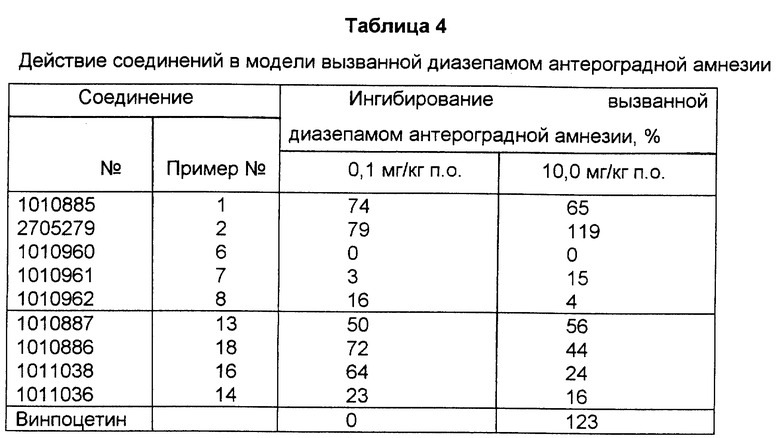
Изобретение относится к новым рацемическим и оптически активным эфирным производным трансаповинкаминовой кислоты формулы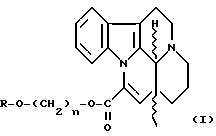
где R обозначает водород или группу 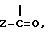 где Z представляет собой C1-4-алкильную, возможно замещенную арильную, аралкильную, гетероарильную или 14-эбурнаменинильную группу; и
где Z представляет собой C1-4-алкильную, возможно замещенную арильную, аралкильную, гетероарильную или 14-эбурнаменинильную группу; и
n - целое число 2, 3 или 4;
а также к их терапевтически приемлемым солям и фармацевтическим композициям, содержащим эти соединения. Кроме того, изобретение относится к способу получения вышеуказанных соединений и композиций.
Соединения по изобретению являются новыми и обладают ценной биологической активностью. В условиях in vitro они проявляют значительное антиоксидантное (ингибирующее перокисление липидов) действие. При исследовании в условиях in vivo они оказывают значительное антиишемическое и антиамнезийное действие.
Соответственно, изобретение относится также к способу лечения, который включает в себя введение терапевтически активного количества соединения формулы I или его терапевтически приемлемой соли пациенту, которому требуется лечение.
В формуле I в значении R: Z как C1-4-алкильная группа может представлять собой насыщенную или ненасыщенную группу с прямой или разветвленной цепью, как например метильную, этильную, н-пропильную, изопропильную, н-бутильную, втор-бутильную, трет-бутильную, изобутильную, винильную, пропенильную группу и тому подобные; как арильная группа Z может быть, например, фенильной группой; как аралкильная группа, она может обозначать бензильную группу, дифенилметильную группу или тому подобные группы; как гетероарильная группа она может представлять собой пяти-, шести- или семичленную циклическую группу, содержащую одинаковые или разные гетероатомы, например атом азота, кислорода или серы, как например пирролильная, фурильная, тиенильная, пиридильная, пиранильная, пиразотилильная, имидазолильная, пиримидинильная, морфолинильная группы и им подобные.
Заместителями вышеуказанных арильной, аралкильной или гетероарильной групп могут быть: галогены, как например фтор, хлор или бром; C1-4-алкильная или C1-4-алкоксигруппы; а также гидроксильная, нитро-, амино-, циано-, трифторометильная группы или им подобные.
Терапевтически приемлемыми солями соединений формулы I по изобретению могут быть соли, полученные присоединением кислот, или четвертичные соли.
Производные аповинкаминовой кислоты формулы II, используемые в качестве исходных веществ, могут быть получены кислотной обработкой подходящего производного гидроксииминооктагидроиндоло[2,3]хинолизина, как указано в описании к патенту Великобритании N GB 2124214.
Из литературы известны соединения, которые структурно родственны соединениям формулы I. Эфиры трансаповинкаминовой кислоты с противовоспалительным, противосудорожным, антипаркинсоновским и антиатеросклеротическим действием описаны в описании к патенту Венгрии N 186891. Антигипоксические или сосудорасширяющие производные аповинкаминовой кислоты, несущие различные боковые цепи, описаны в описании к патенту Венгрии N 187733.
В отличие от вышеуказанных веществ, известных из литературы, новые эфирные производные трансаповинкаминовой кислоты обладают значительным антиоксидантным, антиамнезийным и антиишемическим действием.
Непрерывное сохранение циркуляции крови необходимо для нормального функционирования центральной нервной системы (ЦНС), поскольку это гарантирует достаточное обеспечение повышенных потребностей мозговой ткани в глюкозе и кислороде.
При ишемии и реперфузии нарушаются познавательные функции: в зависимости от тяжести и длительности ишемии могут произойти как обратимые, так и необратимые повреждения. Повреждения структуры и функции мембран нервных клеток могут привести к смерти нейрона.
Различные патологические процессы, такие как образование свободных радикалов, могут возникать как следствие ишемии. Образование свободных радикалов ведет к окислению ненасыщенных жирных кислот (перокисление липидов), которые являются важными компонентами мембран. Это менее специфический процесс разрушения клетки, изменяющий или разрушающий биомолекулы. Таким образом, могут быть повреждены функции различных уровней клеток, органов или целого организма.
Реакции свободных радикалов, видимо, играют роль причины в патогенезе некоторых вызванных ишемией повреждений, таких как например, ишемические кишечные заболевания, ишемия миокарда, геморрагический шок, расстройства цереброваскулярных функций, сопровождаемые ишемией, ишемическое повреждение печени, почечная ишемия и тому подобное.
Благодаря их ингибирующему действию по отношению к перокислению липидов, антиоксидантные соединения гарантируют защиту против повреждений, вызванных свободными радикалами в условиях ишемии. Таким образом, антиоксиданты как антиишемические соединения могут быть полезными в лечении вышеуказанных патологических картин.
Тесты in vitro для исследования антиоксидантного действия
Антиоксидантное действие изучали с использованием двух методов:
1. Действие на перокисление липидов в микросомах мозга, индуцированное NADPH
(J. M. Braughler et al.: Novel 21-Amino Steroids as Potent Inhibitors of Irondependent Lipid Peroxidation (J.Biol. Chem. 262. 10438-10440 (1987)); T. J. Player and A. A. Horton: Enzyme Lipid Peroxidation in Microsomal Fraction of Rat Brain (J. Neurochem. 37, 422-426 (1981)).
Для получения микросом использовали самцов крыс Hannover-Wistar с массой тела 150-250 г. После декапитации мозг целиком удаляли и гомогенизировали в 10-кратном объеме охлажденного на льду 0,25 М раствора сахарозы. Гомогенат центрифугировали в установке Hitachi CR 26Н при 15000 g при 4oC в течение 10 минут, затем супернатант собирали и центрифугировали в установке Hitachi SCP 85Н при 78000 g и 4oC в течение 60 минут. После суспендирования осадка в 0,15 М растворе KCl определяли белковое содержание полученного раствора, и затем доводили его до концентрации 10 мг/мл. Полученные таким образом микросомы замораживали в смеси сухого льда с ацетоном и хранили при -70oC до использования. Компонентами инкубационной смеси являлись: 50 мМ Трис-HCl (pH 6,8), 0,2 мМ FeCl3, 1 мМ KH2PO4, 0,5 мМ ADP, 0,2 мг микросом, а также исследуемое соединение. Инкубацию проводили в конечном объеме 1 мл, времени инкубации 20 минут при температуре 37oC. Перокисление липидов индуцировали добавлением 0,4 мМ NADPH (бланк-образцы не содержали NADPH). Реакцию останавливали добавлением 0,375 мл останавливающего раствора, содержащего 40%-ную трихлоруксусную кислоту и 5 М HCl в соотношении 2:1.
Образование малондиальдегида определяли с использованием тиобарбитуровой кислоты. После остановки реакции, 1 мл 1%-ного раствора тиобарбитуровой кислоты добавляли в каждый из образцов, которые затем помещали в водяную баню при 100oC на 10 минут. Затем образцы центрифугировали при 2000 g в установке Janetzki К70 при 4oC в течение 10 минут. Величины оптической плотности окрашенного супернатанта измеряли при 535 нм на спектрофотометре Hitachi 150-20. В качестве эталонного соединения был использован малондиальдегид бис(диэтилацеталь).
2. Действие на перокисление липидов в гомогенате мозга, индуцированное Fe+2
После декапитации крыс Hannover-Wistar с массой 150-220 граммов каждая целый мозг гомогенизировали в 9 объемах охлажденного на льду буфера Кребса-Рингера (Krebs-Ringer's), содержащего 15 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинилэтансульфоновая кислота) (pH 7,4), 140 мМ NaCl, 3,6 мМ KHl, 1,5 мМ CaCl2, 0,7 мМ MgCl2, 1,4 мМ KH2PO4 и 10 мМ глюкозы. Затем определяли белковое содержание раствора и доводили его до концентрации 10 мг/мл.
После добавления исследуемого ингибирующего агента в объеме от 5 мкл до 200 мкл гомогената инкубационную смесь инкубировали при 37oC в течение 20 минут. Fe+2-индуцированное перокисление липидов осуществляли добавлением 5 мкл 8 мМ раствора Fe2(NH4)2(SO4)2. После окончания инкубационного периода реакцию останавливали добавлением 1 мл останавливающего раствора, содержащего 0,8 М HCl и 12,5% трихлоруксусной кислоты, затем образцы центрифугировали при 2000 g на установке Janetzki К70 при 4oC в течение 10 минут.
1 мл 1%-ного раствора тиобарбитуровой кислоты добавляли к порции супернатанта объемом 0,5 мл, затем пробы помещались в водяную баню при 100oC на 20 минут. Интенсивность развивающейся окраски определяли при 535 нм на спектрофотометре Hitachi 150-20, используя малондиальдегид бис(диэтилацеталь) как эталонное соединение.
На основе корреляций концентрация/действие определяли значения ИК50 для исследуемых соединений, эти результаты показаны в таблице 1 для обоих методов.
Из данных таблицы 1 можно видеть, что каждое из соединений по изобретению при тестировании проявляет антиоксидантную (ингибирующую перокисление липидов) активность. Антиоксидантное действие исследовали как в ферментном (NADPH-индуцированном, АФИ), так и в неферментном (Fe+2-индуцированном, АНИ) тесте на перокисление липидов. Уровень антиоксидантной активности соединений был охарактеризован по значениям их ИК50. Обладающий церебропротективной активностью идебенон, нативный антиоксидант витамин E (DL-α-токоферол) и гепатопротективный силимарин были использованы в качестве эталонных соединений.
Как показывают данные таблицы 1, тестированные соединения показали гораздо большую активность по ингибированию NADPH-(ферментативно) индуцированного перокисления липидов, чем эталонные соединения, о чем свидетельствуют их значения ИК50, гораздо более низкие, чем значения ИК50 для DL-α-токоферола или силимарина. Из соединений, оказавшихся очень эффективными, антиоксидантное действие соединений N 2705279, 1010961, 1011008, 1011037 и 1011047 было сравнимо с действием идебенона. Соединения N 1010885, 1010962, 1010960, 1011005, 1010887, 1010886, 1011038, 1011036 и 2705283 ингибируют NADPH-индуцированное перокисление липидов приблизительно в 20 раз сильнее, чем силимарин.
Подавляющее большинство соединений, перечисленных в таблице 1, показало намного более сильное ингибирующее действие по отношению к Fe+2 (неферментативно) индуцированному перокислению липидов, чем эталонные соединения. Соединения N 2705279, 2705283, 2705284, 1010962, 1010960, 1010961, 1010887, 1010886, 1011038 и 1011036 оказались особенно активными, так как каждое из них было почти в два раза активней, чем идебефенон или DL-α-токоферол. Соединения N 1011005 и 1011037 оказывают действие, аналогичное действию DL-α-токоферола. Соединения N 1010885 и 1011047 ингибируют индуцированное ионами Fe+2 перокисление липидов сильнее, чем идебенон. Антиоксидантная активность соединения N 1011008 также превышала активность силимарина.
Можно также утверждать на основании сравнения данных, полученных в обоих тестах in vitro, что соединения N 2705279, 1010962, 1010960, 1010961, 1010886, 1011038 и 1011036 очень существенно ингибируют перокисление липидов, индуцируемое различными способами (ионами Fe+2 или NADPH), а именно величины ИК50 этих соединений оказались менее 10 мкМ. Так как ни одно из эталонных соединений не могло оказывать такого действия в обоих тестах (а именно, они ингибировали NADPH- или Fe+2 ион-индуцированное перокисление липидов в различной степени), то соединения по изобретению могут считаться более эффективными, чем эталонные соединения.
Каждое из исследуемых соединений обладает значительной антиоксидантной активностью, поскольку они способны ингибировать перокисление липидов, вызванное свободными радикалами, образующимися при реакции Fenton's (катализируемой Fe+2) или в ходе функционирования фермента NADPH-цитохром с редуктазы.
Сокращения:
NADPH - никотинамид-аденин-динуклеотид-фосфат, восстановленный
ТРИС - трис(гидроксиметил)аминометан
ADP - аденозин-5'-дифосфат
Идебенон - 6-(10-гидроксидецил)-2,3-диметокси-5-метил-1,4-бензохинон
DL-α-токоферол - 2,5,7,8-тетраметил-2-(4',8',12'-триметилдецил)-6-хроманол
Эллаговая кислота - 2,3,7,8-тетрагидрокси[1]бензопирано [5,4,3- cde][1] бензопиран-5,10-дион
Силимарин - силибинин + силидианин + силикристин
АФИ - тест на ферментативно (NADPH-) индуцированное перокисление липидов
АНИ - тест на неферментативно (Fe+2) индуцированное перокисление липидов
Применяемые для изучения антиишемического и антиамнезийного действия тесты in vitro
1. Антиишемическое действие (на модели двусторонней перевязки артерий)
(N. Himori et al.: Модель церебральной ишемии у находящихся в сознании мышей с вовлечением активации рецепторов к NMDA и расстройствами способности к обучению и запоминанию [J. Pharmacol. Meth. 23, 311- 327 (1990)])
Сонные артерии мышей (массой 30-32 г каждая) обнажали с двух сторон в условиях анестезии с помощью 450 мг/кг хлоралгидрата внутрибрюшинно (i.p.). Отделяли симпатический и блуждающий нервы, после чего на сонные артерии с обеих сторон накладывали свободные лигатуры. Оба конца каждой лигатуры выводили на спину за ушами. На следующий день, когда мыши были в сознании, лигатуры затягивали, вызывая таким образом прекращение кровотока через сонные артерии. Пережатие артерий продолжали в течение 5 минут, затем лигатуры ослабляли для обеспечения реперфузии. Регистрировали число животных, погибших в течение указанных 5 минут и в течение последующих 24 часов. Проверка достоверности проводилась с помощью теста хи-квадрат. Предварительную обработку исследуемым соединением проводили внутрибрюшинно (i.p.) за 30 минут до лигирования артерий.
Влияние доз соединений в 2 мг/кг на вызванную двусторонним лигированием артерий смертность показано в таблице 2.
Величины ЭД50 эталонных соединений, а также наиболее эффективного из введенных i.p. соединений приведены в таблице 3.
Биологическую активность in vivo соединений, оказывающих in vitro выраженное антиоксидантное действие (ИК50<10 мкМ), исследовали на модели вызванной билатеральным лигированием сонных артерий церебральной ишемии. Преходящее резкое снижение церебрального кровотока, а затем начало реперфузии после прекращения лигирования приводят к образованию чрезвычайно токсичных свободных кислородных радикалов (супероксидного радикала, перекиси водорода). Соединения с антиоксидантными свойствами очень эффективны в плане защиты от токсических эффектов высвобождаемых радикалов. Смертность от 5-минутного билатерального лигирования сонных артерий составляет 70%. Дозы наиболее активных соединений по данному изобретению (2705279, 1010960, 1010887, 1010886) в 2 мг/кг i.p. снижают смертность до 10-30%, в то время как известные антиоксиданты, такие как идебенон или витамин E, менее активны при применении в той же дозе. Превосходная активность соединения N 2705279, оказавшегося наиболее активным, выглядит еще более потрясающей при сравнении величин ЭД50 различных соединений (ЭД50 представляет собой дозу (в мг/кг), уменьшающую смертность на 50% по сравнению с необработанной контрольной группой). Значение i.p. ЭД50 для идебенона составляет 4,5 мг/кг, а для соединения N 2705279 - 0,76 мг/кг. Антиишемичесое действие данного соединения также может быть измерено после перорального введения. В других экспериментах было показано, что данное соединение активно не только на модели глобальной ишемии, затрагивающей все вещество мозга, но также и на моделях так называемой "фокальной" ишемии, вызванной в какой-либо хорошо определенной области мозга.
Антиамнезийное действие соединений по данному изобретению было подтверждено в следующем эксперименте.
2. Вызванная диазепамом антероградная амнезия
C.L. Broekkamp et al: Сравнительные эффекты бензодиазепинов, прогабида и РК 9084 на развитие пассивного избегания у мышей [Psychopharmacology (Berlin) 83, 122-125 (1984)]
Исследование памяти проводили на мышах NMRI массой 25-28 r каждая, с помощью пассивного избегания, способа, основанного на генетически детерминированном никтофильном поведении грызунов. Во время периода обучения предварительно отобранных животных (мышей, переходящих в темное пространство из освещенного в течение 30 секунд) помещали в освещенное пространство. В течение 30 секунд после вхождения в темное пространство животные получали удар электрическим током (1 мА в течение 3 минут) по стопам, после чего регистрировали время до вхождения в темное пространство (латентный период). Через 24 часа животных снова помещали в освещенное пространство и измеряли время до перехода в темное пространство (время удержания = T, с пределом в 300 секунд). Чтобы вызвать антероградную амнезию, животным за 30 минут до обучения вводили внутрибрюшинно диазепам в дозе 3 мг/кг. Исследуемые соединения вводили перорально в дозах 0,1 или 10 мг/кг соответственно, за 1 час до обучения. Процентное выражение защитного эффекта (P%) рассчитывали с помощью следующей формулы и представили в таблице 4.
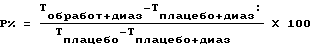
Обладающие антиоксидантной активностью вещества оказывают выраженное защитное действие в случаях церебральной ишемии [приступ транзиторной ишемии (TIA), инсульт] , когда нарушения обучаемости и памяти могут иметь место в сочетании с неврологической симптоматикой различной степени тяжести. Антиамнезийное действие соединений по данному изобретению исследовали на модели вызванной диазепамом антероградной амнезии с использованием в качестве эталонного соединения винпоцетина, имеющего сходную структуру и применяемого в клинической практике. Активные соединения были обнаружены среди как транс-α-этиловых, так и транс-β-этиловых производных аповинкаминовой кислоты. Особенно эффективными оказались соединения NN 2705279, 1010886, 1011038 и 1010887. Наиболее активным было вещество N 2705279, оказывающее значительное защитное действие при применении в обеих используемых дозах и обладающее активностью, значительно превышающей активность винпоцетина. Антиамнезийное действие соединения 2705279 было также подтверждено на других моделях амнезии: оно характеризовалось дозозависимым защитным действием в диапазоне пероральных доз между 0,1 и 10 мг/кг в модели вызванной электрошоком ретроградной амнезии; а также защищало от вызванного ишемией повреждения памяти в диапазоне пероральных доз между 1 и 10 мг/кг. Его антиамнезийная активность также может быть легко измерена на крысах.
Резюме
Новые производные трансаповинкаминовой кислоты по данному изобретению обладают выраженным антиоксидантным и антиишемическими действием. Антиоксидантная активность соединения 2705279, оказавшегося наиболее эффективным, превосходит как in vitro, так и in vivo активность идебенона и винпоцетина, соответственно использовавшихся в качестве эталонных соединений. В добавление к их антиоксидантному и антиишемическому действию соединения по данному изобретению также обладают антиамнезийным действием различной степени выраженности. Соединение N 2705279 является наиболее активным также и в этой области.
Благодаря своему антиоксидантному, антиишемическому и антиамнезийному действию, соединения по данному изобретению являются полезными для лечения патологических процессов, в которых играют роль свободные радикалы, либо в остром периоде болезни, либо при развитии поздних последствий. К ним относятся: цереброваскулярные ишемические повреждения, инсульт, апоплексия, мозговые или спинальные травмы, субарахноидальные или внутримозговые кровоизлияния, а также различные нейродегенеративные заболевания, такие как болезнь Альцгеймера (Aizheimer's). Благодаря своему антиишемическому действию, данные соединения могут оказаться полезными в лечении ишемического повреждения не только мозга, но также и других органов, например печени, сердца или мышц.
Ожидаемые терапевтические дозы соединений по данному изобретению для лечения вышеуказанных клинических синдромов составляют от 0,1 до 40 мг/кг массы тела при их однократном суточном введении или введении в виде разделенных доз пероральным или парентеральным способом.
В соответствии с изобретением новые эфирные производные трансаповинкаминовой кислоты формулы I, так же как и их терапевтически приемлемые соли, могут быть получены трансэтерификацией рацемического или оптически активного эфирного производного трансаповинкаминовой кислоты формулы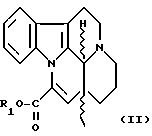
где R1 представляет собой C1-4-алкильную группу, в соответствующем гликоле в присутствии основного катализатора и при желании ацилированием полученного таким образом соединения формулы I, где R является водородом, и/или при желании разделением рацемического соединения формулы I, и/или при желании превращением соединения формулы I в его терапевтически приемлемые соли.
Далее получение новых терапевтически активных соединений по изобретению будет описано в деталях.
В способе по изобретению для получения соединений формулы I, где R обозначает водород, соединение формулы I подвергают трансэтерификации. Эта реакция проходит в присутствии избытка трансэтерифицирующего спирта, например в соответствующем гликоле, предпочтительно этиленгликоле, приемлемо при безводных условиях в присутствии каталитического количества сильного основания. В качестве основания могут использоваться гидриды щелочных металлов или алкоксиды щелочных металлов, приемлемы третичные алкоксиды, лучше так называемый третичный бутоксид калия. Трансэтерификация проводится в интервале температур от 80 до 140oC, преимущественно между 110 и 120oC. При желании продукт, полученный после прохождения реакции, используют на следующей реакционной стадии без выделения, или при желании он может быть выделен способом, при котором реакционную смесь выливают в воду и после фильтрования при желании осадок очищают перекристаллизацией, или после разбавления водой реакционную смесь экстрагируют инертным несмешивающимся с водой органическим растворителем, таким как дихлорметан или хлорбензол, затем продукт выделяют выпариванием или путем образования соли. Соединения формулы I, где R является водородом, могут быть трансформированы ацилированием в соединения формулы I, где R обозначает 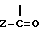 группу.
группу.
Реакция ацилирования может быть проведена с любой органической карбоновой кислотой, содержащей подходящую ацильную группу; или с использованием ее реактивного производного, такого как, например, ацилгалогенид, предпочтительно ацилхлорида, или ангидрида кислоты, или им подобных, известным способом, при желании в присутствии связывающего кислоту агента.
При проведении ацилирования с подходящей карбоновой кислотой реакцию проводят в инертном органическом растворителе, например диполярном апротонном растворителе, таком как диметилформамид (DMF), диметилсульфоксид (DMSO), ацетонитрил или им подобные, в присутствии конденсирующего агента. Используемыми конденсирующими агентами являются, например, производные карбодиимида, такие как дициклогексилкарбодимид или карбонилдиимидазол. Реакция проходит при температуре от 0 до 40oC, предпочтительно при комнатной температуре.
Если ацилирование проводят с помощью ацилгалогенида, приемлемо ацилхлорида, то реакцию проводят в инертном органическом растворителе, таком как алифатический или циклический эфир, например диэтиловый эфир или тетрагидрофуран (THF), или в хлорированном углеводороде, например хлороформе; или в ароматическом углеводороде, например бензоле, хлорбензоле или толуоле; или в органическом основании, например пиридине, предпочтительно в присутствии связывающего кислоту агента. Органические основания, например пиридин, участвующие в реакции, могут одновременно играть роль как растворителя, так и связывающего кислоту агента.
Соединения формулы I, где R представляет собой 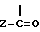 группу, могут быть выделены из растворителя способом, при котором после удаления растворителя, или при желании избытка реагента и связывающего кислоту агента, полученный остаток растворяют в смеси воды и несмешивающегося с водой растворителя, такого как этилацетат, хлороформ, дихлорметан, бензол, диэтиловый эфир или тому подобное, затем при желании величину pH смеси доводят до слабощелочной (pH 8-9) добавлением водного раствора гидроксида аммония или водного раствора гидрокарбоната щелочного металла, а фазы разделяют. Органическую фазу промывают водой, высушивают и удаляют растворитель под пониженным давлением для получения желаемого соединения.
группу, могут быть выделены из растворителя способом, при котором после удаления растворителя, или при желании избытка реагента и связывающего кислоту агента, полученный остаток растворяют в смеси воды и несмешивающегося с водой растворителя, такого как этилацетат, хлороформ, дихлорметан, бензол, диэтиловый эфир или тому подобное, затем при желании величину pH смеси доводят до слабощелочной (pH 8-9) добавлением водного раствора гидроксида аммония или водного раствора гидрокарбоната щелочного металла, а фазы разделяют. Органическую фазу промывают водой, высушивают и удаляют растворитель под пониженным давлением для получения желаемого соединения.
При желании соединения формулы I согласно изобретению могут быть переведены в четвертичные соли. Для образования четвертичной соли предпочтительно используют немного больше, чем эквимолярное количество алкилгалогенида, предпочтительно хлорида, бромида, йодида или алкилсульфата. Эта реакция может проходить в инертном органическом, диполярном апротонном растворителе.
При желании соединения формулы I по изобретению могут быть превращены в их соли, полученные присоединением кислот, известным способом с использованием любой кислоты, пригодной для образования терапевтически приемлемых солей, полученных присоединением кислоты.
При желании соединения формулы I, полученные способом по изобретению, или их соли могут быть подвергнуты дополнительным процедурам очистки, например перекристаллизации. Спектр растворителей, пригодных для перекристаллизации, зависит от свойств растворения и кристаллизации соединений, которые должны быть перекристаллизованы.
Новые эфирные производные трансаповинкаминовой кислоты формулы I согласно изобретению могут быть рацемическими или оптически активными. При использовании оптически активных соединений формулы II в качестве исходных веществ получают оптически активные составы формулы I; в то время как рацемические соединения формулы II, использованные в качестве исходных веществ, дают в результате рацемические соединения формулы I. Из рацематов формулы I оптически активные соединения могут быть получены путем их разделения известным способом.
Новые рацемические или оптически активные эфирные производные трансаповинкаминовой кислоты формулы I или их соли могут быть превращены в фармацевтические композиции путем их смешивания с нетоксичными, инертными твердыми или жидкими носителями и/или другими вспомогательными средствами, обычно используемыми в терапии для парентерального или энтерального введения. Приемлемыми носителями являются, например, вода, желатин, лактоза, крахмал, пектин, стеарат магния, стеариновая кислота, тальк, растительные масла, такие как арахисовое масло, оливковое масло и подобные. Активный ингредиент может быть изготовлен в виде любой обычной фармацевтической композиции, в частности твердой композиции, например таблетки, драже, капсулы, суппозитория и подобных. Количество твердого носителя может изменяться в широких пределах, преимущественно между 25 мг и 1 г. Дополнительно композиции могут содержать также широко используемые другие фармацевтические вспомогательные средства, например стабилизаторы, консерванты, увлажняющие агенты, поверхностно-активные вещества, эмульгирующие агенты и подобные. Композиции могут быть приготовлены известным способом по любой обычной фармацевтической технологии и, при желании, они могут быть подвергнуты любым другим необходимым обработкам, например стерилизации.
Изобретение подробно иллюстрируется с помощью следующих неограничивающих примеров.
Пример 1
Получение 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
После перемешивания 15 г (0,045 моль) метилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 300 мл этиленгликоля в присутствии 1 г трет-бутоксида калия при 120oC в течение 5 часов реакционную смесь охлаждают до комнатной температуры. Смесь выливают в 1 литр охлажденной на льду воды, кристаллический осадок фильтруют, промывают дважды 50 мл воды каждый раз и высушивают с получением 15,9 г (97% выход) указанного в заголовке продукта, т. пл. 82-87oC.
[α]
1Н-ЯМР (CDCl3) δ: 0,65(3H, t, 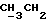 ); 0,66-3,2 (14H, m, скелетные протоны, OH); 3,85 (2H, m, HO
); 0,66-3,2 (14H, m, скелетные протоны, OH); 3,85 (2H, m, HO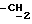 ); 4,43 (2H, m,
); 4,43 (2H, m, 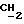 -O-C=O); 6,35 (1H, s, H-15); 7,00-7,55 (4H, m, ароматические протоны).
-O-C=O); 6,35 (1H, s, H-15); 7,00-7,55 (4H, m, ароматические протоны).
Анализ:
Вычислено для C22H25N2O3 (молекулярная масса 366,44), %:
C 72,10; H 7,15; N 7,65.
Найдено, %: C 71,89; H 7,15; N 7,62.
Пример 2
Получение моногидрохлорида 2-ацетоксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору, содержащему 3,66 г (0,01 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 50 мл безводного пиридина, по каплям добавляют 5 мл (0,052 моль) ангидрида уксусной кислоты, реакционную смесь оставляют при комнатной температуре на ночь, затем ее выпаривают до сухого состояния при пониженном давлении. После растворения остатка, полученного после выпаривания, в смеси 100 мл этилацетата, 30 мл воды и 50 мл насыщенного раствора гидрокарбоната натрия, фазы разделяют. Органический слой промывают три раза по 20 мл воды и высушивают над безводным сульфатом натрия.
Раствор остатка, полученного при выпаривании, в 30 мл изопропанола подкисляют до pH 4 добавлением этанольного раствора хлористого водорода, оставляют стоять в холодильнике на ночь, затем кристаллический осадок фильтруют и высушивают для получения 2,8 г (62%) указанного в заголовке продукта, т. пл.: 179-189oC.
[α]
1H-ЯМР (CDCl3) δ: 0,75 (3H, t, 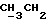 ); 2,1 (3H, s, CH2CO); 4,28 (1H, s, H-3); 6,28 (1H, s, H-15); 4,30-4,70 (4H, m, -OCH2CH2CH2O-); 7,05-7,60 (4H, m, ароматические протоны).
); 2,1 (3H, s, CH2CO); 4,28 (1H, s, H-3); 6,28 (1H, s, H-15); 4,30-4,70 (4H, m, -OCH2CH2CH2O-); 7,05-7,60 (4H, m, ароматические протоны).
Пример 3
Получение моногидрохлорида 2-(2-тиофеноилокси)этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору, приготовленному при комнатной температуре из 3,66 г (0,01 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 20 мл безводного пиридина, добавляют по каплям 4,8 мл (0,045 моль) хлорида тиофен-2-карбоновой кислоты, реакционную смесь выдерживают 24 часа и затем выпаривают до сухого состояния при пониженном давлении. После добавления 100 мл дихлорметана и 20 мл воды к остатку после выпаривания pH доводят до 9 добавлением 20 мл насыщенного раствора гидрокарбоната натрия. После разделения фаз органическую фазу промывают водой и сушат над безводным сульфатом натрия. После фильтрования раствор выпаривают до сухого состояния при пониженном давлении, остаток растворяют в 50 мл изопропанола и pH доводят до 4 прибавлением этанольного раствора хлористого водорода. После выдерживания в холодильнике в течение ночи раствор фильтруют и кристаллический осадок промывают холодным изопропанолом для получения 2,9 г указанного в заголовке продукта (выход 57%), т.пл.: 178-183oC.
[α]
Пример 4
Получение 2-(-4-нитробензоилокси)этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору, приготовленному при комнатной температуре из 3,66 г (0,01 моль) 2-гидроксиэтилового эфира 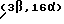 /-/-трансаповинкаминовой кислоты в 20 мл безводного пиридина, добавляют 4,2 г (0,0225 моль) 4-нитробензоилхлорида, реакционную смесь выдерживают в течение ночи. После разведения реакционной смеси 150 мл этилацетата добавляют 50 мл воды и 3 мл концентрированного водного раствора гидрокисида аммония. Фазы разделяют, затем органическую фазу промывают 4 раза по 25 мл воды и высушивают над безводным сульфатом натрия. После фильтрования раствор выпаривают до сухого состояния при пониженном давлении, маслянистый остаток растворяют в 50 мл горячего изопропанола, осветляют активированным углем, и после фильтрования раствор выдерживают несколько часов. Кристаллический осадок фильтруют, промывают 10 мл изопропанола и высушивают до получения 2,6 г (58%) указанного в заголовке продукта, т. пл.: 139-142oC.
/-/-трансаповинкаминовой кислоты в 20 мл безводного пиридина, добавляют 4,2 г (0,0225 моль) 4-нитробензоилхлорида, реакционную смесь выдерживают в течение ночи. После разведения реакционной смеси 150 мл этилацетата добавляют 50 мл воды и 3 мл концентрированного водного раствора гидрокисида аммония. Фазы разделяют, затем органическую фазу промывают 4 раза по 25 мл воды и высушивают над безводным сульфатом натрия. После фильтрования раствор выпаривают до сухого состояния при пониженном давлении, маслянистый остаток растворяют в 50 мл горячего изопропанола, осветляют активированным углем, и после фильтрования раствор выдерживают несколько часов. Кристаллический осадок фильтруют, промывают 10 мл изопропанола и высушивают до получения 2,6 г (58%) указанного в заголовке продукта, т. пл.: 139-142oC.
[α]
Пример 5
Получение дигидрохлорида 2-(никотиноилокси)этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору, содержащему 2,4 г (0,0065 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 20 мл безводного пиридина, добавляют по каплям 2,15 г (0,015 моль) никотиноилхлорида, растворенного в 10 мл пиридина, реакционную смесь перемешивают при комнатной температуре в течение 3 часов, затем выпаривают до сухого состояния при пониженном давлении. После растворения остатка, полученного после выпаривания, в смеси 150 мл этилацетата и 50 мл насыщенного раствора гидрокарбоната натрия фазы разделяют. Органический слой дважды промывают 20 мл воды и сушат над безводным сульфатом натрия. После фильтрования раствор выпаривают до сухого состояния при пониженном давлении, остаток, полученный после выпаривания, растворяют в 30 мл изопропанола и этот раствор подкисляют до pH 4,0 добавлением этанольного раствора хлористого водорода. После выпаривания до сухого состояния при пониженном давлении остаток кристаллизуют из 50 мл этилацетата для получения 1,30 г (36%) указанного в заголовке соединения, т.пл.: 158-164oC.
[α]
Анализ:
Рассчитано для C28H29N3O4 2HCl (молекулярный вес: 544,47), %:
C 61,76; H 5,73; N 7,71; Cl 13,02.
Найдено, %: C 61,73; H 6,05; N 7,69; Cl 12,90.
Пример 6
Получение гидрохлорида 2-(бензоилокси)этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору 4,4 г (0,012 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 60 мл хлорбензола добавляют 1,83 г (0,018 моль) триэтиламина и 2,5 г (0,018 моль) бензоилхлорида. Смесь перемешивают при 40oC в течение 30 минут, затем подщелачивают до pH 8 прибавлением 15 мл 10%-ного раствора гидрокарбоната натрия. После разделения органическую фазу два раза промывают 20 мл воды и высушивают над безводным сульфатом магния. После отфильтровывания осушающего агента и его двукратной промывки по 3 мл хлорбензола каждый раз добавляют раствор гидрохлорида в диоксане до pH 3-4, затем кристаллический осадок отфильтровывают при 0oC, тщательно растирают в порошок в холодном ацетоне и фильтруют для получения 4,8 г (79%) указанного в заголовке продута, т. пл.: 216-217,5oC.
[α]
Пример 7
Получение метансульфоната 2-(4-хлоробензоилокси)этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
После растворения 4,4 г (0,012 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 60 мл дихлорметана добавляют 1,83 г (0,018 моль) триэтиламина и 3,15 г (0,018 моль) 4-хлорбензоилхлорида и реакционную смесь перемешивают при 40oC в течение 30 минут. После охлаждения до 10oC прибавляют 25 мл воды и значение pH смеси доводят до 8 прибавлением 15 мл 10%-ного раствора гидрокарбоната натрия. После разделения органическую фазу дважды промывают 20 мл воды каждый раз и высушивают над безводным сульфатом магния. После отфильтровывания осушающего агента и его двукратной промывки по 5 мл дихлорметана каждый раз, органическую фазу выпаривают до полного удаления растворителя. Остаток растворяют в 35 мл ацетона и подкисляют до pH 4 прибавлением метансульфоновой кислоты. Кристаллический осадок фильтруют при 0oC, промывают холодным ацетоном и высушивают для получения 5,85 г (81%) указанного в заголовке продукта, т. пл.: 197-199oC.
[α]
Пример 8
Получение гидрохлорида 2-(пропионилокси)этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору, содержащему 4,4 г (0,012 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 100 мл хлорбензола, добавляют 5,33 (0,04 моль) ангидрида пропионовой кислоты и 1,4 г (0,014 моль) триэтиламина. После перемешивания реакционной смеси при 80oC в течение 4 часов 40 мл хлорбензола и избыток пропионового ангидрида отгоняют при пониженном давлении. После добавления 25 мл воды к остатку при комнатной температуре pH доводят до 8 добавлением 20 мл 10%-ного раствора гидрокарбоната натрия. После разделения органическую фазу дважды промывают 20 мл воды каждый раз и высушивают над безводным сульфатом магния. Осушающий агент отфильтровывают и дважды промывают 3 мл хлорбензола каждый раз. После выпаривания органической фазы под пониженным давлением до удаления растворителя остаток растворяют в 40 мл ацетона и подкисляют до pH 4 добавлением диоксанового раствора хлористого водорода. Кристаллы отфильтровывают при 5oC, промывают ацетоном и высушивают для получения 4,2 г (76%) гидрохлорида, т. пл.: 219- 220oC.
[α]
Пример 9
Получение гидрохлорида 2-(3,4,5-триметоксибензоилокси)- этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору, содержащему 5,1 г (0,014 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 80 мл дихлорэтана, добавляют 2 г (0,02 моль) триэтиламина и 4,6 г (0,02 моль) 3,4,5- триметоксибензоилхлорида. Смесь перемешивают при 50oC в течение 1 часа, затем добавляют 50 мл воды при комнатной температуре и смесь подщелачивают до pH 8 добавлением 20 мл 10%-ного раствора гидрокарбоната натрия. После разделения органическую фазу дважды промывают 25 мл воды каждый раз и высушивают над безводным сульфатом магния. После отфильтровывания осушающего агента и его промывания дважды по 5 мл дихлорэтана каждый раз фильтрат выпаривают до сухого состояния под пониженным давлением, остаток растворяют в 25 мл этилацетата и подкисляют до pH 4 диоксановым раствором гидрохлорида. Кристаллический осадок фильтруют при 10oC и промывают ацетоном для получения 6,2 г (76%) указанного в заголовке гидрохлорида, т. пл.: 191-194oC.
[α]
Пример 10
Получение дигидрохлорида этиленгликолевого эфира (3β,16α) /-/- бис-трансаповинкаминовой кислоты
К раствору, содержащему 5,1 г (0,014 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 100 мл дихлорэтана, добавляют 6 г (0,06 моль) триэтиламина, затем, при охлаждении, 7,54 г (0,02 моль) гидрохлорида хлорида /-/-трансаповинкаминовой кислоты, затем реакционную смесь перемешивают при комнатной температуре в течение 2 часов. После добавления к смеси 50 мл воды и 20 мл 10%-ного раствора гидрокарбоната натрия до pH 8 и разделения органическую фазу дважды промывают 25 мл воды каждый раз, высушивают над безводным сульфатом магния, фильтруют и дважды промывают 5 мл дихлорэтана каждый раз. Органический слой выпаривают до полного удаления растворителя. Остаток растворяют в 60 мл ацетона и подкисляют до pH 3,5 добавлением хлористого водорода, растворенного в диоксане. Кристаллический осадок фильтруют при 5 oC и промывают ацетоном для получения 6,55 г (63%) указанного в заголовке дигидрохлорида, т. пл.: 222-225oC.
[α]
Пример 11
Получение метансульфоната 2-(ацетокси)этилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты
К раствору 7,3 г (0,02 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты в 100 мл дихлороэтана добавляют 2,2 г (0,022 моль) триэтиламина и 2,42 г (0,03 моль) ацетилхлорида. После перемешивания реакционной смеси при 50oC в течение 15 минут и добавления 50 мл воды при комнатной температуре ее подщелачивают до pH 8 добавлением 20 мл 10%-ного раствора гидрокарбоната натрия. После разделения органическую фазу дважды промывают 25 мл дихлорэтана и высушивают над безводным сульфатом магния. Осушающий агент фильтруют, дважды промывают 5 мл дихлорэтана каждый раз, а фильтрат выпаривают при пониженном давлении до полного удаления растворителя. Остаток растворяют в 40 мл этилацетата и подкисляют до pH 3 метансульфоновой кислотой. После фильтрования кристаллического осадка при 0oC, промывания его этилацетатом и высушивания получают 7,75 г (77%) указанной в заголовке соли, т. пл.: 130-133oC.
[α]
Пример 12
Получение бензосульфоната 2-(ацетокси)этилового эфира (3β,16α) /-/- трансаповинкаминовой кислоты
Следуя процедуре, описанной в примере 11, получение соли проводят с бензолсульфоновой кислотой вместо метансульфоновой кислоты при величине pH 3,5. Бензолсульфонат получают с выходом 7,9 г (70%), т. пл.: 180,5-183oC.
[α]
Пример 13
Получение 2-гидроксиэтилового эфира (3α,16α) /+/-трансаповинкаминовой кислоты
Раствор, содержащий 16,88 г (0,05 моль) метилового эфира (3α,16α) /+/-трансаповинкаминовой кислоты в 335 мл этиленгликоля, перемешивают с 1 г трет-бутоксида калия при 120oC в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь медленно выливают в 1 литр ледяной воды. Осадок фильтруют, промывают 3 раза по 500 мл воды каждый раз и высушивают для получения указанного в заголовке соединения с выходом 17,6 г (96%), т. пл.: 85-90oC.
[α]
Пример 14
Получение гидрохлорида 2-(бензоилокси)этилового эфира (3α,16β) /-/-трансаповинкаминовой кислоты
Следуя процедуре, описанной в примере 6, 4,4 г (0,12 моль) 2-гидроксиэтилового эфира (3β,16α) /+/-трансаповинкаминовой кислоты используют вместо 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты для получения 4,7 г (77 %) указанного в заголовке гидрохлорида, т.пл.: 217-219oC.
[α]
Пример 15
Получение метансульфоната 2-(4-хлоро-бензоилокси)-этилового эфира (3α,16β) /+/-трансаповинкаминовой кислоты
Следуют процедуре, описанной в примере 7, но вместо 4,4 г (0,012 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты используют 4,4 г (0,012 моль) 2-гидроксиэтилового эфира (3β,16α) /+/-трансаповинкаминовой кислоты для получения указанной в заголовке соли с выходом 5,5 г (76%), т. пл. 199-200oC.
[α]
Пример 16
Получение гидрохлорида 2-(пропионилокси)этилового эфира (3α,16β) /+/-трансаповинкаминовой кислоты
Следуют процедуре, описанной в примере 8, но вместо 4,4 г (0,012 моль) 2-гидроксиэтилового эфира (3α,16β) /-/-трансаповинкаминовой кислоты используют 4,4 г (0,012 моль) 2-гидроксиэтилового эфира (3β,16α) /-/-трансаповинкаминовой кислоты, чтобы получить 4,35 г (79%) указанного в заголовке гидрохлорида, т. пл.: 220-221oC.
[α]
Пример 17
Получение 2-(3,4,5-триметоксибензоилокси)этилового эфира (3α,16β) /+/-трансаповинкаминовой кислоты
Следуют процедуре, описанной в примере 9, но вместо 5,1 г (0,014 моль) 2-гидроксиэтилового эфира (3α,16β) /-/-трансаповинкаминовой кислоты используют 5,1 г (0,14 моль) 2-гидроксиэтилового эфира (3β,16α) /+/-трансаповинкаминовой кислоты для получения 6,2 г (76%) указанного в заголовке гидрохлорида, т. пл.: 192-195oC.
[α]
Пример 18
Получение гидрохлорида 2-(ацетокси)этилового эфира (3α,16β) /+/-трансаповинкаминовой кислоты
К раствору 7,3 г (0,02 моль) 2-гидроксиэтилового эфира (3α,16β) /-/- трансаповинкаминовой кислоты в 100 мл дихлорэтана добавляют 2,2 г (0,022 моль) триэтиламина и 2,42 (0,03 моль) ацетилхлорида. После перемешивания при 40o в течение 25 минут с последующим добавлением 50 мл воды при комнатной температуре pH доводят до 8 добавлением 20 мл 10%-ного раствора гидрокарбоната натрия. После разделения органическую фазу дважды промывают 25 мл воды каждый раз и высушивают над безводным сульфатом магния. После фильтрования осушающий агент дважды промывают 5 мл дихлорэтана каждый раз, фильтрат выпаривают при пониженном давлении до полного удаления растворителя. Остаток растворяют в 40 мл этилацетата и подкисляют до pH 4 добавлением диоксанового раствора хлористого водорода. Кристаллический осадок фильтруют при 0oC, промывают этилацетатом и высушивают до получения 6,95 r (78%) указанного в заголовке гидрохлорида, т. пл.: 221-224,5oC.
[α]
Пример 19
Получение гидрохлорида рацемического 2-гидроксиэтилового эфира трансаповинкаминовой кислоты
Следуют процедуре, описанной в примере 13, но вместо 16,8 г (0,05 моль) метилового эфира (3α,16β) /+/-трансаповинкаминовой кислоты используют 16,8 г (0,05 моль) метилового эфира рацемической трансаповинкаминовой кислоты для получения указанного рацемата гидрохлорида с выходом 17,4 г (95%), т. пл.: 97-101oC.
Пример 20
Получение гидрохлорида рацемического 2-(ацетокси)этилового эфира трансаповинкаминовой кислоты
Следуют процедуре, описанной в примере 18, но вместо 7,3 г (0,02 моль) 2-гидроксиэтилового эфира (3α,16β) /+/-трансаповинкаминовой кислоты используют 7,3 г (0,02 моль) рацемического 2-гидроксиэтилового эфира трансаповинкаминовой кислоты для получения 7,1 г (80%) указанного в заголовке гидрохлорида, т. пл.: 187-189oC.
Пример 21
Получение 3-гидроксипропилового эфира (3α,16β) /-/-транс аповинкаминовой кислоты
Следуют процедуре, описанной в примере 1, но вместо 300 мл этиленгликоля используют 300 мл пропиленгликоля. После выливания в воду полученное несмешивающееся с водой масло растворяют в 100 мл дихлорметана и дважды промывают 20 мл воды каждый раз. Органический слой сушат над безводным сульфатом магния и фильтруют. Осушающий агент дважды промывают 5 мл дихлорметана каждый раз, фильтрат выпаривают под пониженным давлением до полного удаления растворителя. Указанное в заголовке соединение получают с выходом 16,8 r в форме светло-желтого вязкого масла. Гидрохлорид соединения плавится при 216-218oC после перекристаллизации из ацетона. \\2 [α]
Пример 22
Получение гидрохлорида 3-(ацетокси)пропилового эфира (3α,16β) /-/-трансаповинкаминовой кислоты
Следуют процедуре, описанной в примере 18, но вместо 7,3 г (0,02 моль) 2-гидроксиэтилового эфира (3α,16β) /+/-трансаповинкаминовой кислоты используют 7,6 г (0,02 моль) 3-гидроксипропилового эфира (3α,16β) /-/-трансаповинкаминовой кислоты для получения в результате указанного в заголовке гидрохлорида с выходом 7,0 г (76%), т. пл.: 205-207oC.
[α]
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АНДРОСТАНА, ЗАМЕЩЕННЫЕ ПО 16-ПОЛОЖЕНИЮ ЧЕТВЕРТИЧНОЙ АММОНИЕВОЙ ГРУППОЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2124021C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2037499C1 |
| ПРОИЗВОДНЫЕ ПРОПАН-2-ОЛА | 1994 |
|
RU2127277C1 |
| ПЕНТАПЕПТИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР ПРОЛИФЕРАЦИИ ЭПИДЕРМАЛЬНЫХ КЛЕТОК | 1991 |
|
RU2018510C1 |
| Способ получения рацематов сложных эфиров цис-и/или транс-аповинкаминовой кислоты или их оптически активных изомеров | 1982 |
|
SU1258326A3 |
| Способ получения рацематов или оптически активных производных цис- или транс-гидроксииминооктагидроиндол @ 2,3-а @ хинолизина или их гидрохлоридов | 1983 |
|
SU1376947A3 |
| ПРОИЗВОДНЫЕ 17-ГАЛОГЕН-4-АЗААНДРОСТЕНА | 1993 |
|
RU2103275C1 |
| СПОСОБ ПОЛУЧЕНИЯ β- ЗАМЕЩЕННОГО 4-АЗААНДРОСТАНА | 1993 |
|
RU2109746C1 |
| ПРОИЗВОДНЫЕ КАРБАЗОЛОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2119914C1 |
| Способ получения оптически активных 9-или 11-замещенных производных аповинкаминовой кислоты или их солей | 1985 |
|
SU1398775A3 |


Изобретение относится к новым рацемическим и оптически активным эфирным производным трансаповинкаминовой кислоты формулы I, где R обозначает водород или группу (а), в которой Z представляет собой C1-4-алкильную, возможно замещенную арильную, аралкильную, гетероарильную или 14-эбурнаменинильную группу, n представляет собой целое число 2, 3 или 4, а также к их терапевтически приемлемым солям. Изобретение дополнительно относится к фармацевтическим композициям, содержащим эти соединения, а также к способу получения вышеуказанных соединений и композиций и к способу лечения. Новые соединения формулы I обладают, в частности, антиоксидантным, антиишемическим, а также антиамнезийным действием и пригодны для ингибирования перокисления липидов и для лечения или защиты от ишемии и амнезии, а также для лечения различных дегенеративных неврологических заболеваний, например болезни Альцгеймера. 4 табл.
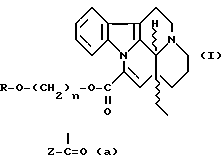
Рацемические или оптические активные эфирные производные трансаповинкаминовой кислоты формулы I
где R обозначает водород или группу  в которой Z представляет собой C1-4алкильную, C6-8арильную группу, возможно, замещенную такими заместителями, как алкокси C1-6, нитро или галоген, арилC6-8алкильную C1-4группу, возможно, замещенную такими заместителями, как нитро или галоген, 5-8-членную гетероарильную группу, имеющую в качестве гетероатома серу или азот, или 14-эбурнаменинильную группу, n представляет собой целое число 2, 3 или 4, а также их терапевтически приемлемые соли.
в которой Z представляет собой C1-4алкильную, C6-8арильную группу, возможно, замещенную такими заместителями, как алкокси C1-6, нитро или галоген, арилC6-8алкильную C1-4группу, возможно, замещенную такими заместителями, как нитро или галоген, 5-8-членную гетероарильную группу, имеющую в качестве гетероатома серу или азот, или 14-эбурнаменинильную группу, n представляет собой целое число 2, 3 или 4, а также их терапевтически приемлемые соли.
| ОБОЛОЧКА ИЗ КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ | 2013 |
|
RU2531108C1 |
| US 4486437, 1984 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1987, ч.2, с.465. | |||

