Изобретение относится к области аналитической химии, а именно к качественному и количественному определению нитропроизводных полициклических ароматических углеводородов (нитро-ПАУ) в сложных смесях и растворах.
Сущность изобретения состоит в том, что анализируемый раствор концентрируют, очищают, отделяют от сопутствующих полициклических ароматических углеводородов (ПАУ) и выделяют фракции, содержащие индивидуальные нитро-ПАУ с последующим измерением низкотемпературной фосфоресценции н-октанового раствора выделенных фракций комбинированным методом добавки и внутреннего стандарта при температуре жидкого азота (77 K) для качественного и количественно определения нитро-ПАУ.
Область применения - предложенный способ может быть использован при контроле продуктов химического синтеза, при анализе загрязнений объектов окружающей среды на содержание канцерогенных и мутагенных нитро-ПАУ.
Цель - упрощение процедуры пробоподготовки и повышение селективности, чувствительности и надежности анализа.
Расшифровка сокращений и терминов
- ppm (parts per million) - частей на миллион;
- ppb (parts per billion) - частей на миллиард;
- sub-ppm (-ppb) - ниже (меньше), чем ppm (ppb) ;
- ВЭЖХ - высокоэффективная жидкостная хроматография; хроматография высокого давления;
- УФ-детектор поглощения света в ультрафиолетовой области;
- EPA (Environment Protection Agency) - агентство по защите окружающей среды (США).
Характеристика аналогов и прототипа.
Качественное и количественное определение нитро-ПАУ важно в связи с тем, что некоторые их них обладают канцерогенной и мутагенной активностью. Нитро-ПАУ широко распространены в окружающей среде: в почве и донных отложениях; они обнаружены в образцах из различных источников, таких как выхлопы транспортных средств, взвеси из окружающего воздуха, экстракты активированного угля, применяемого в копировальной технике и ксерографических тонерах, в конденсате сигаретного дыма.
Аналитические методы для нитро-ПАУ должны быть одновременно высокочувствительными и селективными, во-первых, из-за того, что нитро-ПАУ присутствует в образцах в низких концентрациях (на уровне ppm или даже ppb), причем сами образцы могут иметь вес в несколько сотен миллиграмм; во-вторых, из-за того, что нитро-ПАУ присутствует в образцах наряду с многими другими классами веществ, такими как ароматические карбонитрилы, полициклические кетоны, квиноны и ангидриды, а также ПАУ, которые могут на один - два порядка по концентрации превышать нитро-ПАУ, что зависит от способа сбора образцов и процесса выделения.
Задача различения изомеров нитро-ПАУ чрезвычайно сложна из-за большого количества возможных изомеров и из-за того, что изомеры часто имеют похожие электронные ударные спектры. Проблема идентификации нитро-ПАУ труднее, чем ПАУ.
Из-за того, что нитро-ПАУ присутствуют в следовых количествах на фоне больших количеств накладывающихся веществ, желательно фракционирование образцов, приводящее к относительно чистым фракциям нитро-ПАУ. Для изоляции нитро-ПАУ из сложной матрицы используется метод жидкостной хроматографии (низкого, среднего и высокого давления). Наиболее распространенным вариантом хроматографии, используемой при выделении нитро-ПАУ из сложной матрицы, является нормально-фазовая на силикагеле. Методы различаются преимущественно по элюирующим растворителям и количеством стадий, при использовании ВЭЖХ и хроматографии среднего давления применяют различные способы градиентного элюирования.
Наиболее близким к предлагаемому нами методу является метод, используемый в работе Kamiura T. и др.; близость этого метода к нашему определяется, во-первых, использованием ВЖЭХ для разделения образца и, во-вторых, использованием люминесцентного метода детектирования.
Образцы взвешенных в воздухе частиц экстрагировали в течение ночи дихлорметаном в аппарате Сокслета, концентрировали до 1 мл, очищали на патроне Sep-Pak silica, активированным 2 мл дихлорметана. Элюирование проводили 20 мл дихлорметана. Смыв упаривали досуха под вакуумом, растворяли 0,5 - 1,0 мл 30% дихлорметана в гексане. Раствор вносили на силикагелевую колонку: смыв проводили 40 мл бензол/гексан (40:60) и 60 мл бензола, переводили в дихлорметан, потом в 0,5 мл метанола с 0,5 мл раствора NaHS (концентрация 80 г/мл) для проведения восстановления нитрогрупп до аминов. Смесь грели 1 ч при 70oC. После остывания проводили анализ на хроматографе Shimadzu (ввод составлял 10 - 50 мкл). Для хроматографического разделения использовали обращенно-фазовую колонку Zorbax ODS (250•4,5 мм) с флуоресцентным детектором (возбуждение 392 нм, эмиссия 502 нм). Разделение проводили в градиентном режиме от 60% до 100% метанола в воде. Метод дает хорошие результаты при определении 2-, 3-нитрофлуорантена и 1-, 2-нитропирена. Пределы обнаружения: 2 пг для 1-нитропирена, 4 пг для 2-нитрофлуорена, 40 пг для 1,3-динитропирена [1].
Недостатком этого метода является многоступенчатая процедура пробоподготовки, которая ведет к увеличению вероятности потерь, ограниченность определяемых веществ, связанная с эффективностью дериватизации, на что указывают сами авторы, а также недостаточной чувствительностью для определенных веществ, вызванной использованием ограниченного числа длин волн возбуждения и флуоресценции.
Как известно, квазилинейные спектры люминесценции обладают строгой индивидуальностью для каждого вещества. Предложен способ люминесцентного качественного и количественного определения ПАУ [2, 3] в замороженных растворах нормальных парафинов при температуре жидкого азота (77 K). В работе [4] приводятся данные по спектрам фосфоресценции 22 нитро-ПАУ и пределы их обнаружения. В качестве растворителя использовалась смесь, рекомендованная EPA - эфир/изопропанол (3:1). Данный метод детектирования, обладая преимуществом масс-спектрометра (строгая специфичность), превосходит его по чувствительности, а от метода ВЭЖХ с флуоресцентным детектором отличается исключением стадии дериватизации, большей чувствительностью по целому ряду веществ и возможностью исследовать более широкий спектр веществ.
В заявляемом ниже способе мы предлагаем, используя концентрирующий патрон вместо инфекционной петли и аналитическую колонку с обращенной неподвижной фазой, совместить в одной - три операции: очистки, концентрирования и выделения фракций, содержащих индивидуальные нитро-ПАУ. В случае необходимости накопления вещества операцию можно повторить. Выделенные фракции переводят в октан и анализируются со спектрами фосфоресценции при температуре жидкого азота (77 K). Благодаря специфичности спектров фосфоресценции нитро-ПАУ возможное присутствие во фракции отдельных ПАУ не мешает определению.
Способ определения.
В предлагаемом методе для фракционирования используется высокоэффективный жидкостной хроматограф с инжектором типа Rheodine, с обращенно-фазовой аналитической колонкой с высокой степенью покрытия носителя неподвижной фазой (не ниже 18%) и с УФ-детектором для контроля выхода фракций. Качественный и количественный анализ выделенных фракций осуществляется на люминесцентном спектрометре с низкотемпературной фосфоресцентной приставкой, автоматической разверткой длин волн возбуждения и эмиссии и с двойным монохроматором с разрешением не хуже 0,2 нм (дифракционная решетка с не менее, чем 1200 штрихов/мм).
Перед началом работ необходимо сформировать библиотеку спектров фосфоресценции исследуемых нитро-ПАУ и провести градуировку люминесцентного спектрометра (в приложении приводятся спектральные характеристики и пределы обнаружения некоторого нитро-ПАУ).
1. Подготовка сложных смесей и экстрактов к анализу.
Перед фракционированием экстракт исследуемого образца должен быть переведен в ацетонитрил и, в случае необходимости, профильтрован через мембранный фильтр. Ацетонитрил следует применять потому, что в нем хорошо растворяются нитро-ПАУ. Способ перевода зависит от исходного растворителя, при этом необходимо соблюдать предосторожности, уменьшающие возможность артефактов: разложения нитро-ПАУ или синтез их из ПАУ.
1.1 Процедура фракционирования.
Фракционирование можно проводить в изократическом режиме метанольной подвижной фазой (60 - 70% метанола), при этом осуществляется эффективное разделение нитрофлуоренов и нитропиренов и отделение их от флуорена, пирена и других ПАУ. Если возникает необходимость в разделении более сложных смесей, то более эффективно применять градиентное элюирование, начиная с концентрации метанола 60%. Контроль выхода фракций осуществляют с помощью УФ-детектора на длине волны 255 или 330 нм.
1.1.1 Подготовка хроматографа к работе.
В инжектор хроматографа на место инфекционной петли ставится концентрирующий патрон. Функция концентратора состоит в следующем. Во-первых, он позволяет ввести без потерь большой объем пробы благодаря тому, что удерживающая сила сорбента больше, чем элюирующая у используемого растворителя. Во-вторых, в концентраторе образец сорбируется в виде достаточно узкой полосы на сорбенте, при промывке подвижной фазой концентратора с загруженной пробой происходит предварительное разделение компонент образца на сорбенте концентратора, это позволяет улучшить разделение на колонке по сравнению с инжектированием в инфекционную петлю. В-третьих, концентратор позволяет освободить образец от посторонних примесей и улучшить качество разделения компонента образца. В-четвертых, концентратор, приняв на себя функцию предварительного разделения и предохранения колонки от загрязнения, позволяет отказаться от использования предколонки.
1.1.2. Фракционирование образца.
Исходный ацетонитрильный раствор образца разводят дистиллированной водой в выбранном соотношении и тщательно перемешивают. В случае помутнения раствора или возникновения апполесценции его фильтруют через мембранный фильтр, который затем промывают дважды смесью ацетонитрил - вода, в этом случае получающееся разведение необходимо учесть при расчетах.
Промывают и активируют концентратор, если же предыдущая проба была с большим количеством посторонних примесей, то после нее концентратор можно промыть смесью 30% изопропилового спирта в ацетонитриле.
Перед введением пробы, в режиме загрузки инжектора (Load), производится промывка и активация патрона. В концентратор вводят пробу в растворе, обладающем слабой элюирующей способностью по отношению к исследуемым компонентам, патрон промывают смесью метанол - вода более слабой, чем подвижная фаза. Последнее делается для того, чтобы промыть пробу от слабо удерживаемых на сорбенте примесей. После уравновешивания хроматографической колонки производят ввод пробы (положения инжектора - Inject). Сбор фракций осуществляют в пробирки, ориентируясь по временам удерживания индивидуальных нитро-ПАУ, определенным по хроматограмме стандартной смеси, эта же хроматография может быть использована при оценке концентраций исследуемых веществ, что облегчает правильный подбор условий при фосфоресцентном анализе. Исходная стандартная смесь готовится для ввода в колонку так же, как фракционируемый образец. Повторное хроматографирование стандартной смеси необходимо проводить не более, чем после четырех вводов исследуемых проб. Это тем более важно в связи с возможностью забивания концентратора сильно сорбируемыми компонентами и уменьшения его емкости, т.е. изменения времени удерживания и возникновения проскока при вводе пробы в концентратор.
Собранные фракции содержат индивидуальные нитро-ПАУ, но могут содержать также ПАУ, которые, однако, не мешают определению при люминесцентном анализе. В случае необходимости фракционирование можно повторить, отбирая каждую фракцию в те же пробирки, что и предыдущую. По полученной хроматограмме образца можно оценить концентрации найденных веществ, однако следует отметить, что отсутствие на хроматограмме пика еще не гарантирует отсутствия вещества в этой пробе, поскольку чувствительность УФ-детектора ниже, чем у люминесцентного спектрометра.
1.2. Перевод фракции в н-октан.
Для перевода нитро-ПАУ, содержащихся во фракции, в н-октан предпочтительным следует считать метод переэкстракции, вследствие того, что при этом методе вероятность артефрактов (термическое и фоторазложение нитро-ПАУ, синтез нитро-ПАУ из ПАУ) минимальна. В пробирку с фракцией добавляют от 0,5 до 1,0 мл н-октана и содержимое пробирки перемешивают на ультразвуковой бане до получения эмульсии. После разделения слоев растворителей, отбирают верхний - октановый - слой пастеровской пипеткой. Для наиболее полного извлечения нитро-ПАУ операцию необходимо проводить 3 - 5 раз в зависимости от первоначального объема фракции, при этом достигается 90 - 98% извлечения. Количество необходимых повторов и степень извлечения можно определить предварительно по фракционированию стандартной смеси.
2. Анализ полученных фракций методом низкотемпературной фосфоресценции.
В измерительную кювету вносят равные объемы исследуемого вещества, внутреннего стандарта и растворителя (н-октан), смесь тщательно перемешивают и помещают в сосуд Дьюара с жидким азотом, установленный в держателе измерительного отсека прибора. После замораживания раствора проводят записи спектров фосфоресценции исследуемого вещества и внутреннего стандарта в выбранных для них диапазонах длин волн не менее трех раз.
2.1. Предварительное измерение.
В измерительную кварцевую кювету вносят октановый раствор фракции исследуемой пробы и октан в соотношении 1:2, кювету помещают в сосуд Дьюара с жидким азотом, закрепленный в держателе измерительного отсека прибора, и проводят запись спектра. Запись спектра осуществляется в выбранном диапазоне длин волн от меньшего к большему. Этот диапазон выбирается с таким расчетом, чтобы в него входила длина волны, при которой измеряется уровень фона. Уровень фона измеряется при длине волны меньшей, чем длина волны первого пика спектра фосфоресценции, это обусловливается тем, что при таком выборе уровень фона не зависит от исследуемого вещества. Аналитической считается длина волны первого пика с наименьшей длиной волны даже, если пик не является самым большим. Этот выбор вызван тем, что на высоту первого пика не влияет фон, обусловленный самим веществом. По высоте отклика на длине волны аналитической линии определяют необходимую концентрацию внутреннего стандарта и добавки. Если проведены предварительные оценочные расчеты, то эту операцию можно опустить.
2.2. Измерение с нулевой добавкой.
В измерительную кювету вносят исследуемую пробу, выбранный раствор внутреннего стандарта и октан в соотношении 1:1:1 и проводят запись спектров по п. 2.1.
2.3. Измерение с добавкой.
В измерительную кювету вносят пробу, выбранный раствор внутреннего стандарта и выбранный раствор добавки исследуемого вещества в соотношении 1:1:1 и проводят запись спектров по п. 2.1.
2.4. Определение интенсивности фосфоресценции.
Уровень фона определяют как высоту фона над нулевой линией (темновой ток). Интенсивность фосфоресценции (абсолютную) определяют как высоту отклика для аналитической линии над нулевой линией. Тогда требуемая величина получается вычитанием уровня фона из абсолютной интенсивности фосфоресценции.
2.5. Концентрацию искомого вещества в исследуемом растворе определяют по формуле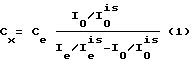 ,
,
где
Cx - концентрация искомого вещества в исследуемом растворе (0 - добавка);
Ce - концентрация эталонного (градуировочного) раствора, используемого в качестве добавки;
I0/I
Ie/I
Замечание: интенсивность фосфоресценции зависит не от концентрации, а от массы веществ. Однако в силу того, что для анализа берутся равные части образца, добавки и внутреннего стандарта, расчет можно проводить, опираясь на концентрацию добавки. Если по каким-либо причинам соотношения эти нарушаются, то в формуле (1) должны использоваться массы вещества.
При расчете окончательного результата (концентрации исследуемого вещества в образце) необходимо учесть промежуточные объемы.
Сравнение предлагаемого способа с прототипом [1] показывает, что если сделать пересчет на концентрации, которые для этого должны быть в вводимой пробе, что, за исключением 1-нитропирена, предлагаемый нами метод оказывается чувствительней (см. таблицу 1); получающийся в результате дериватизации 1-аминопирен имеет сильную флюоресценцию, которая и объясняет столь высокую чувствительность.
Предлагаемый способ иллюстрируется следующими примерами.
Пример 1.
Количественное определение нитро-ПАУ в экстракте сажи выбросов коксохимического завода.
Фильтры с собранной на них сажей выбросов коксохимического завода были проэкстрагированы ацетоном (хч), конечный объем экстракта составил 10 мл. Часть экстракта была использована при определении ПАУ, по результатам определения - бенз[а]пирен - 23,8 мкг/пробу и пирен 166,6 мкг/пробу, - стало очевидно, что для дальнейшего анализа необходимо произвести разведение экстракта, опытным путем было установлено, что экстракт необходимо развести в 100 раз.
200 мкл ацетонового экстракта было упарено при комнатной температуре под вакуумом. Остаток был растворен в 20 мл ацетонитрила (хч, для хроматографии). К 0,6 мл ацетонитрильного экстракта было прибавлено 0,4 мл дистиллированной воды. Полученный раствор был профильтрован через мембранный фильтр для освобождения от появившейся мути, мембранный фильтр был промыт 3•100 мкл раствором ацетонитрила в воде, полученное разведение учитывалось при окончательных расчетах.
Формирование. Для дальнейшей очистки и концентрирования использовали концентрат Элсикон с неподвижной фазой Диасорб C16 (Элсико, Москва), поставленный на место инжекционной петли инжектора высокоэффективного жидкостного хроматографа (Constametric III, LDC, USA, инжектор Rheodine 7125). Для разделения смеси на фракции использовали колонку 4•250 мм с сорбентом Диасорб 130, с неподвижной фазой C16T, с зернением 8 мкм (Элсико, Москва), подвижная фаза - метанол/вода (7:3), скорость протока - 0,4 мл/мин. Для контроля выхода фракций использовали УФ-детектор Spectromonitor III (LDC, USA), контроль осуществлялся при длине волны 255 нм, 0.01 ед. адсорбции на шкалу самописца. Хроматограмма фиксировалась самописцем (Linear1200), скорость ленты 1 мм/мин.
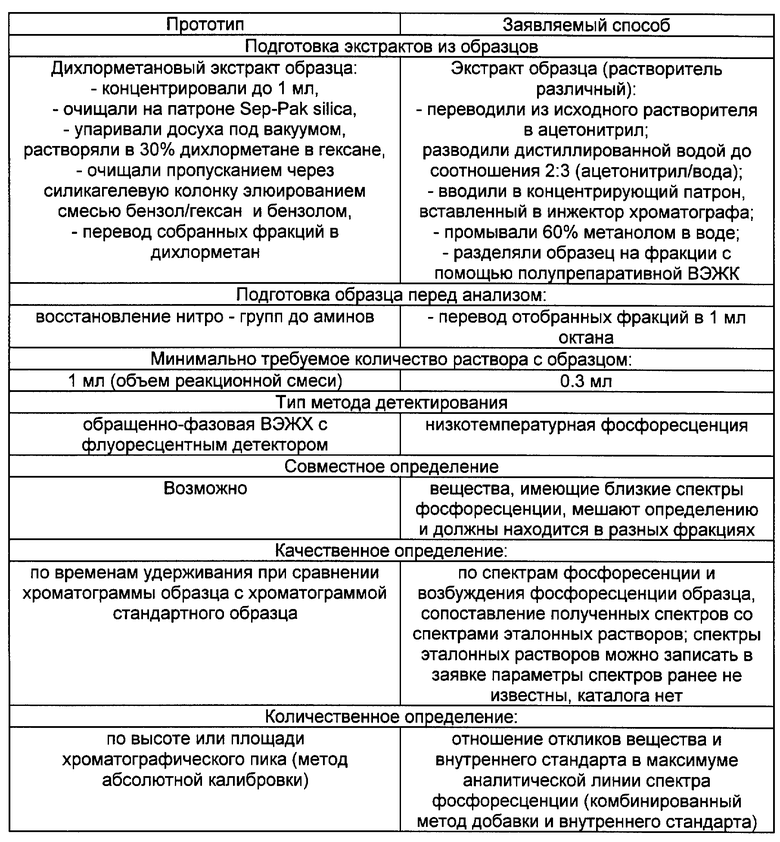
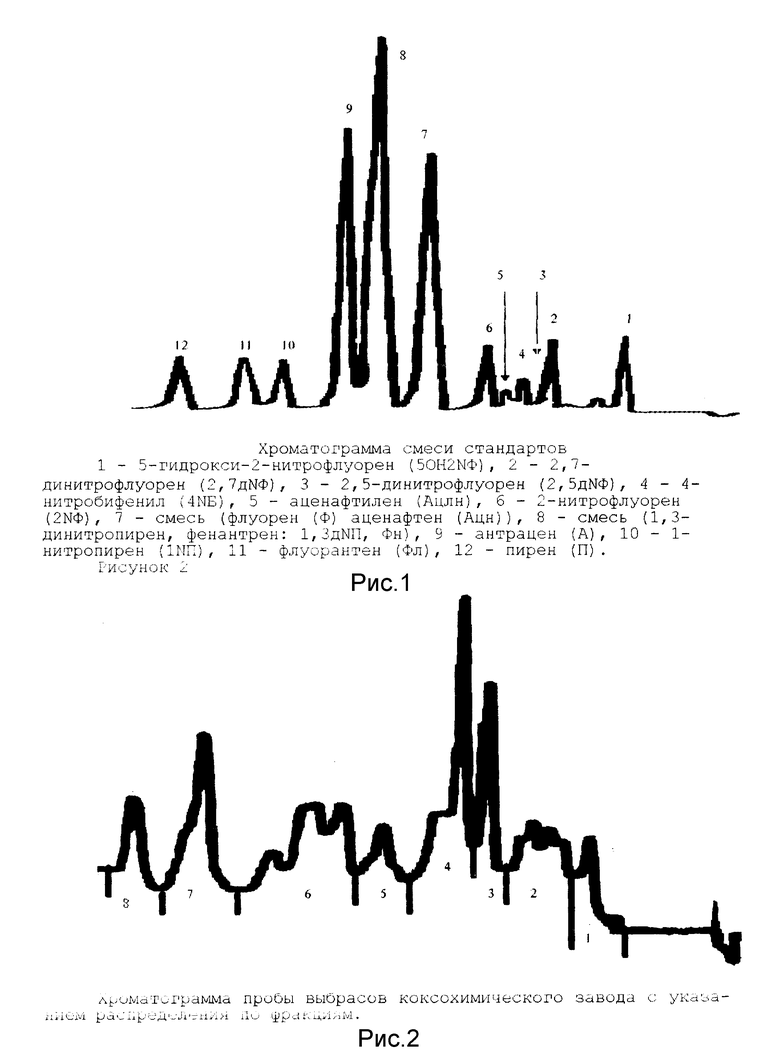
Перед фракционированием пробы для установления ожидаемых времен удерживания интересующих нас фракций провели запись хроматограммы смеси стандартов ПАУ и нитро-ПАУ, приготовленную следующим образом: 0,6 мл смеси стандартов в ацетонитриле разбавлял 0,4 мл дистиллированной воды. В стандартную смесь вводили те ПАУ, которые при хроматографировании выходят вместе с исследуемыми нитро-ПАУ или являются показателями окончания хроматографического разделения. В предварительного промытый и активированный концентратор вводили шприцем 500 мкл приготовленной смеси стандартов, концентратор промывали после этого 350 мкл подвижной фазы с помощью шприца, после чего проводили хроматографирование стандартной смеси. После получения хроматограммы стандартной смеси концентратор опять промывали и активировали. Затем проводили фракционирование пробы, которое заключалось в отборе фракции в мерные пробирки по определенным ранее временам удерживания, условия ввода, очистки и концентрирования пробы такие же, как для стандартной смеси. Хроматограмма смеси стандартов и пробы представлены на рисунках 1,2.
Отобранные фракции переводили в н-октан (хч, для хроматографии) экстракцией 3•0,5 мл н-октана (извлечение 90 - 98%), конечный объем экстракта составил 1,4 мл.
Анализ полученных фракций методом низкотемпературной фосфоросценции проводили на люминесцентном спектрометре М-850 (Hitachi, Japan) с низкотемпературной приставкой.
Измерение с нулевой добавкой: в измерительную кювету вносили по 100 мкл исследуемой фракции выбранного раствора внутреннего стандарта и октана и проводили запись спектров.
Измерение с добавкой: в измерительную кювету вносили по 100 мкл исследуемой фракции выбранного раствора внутреннего стандарта и выбранного раствора добавки исследуемого вещества и проводили запись спектров.
Концентрацию искомого вещества в исследуемом растворе определяли по формуле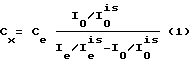 ,
,
где
Cx - концентрация искомого вещества в исследуемом растворе (0 - добавка), г/м;
Ce - концентрация эталонного (градуировочного) раствора, используемого в качестве добавки, г/мл;
I0/I
Ie/I
Значения величин откликов в формуле определяли как среднее по результатам трех измерений. Длины волн аналитических линий приведены в приложении, полученные концентрации приводятся в таблице 2. Окончательный результат получен для массы исследуемого вещества во всей пробе из-за того, что для исследованного образца не была известна масса сажи и объем прокаченного воздуха. Расчет окончательного результата проводили по формуле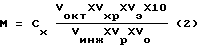 ,
,
где
Cx - концентрация из (1), мкг/мл;
M - масса вещества на пробу, мкг;
V - объемы (мл): октанового раствора - 1,4 (окт), экстракта, взятого на фракционирование - 1,3 (хр), введенный в колонку (инж) - 0.5, взятый из разведенной пробы - 0,6 (р), разведенной пробы - 210 (э), взятый на разбавление - 0,2 (о); 10 - объем пробы (мл).
Обсуждение результатов. На рисунке 1 представлена хроматограмма смеси стандартов ПАУ и нитро-ПАУ, расшифровка хроматограммы дана под рисунком (в скобках даны сокращения). На рисунке 2 приведена хроматограмма образца, полученная при его фракционировании, и приводятся границы отобранных фракций. В таблице 1 приводятся результаты люминесцентного анализа фракций.
Прежде всего надо отметить, что благодаря концентрирующей способности концентратора стало возможным достаточно сильное разбавление образца и за счет этого лучшая очистка образца от неполярных примесей, которые обусловили помутнение раствора при разбавлении и были отделены от образца при фильтрации через мембранный фильтр. При стандартной процедуре введения образца в колонку потребовалось бы предварительное фракционирование на патроне типа Sep-Pak или колонке низкого давления. При этом очистка могла быть менее эффективной, а потери могли быть больше. Косвенным образом это было проведено при фракционировании с помощью тонкослойной хроматографии (правила на другом образце - саже из дизельного двигателя).
Начало и конец отбора фракций определялись таким образом, чтобы во фракцию попадал лишь один представитель нитро-ПАУ. Тот факт, что во фракции могли присутствовать отдельные ПАУ, не мог сказаться на результатах анализа, поскольку их спектрально-люминесцентные характеристики не совпадали с таковыми для нитро-ПАУ из данной фракции.
Следует отметить также отсутствие на хроматограмме фракционирования образца инжекционного пика, который обычно присутствует на хроматограммах реальных проб и связан с наличием в инжектируемом образце посторонних примесей, которые зачастую могут маскировать пики интересующих исследователя веществ. Отсутствие инжекционного пика объясняется, во-первых, вымыванием слабо удерживаемых компонентов образца при введении его в концентратор избытком растворителя (емкость концентратора 0,1 мл, вводится 0,5 мл) и, во-вторых, промывкой образца в концентраторе подвижной фазой. Состав растворителя подбирается таким образом, чтобы обеспечить удерживание в концентраторе интересующих веществ.
При сопоставлении хроматограммы образца (рисунок 2) и результатов люминесцентного анализа следует отметить следующее. Во-первых, во фракции 6 были обнаружены лишь следы 1,3-динитропирена, несмотря на значительную высоту хроматографического пика. Во-вторых, во фракции 7 отсутствует пик, соответствующий 1-нитропирену, но люминесцентный анализ показал его присутствие в этой фракции. Таким образом можно утверждать, что без люминесцентного анализа, опираясь лишь на результаты хроматографического определения, можно было сделать ошибочные выводы относительно 1-нитропирена и невозможно было высказать никаких утверждений о 1,3-динитропирене. В-третьих, при данном уровне концентрации 2,7-динитрофлуорена, возможный уровень концентрации 2,5-изомера находился ниже предела измерения его люминесцентным методом, потому что, согласно литературным данным, выход его приблизительно в три раза ниже, чем 2,7-динитрофлуорена. Таким образом, хотя в данном варианте хроматографирования и отбора фракции эти два изомера попадали в одну фракцию, присутствие 2,5-изомера не могло сказаться на результате анализа.
Сопоставить полученные нами данные по этому типу выбросов с другими данными невозможно из-за отсутствия таких данных в литературе. Однако можно оценить соотношение между разными нитро-ПАУ с помощью данных по сажам различных двигателей. Так, концентрация 1-нитропирена может быть сравнима с концентрацией пирена либо 6 - 10 раз ниже, соответственно 1,3-динитропирена - на порядок меньше, чем 1-нитропирена, наши данные укладываются в эти пределы. Для нитрофлуоренов имеются данные о том, что их концентрации могут превышать концентрации нитропиренов, а соотношения между моно- и динитропроизводными могут быть самые различные. Так что и в этом случае данные, полученные нами, не противоречат литературным данным.
Таким образом, предлагаемый нами метод дает результаты, согласующиеся с результатами, известными из литературы.
Пример 2.
Определение нитро-ПАУ в пробах воздуха из района, загрязненного выбросами промышленных предприятий и автотранспорта.
Проба взвеси из воздуха, отобранная на фильтр, была проэкстрагирована на ультразвуковой бане 3•20 мин 20 мл бензола (хч). Экстракт был упарен до 10 мл. 3 мл было отобрано для анализа на ПАУ. Оставшаяся часть была упарена досуха при комнатной температуре на роторном испарителе под вакуумом, после чего растворена в 1 мл ацетонитрила (хч, для хроматографии). Далее с образцом поступали так же, как в примере 1, фракционирование и люминесцентный анализ проводили на том же оборудовании, что и в примере 1. Распределение образца по фракциям также совпадает. Результаты анализа приведены в таблице 3.
Обсуждение результатов. По данным анализа на ПАУ в пробе было найдено бенз[а] парена - 20 нг/м3, пирена - 80 нг/м3. Из таблицы видно, что для нитропиренов соотношения в целом сохраняются такие же, как в примере 1. В литературе есть сведения о близких уровнях концентраций 1-нитропирена и 2-нитофлуорена, но концентрация первого может быть и выше, чем второго, в нашем случае концентрации сравнимы. Хотя в пробе наблюдается повышенное содержание 2,7-динитрофлуорена, остальные данные согласуются как между собой, так и с данными литературы. По литературным данным загрязнения воздушной среды нитро-ПАУ намного ниже, чем в выбросах двигателей автотранспорта.
Выводы.
Заявляемый способ определения нитро-ПАУ позволяет анализировать нитро-ПАУ в пробах различного происхождения и непосредственно сопоставлять с концентрациями ПАУ, полученными методом квазилинейчатых спектров. Процедура пробоподготовки менее трудоемкая, чем при использовании способа прототипа. В частности, предложенный способ совмещает в одной операции несколько, исключает стадию дериватизации и благодаря этому уменьшает возможные потери. Анализ полученных в результате пробоподготовки фракций, благодаря специфичности спектров фосфоресценции, отличается селективностью и более высокой чувствительностью по сравнению с прототипом. Увеличивается также и число веществ, которые можно проанализировать.
Литература
[1] Kamiura T., Kawaraya T., et al., Determination of 3-nitrofluorantene and 1-nitropyrene in suspended particulate matter by liquid chromatograpgy with fluorescence detection., 1991, Analytica Chimica Acta, 254, 1-2, 27-31. Прототип.
[2] А.Я. Хесина, И.А. Хитрово, Б.З.Геворкян, Возможность количественного определения ПАУ в загрязнениях окружающей человека среды на основе квазилинейчатых спектров люминесценции и возбуждения. 1983, Журнал прикладной спектроскопии, т. XXXVIII, вып. 6, стр. 928-934.
[3] Т.А. Алексеева, Т.А. Теплицкая, Спектрофлуориметрические методы анализа ароматических углеводородов в природных и техногенных средах, Ленинград 1981 г.
[4] O. S. Wolfbeis, W. Posch, G. Gabitz, P.Tritthart, Anal.Chem.Acta, 1983, 147, 405 - 410.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3-OR-ДИ-(2-ХЛОРЭТИЛ)АМИНОСОДЕРЖАЩИЕ αАЦИЛ(ГИДР)ОКСИСТЕРОИДЫ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1998 |
|
RU2139292C1 |
| СПОСОБ ФОРМИРОВАНИЯ КУЛЬТИ БРОНХА | 1994 |
|
RU2076642C1 |
| СРЕДСТВО, ПОДАВЛЯЮЩЕЕ РОСТ ОПУХОЛЕВЫХ КЛЕТОК | 1996 |
|
RU2095066C1 |
| СПОСОБ ЛЕЧЕНИЯ НЕОПЕРАБЕЛЬНЫХ БОЛЬНЫХ ПЛОСКОКЛЕТОЧНЫМ РАКОМ ЛЕГКОГО | 2000 |
|
RU2185206C2 |
| Способ определения состава полициклических ароматических углеводородов | 1986 |
|
SU1425528A1 |
| СПОСОБ ПОЛУЧЕНИЯ N (2-ХЛОРЭТИЛ)- N НИТРОЗО-L-ГОМОЦИТРУЛЛИНА ИЛИ N (2-ХЛОРЭТИЛ)- N НИТРОЗО-2-ГОМОЦИТРУЛЛИНА ДЛЯ ИНЪЕКЦИЙ | 1995 |
|
RU2111742C1 |
| СПОСОБ ИЗМЕРЕНИЯ КОЛИЧЕСТВА ПОЛИСОРБАТА-80 С ПРИМЕНЕНИЕМ ЩЕЛОЧНОГО ГИДРОЛИЗА ОБРАЗЦА С ПОСЛЕДУЮЩЕЙ ВЭЖХ | 2017 |
|
RU2670965C9 |
| СПОСОБ ИДЕНТИФИКАЦИИ НАРКОТИЧЕСКИХ И ПСИХОАКТИВНЫХ ВЕЩЕСТВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ | 2009 |
|
RU2390771C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФРАКЦИОННОГО СОСТАВА СУЛЬФИРОВАННОГО ФТАЛОЦИАНИНА АЛЮМИНИЯ | 2019 |
|
RU2720799C1 |
| СПОСОБ ВЫЯВЛЕНИЯ И ОПРЕДЕЛЕНИЯ ПРОИСХОЖДЕНИЯ НЕИЗВЕСТНЫХ ВЕЩЕСТВ В СПИРТНЫХ НАПИТКАХ | 2009 |
|
RU2392616C1 |

Изобретение относится к аналитической химии, а именно к качественному и количественному определению нитропроизводных полициклических ароматических углеводородов (нитро-ПАУ) в сложных смесях и растворах. Технический результат - упрощение пробоподготовки, повышение селективности, чувствительности и надежности анализа. Сущность изобретения состоит в том, что первоначальный экстракт из исходного растворителя переводят в ацетонитрил, разбавляют дистиллированной водой до получения 50 - 63 %-ного раствора ацетонитрила в воде, концентрируют и очищают от легких и полярных примесей с помощью концентрирующего патрона, вставленного в инжектор хроматографа на место инфекционной петли, разделяют пробу на фракции с помощью ВЭЖХ и после перевода отобранных фракций в октан, определяют в них индивидуальные нитро-ПАУ способом низкотемпературной фосфоресценции по спектрам фосфоресценции, используя комбинированный метод добавки и внутреннего стандарта, а расчет нитро-ПАУ проводят по общепринятой формуле. 2 ил., 3 табл.
Способ определения нитро-ПАУ в экстрактах проб исследуемых образцов путем подготовки пробы для анализа с последующим качественным и количественным определением и расчетом, отличающийся тем, что первоначальный экстракт из исходного растворителя переводят в ацетонитрил, разбавляют дистиллированной водой до получения 55 - 63%-ного раствора ацетонитрила в воде, концентрируют и очищают от легких и полярных примесей с помощью концентрирующего патрона, вставленного в инжектор хроматографа на место инжекционной петли, разделяют пробу на фракции с помощью ВЭЖХ и после перевода отобранных фракций в октан определяют в них индивидуальные нитро-ПАУ способом низкотемпературной фосфоресценции по спектрам фосфоресценции, используя комбинированный метод добавки и внутреннего стандарта, а расчет нитро-ПАУ проводят по общепринятой формуле.
| Kamiura T | |||
| et | |||
| al., Determination of 3-nitrofluorantene and 1-nitropyrene in susprnded particulate matter by liquid chromatograpgy with fluorescence delection, 1991, Analytica Chimica Acta, 254, 1-2, 27-31 | |||
| Хесина А.Я | |||
| и др | |||
| Возможность количественного определения ПАУ в загрязнениях окружающей человека среды на основе квазилинейчатых спектров люминесценции и возбуждения | |||
| Гребенчатая передача | 1916 |
|
SU1983A1 |
| XXXVIII, вып | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Способ определения индивидуальных полициклических ароматических углеводородов в техническом углероде | 1983 |
|
SU1254359A1 |
| HUS 5425916 A, 1995. | |||

