Область техники
Данное изобретение в целом относится к факторам свертывания крови, а более конкретно к экспрессии белков, обладающих биологической активностью по отношению к свертыванию крови.
Уровень техники
Свертывание крови представляет собой процесс, состоящий из сложных взаимодействий различных компонентов крови или факторов, что в результате последовательных реакций приводит к образованию фибринового сгустка. Как правило, компоненты крови, принимающие участие в процессе, называемом "каскад" свертывания, представляют собой проферменты или зимогены, которые представляют собой ферментативно неактивные белки, превращаемые в протеолитические ферменты действием активатора, который в свою очередь является активированным фактором свертывания. Факторы свертывания, подвергшиеся такому превращению, как правило, называют "активированными факторами" и обозначаются добавлением символа "a" (например, VIIa).
Существуют две отдельные системы, инициирующие свертывание крови и вследствие этого участвующие в нормальном гемостазе. Эти системы называют внутренней и внешней системами свертывания крови. Внутренняя система относится к тем реакциям, которые приводят к образованию тромбина с участием тех факторов, которые присутствуют только в плазме крови. Промежуточной стадией внутренней системы свертывания является активирование фактора IX в фактор IXa, и эта реакция катализируется фактором XIa и ионами кальция. Затем фактор IXa принимает участие в активации фактора X в присутствии фактора VIIIa, фосфолипида и ионов кальция. Внешняя система свертывания включает в себя факторы плазмы крови, а также компоненты, присутствующие в тканевых экстрактах. Один из указанных выше проферментов, фактор VII, участвует во внешней системе свертывания крови, превращая (после его активирования в VIIa) фактор X и Xa в присутствии тканевого фактора и ионов кальция. Фактор Xa, в свою очередь, превращает затем протромбин в тромбин в присутствии фактора Va, ионов кальция и фосфолипида. Так как активация фактора X в фактор Xa представляет собой стадию, входящую как во внешнюю, так и во внутреннюю систему свертывания, то фактор VIIa можно использовать для лечения пациентов, страдающих дефицитом ингибиторов фактора VIII (Томас, патент США N 4382083). Есть также некоторые основания предполагать, что фактор VIIa может принимать участие во внутренней системе свертывания, играя роль также в активации фактора IX (Зюр и Немерсон, J. Biol chem 253: 2203-2209, 1978).
Экспериментальный анализ показывает, что человеческий фактор VII представляет собой одноцепочечный гликопротеин с молекулярным весом около 50000 дальтон. В этой форме фактор, циркулирующий в крови, является неактивным зимогеном. Активация фактора VII в VIIa может катализироваться несколькими различными протеазами плазмы, такими, как фактор XIIa. Активация фактора VII приводит к образованию двух полипептидных цепочек, а именно тяжелой цепочки (с молекулярной массой 28000) и цепочки меньшего размера (с молекулярной массой 17000), которые связаны друг с другом по меньшей мере одной дисульфидной связью. Фактор VII может также активироваться в VIIa in vitro, например по методике, описанной Томасом в патенте США N 4456591.
Фактор IX циркулирует в крови в форме одноцепочечного предшественника фермента и имеет молекулярный вес 57000, превращаясь в активную сериновую протеазу (фактор IXa) под действием фактора XIa в присутствии фактора VIII. Фактор IXa состоит из маленькой и большой цепей молекулярного веса соответственно 16000 и 29000.
Обычное лечение пациентов, страдающих нарушениями свертывания (например, дефицитом фактора VIII и IX), как правило, включает в себя замещающую терапию с использованием криопреципитата или других фракций плазмы человека, обогащенных нужным фактором. Эти препараты могут быть получены из сборной человеческой плазмы, однако получение криопреципитатов требует использования относительно большого количества плазмы человека в качестве сырья.
Фактор VII терапевтически используется при лечении индивидуумов, страдающих дефицитом фактора VII, а также групп людей, страдающих дефицитом фактора VIII и IX и пациентов с болезнью Виллебранда. Если говорить подробнее, то пациенты, подвергающиеся терапевтическому лечению введением факторов VIII и IX, часто вырабатывают антитела на эти белки. Длительное лечение становится крайне сложным из-за присутствия этих антител. Пациентов, у которых наблюдается это осложнение, обычно лечат активированным протромбиновым комплексом, который, как известно, состоит из смеси активных и неактивных свертывающих ферментов, включающих фактор VIIa. Кроме того, последние исследования указывают на то, что небольшие количества (40-50 мкг) введенного фактора VIIa эффективны при подавлении серьезных случайных кровотечений у пациентов, страдающих дефицитом фактора VIII и имеющих в крови высокую концентрацию антител (Гендер и Кисиел, J. Clin Invest, 71: 1836-1841, 1983).
Так как при получении криопреципитатов используются различные источники плазмы, то трудно проводить испытания препаратов на отсутствие в них вирусных загрязнений. Например, практически у всех пациентов, принимающих криопреципитат, обнаруживается положительная реакция при испытании на гепатит. Последние сообщения указывают на то, что некоторые страдающие гемофилией пациенты, принимающие преципитат, имеют развивающийся приобретенный синдром иммунодефицита (СПИД). Кроме того, очистка больших количеств этих факторов крайне сложна и дорога.
Следовательно, в данной области существует потребность в разработке способа получения относительно больших количеств очищенных препаратов фактора VIIa и фактора IX. Данное изобретение удовлетворяет эту потребность за счет использования технологии рекомбинантной ДНК, которая с успехом устраняет проблему вирусного загрязнения и одновременно представляет концентрированный и гомогенный источник активного фактора VIIa для лечения пациентов, страдающих дефицитом фактора VIII и фактора IX и болезнью Виллебранда, а также дает источник очищенного фактора IX для использования в замещающей терапии.
Описание изобретения
Говоря коротко, в данном изобретении описывается ДНК-конструкиця, содержащая нуклеотидную последовательность, по крайней мере частично кодирующую фактор VII. Нуклеотидная последовательность включает в себя первую нуклеотидную последовательность, кодирующую домен связывания кальция, соединенную со второй нуклеотидной последовательностью, расположенную за первой последовательностью. Вторая нуклеотидная последовательность кодирует каталитический участок, обеспечивающий активность фактора VIIa как сериновой протеазы. Соединенные последовательности кодируют белок, который после активации обладает практически такой же биологической активностью по отношению к свертыванию крови, как фактор VIIa. Первая нуклеотидная последовательность может быть практически такой же, как ген, кодирующий фактор VII, фактор IX, фактор X, протеин C, протромбин или протеин S. Кроме того, первая нуклеотидная последовательность может также кодировать главный пептид, кодируемый соответствующим геном.
В частности, первая нуклеотидная последовательность может быть получена из геномного клона или клона к-ДНК или фактора VII, и может кодировать главный пептид и аминоконцевую последовательность фактора VII. Первая нуклеотидная последовательность может также включать в себя двунитевый олигонуклеотид. Особенно предпочтительна такая первая нуклеотидная последовательность, которая кодирует лидерный белок и аминоконцевую последовательность фактора IX.
Кроме того, в данном изобретении описываются рекомбинантные плазмиды, способные интегрироваться в ДНК клетки-хозяина млекопитающего. Одна из плазмид включает в себя промотор, расположенный после набора расщепляющих РНК центров, и за центрами расщепления РНК расположена нуклеотидная последовательность, по крайней мере частично кодирующая фактор VII. Нуклеотидная последовательность включает в себя первую нуклеотидную последовательность, кодирующую домен связывания кальция, соединенную со второй нуклеотидной последовательностью, расположенной за первой последовательностью. Вторая нуклеотидная последовательность кодирует участок, отвечающий за активность фактора VIIa как сериновой протеазы. Соединенные последовательности кодируют блок, который после активации обладает практически такой же биологической активностью по отношению к свертыванию крови, как фактор VIIa. После нуклеотидной последовательности расположен сигнал полиадениляции.
Аналогично указанной выше рекомбинантной плазмиде в данном изобретении описывается также вторая плазмида, включающая в себя промотор, за которым следует набор сайтов расщепления РНК, а за сайтами расщепления РНК идет нуклеотидная последовательность, кодирующая, по крайней мере, частично, фактор IX. Нуклеотидная последовательность включает в себя первую нуклеотидную последовательность, кодирующую домен связывания кальция, соединенную со второй нуклеотидной последовательностью, расположенной за первой последовательностью. Вторая нуклеотидная последовательность кодирует участок, ответственный за активность фактора IX как серийной протеазы. Соединенные последовательности кодируют блок, обладающий практически такой же биологической активностью по отношению к свертыванию крови, как фактор IX. За нуклеотидной последовательностью следует сигнал полиадениляции.
Третий аспект данного изобретения касается клеток млекопитающих, стабильно трансформированных таким образом, что они вырабатывают белок, обладающий после активации практически такой же биологической активностью, как фактор VIIa. Клетки трансформируют с помощью ДНК-конструкции, содержащей нуклеотидную последовательность, по крайней мере частично кодирующую фактор VII. Нуклеотидная последовательность включает в себя первую нуклеотидную последовательность, кодирующую домен связывания кальция и соединенную со второй нуклеотидной последовательностью, расположенной после первой последовательности. Вторая нуклеотидная последовательность кодирует участок, ответственный за активность фактора VIIa как сериновой протеазы. Соединенные последовательности кодируют белок, который, после активации, проявляет практически такую же биологическую активность по отношению к свертыванию крови, как фактор VIIa.
В данном изобретении описываются также клетки млекопитающих, стабильно трансформированные таким образом, чтобы вырабатывать белок, имеющий практически такую же биологическую активность, как фактор IX. Клетки трансформируют с помощью ДНК-конструкции, содержащей нуклеотидную последовательность, по крайней мере частично кодирующую фактор IX. Нуклеотидная последовательность включает в себя первую нуклеотидную последовательность, кодирующую участок связывания кальция, и соединена со второй нуклеотидной последовательностью, расположенной за первой. Вторая нуклеотидная последовательность кодирует участок, ответственный за каталитическую активность фактора IX как сериновой протеазы. Соединенные последовательности кодируют белок, имеющий практически такую же биологическую активность по отношению к свертыванию крови, как фактор IX.
В данном изобретении предлагается также способ получения белка, обладающего кровосвертывающей активностью фактора VIIa, путем создания клетки-хозяина млекопитающего, содержащей ДНК-конструкцию, содержащую в свою очередь нуклеотидную последовательность, по крайней мере частично кодирующую фактор VII. Нуклеотидная последовательность включает в себя первую нуклеотидную последовательность, кодирующую домен связывания кальция и соединенную с расположенной за ней второй последовательностью. Вторая последовательность кодирует участок, ответственный за каталитическую активность фактора VIIa как сериновой протеазы. Соединенные последовательности кодируют белок, который, после активации, обладает практически такой же биологической кровосвертывающей активностью, как фактор VIIa. Последовательные операции состоят в том, что клетку-хозяина млекопитающего выращивают в подходящей среде и выделяют белковый продукт, кодируемый ДНК-конструкцией и вырабатываемый клеткой-хозяином млекопитающего. Белковый продукт затем активируют с получением фактора VIIa.
Еще одним аспектом данного изобретения является способ получения белка, обладающего биологической кровосвертывающей активностью фактора IX. Способ включает в себя создание клетки-хозяина млекопитающего, содержащей ДНК-конструкцию, включающую в себя нуклеотидную последовательность, кодирующую, по крайней мере, частично, фактор IX. Нуклеотидная последовательность состоит из первой нуклеотидной последовательности, кодирующей участок связывания кальция, соединенной со второй нуклеотидной последовательностью, расположенной за первой. Вторая нуклеотидная последовательность кодирует участок, обеспечивающий каталитическую активность фактора IX как сериновой протеазы. Соединенные последовательности кодируют белок, обладающий практически такой же биологической кровосвертывающей активностью, как фактор IX. Клетку-хозяина млекопитающего сначала выращивают в подходящей среде и затем выделяют белковый продукт, закодированный в клетке-хозяине. Описываются также белковые продукты, получаемые описанными выше способами.
В данном изобретении описывается также ДНК-конструкция, содержащая последовательность ДНК, кодирующую фактор VII. В предпочтительном варианте последовательность ДНК включает в себя последовательность к-ДНК по рисунку 1b, от по (пары оснований) 36 до по 1433. В другом предпочтительном варианте последовательность ДНК содержит последовательность к-ДНК по рисунку 1b от по 36 до по 99, расположенную перед последовательностью, соответствующей участку от по 166 до по 1433. Описываются также рекомбинантные плазмы, способные интегрироваться в ДНК клетки-хозяина млекопитающего, включающие в себя только что описанные последовательности ДНК.
Описаны также клетки млекопитающих, стабильно трансформированные с помощью рекомбинантной плазмиды, содержащей последовательность ДНК, кодирующую фактор VII. В предпочтительных вариантах последовательность ДНК содержит последовательность к-ДНК по рисунку 1b от по 36 до по 1433 или последовательность к-ДНК по рисунку 1b от по 36 до по 99, расположенную перед последовательностью от по 166 до по 1433.
Описан также способ получения белка, обладающего биологической кровосвертывающей активностью фактора VIIa путем стабилизации клетки-хозяина млекопитающего, содержащей ДНК-конструкцию, описанную выше. Клетку-хозяина млекопитающего выращивают в подходящей среде и выделяют белковый продукт, кодируемый ДНК-конструкцией. Белковый продукт активируют для получения фактора VIIa.
Другие аспекты данного изобретения поясняются с помощью представленного ниже подробного описания и прилагаемых рисунков.
Краткое описание рисунков
Рисунок 1a иллюстрирует частичную последовательность к-ДНК фактора VII, получаемую соединением к-ДНК клонов λ VII2115 и λ VII1923.
Рисунок 1b иллюстрирует к-ДНК последовательность фактора VII λ VII2463. Стрелки обозначают размер делеции в последовательности λ VII565. Цифры над последовательностью обозначают аминокислоты. Числа, расположенные ниже, обозначают нуклеотиды.
На рисунке 2a иллюстрируются аминоконцевые участки нескольких факторов свертывания.
На рисунке 2b представлено сравнение аминокислотной последовательности фактора VII, полученной из деструктивного анализа белка, с последовательностью, кодируемой к-ДНК.
На рисунке 3 представлено соединение основных последовательностей фактора IX в последовательность, кодирующую домен связывания кальция.
На рисунке 4 показано соединение гибридных подобных фактору IX последовательностей в частичную к-ДНК фактора VII с получением структурной кодирующей последовательности.
На рисунке 5 показано конструирование плазмиды, содержащей последовательность, кодирующую слитый белок фактора IX/фактор VII.
На рисунке 6 представлен экспрессивный вектор F IX/VII/pD2. Использованы следующие обозначения: Ad2 MLP - основной промотор из аденовируса 2; L 1-3 - состоящая из трех частей лидерная последовательность аденовируса 2; 5' SS - сайт 5' расщепления; 3'SS - сайт 3' расщепления; pA - сигнал полиадениляции из SV 40.
На рисунке 7 показана нуклеотидная последовательность слитой к-ДНК фактор IX/фактор VII.
На рисунке 8 показан экспрессивный вектор pM7135. Использованы следующие обозначения: E - усилитель SV 40; ori - единицы структуры 0-1 Ad5; pA - сигнал ранней полиадениляции из SV 40;
Δ - участок делеции "отравляющей" последовательности pBR322; другие обозначения такие же, как в рисунке 6.
На рисунке 9 показано субклонирование 2463 по к-ДНК фактора VII.
На рисунке 10 показано субклонирование 565 по к-ДНК фактора VII.
На рисунке 11 показано соединение 5'-окончания pVII565 и 3'-участка pVII2463 в p C18 с образованием pVII2397.
На рисунке 12 иллюстрируется конструирование экспрессионных плазмид F VII/2463/ /pDX и FVII/565+2463//pDX. pA обозначает сигнал полиадениляции из SV 40 в ранней или поздней ориентации, как описано в примере 9. Другие обозначения такие, как описано в рисунке 8.
Лучший вариант осуществления изобретения
Перед рассмотрением данного изобретения может быть полезно ознакомиться с обозначениями некоторых терминов, используемыми ниже.
Комплементарная ДНК или к-ДНК: молекула или последовательность ДНК, ферментативно синтезированная из последовательностей, присутствующих в матрице м-РНК.
ДНК-Конструкция: молекула ДНК или клон таких молекул, как однонитевых, так и двунитевых, которые в частичной форме могут быть выделены из природного гена или модифицированные и содержащие сегменты ДНК, скомбинированные и соединенные таким способом, который отсутствует в природе.
Плазмида или вектор: ДНК-конструкция, содержащая генетическую информацию, которая вызывает репликацию при введении в клетку-хозяина. Плазмида, как правило, содержит по крайней мере одну генную последовательность, экспрессия которой происходит в клетке-хозяине, а также последовательности, облегчающие экспрессию такого гена, включая промоторы и инициирующие транскрипцию сайты. Они могут быть линейными или замкнутыми в кольцо молекулами.
Соединенный: имеются в виду последовательности ДНК, соединяемые при присоединении 5'- и 3'-окончаний одной последовательности соответственно к 33- и 5'-окончаниям присоединяемой последовательности через фосфодиэфирные связи. Соединение может достигаться такими способами, как связывание липких концов, синтезом соединенных последовательностей клонированием к-ДНК или удалением или введением последовательностей способом прямого мутагенеза.
Лидерный пептид: аминокислотная последовательность, имеющаяся на N-окончаниях некоторых белков и, как правило, отщепляемая от белка в ходе последующей переработки и секретирования. Лидерные пептиды включают в себя последовательности, направляющие белок в клетке на секретирование. Используемое здесь выражение "Лидерный пептид" может также обозначать часть природного лидерного пептида.
Домен: трехмерная, самоорганизующаяся структура из особых аминокислот в белковой молекуле, содержащая все или часть структурных элементов, необходимых для обеспечения определенной биологической активности белка.
Биологическая активность: функция или набор функций, осуществляемых молекулой в биологическом процессе (например, в организме или in vitro). Биологические активности белков могут подразделяться на каталитическую и эффекторную активности. Каталитические активности факторов свертывания, как правило, включают в себя активацию других факторов путем специфического расщепления предшественников. Эффекторные активности включают в себя специфическое связывание биологически активных молекул кальцием или другими малыми молекулами, макромолекулами, такими как белки, или клетками. Эффекторная активность часто необходима или существенна для каталитической активности в биологической среде. Каталитическая и эффекторная активности могут быть в некоторых случаях обеспечиваться одним и тем же доменом белка.
Биологическая активность фактора VIIa характеризуется участием во внешней системе свертывания крови. Фактор VIIa активирует фактор X с образованием фактора Xa, который в свою очередь превращает протромбин в тромбин, инициируя образование фибринового сгустка. Так как активация фактора X обычно происходит как во внешней, так и во внутренней системе свертывания крови, то фактор VIIa может быть использован для лечения пациентов с сильной недостаточностью активных факторов IX и VII или больных болезнью Виллебранда.
Биологическая активность фактора IX характеризуется участием в свертывании крови во внутренней системе свертывания. Фактор IXa активируется в фактор IXa фактором XIa. Затем фактор IXa активирует фактор X в фактор Xa в присутствии фактора VIIIa, фосфолипида и ионов кальция. Затем фактор Xa участвует в превращении протромбина в тромбин, инициируя образование фибринового сгустка.
Как отмечалось выше, выделение фактора VII из человеческой плазмы занимает много времени и дорого стоит, так как фактор является редким белком, присутствующим в концентрации лишь около 300 мкг на литр крови. Кроме того, его тяжело отделять от протромбина, фактора IX и фактора X и он чувствителен к протеолитическому действию в ходе очистки (Кисиел и Мак-Маллен). Хотя одноцепочечный человеческий фактор VII и был очищен до гомогенного состояния (Кисиел и Мак-Маллен), однако опубликованные способы очистки ограничены, как правило, низким выходом и/или загрязнением другими факторами свертывания.
Факторы VII и IX вырабатываются в печени и требуют витамина K для своего биосинтеза. Витамин K необходим для образования особых остатков гамма-карбоксиглутаминовой кислоты в факторах. Эти необычные аминокислотные остатки, образующиеся пост-трансляционной модификацией, связываются с ионами кальция и ответственны за взаимодействие белка с фосфолипидными носителями. Кроме того, каждый из факторов VII и IX содержит один остаток бета-оксиаспарагиновой кислоты, который также образуется после трансляции белков. Однако роль этой аминокислотной последовательности неизвестна.
Учитывая тот факт, что активности факторов VII и IX зависят от пост-трансляционных модификаций, включающих в себя гамма-карбоксилирование особых остатков глутаминовой кислоты и могут также зависеть от гидроксилирования особого остатка аспарагиновой кислоты, маловероятно, что активный продукт может быть получен клонированием и экспрессией факторов VII и IX в микроорганизме.
В данном изобретении, соответственно, предлагается способ получения белка, имеющего кровосвертывающую биологическую активность фактора VIIa, с использованием стабильно трансформированных клеток млекопитающих. Кроме того, в данном изобретении предлагается также способ получения белка, обладающего биологической активностью по отношению к свертыванию крови, аналогичной активности фактора IX.
Как отмечалось выше, факторы VII и IX требуют для своего биосинтеза витамина K. Кроме того, такие плазменные белки, как протромбин, фактор X, Протеин C и Протеин S также требуют для своего биосинтеза витамин K. Аминоконцевые участки этих белков, содержащие остатки гамма-карбоксиглутаминовой кислоты, аналогичны как по аминокислотной последовательности, так и по биологической функции (рисунок 2a). Кроме того, карбоксиконцевые участки фактора VII, протромбина, фактора IX, фактора X и Протеина C определяют их специфическое действие как сериновых протеаз.
Фактор VII является белком, присутствующим в плазме в следовых количествах, и считается, что редкой является м-РНК, кодирующая фактор VII. Следовательно, очистка фактора VII из плазмы в количествах, достаточных для проведения подробного анализа последовательности и определения свойства, остается трудной. Разложение фактора VII в ходе очистки, даже в присутствии ингибиторов протеаз, было отмечено Кисиелом и Мак-Малленом (см.). В силу этих трудностей фактор VII был плохо охарактеризован по сравнению с другими компонентами свертывающей системы крови, присутствующими в больших количествах. Действительно, в работе Кисиела и Мак-Маллена (см.) была получена информация о последовательности только для 10 остатков каждой цепи фактора VII и в каждой последовательности идентификация двух остатков является предварительной. Данные по частичной аминокислотной последовательности бычьего фактора VII также были опубликованы (Дискипио и др., см.).
Предполагаемая редкость м-РНК фактора VII является одной из причин плохого значения гена фактора VII. Успех обычной техники клонирования к-ДНК зависит от наличия достаточного количества м-РНК, используемой в качестве матрицы. Преждевременная терминация обратной транскрипции приводит к получению клонов к-ДНК, лишенных 5'-окончания, и этому способствует низкая концентрация м-РНК. Были разработаны несколько общих подходов для клонирования к-ДНК веществ, имеющихся в низкой концентрации (Maniatis и др., Molecular Cloning: A Laboratory Manual, lold Spring Harba Laboratory, 1982), но недостаток знания аминокислотной последовательности интересующего продукта делает невозможным предсказание ДНК-последовательности и разработку подходящих олигонуклеотидных проб. Хотя можно относительно прямо получать частичный клон к-ДНК гена, кодирующего редкий белок, с помощью этих разработанных подходов, однако клоны к-ДНК полной длины гена, кодирующего редкие белки, такие как фактор VII, получить по-прежнему исключительно трудно.
По сравнению с фактором VII фактор IX является относительно нередким белком и известна последовательность клона к-ДНК гена человеческого фактора IX (Kurachi, Davie Proc. cvatl Acad Sci USA, 79: 6461-6464, 1982; и Anson и др. , EMBO J, 3: 1053-1060, 1984). Была охарактеризована структура гена фактора IX и была определена аминокислотная последовательность белка на основании известной нуклеотидной последовательности. Были опубликованы также некоторые данные по белковой последовательности для человеческого и бычьего факторов IX и анализируемые последовательности (Дискипио и др., см.). Аминоконцевой участок белка содержит 12 остатков глутаминовой кислоты, которые превращаются в зрелом белке в остатки гамма-карбоксиглутаминовой кислоты (GIa). Были также определены сайты расщепления, участвующие в активации фактора IX (Kurachi и Davie, см.). Последовательность 5'-окончания клона к-ДНК фактора IX кодирует сигнальный пептид, типичный для сигнальных пептидов, обнаруженных в большинстве секретируемых белков (Kurachi и Davie, см.). Ранее описывалась экспрессия гена фактора IX методами рекомбинантной ДНК.
Как трудно получить клон к-ДНК полной длины гена фактора VII, то были разработаны три новых подхода для получения 5'-окончания кодирующей последовательности, включая участок, кодирующий лидерный пептид. По первому способу частичный клон к-ДНК фактора VII соединяют с фрагментом, кодирующим лидерный пептид и 5 - окончание фактора IX. Этот подход основан на том наблюдении, что аминоконцевые участки двух молекул ответственны за связывание кальция соответствующими белками, и на открытии того, что относящаяся к связыванию кальция активность фактора IX аналогична активности фактора VII. Результирующий полипептид сохраняет биологическую активность нативного фактора VII, так как за специфическую активность факторов свертывания как сериновых протеаз отвечают карбоксиконцевые участки молекул. Второй подход сочетает клон частичной к-ДНК с последовательностью ДНК, кодирующей лидерную и аминоконцевую последовательности фактора VII. Частичная последовательность к-ДНК и аминокислотная последовательность фактора VII, описанные здесь, способны выбирать из библиотеки геномной ДНК или библиотеки к-ДНК клоны, включающие 5'-окончание гена фактора VII. В третьем способе соединяют клон частичной к-ДНК с гибридными кодирующими последовательностями, содержащими фрагмент к-ДНК, кодирующий лидерный пептид фактора IX и сегмент синтетического гена, кодирующий согласованный домен связывания кальция или предсказанную аминоконцевую последовательность фактора VII. Последовательность, кодирующая аминокончание фактора VII, была установлена с помощью описываемых здесь ранее неопубликованных данных по аминокислотной последовательности. Согласованную последовательность определили из данных по фактору VII и из данных, опубликованных по последовательности других плазменных белков, зависящих от витамина K.
В соответствии с описанным выше способом отбора клонов, содержащих 5'-окончание гена фактора VII, авторы данного изобретения успешно получили полномерную, правильную к-ДНК, пригодную для экспрессии.
Среди полученных клонов к-ДНК клон, обозначенный "λ VII2463", содержит наибольшую вставку к-ДНК фактора VII. Было установлено, что она содержит полную кодирующую последовательность фактора VII. Этот клон содержит 35 нуклеотидов нетранслированного участка 5', 180 нуклеотидов, кодирующих лидерный участок из 60 аминокислот, 1218 нуклеотидов, кодирующих 406 аминокислот зрелого белка, стоп-кодон, 1026 нуклеотидов нетранслируемой 3'-последовательности и 20 оснований конца poly /A/ /начиная с позиции 2463/. Две нити этой к-ДНК не подвергались полному анализу последовательности. Сравнение ее с двумя вставками к-ДНК, выделенными ранее из клонов λ VII2115 и λ VII1923, свидетельствует, что λ VII2463 содержит, в индивидуальном фрагменте Eco R1, к-ДНК фактора VII, кодирующую лидерную последовательность фактора VII и последовательности зрелого белка.
Был выделен второй клон, λ VII565, содержащий вставку к-ДНК, идентичную к-ДНК клона λ VII2463 от нуклеотида 9 до нуклеотида 638, за исключением того, что отсутствуют нуклеотиды от 100 до 165 (рисунок 1b). При сравнении к-ДНК с геномной ДНК фактора VII установлено, что отсутствующая последовательность точно соответствует одному экзоноподобному участку. Следовательно, получены две к-ДНК фактора VII, отражающие различные стадии сплайсинга м-РНК.
Лидерная последовательность, кодируемая λ VII2463, является исключительно длинной (60 аминокислот) и имеет гидрофобный характер, отличающийся от фактора IX, протеина C и протромбина. Эта лидерная последовательность содержит два метионовых остатка, в положениях -60 и -26. Инициирование наиболее вероятно начинается на первом met, так как гидрофобный участок, типичный для сигнальных пептидов, следует за met в положении -60, а не met в положении -26. Интересно, что отсутствующая последовательность в λ VII565, соответствующая точно экзоноподобному участку в геномном клоне, приводит к аминокислотной лидерной последовательности из 38 аминокислот, гидрофобные характеристики которой более похожи на фактор IX, протеин C и протромбин.
Так как неясно, насколько описанные выше лидерные последовательности соответствуют нативным, то были приложены усилия для анализа 5'-концевой последовательности. Вкратце этот способ включал в себя конструирование и отбор библиотеки человеческой геномной ДНК и идентифицирование геномных клонов, содержащих последовательности гена фактора VII. Участок 5' геномной последовательности присоединяли затем к к-ДНК для конструирования полномерного клона.
При дополнительном конструировании 5-фрагмент к-ДНК фактора VII λ VII565, содержащий участок, полностью кодирующий лидерный участок, и последовательность, кодирующую 29 аминокислот зрелого белка, связывали с фрагментом к-ДНК λ VII2463 (содержащим остальную часть зрелого белка и нетранслируемые 3 - последовательности). Эта "565-2463" - последовательность кодирует полный фактор VII, к-ДНК-последовательность является индивидуальным Eco RI-фрагментом.
Описанные выше последовательности ДНК вставляли затем в подходящий вектор экспрессии, который в свою очередь использовали для трансформирования клетки млекопитающего. Вектор экспрессии, используемый для осуществления данного изобретения, включает в себя промотор, способный направить транскрипцию чужеродного гена в трансформированной клетке млекопитающего. Предпочтительны вирусные промоторы из-за их эффективности при прямой транскрипции. Таким особенно предпочтительным промотором является основной поздний промотор аденовируса 2. Такой вектор экспрессии будет также содержать набор сайтов сплайсинга РНК, расположенный за промотором и выше сайта вставки гена, кодирующего белок, имеющий биологическую кровосвертывающую активность. Предпочтительные последовательности сайта сплайсинга РНК могут быть получены из аденовируса и/или из генов иммуноглобулина. В векторах экспрессии содержится также сигнал полиадениляции, расположенный за сайтом вставки. Предпочтительны вирусные сигналы полиадениляции, такие как сигналы ранней или поздней полиадениляции из SV 40 или сигнал полиадениляции из участка аденовируса 5: EIo. В особенно предпочтительном варианте вектор экспрессии содержит также вирусную лидерную последовательность, такую как состоящий из трех частей лидер аденовируса 2, расположенную между промотором и сайтами сплайсинга РНК. Предпочтительные векторы могут также содержать усиливающие последовательности, такие как усилитель SV 40.
Последовательности клонируемой ДНК можно также вводить в культивируемые клетки млекопитающих с помощью трансформации, вызываемой фосфатом кальция (Wiglir и др. , Cell, 14: 725, 1978; Corsaro и Pearson, Somatic Cell Genetics, 7: 703: 1981; Graham и van der Eb, Virology 52:456, 1973.) Получают осадок, образованный ДНК и фосфатом кальция, и этот осадок наносят на клетки. Часть клеток поглощает ДНК и удерживает ее внутри клетки в течение нескольких дней. Небольшая часть клеток (обычно 10-4) стабильно интегрирует ДНК в геном. Для идентификации этих стабильных интегрантов вместе с интересующим геном обычно вводят ген, придающий отличительный фенотипический признак (отличительный маркер). Предпочтительными отличительными маркерами являются гены, придающие устойчивость к лекарствам, таким как G-418 и метотрексат. Отличительные маркеры можно вводить в клетки на отдельной плазмиде одновременно с интересующим геном или их можно вводить в той же самой плазмиде. Предпочтительным отличительным маркером является ген устойчивости к лекарству G-418, содержащийся в плазмиде pKO-neo (Southern и Birg J. Mol. Appl.Genet. , I: 327-341, 1982). Может оказаться желательным прибавление дополнительной ДНК, известной как "несущая ДНК", в смесь, вводимую в клетки. После того, как клетки поглотили ДНК, их оставляют расти в течение определенного времени, обычно 1-2 дней, для начала экспрессии интересующего гена. Затем проводят отбор лекарством для отбора тех клеток, которые обнаруживают стабильную экспрессию отличительного маркера. Для экспрессии интересующего белка могут быть отобраны клоны таких клеток.
Факторы VII и IX, вырабатываемые трансформированными клетками, могут быть выделены из клеточной культуральной среды адсорбцией на цитрате бария. Среду перемешивают с цитратом натрия и хлоридом бария и собирают осадок. Осадок затем можно проанализировать на присутствие соответствующего фактора свертывания. Дальнейшую очистку можно провести с помощью иммуноадсорбции. Предпочтительно, чтобы иммуноадсорбционная колонка содержала высокоспецифическое моноклональное антитело. Кроме того, очистка адсорбированного на цитрате бария продукта может проводиться более общепринятыми биохимическими способами или с помощью жидкостной хроматографии высокого давления.
Превращение одноцепочечного фактора VII в активный двухцепочечный фактор VIIa может проводиться с помощью фактора XIIa, как описано Hender и Kisiel (J. Clin Invest, 71: 1836-1841, 1983) или другими протеазами, имеющими трипсиноподобную специфичность (Kisiel и Fujikawa, Bchring. Inst. Mitt, 73: 29-42, 1983).
Итак, в данном изобретении предлагается способ получения белков, имеющих активность зависящих от витамина K факторов свертывания крови, с помощью трансформированных клеток млекопитающих. Генные последовательности, кодирующие специфические домены сериновых протеаз факторов свертывания, выделяют из библиотек к-ДНК. Последовательности, кодирующие лидерные пептиды и домены связывания кальция, выделяют из библиотек к-ДНК или генома или конструируют из синтезированных олигонуклеотидов. Затем последовательности соединяют с получением соответствующего вектора экспрессии, кодирующего белок, имеющий нужную биологическую кровосвертывающую активность. Результирующий вектор и плазмиду, содержащую маркер устойчивости к лекарству, совместно вводят в культивируемые клетки ткани подходящего млекопитающего. Трансформированные клетки отбирают затем добавлением соответствующего лекарства, такого как G-418. Затем белковые продукты очищают из среды клеточного роста и оценивают на биологическую активность анализом на свертывание крови и перекрестным иммунологическим анализом с использованием антител, полученных против аутентичных факторов свертывания.
Кратко суммируем представленные ниже примеры: в примере 1 описывается клонирование полномерной последовательности к-ДНК фактора VII. В примере 2 описывается частичная аминокислотная последовательность человеческого фактора VII, включающая в аминоокончании последовательность из приблизительно 30 аминокислот. В примере 3 описывается конструирование и отбор библиотеки человеческой геномной ДНК и идентификация геномных клонов, включающих последовательности гена фактора VII. В примере 4 описано конструирование двух сегментов гибридного гена, каждый из которых содержит фрагмент к-ДНК, кодирующий лидерный пептид фактора IX и синтезированный двунитевый фрагмент, кодирующий согласованный домен связывания кальция. Гибридные последовательности соединяют затем в клоны частичной к-ДНК фактора VII. С использованием мутагенеза in vitro согласованную последовательность затем изменяют так, чтобы она соответствовала данным по белковой последовательности фактора VII. В примере 5 описывается конструирование генной последовательности, кодирующей слитый белок, включающий связывающий кальций домен фактора IX и специфический домен сериновой протеазы фактора VII. В примере 6 описано конструирование вектора pD2 для использования в экспрессии белков, обладающих биологической кровосвертывающей активностью, в трансформированных клетках млекопитающих. Слитый ген, описанный в примере 5, экспрессируют с использованием этого вектора. В примере 7 описано использование вектора pD2 для экспрессии гена фактора IX в линии трансформированных клеток млекопитающих. В примере 8 описывается конструирование вектора pM7135, содержащего последовательности ДНК, кодирующие продукт первичной трансляции, содержащий лидерную последовательность фактора IX, слитую с фактором VII. Этот вектор может быть использован для получения белка, имеющего активность фактора VII, в линии трансформированных клеток млекопитающего. В примере 9 описана экспрессия фактора VII с использованием последовательностей к-ДНК и экспрессии фактора VII из гибридной последовательности геномной к-ДНК.
Представленные ниже примеры служат для иллюстрации и не ограничивают данного изобретения.
Примеры
Расщепляющие ферменты были получены из Bithisda Research Laboratories /BRI/ и New England Biolab, за исключением специально указанных случаев, использовались так, как рекомендует поставщик. Олигонуклеотиды синтезировали на ДНК-синтезаторе Applied Biosystems Model 380 A и очищали на денатурированных гелях электрофорезом на полиакриламидном геле. E.coli -Клетки трансформировали так, как описано Maniatis и др. (Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory 1982). Клонирующие векторы M13 и pUC и штаммы - хозяева получали из BRI. Фактор VII получали из человеческой плазмы так, как описано Kisiel и McMullen (см.).
Пример 1. Клонирование частичной к-ДНК- фактора VII
A. Конструирование библиотеки к-ДНК человеческой печени.
Библиотеку к-ДНК получали из м-PHK печени человека в соответствии со стандартными способами. Препарат к-ДНК седиментировали через градиент щелочной сахарозы, и собирали фракции, содержащие частицы большие, чем приблизительно 1000 нуклеотидов. Первый препарат делали двунитевым с помощью ревертазы, обрабатывали SI - нуклеазой и оставшиеся концы заполняли с помощью ДНК-полимеразы I (фрагмент Klenow) в присутствии всех четырех дезоксирибонуклеотидтрифосфатов. к-ДНК с тупыми концами обрабатывали Eco RI-метилазой и связывали с фосфорилированными линкерами Eco RI с помощью T -ДНК - лигазы. Связанный препарат ДНК подвергали интенсивному расщеплению с помощью Eco RI для удаления избыточных линкерных последовательностей, и двунитевые ДНК большие, чем приблизительно 1000 пар оснований по длине, очищали центрифугированием в градиенте нейтральной сахарозы. Нативную ДНК λgt II связывали в конкатемеры, расщепляли для завершения с помощью Eco RI и 5'-концевые фосфаты удаляли обработкой бактериальной щелочной фосфатазой. Соединенные к-ДНК печени человека связывали с ДНК фага, наносили in vitro и использовали для заражения E. coliy 1088. В этой библиотеке генерируется приблизительно 14 • 106 первичных фаговых пятен, состоящих из 7 библиотек по приблизительно 2 • 106 пятен каждая. Более чем 90% из них являются рекомбинантами, содержащими вставки человеческой ДНК, на что указывает потеря у них бета-галактозидазной активности и анализ 20 случайных клонов расщеплением с помощью Eco RI с последующим электрофорезом на агарозном геле. Библиотеку к-ДНК в форме частиц фага очищают центрифугированием в градиенте хлорида цезия и хранят в буфере SM.
B. Отбор библиотеки к-ДНК печени человека для клонов фактора VII
Описанную выше библиотеку экспрессивной к-ДНК из печени человека отбирают на специфический антиген, с использованием 125 I-меченого моноклонального антитела фактора VII, полученного с использованием очищенного фактора VII. Отбор 6 • 10 пятен фага идентифицирует один изолят, обозначаемый как λ VII2115, дающий положительную реакцию на антитело.
Клон фага λ VII2115 испытывают по отношению к двум другим моноклональным антителам анти-фактор VII и кроличьему поликлональному антителу к фактору VII. Изолят λ VII2115 дает положительную реакцию на все эти антитела анти-фактор VII.
ДНК получают из лизата пятен λ VII2115. Расщепление этой ДНК с помощью EcoRI освобождает вставку длиной 2139 по. Эту вставку субклонируют в векторы фага M13 для терминации цепи дидезокси - ДНК секвентирования. Эта вставка к-ДНК содержит сайты Pst I в положениях 214, 839 и 1205 (обозначены на рисунке 1a соответственно Pst Ia, Pst Ib и Pst Ic) и сайт Sma I, расположенный в положении 611. Секвентируют следующие матрицы M13:
1) полномерный /2139 оснований/ фрагмент EcoRIa RIa _→ Eco R Ib в M3mp18 (обозначает клон F Г-1),
2) фрагмент Pst Ia _→ EcoRIa длиной 214 оснований, в M13mp19 (F 7-2),
3) фрагмент в M13mp18 (F7-3) длиной 625 оснований, Pst Ia _→ Pst I,
4) фрагмент в M13mp18 (F7-7), Pst Ib Pst Ia, 625 оснований,
5) фрагмент в M13mp10 (F7-8), SmaI _→ Pst Ib, 228 оснований,
6) фрагмент в M13mp18 (F7-9), Pst Ic _→ Pst Ic, 366 оснований,
7) фрагмент в M13mp18 (F7-10), Pst Ic _→ Pst Ib, 366 оснований,
8) фрагмент в M13mp19 (F7-11), Pst Ic _→ Eco RIb, 950 оснований,
9) полномерный фрагмент в M13mp18 (F7-12), Eco RIb Eco RIa,
(рестрикционные сайты обозначены также, как на рисунке Ia). Данные подтверждают последовательность у обеих нитей для 91% кодирующего участка и 15% некодирующего 3' - участка и дают информацию по одной нити для остальных 9% кодирующего участка и 85% некодирующего участка.
Сравнение аминокислотной последовательности, предсказанной из последовательности к-ДНК, с известной аминокислотной последовательностью и с представленной ниже аминокислотной последовательностью (пример 2) указывает на наличие аномалии, которую можно объяснить отсутствием трех нуклеотидов в последовательности ДНК около положения 400. Для получения дополнительных данных по последовательности VII2115 расщепляют EcoRI и фрагмент, кодирующий фактор VII, вводят в pU/C 13, расщепляемый EcoRI. Результирующую рекомбинантную плазмиду, обозначенную pUC V112115, расщепляют XbaI по положению 328. Расщепленный образец делят пополам: половину метят с помощью α32 Pol СТР и ДНК-полимеразы I (фрагмент Klenow), другую половину метят с помощью γ32 p АТР и полинуклеотидкиназы. Меченые плазмиды расщепляют затем с помощью Pst I и получают фрагменты длиной 113 и 509 пар оснований. Обе нити каждого из этих фрагментов секвентируют по методу Manam и bilbert. Фрагмент длиной 113 пар оснований секвентируют полностью, а в фрагменте длиной 509 пар оснований секвентируют 210 пар оснований. В результате секвентирования обнаруживают три дополнительных основания (один C и два G), что приводит данные по последовательности ДНК в согласие с данными по последовательности белка и указывает на то, что предыдущие аномальные результаты возникают в результате сжатия в секвентирующем геле, обусловленного вторичной структурой, включающей в себя G и C. Подтверждается также последовательность последних 9% кодирующего участка для обеих нитей.
Дополнительный анализ вставки PU CV112115 подтверждает, что часть этого клонированного фрагмента кодирует последовательность из II аминокислот, которая известна как расщепляющий сайт фактора VII. Сравнение этой последовательности с аминокислотными последовательностями фактора IX и фактора X свидетельствует о том, что клон, содержащий последовательность фактора VII, начинается (приблизительно) с нуклеотидов, кодирующих аминокислоту 36 природного белка фактора VII и продолжается через приблизительно 1000 кодирующих и 1100 некодирующих нуклеотидов и последовательность poly A. Кроме того, было установлено, что этот клон имеет мутационный сдвиг структуры в кодирующем 3'-участке.
Для получения правильного кодирующего 3'-участка все 14 миллионов клонов семи библиотек к-ДНК λgt II подвергают отбору гибридизацией пятен с помощью транслируемой с меткой к-ДНК λ VII2115.
Семь положительных изолятов отбирают затем с помощью дидезокси-секвентирования плазмид pUC, в которых субклонированы вставки к-ДНК. Клоны λgt II расщепляют с помощью EcoRI и фрагменты фактора VII вставляют в pUC13, которую расщепляют EcoRI. Как было установлено, за одним исключением все остальные начинают с позиции, соответствующей основанию 212 вставки и λ VII2115, в одном случае - исключении имеется только некодирующая 3'-последовательность. Один из клонов, начинающий с основания 212, был выбран для анализа и обозначен как клон pUCVII1923.
Так как анализ pUCVII2115 указывает на присутствие мутационного сдвига между позициями 657 и 815, то pUCVII1923 сначала анализировали по этому участку секвентированием по Makam-gilbert. Плазмиду pUCVII1923 расщепляют с помощью Nar I (позиция 779 на рисунке 1a). Расщепленную ДНК метят 32p dCTP с использованием ДНК-полимеразы 1 (фрагмент Klenow) и последовательно расщепляют с помощью Ava I (проводящей расщепление по тому же сайту, что и Sma1 на рисунке 1) и Taq1 (сайт при 1059), получая фрагмент Nar I-AvaI длиной 166 и по и фрагмент Nar I-Taq I длиной 200 по. Каждый из них секвентируют. C, недостающий в pUCVII2115, обнаруживается в положении 697 и другой C, также отсутствующий в pUCVII2115, обнаруживается в положении 798.
Было установлено, что остальная последовательность кодирующего участка pUCVII1923 является правильной по данным секвентирования по дидезокси-методу субклона М13 полной вставки pUCVII1923. Праймер dac ZC87 (таблица 1) используют для последовательности от положения 212 (рисунок 1a) до 512, праймер ZC218 (CTCTGCCTGCCGAAC) используют для последовательности от 715 до 1140 и праймер ZC217 (ATGAGAAGCGCACGAAG) используют для последовательности от 720 до 350. Так как вставка pUCVII2115 правильна от положения 13 (позиции 1-12 содержит искусственный линкер), а pUCVII1923 правильна от позиции 212 до конца, то соединенные вместе они дают молекулу с правильной структурой начиная с позиции 13 (рисунок 1a) и до конца. Обычно используемый для этого соединения участок представляет собой сайт XbaI в позиции 328. Последовательность правильной составной молекулы показана на рисунке 1a.
Так как клонированием к-ДНК трудно получить полномерный клон фактора VII, то использовали три подхода для получения отсутствующей кодирующей последовательности и необходимых лежащих выше процессорной и сигнальной последовательностей. Первый подход заключается в том, что необходимую последовательность получают из библиотеки человеческой геномной ДНК или дополнительным отбором библиотек к-ДНК. Второй подход заключается в синтезировании необходимой кодирующей 5'-последовательности на основе данных по аминокислотной последовательности фактора VII (пример 2) и опубликованных последовательностей генов, кодирующих зависимые от витамина K факторы свертывания (Kurachi и Davie см.; Davie и др., см.) в присоединении ее к части последовательности фактора IX. Третий подход основан на функциональной гомологичности аминоконцевых участков фактора VII и фактора IX. Конструируют последовательность, содержащую кодирующие участки лидерной последовательности и аминоконцевой участок фактора IX. Затем ее в нужной ориентации присоединяют к частичной к-ДНК фактора VII.
Для получения последовательностей ДНК, содержащих полную последовательность ДНК фактора VII, были предприняты попытки выделить оставшуюся 5'-последовательность ДНК. Это достигается использованием 5'-концевого фрагмента EcoRI-XbaI длиной 0,3 килобазы из к-ДНК вставки λ VII2115 для отбора библиотеки к-ДНК, содержащей 2 х 10 фагов. Библиотеку конструируют с использованием poly /A/ -м- РНК из клеток Hep G2 с последующим применением вышеописанных способов. РНК Ретранскрибируют для получения первой нити к-ДНК, а вторую нить синтезируют с помощью ДНК-полимеразы I и РНК-азы Н. После метилирования EcoRI и пропускания через колонку с сефарозой 6B концы ДНК затупляют с помощью T4 - ДНК-полимеразы. Добавляют линкеры EcoRI и избыточные линкеры удаляют отщеплением с помощью EcoRI и хроматографией на Сефарозе Ch 2B. ДНК, находящуюся в метровом объеме, собирают и связывают с λgt 11, расщепленной EcoRI и обработанной кишечной бычьей фосфатазой. ДНК наносят и вводят в E. coli Y1088. Обнаруживают несколько положительных результатов и фрагменты EcoRl затем субклонируют в векторы фага М13 для дидезокси-секвентирования с использованием либо универсального праймера М13, либо специфических олигонуклеотидов фактора VII.
Таким образом, получают три новых к-ДНК-клона фактора VII с полностью определенными последовательностями. Как было установлено, большая часть этих к-ДНК из клона, обозначенного λ VII2463, содержит полную кодирующую последовательность фактора VII. Этот клон включает в себя 35 нуклеотидов нетранслируемого 5'-участка, 180 нуклеотидов, кодирующих 60 аминокислот лидерного участка, 1218 нуклеотидов, кодирующих 406 аминокислот нативного белка, стоп-кодон, 1026 нуклеотидов нетранслируемой 3'-последовательности и 20 оснований poly /A/-конца (начиная с позиции 2463). Обе нити этой к-ДНК полностью секвентировали. Сравнение ее с двумя к-ДНК, выделенными ранее из клонов λ VII2115 и λ VII1923, показывает, что клон λ VII2463 содержит дополнительно 321 нуклеотид выше вставки в λ VII2115 и 519 нуклеотидов выше вставки в λ VII1923. Перекрывающиеся последовательности λ VII2463 фактора VII и эти две предыдущие к-ДНК совпадают, за исключением того, что к-ДНК λ VII2463 не содержит делецию одного основания в позиции 1005 и 1106, обнаруженную в к-ДНК, λ VII2115. Таким образом, λ VII2463 содержит в одном фрагменте EcoRI к-ДНК фактора VII, кодирующую лидерный участок фактора VII и последовательности зрелого белка.
Была выделена дополнительная к-ДНК, λ VII565, которая, как было установлено, содержит 5'-концевые последовательности фактора VII, но усечена в кодирующих последовательностях. Ее 5 - окончание соответствует нуклеотиду 9 (рисунок 1).
По сравнению с полимерной λ VII2463, λ VII565 лишена, как было установлено, последовательности, соответствующей одному экзоноподобному участку в лидерной последовательности. В λ VII565 отсутствуют основания 100-165 (рисунок 1b). Отсутствующие последовательности точно соответствуют одному экзоноподобному участку по данным геномной последовательности (как описано в примере III). Таким образом, структура λ VII565 может быть следствием альтернативных стадий сплайсинга в лидерной последовательности.
Лидерная последовательность, закодированная в λ VII2463, является исключительно длинной (60 аминокислот) и имеет совершенно другой гидрофобный профиль, чем фактор IX, протеин C и протромбин. Эта лидерная последовательность содержит два остатка метионина, в положениях - 60 и - 26. Наиболее вероятно, что инициирование начинается с первого остатка метионина, так как гидрофобный участок, типичный для сигнальных пептидов, начинается с метионина в положении-60, а не с метионина в положении - 26. Интересно, что последовательности, отсутствующие в λ VII565 и точно соответствующие экзоноподобному участку в геномном клоне, дают лидерную последовательность из 38 аминокислот с гидрофобной структурой, более близкой к структурам вышеуказанных белков.
Пример 2: Аминокислотная последовательность человеческого фактора VII.
Расшифровка аминокислотной последовательности человеческого фактора VII нужна для подтверждения идентичности полученных к-ДНК-клонов, правильности последовательности к-ДНК фактора VII, для получения информации, позволяющей синтезировать специфические олигонуклеотидные пробы для отбора к-ДНК и геномных библиотек для клонов, содержащих 5'-последовательностей для конструирования синтетического фрагмента, кодирующего аминоконцевой участок фактора VII.
Очищенный человеческий фактор VIIa восстанавливают и карбоксиметилируют. Малую и большую полипептидные цепи карбоксиметилированного фактора VIIa разделяют жидкостной хроматографией высокого давления (ЖХВД) на колонке с обращенной фазой Micro Pack C18 (Varian Corp), создавая градиент 0,1% TFA в дистиллированной воде (A) и 0,1% TFA в ацетонитриле (B), 0-40% B в течение 5 мин, 40-80% B в течение 25 мин и 80-100% B в течение 5 мин. Приблизительно 300 пикмолей каждой пептидной цепи анализируют автоматизированным разложением по Эдману с использованием bas-Phase Protein Sequencer (Applied Biosystems Ink).
В аминоконцевом участке большой и малой полипептидных цепей идентифицируют соответственно 18 и 29 остатков. Аминоконцевая последовательность большой цепи фактора VIIa согласуется с последовательностью, кодируемой клоном к-ДНК pUCVII2115 (рисунок 2b). Аминокислотные остатки обозначены в рисунках 2a и 2b одной буквой следующим образом: A - аланин, C - цистеин, D - аспарагиновая кислота, E - глутаминовая кислота, F - фенилаланин, M - метионин, N - аспарагин, P - пролин, O - глутамин, R - аргинин, S - серин, T- треонин, V - валин, W - триптофан, Y - тирозин, X - неизвестный остаток, G - глицин, H - гистидин, I - изолейцин, K - лизин, L - лейцин, * указывает на то, что остатки Gla(γ) предполагаются на основании гомологичности структур других известных факторов свертывания и отсутствия каких-либо других фенилтиогидрантоинаминокислот в этих позициях. Прочерки (-) введены для лучшего сопоставления последовательностей. Кроме того, информация указывает на то, что аминокислоты в положениях 5 и 9 являются лизинами, а не треонином и аргинином, соответственно, как докладывалось ранее (Кисиел и Мак-Маллен, см). Анализ последовательности малой цепи фактора VIIa, происходящей из аминоконцевого участка фактора VII, ограничен приблизительно 6 остатками до перекрывания со структурой, кодируемой 5'-окончанием к-ДНК клона pUCVII2115.
Для получения дополнительных данных по последовательности, два наномоля карбоксиметилированной малой цепи расщепляли 12 ч бычьим химотрипсином (1: 100 вес/вес, фермент: субстрат) в 0,1 М бикарбонате аммония при pH 7,8 и 37oC. Полученные фрагменты очищают ЖХВД на колонке с обращенной фазой Micro Pack C18 с использованием указанных выше растворителей в градиенте 0 - 30% в течение 5 мин, 30 - 60% B в течение 25 мин и 60 - 80% B в течение 10 мин. Пептиды идентифицируют по их УФ-поглощению при 220 и 280 нм. Лиофилизованные пептиды (приблизительно по 1 наномолю каждого) анализируют разложением по Эдману. Результаты (рисунок 2b) подтверждают большую часть к-ДНК последовательности в соответствующем участке клоне pU CVII2115. В целом идентифицировано 113 из 152 остатков (75%) малой цепи Фактора VIIa. Эта последовательность идентична последовательности, кодируемой известной структурой к-ДНК. Косвенные признаки указывают на то, что Asu 145 является сайтом присоединения углевода.
Пример 3. Клонирование геномной последовательности Фактора VII
В одном подходе получения 5'-концевой последовательности из к-ДНК, библиотеку лямбда-фага, содержащую ДНК плодной печени человека, отбирают с помощью меченно-транслируемой к-ДНК Фактора VII. Часть геномной библиотеки помещают на E. coli E392 (АТСС 33562) с получением общего количества пятен 7,2 • 10. Пятна фага адсорбируют с пластинок на нитроцеллюлозу и гибридизуют 32p меченной к-ДНК. Получают 8 клонов и пятна очищают.
С использованием фрагмента ДНК (EcoRIa-XbaI, рисунок 1) из 5'-окончания к-ДНК фактора VII (λ VII2115) и стандартных методик идентифицируют эти геномные клоны, содержащие 5-концевые последовательности. Эти фаги обозначают как 7 m 1, 7 m 2 и 7 m 3. Из этих рекомбинантных фагов получают ДНК и проводят предварительный анализ последовательности рестрикционными эндонуклеазами. Фаг 7m 1, имеющий самый сильный сигнал гибридизации, используют для получения более подробной рестрикционной карты и помещения на эту карту последовательностей к-ДНК EcoRI-XbaI внесением по Southern.
Для того, чтобы определить, содержит ли фаг 7mI последовательности ДНК, кодирующие аминоконцевые аминокислоты белка Фактора VII, пятна Southern ДНК фага после рестрикции гибридизуют смесями олигонуклеотидов, чьи последовательности рассчитывают из аминоконцевой аминокислотной последовательности фактора VII. Олигонуклеотиды ZC188, ZC360 и ZC401 (таблица 1) радиоактивного метят T4-полинуклеотидкиназой и гибридизуют в пятна ДНК фага при температуре, на несколько градусов более низкой, чем их Tm. Результаты этого анализа показывают, что фрагмент 7mISst I длиной 3,7 килобазы содержит последовательности, гибридизованные этими олигонуклеотидами. Этот фрагмент SstI субклонируют в M13 для анализа последовательности ДНК. Результаты, полученные с использованием ZC360 в качестве праймера секвентирования, идентифицируют область длиной приблизительно 60 нуклеотидов, что соответствует данным по аминоконцевой последовательности белка.
Так как известно, что геномный клон 7mI содержит последовательности длиной 7 килобаз выше экзона 2, то этот клон участвует в кодировании нетранслируемых 5'-последовательностей фактора VII и лидерных последовательностей до аминокислотной позиции - 17. Для подтверждения того, что экзон I закодирован в геномном клоне 7mI, используют информацию о лидерной последовательности из клонов λ/ VII2463 и λ VII5656 для расчета олигонуклеотидов ZC528 и ZC529 (см. в конце текста).
Это используют для пробы ДНК 7mI и, как установлено, субклон 7SD гибридизован обоими этими олигонуклеотидами. Устанавливают, что экзон 1 состоит из двух экзонных последовательностей: экзона 1a, гибридизованного ZC528 (соответствует нуклеотидам 1 - 30 в λ VII2463), и экзона 1b, гибридизованного ZC528 (соответствует нуклеотидам 119 - 148 в λ VII2463). Секвентируют последовательности интрона, примыкающие к обоим экзонам 1a и 1b: 1a содержит согласованную донорную последовательность сплайсинга в 3'-окончании экзона, а к каждому концу 1b примыкает акцептор согласованного сплайсинга (выше 1b) или донорная последовательность (ниже 1b). Точно определяют положение экзона 1a в геномном клоне 7mI и определяют наличие экзона 1b в определенном участке. Последовательности экзона 1b присутствуют в λ VII2463, а λ VII565, как оказалась, происходит из РНК, помещенной между экзоном 1a и экзоном 2, с внешней петлей последовательности экзона 1b.
Получают различные 7mI -субклоны в puC и M13-векторах для облегчения секвентирования остальных экзонов. Соответствующие олигонуклеотиды получают из последовательностей к-ДНК, соответствующих экзонам 1-7, и используют их для последовательностей всех экзонов кроме последнего экзона. Геномная последовательность точно соответствует последовательностям к-ДНК на этих участках. Кроме того, были определены связывающие участки интрон/экзон для экзонов 1-7, и в настоящее время для большинства из них точно определено место в клоне 7mI. Размеры и положение интронов в гене фактора VII перечислены в таблице 2.
Как известно, фаг 7mI лишен 3'-окончания фактора VII, включающего в себя экзон 8. Для получения этих последовательностей в 12-13 килобаз - обогащенную библиотеку BamH1 в Xh 47,1, полученную из первичных фибробластов кожи человека, вводят пробу с двумя транслируемыми с меткой фрагментами PstI к-ДНК фактора VII (соответствует последовательностям экзона 7 и нетранслируемым 3'-последовательностям). Клон, обозначенный как 7DCI, определяют по обеим пробам. Последующим анализом с помощью рестрикционных эндонуклеаз и анализом пятен по Southern устанавливают, что клон 7DCI перекрывается и простирается на приблизительно 3 килобазы за окончание клона 7 m 1 и что он содержит экзон 3. Фрагмент длиной 3,9 килобазы (XbaI-BamH1) из ДНК 7DCI, содержащей экзон 8, субклонируют в M13 и проводят анализ последовательности с помощью олигонуклеотидов, комплементарных окончаниям 5' и 3'. В этом клоне присутствует полная экзонная последовательность.
Пример 4. Гибридные гены фактор IX-фактор VII, содержащие синтезированную кодирующую последовательность
A. Конструирование последовательности гибридный лидерный участок фактора IX - синтетическая последовательность, кодирующая 5'-участок фактора VII.
Вторым подходом для получения 5'-кодирующей последовательности фактора VII является синтез соответствующего двунитевого фрагмента с использованием нуклеотидной последовательности, рассчитанной на основании аминоконцевой аминокислотной последовательности фактора VII, аминокислотных последовательностей других зависимых от витамина K факторов свертывания и известных нуклеотидных последовательностей генов других зависимых от витамина K факторов свертывания. Для получения нужных сигналов секретирования и переработки для секреции аналога зрелого фактора VII, этот синтетический фрагмент (согласованная последовательность) соединяют с одним из двух лидерных участков, полученных из клона к-ДНК фактора IX. Этот подход иллюстрируется на рисунке 3.
к-ДНК, кодирующую человеческий фактор IX, получают из библиотеки м-РНК из печени человека. Последовательность фактора IX выделяют из вектора pBR-322 расщеплением PstI и вводят в сайт PstI pUC13. Эту плазмиду обозначают FIX-pUC13. Для удаления G-обогащенного участка, присутствующего в 5'-окончании вставки фактора IX в результате клонирования к-ДНК, в 5'-окончание клонированного фрагмента вводят синтетический олигонуклеотидный адаптер. Олигонуклеотиды ZC212 и ZC213 (таблица 1) синтезируют и ренатурируют с получением перекрывания в 22 пары оснований, концы фрагмента заполняют и разрезают подходящими рестрикционными эндонуклеазами, и результирующий фрагмент присоединяют к последовательности фактора IX.
Для конструирования адаптера по 100 микомолей каждого из ZC212 и ZC213 лиофилизуют и повторно суспендируют в 10 мкл 10 х киназа / лигаза буфера (600 мМ Трис, pH 8,0 100 мМ хлорида магния, 100 наномолей DTT) плюс 86 мкл воды. Реакцию ренатурации проводят при 65oC в течение 10 мин, смесь медленно охлаждают до комнатной температуры и помещают на лед. К этой смеси прибавляют 4 мкл смеси 2,5 мМ dNTR и 1 мкл (8 единиц) T4 - ДНК полимеразы. Реакцию проводят в течение 45 мин при 14oC. Прибавляют 10 мкл 5М ацетата аммония и ДНК экстрагируют один раз смесью фенол/хлороформ и дважды хлороформом и осаждают этанолом. ДНК центрифугируют и суспендируют в 100 мкл среднего солевого буфера (Maniatis и др., см., p. 100), расщепляют 9 ед. PstI и 8 ед. CfoI и экстрагируют так, как описано выше.
Затем конструируют модифицированную последовательность фактора IX соединением 0,16 пикомоля синтетического адапторного фрагмента PstI-CfoI, 0,414, пикомоля фрагмента CfoI-Ba H1 фактора IX длиной 1,4 килобазы из FIX-pu C13 и 0,14 пикомоля векторного фрагмента Bam H1-PstI pUC13 длиной 2,7 килобазы в 20 мкл реакционной среды, содержащей 60 мМ трис-TCI, pH 7,5; 10 мМ хлорида магния, 10 мМ DTT и 0,9 ед. T4-лигазы. Реакционную смесь инкубируют 3 час при комнатной температуре и используют для трансформирования E. coli J M83. Клетки помещают на пластинки с 50 мкл 2% X gal (5-бром-4-хлор-3-индонил-β-D-галактозид) в h-бульсы, содержащий 40 мкг/мл ампициллина, и инкубируют при 37oC в течение ночи. Белые колонии отбирают на другую пластинку, содержащую ампициллин, и выращивают при 37oC в течение ночи. Колонии помещают на бумагу Ватман 540, и бумагу готовят для гибридизации. Бумагу инкубируют при 44oC в течение 2 час в 0,9 M NaCl, 0,09 M Трис-HCl, pH 7,5 6 мM ЭДТУК, 0,5% Nonidit P-40, 150 мкг/мл т-РНК. E.Coli Бумагу метят 32P меченным C235 (Таблица I), 14-мером, специфическим для измененной 5'-концевой последовательности. Гибридизацию с 1 - 2•106 имп/мин на фильтр проводят при 44oC в течение ночи в буфере предгибридизации. Затем фильтры промывают 3 раза в 6 х SSC, 0,1% SDS при 4oC и 3 раза в 2 х SSC, 0,1% SDS при 44oC и помещают на рентгеновскую пленку. Обнаруживают два положительных клона. Один из этих клонов обозначают FIX (-G) _→ pUC13.
Для подтверждения последовательности измененного участка фактора IX, входящего в конструкцию FIX (-G) _→ pUC13, проводят секвентирование непосредственно плазмиды с использованием обратного праймера BRh и с использованием конца праймера, меченного полинуклеотидкиназой и γ32 P АТР. Последовательность соответствует предсказанной.
Результирующая рекомбинантная плазмида содержит три сайта расщепления Hae III, первый в положении 39 последовательности фактора IX (нумерация основана на последовательности, опубликованной Anson и др., (см), начиная с первого ATG), второй в положении 130 и третий в полилинкере pUC13. Сайт в положении 130 является одной парой оснований выше кодонов для сайта расщепления hys-Arg молекулы фактора IX. В результирующей конструкции гибрида фактор IX-фактор VII лидерная последовательность Фактора IX, оканчивающаяся в положении 39 или 130, соединена с синтетическим двунитевым фрагментом, содержащим предсказанную согласованную последовательность и последние 3 кодона лидерной последовательности фактора IX.
Синтетический согласованный фрагмент получают соединением олигонуклеотидов ZC286 - ZC289 (таблица I) с образованием двунитевого фрагмента. По 100 пикомолей каждого олигонуклеотида лиофилизуют и суспендируют в 10 мкл 1 х киназного буфера и инкубируют в течение ночи при 4oC, затем нагревают при 65oC в течение 10 мин. Обработанные киназой олигонуклеотиды делят на две части. Первая часть содержит ZC286 + ZC287, часть 2 содержит ZC288 + ZC289. Соединенные пары ренатурируют 10 мин при 65oC, охлаждают до комнатной температуры в течение 2 час помещают на лед на 30 мин.
Фрагмент модифицированного фактора IX удаляют из FIX3 (-G) pU C13 в виде фрагмента Hind III-EcoRI. Приблизительно 20 мкг плазмиды расщепляют 30 ед каждого из Hind III и EcoRI в 100 мкл буфера Hind III (BRh), содержащего 4 мкг РНК-азы A, при 37oC, в течение ночи. Реакцию обрывают нагревом при 65oC в течение 10 мин, и вектор и фрагменты фактора IX подвергают электрофорезу на 10% агарозном геле и очищают электроэлюированием. Фрагмент фактора IX осаждают этанолом, суспендируют в буфере, содержащем РНК-азу A в концентрации 400 нкг/мкл и расщепляют 9 ед. Hae III в течение ночи при 37oC. Фрагмент Hind III - Hae III Фактора IX длиной 39 пар оснований выделяют из этого перевара электрофорезом на 1,5% агарозном геле с последующим электроэлюированием. Для получения фрагмента фактора IX Hind III-Hae III длиной 130 пар оснований FIX - pUC13 расщепляют с помощью EcoRI и Hind III и фрагмент фактора IX выделяют так, как описано выше. Приблизительно 3 мкг этого фрагмента Hind III - EcoRI расщепляют 6 ед. Hae III при 37oC и в раствор, содержащий 50 мM ЭДТУК, отбирают аликвоты с интервалами 5 мин в течение 30 мин. Аликвоты соединяют и фрагмент Hind III - Hae III длиной 130 пар оснований очищают электрофорезом на 5% акриламидном геле с последующим электроэлюированием.
Результирующие гибридные согласованные последовательности фактора IX получают соединением четырех частей, олигонуклеотидных частей 1 и 2, фрагмента Hind III-Hae III фактора IX (длиной 39 или 130 пар оснований) и фрагмента Hind III-EcoRI и pUC13. Результирующие плазмиды используют для трансформирования E. coli HB101 (АТСС 33684). Колонии отбирают расщеплением ДНК с помощью EcoRI и Hind III. Последовательность, содержащую участок фактора IX длиной 39 пар оснований, соединяют с синтетической согласующей последовательностью и эту конструкцию далее называют мини-FIX-FVII. Плазмиду, содержащую такую конструкцию, обозначают pM7200 (-C). Последовательность, содержащую последовательность фактора IX длиной 130 по, соединенную с синтетической согласующей последовательностью, обозначают м макси-FIX-FVII. Плазмиду, содержащую эту конструкцию, обозначают pM7100(-C). Согласующая последовательность кодирует полипептид, имеющий следующую аминокислотную последовательность:
Ala-Asn-Ala-Phe-Leu-Gln-Arg-Pro-Gly-Sir-Ley-Gla-Arg-Gla-Cys-hys-Gla-Gln-Cys-Ser-Phe-Gla-Aca-Arg-Gla-Ile-Phe-Gla-Gly-Ley-Asn-Arg-Thr-hys-Ley
B. Присоединение гибридного фрагмента согласованной последовательности фактора IX к клону к-ДНК фактора VII.
Гибриды согласованного участка Фактора IX (либо мини, либо макси) присоединяют к 5'-участку к-ДНК фактора VII и вектору pUC13 связыванием трех частей (рисунки 4 и 5). Фрагмент вектора получают расщеплением 6 мкг pUC13 10 ед. каждого из Xba I и Hind III в Hind III-буфере, содержащем РНК-азу A (400 нг/мкл). Фрагмент мини- FIX-FVII получают расщеплением 2 мкг pM7200 (-C) 10 ед. каждого из Hind III и EcoRI, как указано выше. Фрагмент макси-FIX-FVII аналогичным образом получают из pM7100 (-C). 5'-Участок к-ДНК фактора VII получают из плазмиды (pu CC705), содержащей 5'- фрагмент EcoRI- Xba I pUCVII2115, субклонированной в pUC13 расщеплением с помощью XbaI и EcoRI. Расщепление проводят при 37oC в течение 2 час и продукты разделяют электрофорезом на 1,5% агарозном геле. Целевые фрагменты электроэлюируют, экстрагируют смесью фенол/ хлороформ и хлороформом и осаждают этанолом. Три фрагмента, а именно pUC13 / Xba I-Hind III, фактор IX - фактор VII (мини или макси) (Hind III-EcoRI) и 5' - фактор VII-/EcoRI-XbaI связывают, затем в 20 мкл лигазного буфера, содержащего 2 мкл 20 мM АТР и 0,9 ед T4-ДНК-лигазы в течение ночи при 4oC. Колонии отбирают рестрикционным анализом с помощью Hind III и Xba I. Рекомбинантные плазмиды, содержащие мини- и макси- FIX - FVII последовательности, обозначают, соответственно, pM7200 и pM7100 (рисунок 4).
Так как при получении к-ДНК фактора VII использовалось прибавление линкера, то нужно провести модификации слитых последовательностей для получения правильной структуры кодирующих последовательностей. Как мини-, так и макси- стыки содержат сайт EcoRI в соединении между гибридом согласованной последовательности фактора IX и к-ДНК фактора VII, и это артефакт реакции клонирования к-ДНК. Кроме того, мини-стык требует прибавления C для изменения последовательности в сайте Hae III от 5′AGG CCA3′ до 5′AGG CCCA3′ и установления правильной структуры считывания ниже этой последовательности. Эти корректирующие изменения проводят нуклеотиднаправленным сайт-специфическим мутагенезом. Фрагмент мини-FIX-FVII удаляют из pM7200 расщеплением с помощью Hind III и XbaI и вставляют в M13 m p19. Фрагмент макси-FIX-FVII очищают от pM 7100 и субклонируют аналогичным образом. Мутагенные праймеры ZC333 и ZC336 (см. таблицу I) используют для удаления сайта EcoRI и, соответственно, вставки основания. В каждом случае используют универсальный праймер ZC87 в качестве второго праймера. Мутагенные праймеры фосфорилируют, соединяя 40 пикомолей праймера и 60 пикомолей АТР с 1 ед. T4-ДНК-киназы, на ночь при 60oC. Для удаления сайта Eco RI из гибрида макси-FIX-FVII, 1 мкг однонитевой матрицы M13 соединяют с 20 пикомолями каждого из ZC333 и ZC87 в общем объеме 10 мкл. Праймеры ренатурируют на матрице в течение 10 мин при 65oC, охлаждают до комнатной температуры в течение 5 мин, затем помещают на лед на 5 мин. Праймеры увеличивают с помощью ДНК-полимеразы I (фрагмент Klenow). Для удаления сайта EcoRI и корректировки структуры считывания в гибриде мини- FIX-FVII, 1 мкг подходящей однонитевой матрицы M13 соединяют с 20 пикомолями каждого из ZC333, ZC336 и ZC87. Реакции ренатурации и увеличения праймера проводят так, как описано выше. Пятна отбирают с помощью 32P - меченного праймера (ZC333 или ZC336) при 60oC и подтверждают последовательности дидезокси-секвентированием. Результирующие конструкции, содержащие макси- и мини-FIX-FVII последовательности, обозначают соответственно pM7111 и pM7211.
Согласованная последовательность содержит несколько участков, не совпадающих с данными по последовательности белка, полученными для фактора VII (рисунок 2). Для получения последовательности, кодирующей полипептид, обладающий большей гомологичностью к аминоконцевому участку Фактора VII, согласованную последовательность изменяют олигонуклеотиднаправленным сайт-специфическим мутагенезом. Вводят изменения, заключающиеся во введении лейцина в положение 8, замена лизина на изолейцин в положении 18 (цифры соответствуют позиции аминокислоты после введения в позицию 8), аланина на аспарагин в позиции 26 и последовательности Gly-Leu-Asn на Ala-Ser-Asp в позициях 32 - 34 (на основании предварительных данных по аминокислотной последовательности).
Изменения последовательности в положениях 8 и 18 проводят с использованием PM7111 (принимающая нить) в качестве матрицы. Праймеры ZC352 (5′CCC AGG TCT CAG CTC CTC CAG3′) и ZC353 (5′CTG CTC CTC CTT ACA CTC TCT3′) ренатурируют на матрице и расширяют так, как описано выше. Результирующий клон фага обозначают pM7114. Последовательность вставки в pM7114 подтверждают дидезокси-секвентированием.
Аналогичным образом вносят изменения в положения 26 - 34 в матрице pM7114 (принимающая нить) с использованием мутагенного праймера Z 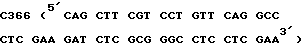 и ZC87 (таблица 1) в качестве второго праймера. Результирующую конструкцию обозначают pM7115. Последовательность всех 550 по вставки в векторе M13 определяют дидезокси - секвентированием устанавливают, что она является правильной.
и ZC87 (таблица 1) в качестве второго праймера. Результирующую конструкцию обозначают pM7115. Последовательность всех 550 по вставки в векторе M13 определяют дидезокси - секвентированием устанавливают, что она является правильной.
Пример 5. Конструирование слитой к-ДНК фактор IX - Фактор - VII
Слитую к-ДНК фактор VII получают с использованием к-ДНК фактора IX, полученной из библиотеки к-ДНК печени человека и последовательности к-ДНК фактора VII, описанной в примере I.
Точка слития, выбранная для гибридного белка, находится между аминокислотой +38 (треонин) фактора IX и первым лизином, закодированным последовательностью к-ДНК Фактора VII. Такой белок может быть закодирован последовательностью, состоящей из первых 252 по последовательности к-ДНК фактора IX и всей последовательности к-ДНК фактора VIIpUCVII2115, за исключением первых двух кодонов. Для конструирования этой гибридной последовательности последовательность фактора IX сначала сливают с pUCVII2115 с использованием обычных рестрикционных сайтов. Это связывание приводит к плазмиде FIX/VII/12 (описано выше), содержащей первые 310 по к-ДНК фактора IX, соединенные с полной к-ДНК фактора VII. Для достижения точной стыковки, нужной для гибридного белка, инвертированные пары оснований удаляют олигонуклеотиднаправленным мутагенезом.
Соединение последовательности к-ДНК фактора IX с последовательностью к-ДНК фактора VII достигается связыванием фрагмента Hind V III-A а III длиной 0,3 килобазы из FIX (-G) _→ pU C13 (пример 4) с фрагментом Sma - Hind III длиной 4,7 килобазы из pUCVII2115 (рисунок 5). Фрагмент Hind III - Aha III получают расщеплением 3 мкг FIX (-G) _→ pUC13 40 ед. Hind III в 40 мкл среднего солевого буфера (Maniatis и др., см) при 37oC в течение 4 час. Объем затем увеличивают до 100 мкл солевого буфера и прибавляют 5 ед. Aha III и инкубирование продолжают при 37oC в течение 18 час. Фрагменты ДНК отделяют электрофорезом на 1% агарозе и полосу 0,3 килобазы выделяют так, как описано выше. Частичное Sma I - расщепление pUCVII2115 проводят инкубированием 3 мкг pUCVII2115 при 25oC в течение 1 час с 4,8 ед. Sma I в реакционном объеме 30 мкл. Реакцию обрывают инкубированием в течение 15 мин при 65oC. Затем образец экстрагируют один раз равным объемом фенола и осаждают этанолом.
Осадок собирают 10 мин микроцентрифугированием, промывают 70% этанолом и сушат на воздухе. ДНК снова растворяют в 30 мкл среднего солевого буфера и разлагают 30 ед. Hind III при 37oC в течение 3 ч. ДНК подвергают электрофорезу на 0,7% агарозе и фрагмент Hind III - Sma I длиной 4,7 килобазы выделяют так, как описано выше. Эквимолярные количества двух фрагментов (0,048 пикомоля) связывают в 10 мкл реакционной среды, содержащей 50 мM Трис-HCl, pH 7,5, 10 мM хлорида магния, 1 мM DTT, 1 мM АТР и 3 ед. T4-ДНК-лигазы при 14oC в течение 3,5 ч и затем используют для трансформирования E. coli RRI (АТСС 31343).
Клетки выращивают на ампициллиновых пластинках и 12 из результирующих колоний отбирают рестрикционным ферментативным расщеплением на присутствие целевой плазмидной конструкции. ДНК из колонии 12 (FIX/VII/12) дает ожидаемую модель рестрикционного ферментативного расщепления и используется в следующей стадии конструирования гибридного гена.
Олигонуклеотиднаправленный мутагенез проводят на матрице однонитевой ДНК. Таким образом, необходимо клонировать слитые последовательности фактора IX/ Фактор VII в M13 р19.
Для получения удобного небольшого фрагмента ДНК фрагмент Hind III - Xba I длиной 640 пар оснований выделяют из FIX-VII/12. Этот фрагмент содержит 310 пар оснований 5'- конца - к-ДНК фактора IX и 330 пар оснований последовательности фактора VII. Получают вектор расщеплением 1 мкг ДНК M13mp19 RF с помощью 20 ед. Hind III и 20 ед. Xba I в 40 мкл среднего солевого буфера при 37oC в течение 18 час. ДНК подвергают электрофорезу на 1,2% агарозе и из геля выделяют фрагмент длиной 6,4 килобазы, как описано выше. 5 мкг ДНК FIX/VII/12 расщепляют 10 ед. Xba 1 в 40 мкл среднего солевого буфера при 37oC в течение 18 час. Добавляют 12 ед. Hind III и расщепление продолжают при 37oC еще в течение 7 час. Результирующие фрагменты разделяют электрофорезом на 1,2% агарозе и фрагмент 640 по элюируют так, как описано выше. 10 нг линеаризованного M13 m p19 и 1 нг фрагмента 640 по связывают при 14oC в течение 1 час и затем используют для трансформирования E. coli YM101. Клетки покрывают X - gal и IPTG, и 8 синих пятен отбирают и используют для заражения 2,5 мл культуры E. coli JM103 при A = 0,3. После 18 час выращивания при 37oC клетки собирают центрифугированием в клинической центрифуге при комнатной температуре и 20 мкл супернатанта, содержащего фаг M13, смешивают с 10 мкг/л этидий бромида. По сравнению с известными стандартами каждый из 8 клонов имеет вставку примерно нужного размера. Затем получают однонитевую ДНК из 1,5 мл супернатанта. Эту конструкцию затем секвентируют дидезоксиспособом с использованием олигонуклеотида ZC87 в качестве праймера для подтверждения того, что соединение вставки правильно. Один из правильных клонов (# 4) используют в качестве матрицы в олигонуклеотиднаправленном мутагенезе для получения функционального слития фактор IX - фактор VII.
Олигонуклеотид ZC249, 20-мер, состоящий из 10 по целевой последовательности фактора IX и 10 по целевой последовательности фактора VII (таблица I) используют в качестве мутагенного праймера. В качестве второго праймера используют олигонуклеотид ZC87, гибридизующий последовательность M13mp19.
Использованная методика мутагенеза является модификацией методики Zoller и Smith (см). Для ренатурации 20 пикомолей ZC249 фосфорилируют инкубированием в течение ночи при 4oC в 20 мкл 60 мК Трис-HCl, pH 8,0, 10 мM хлорида магния, 1 мM DTT, 1 мM АТР, 1 ед. T4-киназы. Реакцию обрывают инкубированием при 65oC в течение 15 мин и образец лиофилизуют. Один пикомоль однонитевой матрицы клона # 4 и 20 пикомолей ZC87 прибавляют в 10 мкл ренатурирующего буфера (200 мM Трис-HCl, pH 7,5, 100 мM хлорида магния, 500 мM хлорида натрия, 10 мM D TT). Образец нагревают при 65oC в течение 10 мин, инкубируют при комнатной температуре в течение 5 мин и затем помещают на лед. К образцу прибавляют 10 мкл следующего свежеприготовленного раствора: 20 мM Трис-HCl, pH 7,5, 10 мM хлорида магния, 10 мM DTT, 1 мM diTPS, 1 мM АТР, 0,15 ед/мкл T4-ДНК-лигазы, 0,25 ед/мкл ДНК-полимеразы I E. coli (фрагмент Klenow). Затем реакционную смесь инкубируют при 15oC в течение 3 час и образец используют для трансформирования E. coli JM101.
Результирующие пятна наносят на нитроцеллюлозу и отбирают гибридизацией на 32P-меченном ZC249. Сухие фильтры BA85 (0,45 мкм) наносят на пластинки агара, и фагу позволяют абсорбироваться в течение 5 мин. Фильтры удаляют, сушат в течение 5 мин, помещают на бумагу Ватман 3 мM, насыщают в 0,5 M NaOH, 1,5 M NaCl в течение 5 мин, сушат на воздухе 3 мин, помещают на бумагу Ватман, насыщают в 1 м Трис-HCl, pH 8,1, 5 M NaCl в течение 5 мин и сушат на воздухе 3 мин. Стадию с Трис-HCl повторяют и фильтры промывают в 100 мл 6 х SSC в течение 2 мин при комнатной температуре. После высушивания на воздухе фильтры выдерживают при 80oC в течение 2 час и предгибридизируют при 47oC (Tm - 4 для ZC249) в течение ночи в 6,7 х SSC, pH 6,5, 2 мг/мл т-РНК E. Coli и 0,2% (вес./объем) каждого из БСА Ficoll и поливинилпиролидина.
После предгибридизации фильтры инкубируют при 2,5•106 имп/ фильтр меченным ZC249 в том же самом буфере гибридизации SSC при 47oC в течение ночи. После гибридизации фильтры промывают 3 раза по 5 - 10 мин при комнатной температуре в 6 х SSC и помещают на рентгеновскую пленку. Пятна с предварительным положительным результатом снова помещают на пластинки и отбирают так, как описано выше. Затем отбирают индивидуальные пятна и получают однонитевую ДНК и секвентируют с использованием ZC275 в качестве праймера. Олигонуклеотид ZC275 соответствует последовательности 40 по в 5'-направлении ZC249 в той же самой нити (таблица I).
Идентифицируют 4 положительных пятна. Полную вставку в M13mp19 для одного клона (FIX/VII - 9) секвентируют дидезокси-способом с использованием олигонуклеотидов ZC87 и ZC275 и устанавливают ее правильность. Подтвержденная последовательность представлена основаниями 1 - 567 на рисунке 7. Затем PF ДНК из этого клона используют для заключительной стадии конструирования гибридного гена.
Используют три фрагмента для изготовления финальной конструкции: фрагмент 0,6 килобазы Hind III - XbaI из FIX /VII-9, содержащий слитые последовательности FIX/VII; фрагмент к-ДНК фактора VII XbaI-BamH1 длиной 1,7 килобазы из pUCVII1923, и фрагмент 2,7 килобазы Bam H1-Hind III из pUC13.3 мкг FIX/VII-9 (PF -ДНК) расщепляют при 37oC в течение 6 час с помощью 45 ед. XbaI в объеме 50 мкл. ДНК осаждают этанолом, суспендируют и расщепляют при 37oC в течение 4 час 50 ед. Hind III. Образец подвергают электрофорезу на 1% агарозе и полосу 0,6 килобазы электроэлюируют на бумаге NA 45 (Schliecher Schuell). ДНК элюируют из бумаги 1,5 M NaCl, 1,50 мM Трис- HCl pH 8, 1 мM ЭДТУК, экстрагируют фенолом и осаждают этанолом.
Для получения оставшейся последовательности к-ДНК фактора VII 5 мкг pUCVII1923 расщепляют при 37oC в течение 3 час 36 ед. XbaI в 40 мкл среднего солевого буфера. Затем прибавляют 8 мкл 10 x крепкого солевого буфера, 28 мкл воды и 4 мкл (40 ед) BamHI в реакционную смесь инкубируют при 37oC в течение 3 час. Фрагменты ДНК разделяют электрофорезом в 1% агарозе и выделяют фрагмент 1,7 килобазы, так как описано выше.
Фрагмент вектора получают расщеплением 1 мкг pUC13 10 ед. Hind III в 20 мкл среднего солевого буфера при 37oC в течение 1 час. Затем прибавляют 1 мкл 10 x крепкого солевого буфера и 10 ед Bam HI и инкубирование продолжают еще в течение 2 час, ДНК очищают на 1% агарозном геле так, как описано выше.
Эквимолярные количества (приблизительно 0,56 пикомоля) трех фрагментов связывают при комнатной температуре в течение 45 мин в 10 мкл 50 мM Трис-HCl, pH 7,5, 10 мM хлорида магния, 1 мM D TT, 1 мM АТР и 3 ед. T4-ДНК-лигазы.
Реакционную смесь используют для трансформирования E. coli JM 83. Клетки помещают на среду, содержащую 40 мкг/мл ампициллина с 50 мкл 2% X-gal, добавленными в каждую выемку. ДНК получают из 7 белых колоний и затем отбирают расщеплением рестрикционными ферментами. Один из клонов, обнаруживающий правильную структуру, обозначается FIX/VII _→ pUC13.
Пример 6: Экспрессия биологически активных аналогов Фактора VII
Вектор pD2 экспрессии клеток млекопитающих был выбран для экспрессии гена FIX/VII в трансформированных клетках животных. Его конструируют из плазмиды pDHFR-III следующим образом. Сайт Pst/1, примыкающий к DHFR-к-ДНК в pUHFR III превращают в сайт BamH1 обычным связыванием. ДНК p D HFR III инкубируют с 10 мM трис с pH 7,6, 6 мM β_MSH, 6 мM NaCl, 10 мM хлорида магния, 2,5 ед. Pst I в течение 10 мин при 37oC с последующей экстракцией фенолом и осаждением этанолом. Липкий конец Pst I затупляют с использованием T4-ДНК- полимеразы. После экстракции фенолом и диализа против 10 мM трис-, pH 8,0, 1 мM ЭДТУК, 0,3 M NaCl, ДНК, осаждают этанолом. ДНК суспендируют в 20 мкл 1,4 мM АТР, 50 мM Трис, pH 7,6, 10 мM хлорида магния, 1 мM дитиореитола и затем инкубируют с 5 нг линкеров BamH1, обработанных T4-полинуклеотидкиназой (New England Biolabl.) и 200 ед. T4-полинуклеотидлигазы в течение 12 час при 12oC с последующей экстракцией фенолом и осаждением этанолом. ДНК расщепляют 90 ед. BamH1 при 37oC в течение 1 час и подвергают электрофорезу на 1,4% агарозном теле. Фрагмент ДНК 4,9 килобазы (соответствует ДНК p D HFR III, лишенной к-ДНК DHFR и сигнала полиадениляции SV40) электроэлюируют и рециркуляризуют с помощью полинуклеотидлигазы, после чего заражают им E. coli HB101. Колонии, чувствительные к ампициллину, отбирают экспресс-анализом, и правильный клон выращивают для получения большого количества препарата плазмидной ДНК.
Результирующую плазмиду расщепляют 20 ед. Bam H1 и обрабатывают 2,5 мкг бычьей кишечной фосфатазы, и подвергают электрофорезу на 1,4% агарозном геле. 25 мкг pSV40 (клон ДНК SV 40, введенный в сайт Bam H1 pBR322) расщепляют 25 ед. Bcl I в течение 1 час при 50oC, затем дополнительно 25 ед. Bam H1 и инкубируют еще в течение 1 час при 37oC. Эту ДНК подвергают электрофорезу на 1,4% агарозном геле. Расщепленный Bam H1 вектор (т.е. лишенный сигнала полиадениляции) соединяют с фрагментом ДНК SV 40 (0,14 - 0,19 единиц карты), содержащим сигнал поздней полиалениляции инкубированием очищенных на геле фрагментов (0,1 мг каждого) в 20 мкл 50 мM Трис, pH 7,6, 10 мM хлористого магния, 1 мM дитиотреитола, 1,4 мM АТР и 100 ед. T4-полинуклеотидлигазы в течение 4 час при 12oC, с последующим трансформированием в E. coli RRl. Положительные колонии идентифицируют экспресс-анализом и получают большое количество плазмидного препарата правильной ДНК, pD2.
Для изготовления конструкции экспрессии фактора IX/VII, 1 мкг pD2 расщепляют при 37oC в течение 1 ч с 20 ед. BamH1 в 20 мкл крепкого солевого буфера. Добавляют 20 мкл 10 мM Трис- HCl, pH 8, 1 мM ЭДТУК и 0,1 ед. бычьей щелочной фосфатазы (Boeringer). Реакционную смесь инкубируют при 37oC в течение 1 часа и реакцию обрывают нагревом до 75oC в течение 10 мин 10 мкг FIX/VII _→ pUC13. Расщепляют при 37oC в течение 2 ч с 150 ед. BamH1 в 150 мкл крепкого солевого буфера. Фрагменты ДНК разделяют электрофорезом на 1,2% агарозе и выделяют фрагмент 2,3 килобазы. Эквимолярные количества (0,015 пикомоля) фрагмента BamH1,2,3 килобазы и фрагмента вектора p D 2 связывают при 14oC в течение 2,5 ч так, как описано выше. Реакционную смесь используют для трансформирования клеток E. Coli RRl, которые затем помещают в среду, содеожащую 10 мкг/мл ампициллина. Плазмидную ДНК получают из 12 результирующих колоний и отбирают расщеплением рестрикционными ферментами. Один из клонов, имеющий правильную карту рестрикции, обозначается FIX/VII/pD2 (рисунок 6) E.coli RRl, трансформированные FIX/VII/pD2, депонированы в АТСС под номером 53068.
Методика, использованная для трансформирования клеток почек молодых хомяков (ВНК) (получено из Американской Коллекции типовых Культур) (номер CCL 10) с помощью FIX/VII/pD2, аналогична стандартным методам. Клетки ВНК выращивали при 37oC, 5% CO2, в среде Дульбекко (с добавкой 10 термоинактивированной бычьей зародышевой сыворотки, глутамина и пенициллина-стрептомицина) в чашке Петри диаметром 60 мм для тканевых культур до заполнения 20%. В целом 10 мкг ДНК используют для заряжения одной 60 мм чашки, 3,75 мкг FOX/VII/pD2 2, 1,25 мкг pK0 - neo и 5 мкг ДНК из спермы лосося. ДНК осаждают в 0,3 М ацетата натрия, 75% этаноле, промывают 70% этанолом и снова растворяют в 20 мл 10 мМ Трис-HCl, pH 8, 1 мМ ЭДТУК, ДНК соединяют с 440 мкл воды, 500 мкл 250 мМ NaCl, 1,5 мМ NaHPO4. 12 мМ декстрозой и 50 мМ HEPES pH 7,12, 60 мкл 2 М CaCl2 по каплям добавляют при комнатной температуре в течение 30 мин. Затем раствор прибавляют к клеткам и клетки выдерживают 4 часа при 37oC. Среду удаляют и в течение 2 мин при комнатной температуре прибавляют 5 мл 20% ДМСО в среде Дульбекко с сывороткой. Затем чашку быстро промывают двумя сменами среды и инкубируют в свежей среде в течение ночи. Через 24 часа после прибавления ДНК среду удаляют и прибавляют селективную среду (10 мг/мл G418, 498 мкг/мг, Gibco в среде Дульбекко с сывороткой), через 10 и 13 дней индивидуальные клоны, представляющие собой клетки, имеющие введенный pK0-nеоген и вследствие этого устройства к G 418, переносят на пластинки с 96 углублениями (или с 24 углублениями) и выращивают для анализа на белок.
Клетки выращивают в среде Дульбекко плюс 10% плодной телячьей сыворотки, содержащей 5 мкг/мл витамина K (Phytonadion Merck). Среду отделяют от клеток и клеточных остатков центрифугированием и анализируют на полипептид Фактора VII (с помощью ELISA) и на биологическую активность. Клетки удаляют с пластинок трипсином, промывают свежей средой, центрифугируют и замораживают при -20oC. Клеточные гранулы затем оттаивают в PBS, гранулируют и суспендируют в PBS, содержащей 0,25% Тритона X-100. Образцы разбавляют и анализируют на полипептид и активность.
ELISA-Анализ на Фактор VII проводят следующим образом. 200 мкл моноклонального антитела против человеческого фактора VII (5 мкл/мл в 0,1 M карбонате натрия, pH 9,6) инкубируют в каждом углублении микротитровальной пластины с 96 углублениями в течение 2 ч при 37oC. Затем углубления инкубируют с 220 мкл 1% бычьего сывороточного альбумина (БСА) и 0,05% Tween в PBS, pH 7,2, 2 ч при 37oC. Пластинки промывают водой, сушат на воздухе и хранят при 4oC. Для анализа образцов 200 мкл образца инкубируют 1 час при комнатной температуре в углублениях, покрытых антителом. Затем углубления промывают 4 раза 200 мкл PBS, содержащего 0,05 Tween 20. Затем углубления инкубируют в течение 1 ч при комнатной температуре с 200 мкл IgG фракции кроличьей поликлональной антисыворотки против Фактора VII ( 5 мкг/мл в PBS, содержащем 1% БСА и 0,05% Tween 20). Затем проводят инкубирование с козьими антикроличьими IgG вместе с щелочной фосфатазой. Эти углубления промывают затем четыре раза PBS, содержащей 0,05% Tween 20. В углубления добавляют 200 мкл пара-нитрофенилфосфата (30 мг), растворенного в диэтаноламиновом буфере (96 мл в литре), pH 9,8, содержащем 56 мг/л хлористого магния. Ферментативную реакцию проводят при 37oC и появление желтого цвета регистрируют при 405 нм с использованием монитора пластины ELISA. Результаты, полученные для клеточных сред, представлены в таблице 3.
Фактор VII, полученный трансфецированными клетками, может быть извлечен из клеточных культуральных сред путем адсорбции к цитрату бария. Истощенную среду смешивают с цитратом натрия и хлоридом бария, и преципитат собирают. Осажденное вещество затем можно исследовать на присутствие подходящего коагулирующего фактора. Дальнейшую очистку можно осуществить посредством иммуноадсорбции. Предпочтительно, чтобы иммуноадсорбционная колонка содержала высокоспецифическое моноклональное антитело. Альтернативно, очистку барий-цитрат-осажденного вещества можно осуществить при помощи более традиционных биохимических методов или путем высокоэффективной жидкостной хроматографии (ВЭЖХ).
D) Конверсию одноцепочечного фактора VII в активный двухцепочечный фактор VIIa можно осуществить с использованием фактора XIIa, что приводит к получению белка, который имеет такую же или, в основном, такую же биологическую активность относительно коагуляции крови, что и человеческий фактор VIIa или другие протеазы, имеющие трипсин-подобную специфичность.
Биологическую активность фактора VII анализируют с использованием одноэтапного анализа коагулирующей активности в соответствии с обычными способами. Результаты, полученные для клеточных сред, приведены в Таблице 3.
Пример 7: Экспрессия фактора IX
14 мкг FIX (G) _→ pUC13 расщепляют 30 ед. BamHI в 30 мкл крепкого солевого буфера в течение 3 ч при 37oC. Затем ДНК подвергают электрофорезу на 1% агарозе и из геля выделяют полосу 1,4 килобазы, содержащую последовательность Фактора IX.
3 мкг Вектора pD2 расщепляют 30 ед. BamHI в 30 мкл крепкого солевого буфера в течение 3 ч при 37oC, ДНК подвергают электрофорезу на 1% агарозе и выделяют линейный фрагмент 1,5 килобазы. Затем ДНК обрабатывают 0,12 ед. бычьей щелочной фосфатазы в 30 мкл 10 мМ Трис-HCl с pH 8 N 1 мМ ЭДТУК в течение 30 мин при 37oC. Концентрацию соли доводят до 0,3 М ацетата натрия и образец дважды экстрагируют фенолом, один раз хлороформом и осаждают ДНК этанолом. Осадок промывают 70% этанолом, сушат и растворяют в 20 мкл 10 мМ Трис-HCl, pH 8, 1 мМ ЭДТУК. Эквимолярные количества (0,02 пикомоля) двух фрагментов с помощью 10 ед. T4-ДНК-лигазы, как описано выше. Реакционную смесь используют для трансформирования клеток E.coli RRl. ДНК из 12 результирующих устойчивых к ампициллину колоний отбирают разложением рестрикционными ферментами. Один из клонов, с фрагментом 1,4 килобазы, введенным в нужной ориентации, обозначают как FIX/-G//pD2. Клетки E.coli RRl, трансформированные FIX/-G//pD2 депонируют в АТСС под номером 53067.
Клетки ВНК совместно трансформируют FIX/-G//pD2 и pKOeo как описано выше. Устойчивые к лекарству клетки выбирают и готовят для анализа ELISA и активности так, как описано в примере 6.
Анализ на биологическую активность основан на способности Фактора IX уменьшать время свертывания плазмы от пациентов с дефицитом фактора IX до нормального значения. Это осуществляют так, как описано Proctor и Rapaport Amer. J. Clin Path, 36: 212, 1961. Результаты представлены в таблице 4.
Количество полипептида Фактора IX определяют с помощью ELISA в основном так же, как описано в примере 6, с использованием поликлональной кроличьей антисыворотки к Фактору IX. После инкубирования углублений с образцами, содержащими Фактор IX, углубления промывают и инкубируют 1 час при комнатной температуре с 200 мкл аффинно очищенного кроличьего поликлонального анти-Фактора IX вместе с щелочной фосфатазой, с разбавлением 1 : 1000 в PBS, содержащем 1% БСА и 0,05% Tween 20. Затем углубления промывают четыре раза с PBS, содержащим 0,05% Tween 20, и прибавляют субстрат фермента так, как описано выше. Инкубирование проводят при 4oC в течение ночи или 2 ч при 37oC.
Как показано в таблице 4, 70 - 80% Фактора IX (полипептида) выделяется в среду, и приблизительно 50% этого количества биологически активно. В клеточном осадке активность Фактора IX отсутствует.
Наибольшие уровни активности достигаются при прибавлении к клеточной культуральной среде витамина K (фитонадион, Merck) к концентрации 1 - 10 мг/мл.
Несколько дополнительных анализов проводят для демонстрации того, что клетки вырабатывают аутентичный Фактор IX. Образцы, содержащие по данным описанного выше анализа активность Фактора IX, инкубируют с плазмой, лишенной Фактора VIII, при этом не наблюдают изменения времени свертывания, и это указывает на то, что активность обусловлена аутентичным Фактором IX, а не неспецифическим свертывающим агентом. Этот вывод может быть дополнительно подтвержден удалением активности Фактора IX из образцов с помощью специфического антитела. От 97 до 98% активности Фактора IX иммуноосаждается из клеточных супернатантов кроличьим поликлональным антителом против Фактора IX. Это антитело также осаждает свыше 99% активности Фактора IX из нормальной плазмы. Активность Фактора IX не снижается в супернатантах прибавлением кроличьего поликлонального антитела на эритропротеин.
Пример 8: конструирование вектора экспрессии Фактора VII.
Конструируют вектор экспрессии, содержащий 5'-кодирующий участок синтетического Фактора VII, соединенный с частичной к-ДНК фактора VII. Вектор, обозначенный как pM7135, получают вставкой последовательности лидерный участок Фактора IX-5'-участок Фактора VII из pM7115 и 5'-последовательности Фактора VII из FIX/VII/pD2 в плазмиду pD3, содержащую расширитель SV40 и основной поздний промотор аденовируса 2, а также трехчленный лидерный участок.
Плазмиду pD3 получают из плазмиды pDHFR III. Сайт Pst I, расположенный непосредственно выше последовательности DHFR в pDHFR III, превращают в сайт Bcl I расщеплением 10 мкг плазмиды 5 ед. Pst I в течение 10 мин при 37oC в 100 мкг буфера A (10 мМ Трис, pH 8, 10 мМ хлорида магния, 6 мМ NaCl, 7 мМ β_MSH). ДНК экстрагируют фенолом, осаждают этиловым спиртом и суспендируют в 40 мкл буфера B (50 мМ Трис pH 8, 7 мМ хлорида магния, 7 мМ β_MSH), содержащего 10 мМ dCTP и 16 ед. T4-ДНК-полимеразы и инкубируют при 12oC в течение 60 мин. После осаждения этанолом ДНК связывают с обработанными киназой линкерами Bcl I (2,5 мкг) в 14 мкл буфера C (10 мМ Трис pH 8, 10 мМ хлорида магния, 1 мМ DTT, 1,4 мМ АТР), содержащего 400 ед. T4-полинуклеотидлигазы в течение 12 ч при 12oC. После экстракции фенолом и осаждения этанолом ДНК суспендируют в 120 мкл буфера D /75 мМ KCl, 6 мМ Трис pH 7,5, 10 мМ хлористый магний, 1 мМ DTT/, расщепляют 80 ед. Bcl I в течение 60 мин при 50oC и подвергают электрофорезу через агарозу. Форму III плазмидной ДНК (10 мкг) выделяют из геля и связывают в 10 мкл буфера C, содержащего 50 ед. T4-полинуклеотидлигазы в течение 2 ч при 12oC и используют для трансформирования E. coli HB101. Положительные колонии идентифицируют быстрым ДНК-препаративным анализом и плазмидную ДНК (обозначаемую pDHFR') получают из положительных колоний и трансформируют в dAM E. coli.
Затем получают плазмиду pD 2' расщеплением pDHFR' (15 мкг) и p SV40 (25 мкг) в 100 мкл буфера D с 25 ед. Bcl I в течение 60 мин при 50oC, с последующим добавлением 50 ед. BamH1 и дополнительным инкубированием при 37oC в течение 60 мин. Фрагменты ДНК разделяют электрофорезом на агарозном геле и выделяют 4,9 килобазы фрагмент pDHFR и фрагмент SV40, 0,2 килобазы. Эти фрагменты (200 нг ДНК pDHFR и 100 нг ДНК SV40) инкубируют в 10 мкл буфера C, содержащего 100 ед. T4-полинуклеотидлигазы в течение 4 ч при 12oC и результирующую конструкцию (pD2) используют для трансформирования E. coli RRI.
Плазмиду pD2 модифицируют, удаляя "отравляющие" последовательности в участке pBR 322 (Lusky и Botcham, Nature, 293: 79 - 81, 1981). Плазмиды pD2 (6,6 мкг) и pM -1 (Lusky и Botcham, см.) (4 мкг) инкубируют в 50 мкл буфера A с 10 ед. каждого из EcoRI и NruI в течение 2 ч при 37oC, после чего подвергают электрофорезу на агарозном геле. Фрагмент 1,7 килобазы pD2' и фрагмент pML-I 1,8 килобазы выделяют и связывают друг с другом (по 50 нг каждого) в 20 мкл буфера C, содержащего 100 ед. T4-полинуклеотидлигазы в течение 2 ч при 12oC и затем трансформируют в E. coli HB101. Колонии, содержащие целевую конструкцию (обозначенную Δ pD2), идентифицируют быстрым препаративным анализом. Затем 10 мкг Δ pD2 расщепляют 20 ед. каждого из EcoRI и Bgl II в 50 мкл буфера A в течение 2 ч при 37oC. ДНК подвергают электрофорезу на агарозе и выделяют целевой фрагмент 2,8 килобазы (фрагмент C), содержащий pBR 322, сайт сплайсинга 3' и последовательности poly A.
Для получения остальных фрагментов, используемых для конструирования pD3, pDHFR III модифицируют для превращения сайта Sac II (SStII) либо в сайт Hind III, либо в сайт Kph I. Десять мкг pDHFR III расщепляют 20 ед. Sst II в течение 2 ч при 37oC, затем экстрагируют фенолом и осаждают этанолом. Ресуспендированную ДНК инкубируют в 100 мкл буфера B, содержащего 10 мМ dCTP и 16 ед. T4-ДНК-полимеразы в течение 60 мин при 12oC, экстрагируют фенолом, диализуют и осаждают этанолом. ДНК (5 мкг) связывают с 50 нг обработанных киназой линкеров Hind III или Kpn I в 20 мкг буфера C, содержащего 400 ед. T4-ДНК-лигазы, в течение 10 ч при 12oC, экстрагируют фенолом и осаждают этанолом. После суспендирования в 50 мкл буфера A результирующие плазмиды расщепляют 50 ед. Hind III или Kpn I и подвергают электрофорезу на агарозе. Выделенную из геля ДНК (250 нг) связывают в 30 мкл буфера C, содержащего 400 ед. T4-ДНК-лигазы, в течение 4 ч при 12oC и используют для трансформирования E. coli PPI. Результирующие плазмиды обозначают как pDHFR III (Hind III) и pDHFR III (Kpn I). Затем фрагмент KpnI-Bgl 11,700 по (фрагмент A) очищают от pDHFR III (KpnI) расщеплением с помощью BglII и KpnI электрофорезом на агарозном геле.
Расширяющую последовательность SV40 вставляют затем в pDHFR III (Hind III) следующим образом: 50 мкг ДНК SV40 инкубируют в 120 мкл буфера A с 50 ед. Hind III в течение 2 ч при 37oC и фрагмент Hind III C SV40 (5089-968 по) очищают на геле. Плазмиду pDHFR III (Hind III) (10 мкг) обрабатывают 250 нг бычьей кишечной фосфатазы в течение 1 ч при 37oC, экстрагируют фенолом и осаждают этанолом. Линеаризованную плазмиду (50 нг) связывают с 250 нг Hind III CSV40 в 16 мкл буфера C в течение 3 ч при 12oC, с использованием 200 ед. T4-полинуклеотидлигазы и трансформируют в E. coli HB101. Из этой плазмиды выделяют затем фрагмент EcoRI-KpnI (фрагмент B), 700 пар оснований.
Для результирующего конструирования pD3 фрагменты A и B (по 50 нг каждого) связывают с 10 нг фрагмента C с помощью T4-полинуклеотидлигазы в течение 4 ч при 12oC, и затем трансформируют E.coli PP1. Положительные колонии отбирают быстрым препаративным анализом и получают большое количество препарата pD3.
Затем конструируют вектор экспрессии pM7135. Репликативную форму pM7115 расщепляют BamH1 и XbaI и на геле очищают фрагмент 550 по, содержащий лидерный участок фактора IX и 5'-последовательность Фактора VII. Плазмиду FIX/VII/pD2 расщепляют с помощью XbaI и BamH1 и на геле очищают фрагмент, 1700 по, содержащий 3'-участок к-ДНК Фактора VII. Плазмиду pD3 расщепляют BclI, обрабатывают бычьей щелочной фосфатазой и соединяют три фрагмента тройным связыванием. Результирующие конструкции отбирают на присутствие фрагмента Xba1 длиной 2000 пар оснований. Отбирают плазмиду, имеющую правильную ориентацию, и обозначают ее как pM7135 (рисунок 8).
Пример 9: Экспрессия Фактора VII из клонов к-ДНК.
Для экспрессии к-ДНК Фактора VII, содержащей лидерную последовательность Фактора VII, ДНК из λ VII2463 или λ VII565 и λ VII2463 клонируют в вектор экспрессии, содержащий основной поздний промотор Ad2, расширитель SV40, трехчленную лидерную последовательность Ad2, набор сплайсинга и полиадениляционный сигнал SV40. Этот вектор содержит единственную последовательность EcoRI как сайт к-ДНК-вставки. Устанавливают экспрессию последовательностей из λ VII2463, кодирующей лидерную последовательность из 60 аминокислот и из λ VII565 и λ VII2463, которые лишены кодонов для аминокислот от -18 до -39 и кодируют лидерную последовательность длиной 38 аминокислот. Так как структура лидерного участка Фактора VII идентифицируется только клонированием к-ДНК и так как имеется неясность, обусловленная получением двух различных 5'-концевых к-ДНК, то авторы сконструировали также последовательность геномной к-ДНК Фактора VII. 3'-Участок λ VII2463 (от сайта Bgl II в экзоне 2 до сайта EcoRI, связывающего 3' с окончанием /A/) соединяют с субгеномным фрагментом клона 7 1, кодирующим экзоны Ia, Ib и оставшующся часть эксзона 2. Этот субгеномный фрагмент, реконструированный как фрагмент EcoRI-Bg III, 4,4 килобазы, соединяют с к-ДНК λ VII2463 и клонируют в вектор экспрессии млекопитающих.
Говоря коротко, для конструирования субклона фрагмент EcoRI к-ДНК Фактора VII λ VII2463 клонируют в сайт EcoRI pUC18 и обозначают pVII2463. Аналогичным образом вставку к-ДНК EcoRI λ VII565 субклонируют в pUC18 и обозначают pVII565. Конструируют гибрид между 5'-участком последовательности Фактора VII клона pVII565 и 3'-сегментом ДНК Фактора VII pVII2463 клонированием наибольшего 5'-фрагмента EcoRI-Bgl II Фактора VII pVII565 и фрагмента Bgl II-Hind III Фактора VII (сайт Hind III в полилинкере pVII2463) pVII2463 в pUC18, расщепленный EcoRI и Hind III. Эту конструкцию обозначают pVII2397. Вставки pVII2463 и pVII2397 удаляют расщеплением EcoRI и очищают на геле для вставки в векторы экспрессии в клетках млекопитающих, как это описано ниже.
A. Экспрессия полимерной к-ДНК Фактора VII.
Экспрессия фактора VII достигается в векторе pDX. Этот вектор получают из pD3 (описано выше в примере 8) и pD3', вектора, идентичного pD3, за исключением того, что сигнал полиадениляции SV40 (т. е. фрагмент от BamH1 SV40/2533 по до BclI/2770 по) находится в поздней ориентации. Таким образом pD3' содержит сайт BamH1 в качестве сайта вставки гена.
& Для получения pDX сайт EcoRI в pD3' превращают в сайт Bcl I расщеплением EcoRI, инкубированием с нуклеазой S1 и связыванием с линкерами Bcl I. Получают ДНК из положительно идентифицированной колонии и получают фрагмент XhoI-Pst 1, 1,9 килобазы, содержащий измененный рестрикционный сайт, электрофорезом на агарозном геле. Во второй модификации pD3, расщепленный BclI, связывают с обработанными киназой адапторами EcoRI-BclI (сконструированными из олигонуклеотидов ZC525, 5'GGAATTCT3' и ZC526, 5'GATCAGAATTCC3') для получения сайта EcoR1 как положения вставки гена в вектор экспрессии. Идентифицируют положительные колонии рестрикционным анализом и используют ДНК из них для выделения фрагмента XhoI-Pst1, 2,3 килобазы, содержащего модифицированный рестрикционный сайт. Два описанных выше фрагмента ДНК инкубируют вместе с Т4-ДНК-лигазой, трансформируют в E.coli HB101 и положительные колонии идентифицируют рестрикционным анализом. Получают препарат такой ДНК, названной pDX (рисунок 12). Эту ДНК расщепляют EcoRI и затем инкубируют с бычьей кишечной фосфатазой. Затем очищенную ДНК инкубируют с Т4-ДНК-лигазой и фрагментом к-ДНК EcoRI фактора VII из pVII2463 или с фрагментом к-ДНК EcoR1 фактора VII из pVII2397. Результирующие клоны обозначают соответственно FVII/2463//pDX и FVII/565+2463//pDX (рисунок 12). После трансформирования в E. coli M83 и последующего идентифицирования рестрикционным анализом получают препараты плазмидной ДНК и проверяют усиленным рестрикционным расщеплением эндонуклеазами. Плазмиды FVII/2463//pDX и FVII/565+2463//pDX депонируют в Коллекцию культур американского типа под номерами соответственно 40206 и 40205.
Каждую из FVII/2463//pDX и FVII/565+2463//pDX /10 мкг каждой/ используют для трансформирования, вместе с 10 мкг несущей ДНК из спермы лосося, либо клеток ВНК tk-t13/Floros и др., Exper-Cell. Res, 132: 215 - 223, 1981/, либо клеток COS, с использованием стандартного осаждения фосфата кальция. После заражения клетки культивируют в подходящей среде, содержащей 5 мкг/мл витамина K, в течение 2 дней. В это время супернатанты анализируют на ELISA-положительные соединения с использованием моноклонального антитела против фактора VII. Как FVII/2463//pDX, так и FVII/565+2463//pDX приводят к выработке полипептида фактора VII, обнаруживаемого в супернатанте клеток COS, а в супернатанте клеток ВНК обнаруживается фактор VII в результате FVII-565+2463//pDX. Sham-Трансформированные клетки ВНК или COS не вырабатывают заметных количеств фактора VII (таблица 5).
Явная экспрессия фактора VII наблюдается также у нескольких других линий клеток, перечисленных в таблице 6.
Клетки совместно заражают 10 мкг либо FVII/2463//pDX, либо FVII/565+2463//pDX вместе с 1 мкг плазмиды, содержащей хлорамфениколацетилтрансацетилазный ген (для идентификации совместно зараженных клеток) и 10 мкг ДНК спермы лосося. В качестве контроля используют Моск-трансформированные клетки. Истощенную среду анализируют через 6 дней с помощью ELISA. Результаты представлены в таблице 7.
Проводят совместную трансформацию либо FVII/2463//pDX /10 мкг/, либо FVII/565+2463//pDX /10 мкг/ с 10 мкг ДНК спермы лосося и 1 мкг плазмиды, кодирующей устойчивую форму дигидрофолатредуктазы (Simonsen и Levinson, Proc. Nutl. Acad. Sci. USA, 80: 2495 - 2499, 1983) в векторе экспрессии млекопитающих клеток ВНК tk-t 13. Через 2 дня клетки разделяют 1:14 и помещают в селективную среду, содержащую 250 нМ либо 1000 нМ метотрексата (МТХ) и 5 мкг/мл витамина K (фитадион, Merek). Через две недели колонии выделяют и выращивают до заполнения 50 - 90%. Затем супернатантную среду анализируют на полипептид фактора VII с помощью ELISA. Из 25 положительных клонов подвергают дальнейшему анализу. Клетки в количестве 5 • 104 (группа I) или 1 • 105 (группа II) помещают в 10 см-чашки, содержащие 5 мкг/мл витамина K, и либо 250 нМ, либо 1000 Нм метотрексата. Через 5 дней быстрее растущие клоны (помеченные звездочкой в таблице 8) делят 1:2 и через 24 ч среду меняют во всех чашках. Через 23 ч (группа I) или 20 ч (группа II) собирают супернатантную среду и считают клетки в каждом клоне. Среду анализируют как с помощью ELISA, так и одностадийным испытанием на свертывание. Результаты представлены в таблице 8.
B. Экспрессия гибрида геномной к-ДНК фактора VIII.
Вектор экспрессии, содержащий геномные последовательности, являющиеся геномным 5'-окончанием фактора VII, и последовательности к-ДНК из 3'-окончания гена фактора VII, получают следующим образом. Для реконструирования 5'-окончания используют три субклона геномной плазмиды 7mI:7Bam, 7SD и 7SE. Плазмида 7Ba представляет собой фрагмент EcoRI-BamH1 3,6 килобазы, содержащий экзон 1a, субклонированный в pUC12. Из этого субклона выделяют фрагмент EcoRI-XbaI, 0,7 килобазы, содержащий экзон 1a, и называют его фрагментом a. Плазмида 7SE представляет собой фрагмент Sst 1, 3,7 килобазы, содержащий экзон Ib, субклонированный в pUC18. Из этого субклона выделяют фрагмент Xba1-Sst 1, 3,1 килобазы, содержащий экзон 1b, и называют его фрагментом b. Плазмида 7SE представляет собой фрагмент Sst 1, 3,9 килобазы, содержащий экзоны 2 - 4, субклонированный в M13 mp19. Из геля выделяют фрагмент Sst1-Bgl II (0,6 килобазы), содержащий 5'-участок экзона 2, и называют его фрагментом c. Остальную часть к-ДНК 3'-фактора VII (фрагмент d) получают как фрагмент Bgl II-EcoR1 из pUVII2463. Фрагменты a - d связывают с pDX, расщепленной EcoR1 и обработанной бычьей кишечной фосфатазой, и затем трансформируют в клетки E. coli J М83 и HB101. Положительные колонии идентифицируют рестрикционным анализом и получают из них плазмидную ДНК.
Для экспрессии фактора VII плазмидную ДНК используют для совместного заражения клеток ВНК или COS, как описано выше. Трансформированные клетки культивируют в среде, содержащей витамин K, в течение 2 дней, и среду анализируют на присутствие фактора VII с помощью ELISA.
Из изложенного выше можно сделать вывод о том, что хотя с иллюстративной целью здесь и описаны конкретные варианты осуществления данного изобретения, однако могут быть разработаны различные модификации, не выходящие за рамки объема и сути данного изобретения. В соответствии с этим данное изобретение не ограничено ничем, кроме прилагаемой формулы изобретения.



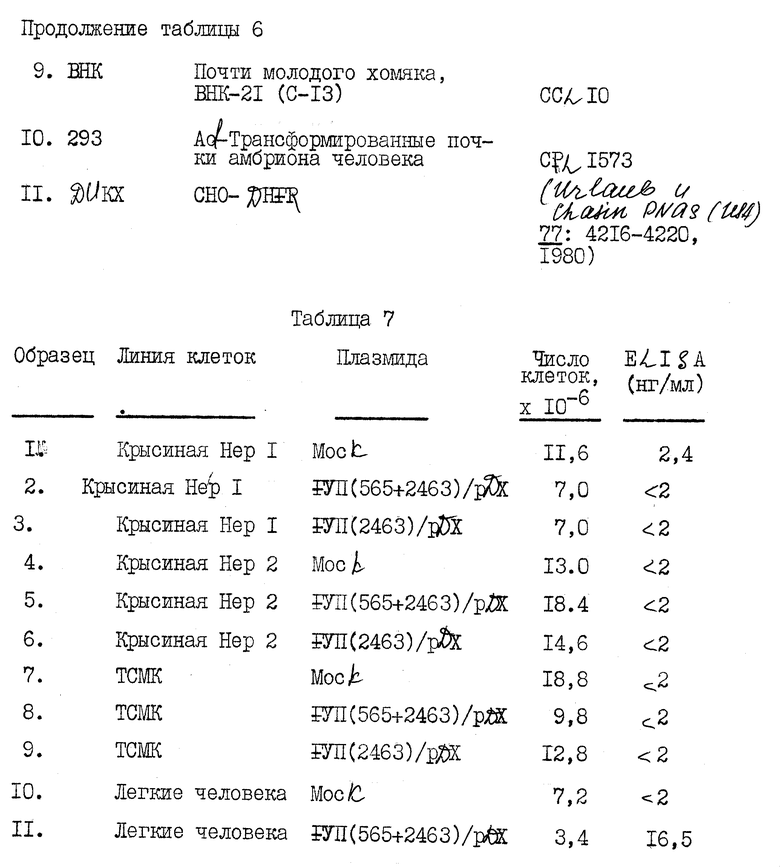
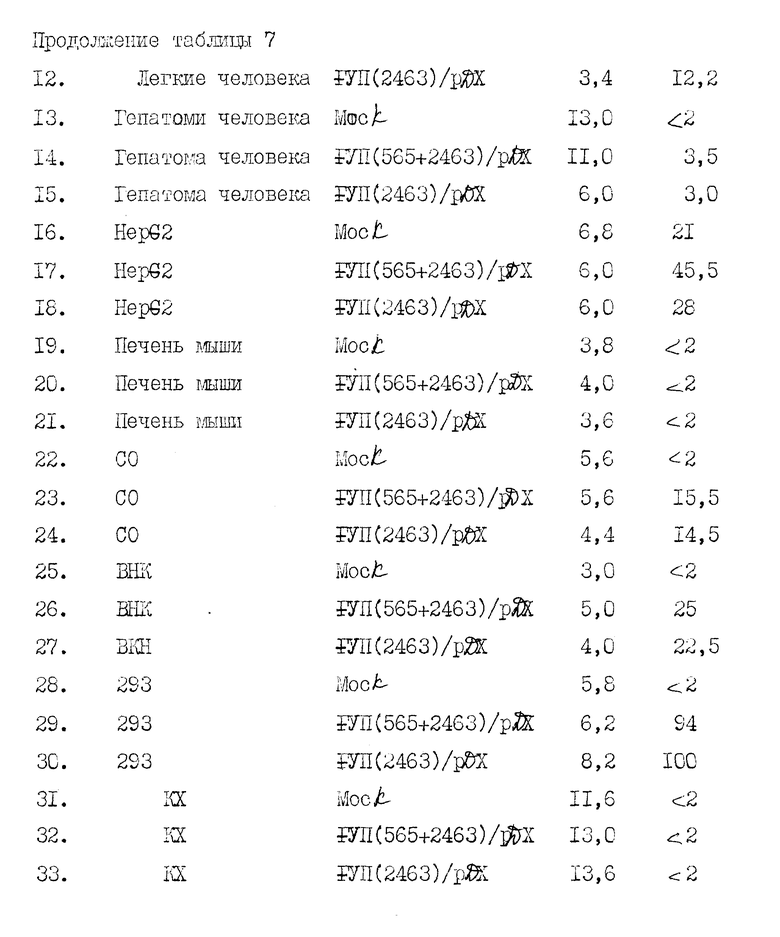
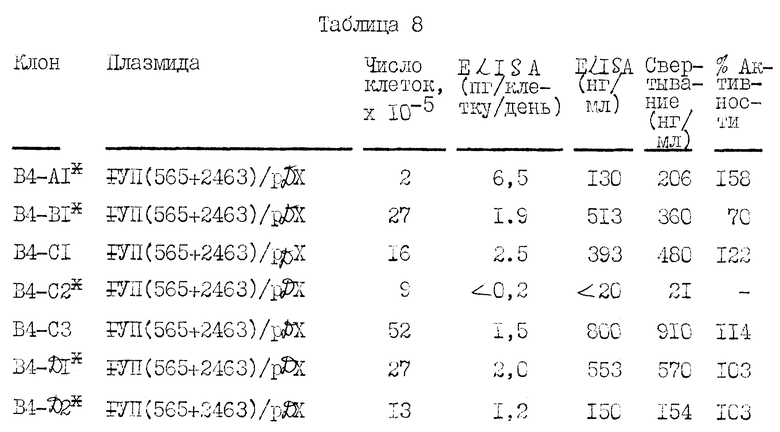
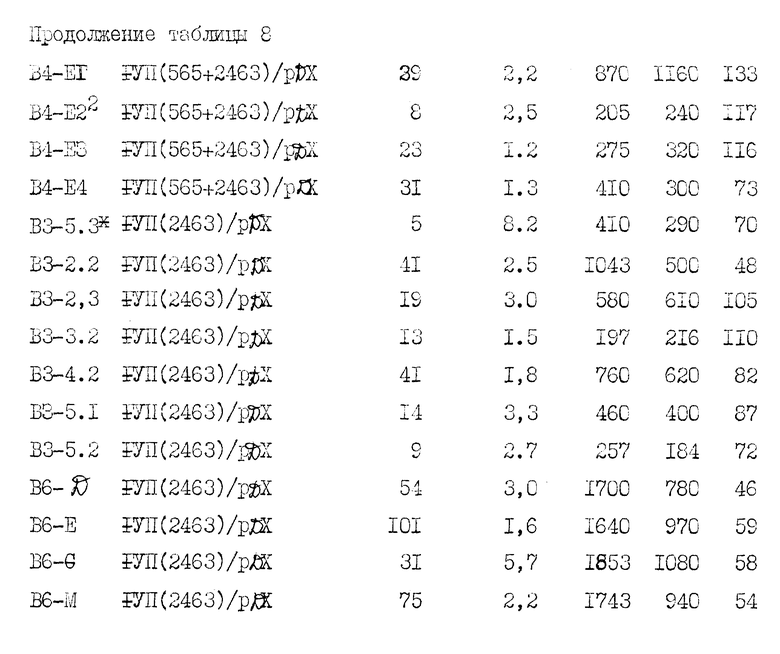
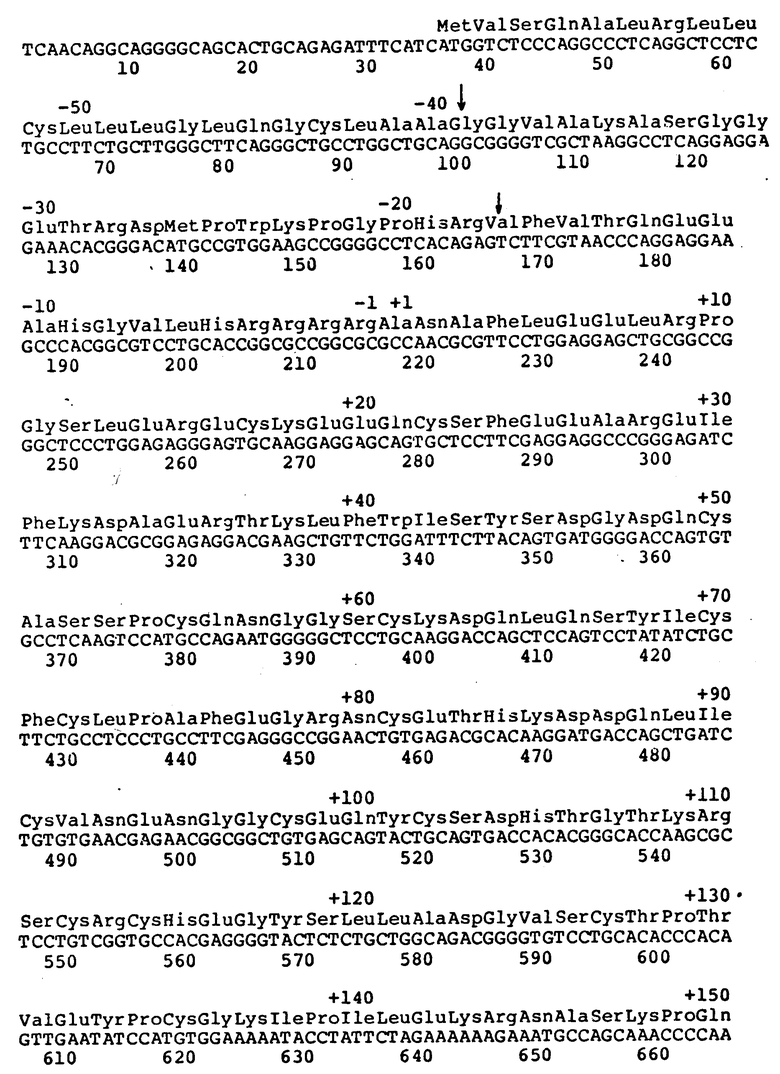







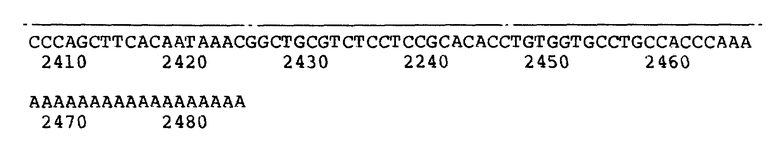

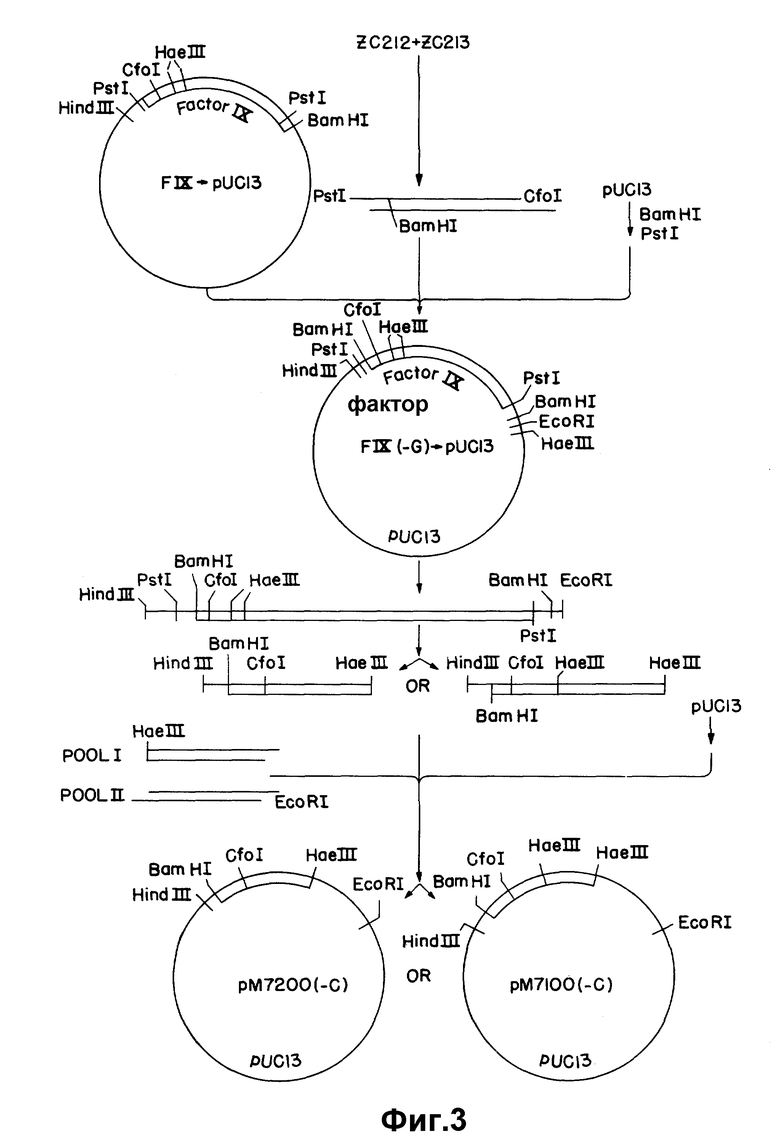
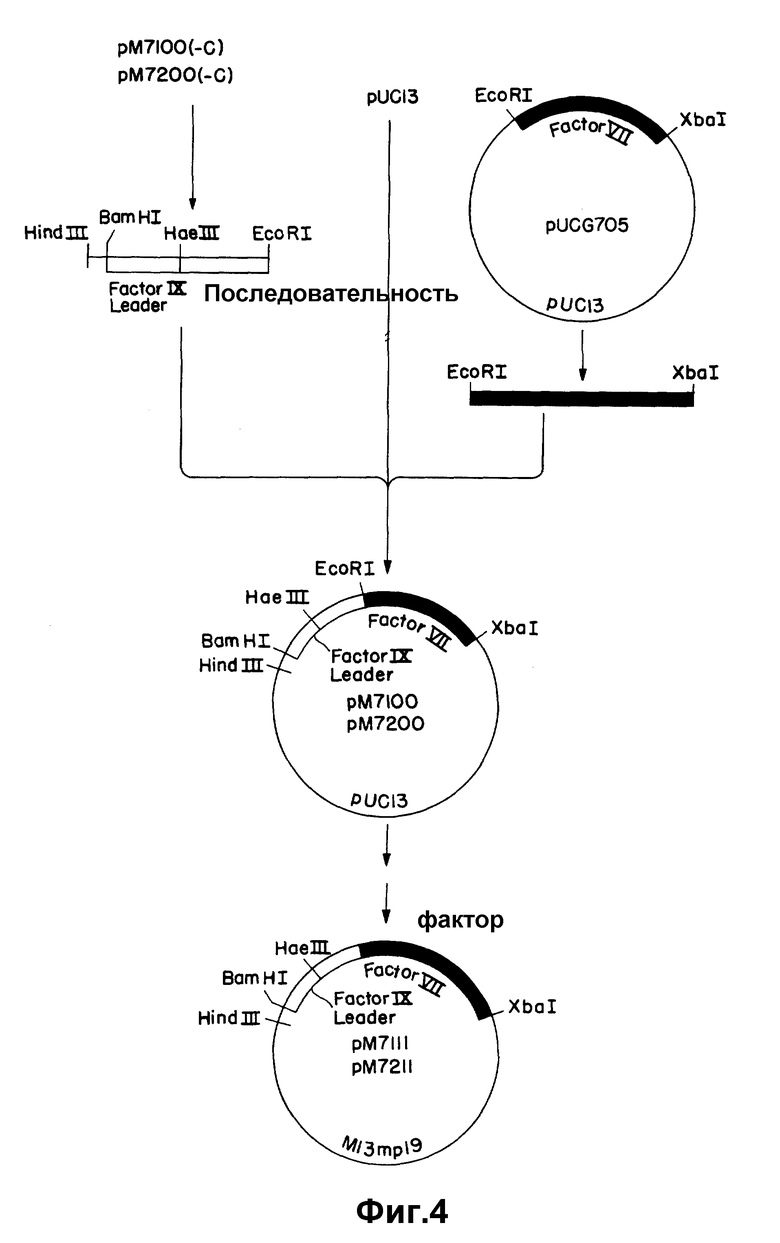
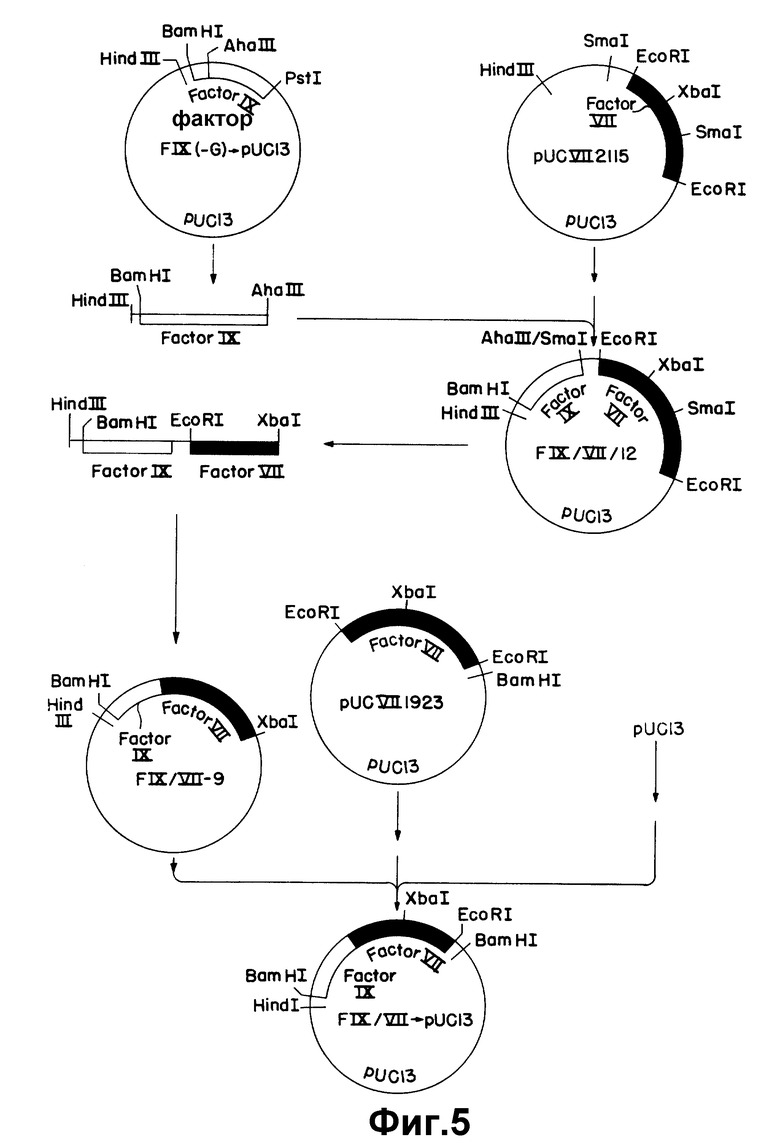
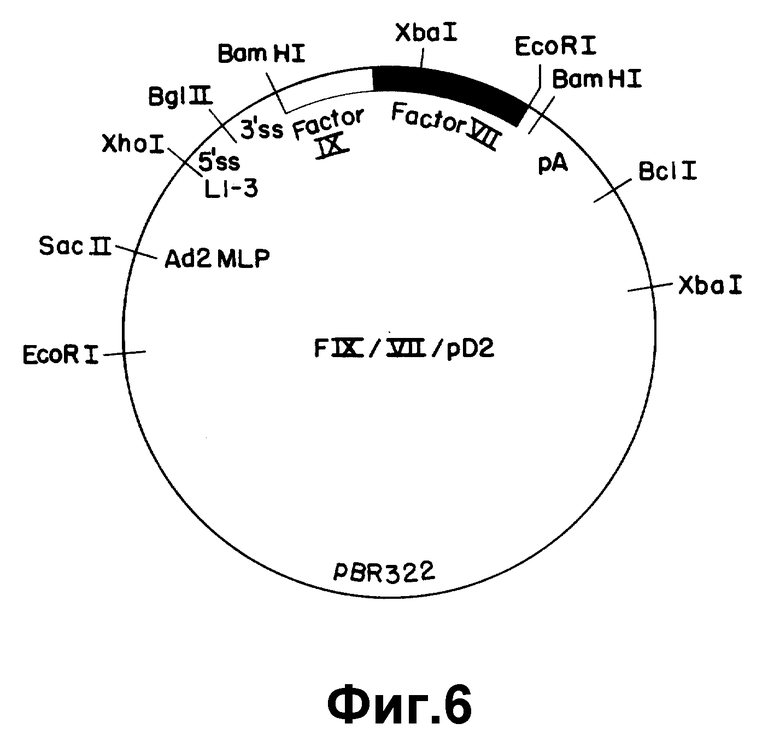

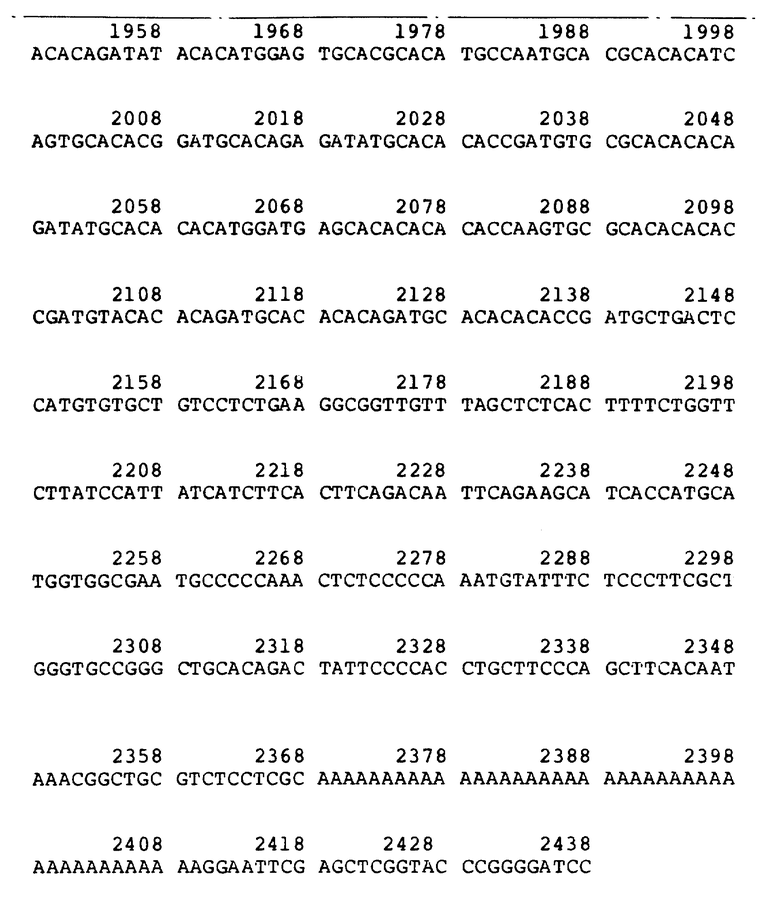

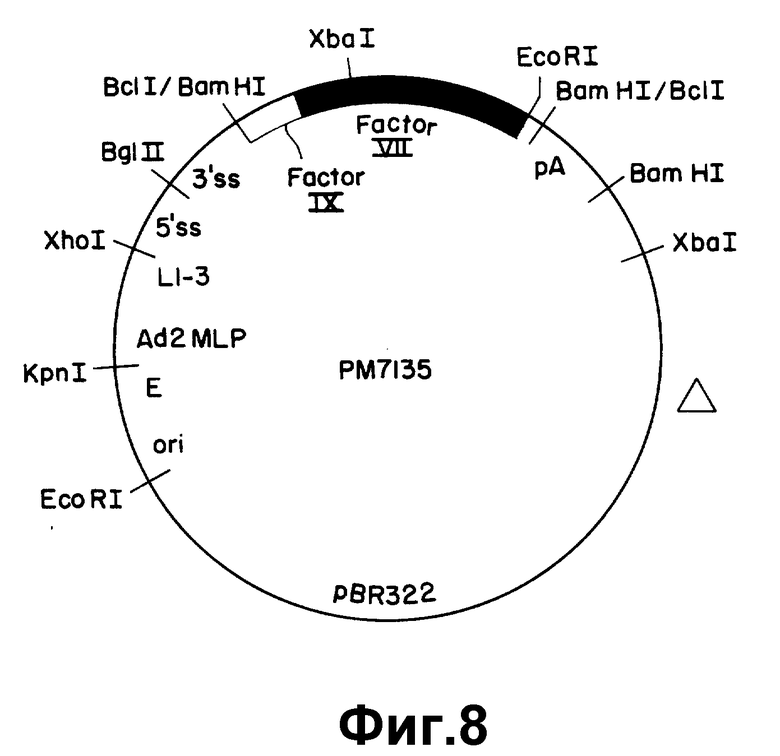
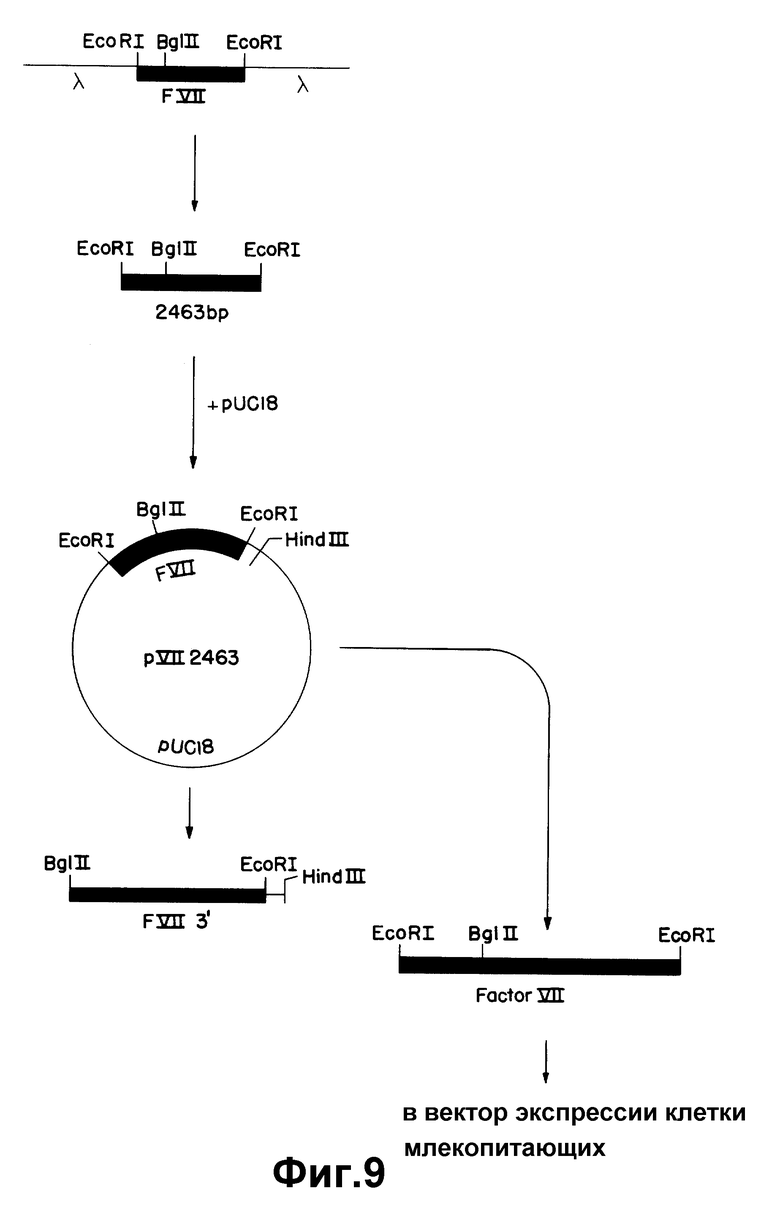
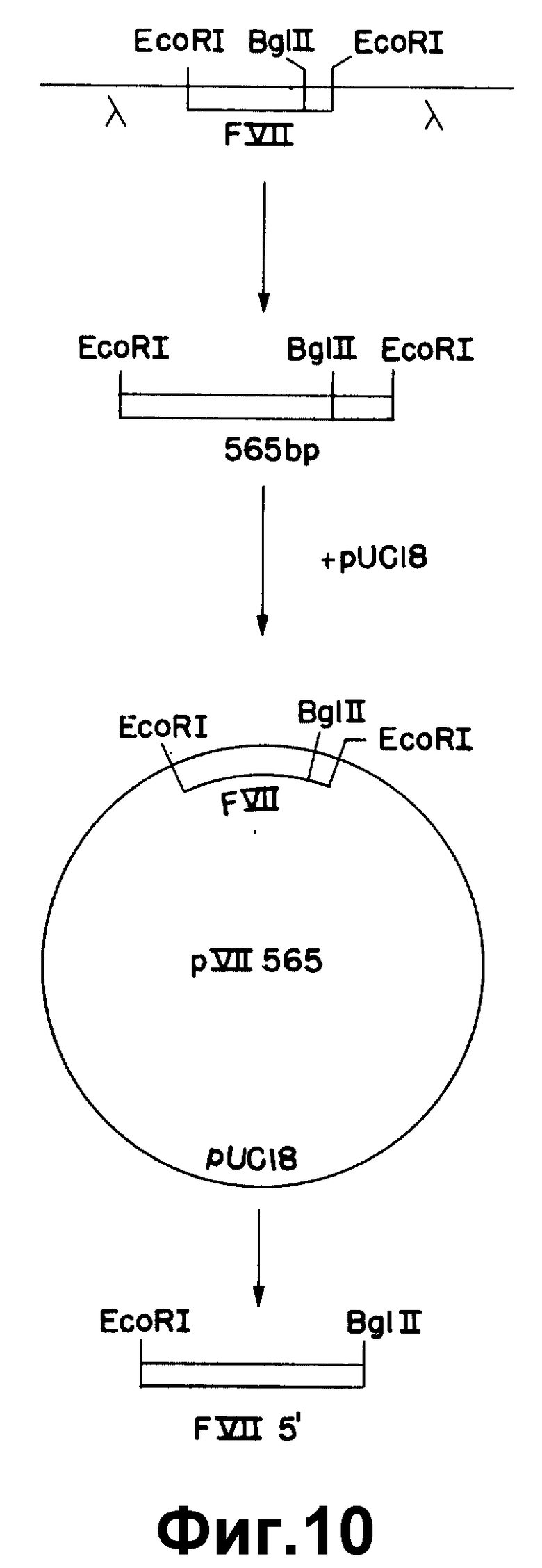
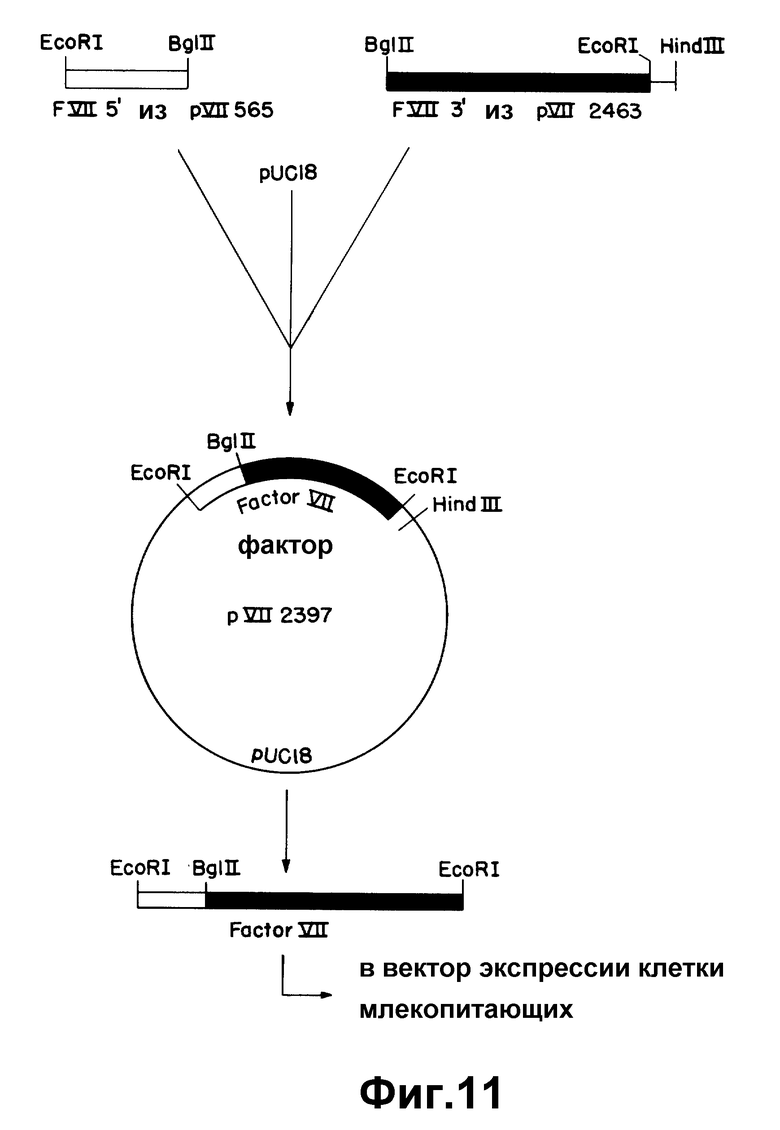
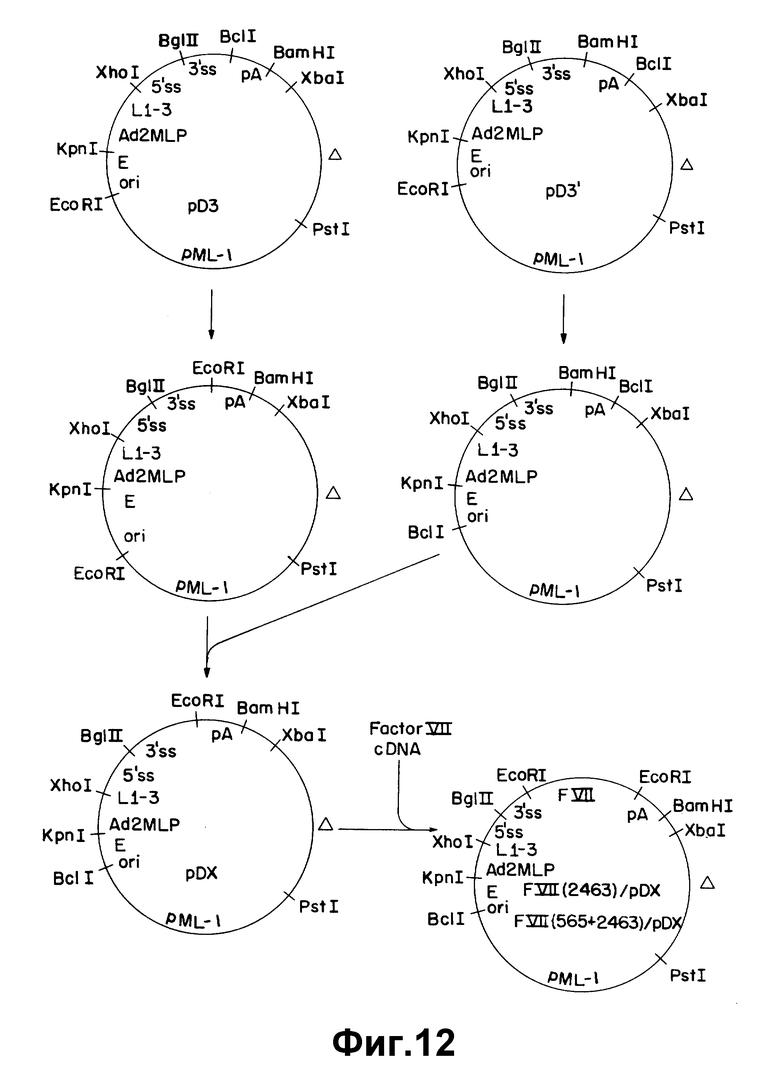
Предлагаемый способ относится к биотехнологии. Для его осуществления культивируемые клетки млекопитающих, в частности HepG2, COS, BHKtk-t 13,293 и др. , трансформируют рекомбинантным вектором экспрессии, который включает фрагмент ДНК, кодирующий зрелый фактор VII человека с предшествующей ему полной или частью сигнальной последовательности, стабильно трансформированные клетки отбирают и культивируют, затем из культуральной среды изолируют рекомбинантный белок, который после активации подходящей протеазой проявляет активность в процессе свертывания крови, аналогичную таковой природного фактора VIIа. Изобретение обеспечивает возможность получения значительных количеств рекомбинантной формы фактора VII с достаточно высокой степенью очистки. 2 з.п. ф-лы, 12 ил., 8 табл.
R1-R2-R3,
где R1 - аминокислотные остатки (-60)-(-40);
R2 отсутствует или аминокислотные остатки (-39)-(-18);
R3 - аминокислотные остатки (-17)-406,
в следующей аминокислотной последовательности фактора YII человека:
полученные клетки культивируют в условиях, обеспечивающих накопление полипептидного продукта, а выделение и очистку осуществляют из культуральной среды путем осаждения цитратом бария, иммуноадсорбцией на антителах к фактору YII или хроматографией.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Journal of Biological Chemistry, v | |||
| Гудок | 1921 |
|
SU255A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Biochemistry, v | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ МЕЖДУ СТЕКЛОМ И МЕТАЛЛОМ, В ОСОБЕННОСТИ ДЛЯ ВВОДОВ ПРОВОДНИКОВ СИЛЬНОГО ТОКА В ПУСТОТЕЛЫЕ ТРУБКИ И Т.П. ПРИБОРЫ | 1925 |
|
SU4189A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US, патент, 4456591, A 61 K 35/14, 1984. | |||
