющееся в течение нескольких часов в 2 мл воды для инъекционных целей без помутнения и свободное от частиц.
Для составленной таким образом композиции показан терапевтический эффект.
Источники информации
1. Jacobson, L. O., Goldwasser, E. Fried, W., and Pizak, L. F., Trans. Assoc. Am. Physicians TO:305-317 (1957).
2. Krantz, S. B. and Jacobson, L. O. Chicago; University of Chicago Press 1970, pp. 29-31.
3. Hammond, D and Winnick, S. Ann. N.Y. Acad. Scl. 230:219-227 (1974).
4. Sherwood, J. B. and Goldwasser, E., Endocrinology 103:866-870 (1978).
5. Fried, W. Blood 40:671-677 (1972).
6. Fisher, J. J. Lab. and Clin. Med. 93:695-699 (1979).
7. Naughton, B. A., Kaplan, S. M., Roy, M., Burdowski, A. J., Gordon, A. S., and Piliero, S. J. Science 196:301-302.
8. Lucarelli, G. P., Howard, D., and Stohlman, F., Jr. J. Clin. Invest 43:2195-2203 (1964).
9. Zanjani, E. D., Poster, J., Burlington, H., Mann, L. I., and Wasserman, L. R. J. Lab. Clin. Med. 89:640-644 (1977).
10. Krantz, S. B. , Gallien-Lartigue, O., and Goldwasser, E. J. Biol Chem. 238:4085-4090 (1963).
11. Dunn, C. D., Jarvis, J. H. and Greenman, J. M. Exp. Hematol. 3:65-78 (1975).
12. Krystal, G. Exp. Hematol. 11:649-660 (1983).
13. Iscove, N.N. and Guilbert, L.J., M.J. Murphy, Jr. (Ed.) New York : Sprinqer-Verlaq, pp. 3 - 7 (1978).
14. Goldwasser, E., ICN UCLA Symposium, Control of Cellular Division and Development, A. R. Liss, Inc., pp. 487 - 494 (1981).
15. Cline, M. J. and Golde, D. W. Nature 277:177-181 (1979).
16. Metcalf, D. , Johnson, G. R. , and Burgess, A. W. Blood 55:138- (1980).
17. Krane, N. Henry Ford Hasp. Med. J. 31:177-181 (1983).
18. Eschbach, J., Madenovic, J., Garcia, J., Wahl, P., and Adamson, J. J. Clin. Invest. 74:434-441 (1984).
19. Anagnostou, A., Barone, J., Vedo, A., and Fried, W. Br. J. Hematol 37:85-91 (1977).
20. Miyake, T., Kung, C., and Goldwasser, E. J. Biol. Chem. 252:5558-5564 (1977).
21. Yanagawa, S., Hirade, K., Ohnota, H., Sasaki, R., Chiba, H., Veda, M., and Goto, M. J. Biol. Chem. 259:2707-2710 (1984).
22. Lawn, R. M., Fritsch, E. F., Parker, R. C., Blake, G., and Maniatis, T. Cell 15:1157- (1978).
23. Sanger, F., Nicklen, S., and Coulson, A. R. Proc. Nat'l. Acad. Sci., U.S.A. 74:5463 - (1977).
24. Zanjanc, E. D. , Ascensao, J.L., McGlave, P.B., Banisadre, M., and Ash, R. C. J. Clin. Invest. 67:1183- (1981).
25. Toole, J.J., Knopf, J.L., Wozney, J.M., Sultzman, L.A. Buecker, J. L. , Pittman, D. D., Kaufman, R. J., Brown, E., Shoemaker, C., Orr, E. C., Amphlett, G. W., Foster, W. B., Coe, M. L., Knutson, G. J., Fass, D. N., and Hewick, R. M. Nature in Press.
26. Goldwasser, E. Blood Suppl. 1, 58, xlii (abstr) (1981).
27. Sue, J. M. and Sytkowdki, A. J. Proc. Nat'l Acad. Sci U.S.A. 80: 3651-3655 (1983).
28. Bersch, N. and Golde, D.W., In Vitro Aspects of Erythropoiesis, M. J. Murphy (Ed.) New York : Springer-VerIag (1978).
29. Cotes, P. M. and Bangham, D. R. Nature 191:1065 - (1961).
30. Goldwasser, E. and Gross, M. Methods in Enzymol 37:109-121 (1975).
31. Nabeshima, Y. -i, Fujii-Kuriyama, Y., Muramatsu, M., and Ogata, K. Nature 308:333-338 (1984).
32. Young, R. A. , Hagencuhle, O. and Schibler, U. Cell 23:451-558 (1981).
33. Medford, R. M., Nguyen, H. T., Destree, A. T., Summers, E. and Nadal-Ginard, B. Cell 38:409-421 (1984).
34. Ziff, E. B. Nature 287:491-499 (1980).
35. Early, P. Cell 20:313-319 (1980).
36. Sytkowski, A. Bio. Biop. Res. Comm. 96:143-149 (1980).
37. Murphy, M. and Miyake, T. Acta. Haematol. Jpn. 46:1380-1396 (1983).
38. Wagh, P. V. and Bahl, O. P. CRC Critical Reviews in Biochemistry 307-377 (1981).
39. Wang, F. F., Kung, C. K. -H. and Goldwasser, E. Fed. Proc. Fed. Am. Soc. Exp. Biol. 42:1872 (abstr) (1983).
40. Lowy, P., Keighley, G. and Borsook, H. Nature 185:102-103 (1960).
41. VanLenten, L. and Ashwell, G. J. Blol. Chem. 247:4633-4640 (1972).
42. Lee-Huang, S. Proc. Nat'l Acad. Sci. U.S.A. 81:2708-2712 (1984).
43. Fyhrquist, F., Rosenlof, K., Gronhagen-Riska, C., Hortling, L. and Tikkanen, I. Nature 308:649-562 (1984).
44. Ohkubo, H., Kageyama, R., Vjihara, M., Hirose, T., Inayama, S., and Nakanishi, S. Proc. Nat'l Acad. Sci. U.S.A. 80:2196-2200 (1983).
45. Suggs, S.V., Wallace, R. B., Hirose, T., Kawashima, E. H. and Itakura, K. Proc. Nat'l. Acad. Sci. U.S.A. - 78:6613-6617 (1981).
46. Woo, S. L. C., Dugaiczyk, A., Tsai, M. -J., Lai, E. C., Catterall, J. F. and O'Malley, B. W. Proc. Nat'l. Acad. Sci. U.S.A. 75:3688- (1978).
47. Melchior, W. B. and VonHippel, P. H. Proc. Nat'l Acad. Soc.U.S.A. 70:298-302 (1973).
48. Orosz, J. M. and Wetmis, J. G. Biopolymers 16:1183-1199 (1977).
49. Anderson, S and Kingston, I. B. Proc. Nat'l Acad. Sci.- U.S.A. 80: 6836-6842 (1983).
50. Ullrich, A. , Coussens, L., Hayflick, J. S. Dull, T. J., Gray, A., Tam, A. W., Lee, J., Yarden, Y., Libermann, T. A., Schlessinger, J., Downward, J., Mayes, E. L. V., Whittle, H., Waterfield, M.D. and Seeburg, P. H. Nature 309: 418-425 (1984).
51. Fisher, J. Proc. Soc. Exptl. Biol. and Med. 173:289-305 (1983).
52. Kozak, M. Nuc. Acid Res. 12:857-872 (1984).
53. Benton, W.D. and Davis, R.W. Science 196:180-182 (1977).
54. Sherwood, J.B. and Goldwasser, E. Blood 54:885-893 (1979).
55. Derman, E., Krauter, K., Walling, L., Weinberger, C., Ray, M., and Darnell, J. T., Cell 23:731- (1981).
56. Gluzman, Y., Cell 23:175-182 (1981).
57. Hewick, R. M., Hunkapiller, M. E., Hood, L. E., and Dreyer, W. J. J. Biol. Chem. 256:7990-7997 (1981).
58. Towbin, H. , Stachelin, T., and Gordon, J., Proc. Nat'l Acad. Sci. 76:4380- (1979).
59. Carnott, P., DeFlandre, C. C. R. Acad. Sci. Paris 143:432- (1960).












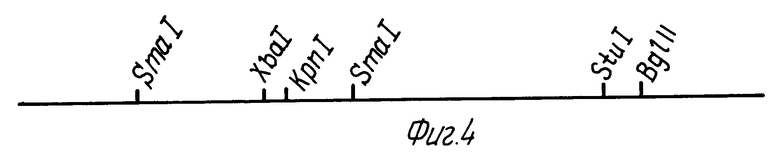
Использование: биотехнология, медико-биологическая промышленность, медицина. Сущность изобретения: клонированы фрагменты геномной ДНК из фетальной печени человека и соответствующие фрагменты кДНК, обеспечивающие высокие уровни экспрессии эритропоэтина в клетках млекопитающих, получена линия клеток СНО продуцент рекомбинантного ЭПО (CHO EPO Cla 44.02-7); в результате экспрессии клонированных фрагментов ДНК в клетках млекопитающих получен О-гликозилированный ЭПО человека с удельной активностью не менее 200000 ед/мг и фармацевтическая композиция, использующая указанный рекомбинантный ЭПО в качестве активного начала. 10 с. и 2 з.п. ф-лы, 12 ил., 11 табл.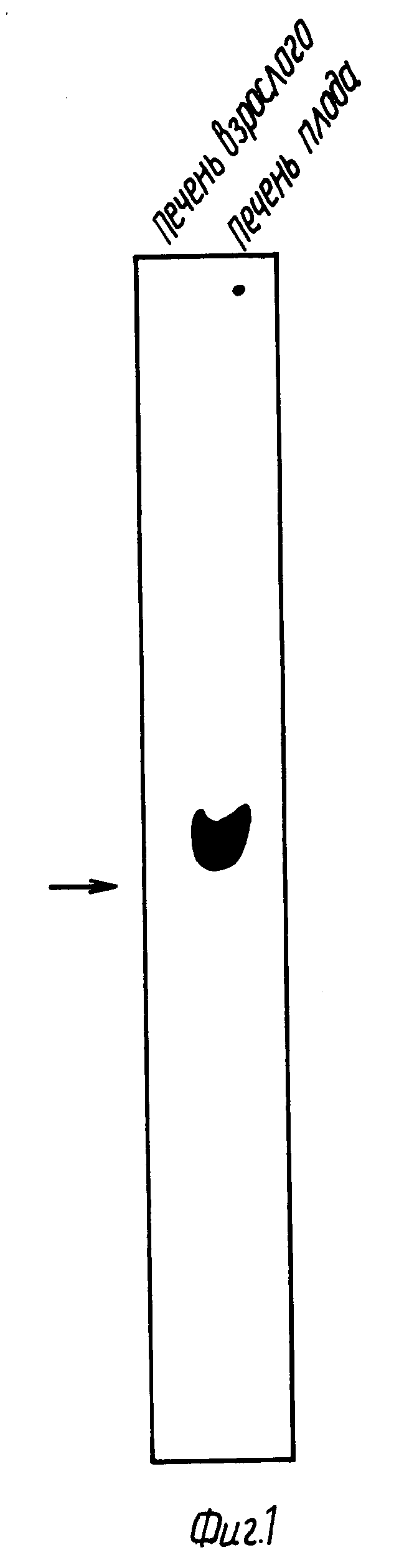
| US, патент N 4377513, A 61 K 37/24, 1983. |
