Изобретение относится к области получения олефиновых углеводородов, в частности, олефиновых углеводородов C2-C20, дегидрированием парафиновых углеводородов.
Олефины являются важными промежуточными веществами для производства химических продуктов, имеющих широкое распространение, таких как полипропилен, высокооктановые добавки (метил-третично-бутиловый эфир-МТБЭ), топлива с высоким октановым числом, алкилатпроизводные и множество других продуктов.
Несмотря на растущий спрос на эти продукты, расширение их производства часто невозможно из-за ограниченного наличия олефинов, например, изобутилена для МТБЭ.
Это привело к установлению других источников поставки олефинов наряду с традиционными (например, крекинг). Среди них источник, который становится все более и более важным - это реакция дегидрирования легких парафиновых углеводородов. Данная реакция, простая с точки зрения стехиометрии, имеет проблемы в плане термодинамики и кинетики. Реакция эндотермическая и регулируется термодинамическим равновесием. Это приводит к необходимости создания температур выше 500oC при дегидрировании C2-C20 парафинов для получения экономически выгодных конверсий за проход. Кроме того, возникает необходимость снабдить систему теплом.
Несмотря на высокие рабочие температуры, скорость дегидрирования низкая и поэтому необходимо использовать соответствующий катализатор. Последний должен быть термически стабильным и способным гарантировать высокие селективности в требуемые олефины, сводя при этом к минимуму побочные реакции изомеризации крекинга, коксообразования и ароматизации и обеспечения промышленно пригодные значения конверсии.
Неизбежное образование кокса на катализаторе вызывает прогрессирующее снижение каталитической активности, и поэтому необходимо проводить периодическую регенерацию.
И, как результат, каталитический состав должен иметь высокую стабильность в условиях, которым он подвергается во время реакции и в периоды регенерации.
Известны способы получения олефиновых углеводородов дегидрированием при повышенных температурах соответствующих парафиновых углеводородов в присутствии каталитических составов на основе благородных металлов (Патенты США N 3531543, 4786625, 4886928 и Европ. патент N 351067), а также на основе оксидов металлов в присутствии промоторов, в большинстве случаев это нанесенный Cr2O3 (Патенты США N 2945823, 2956030, 2991255 и патент Великобритании N 2162082).
Однако обе группы указанных составов имеют недостатки. Составы на основе благородных металлов требуют особой обработки на стадии регенерации с целью сохранения дегидрирующей активности металлических компонентов, например пост-обработки хлор-содержащими веществами с последующим восстановлением (Патент США N 4438288). Составы на основе оксида хрома, нанесенного на оксид алюминия, оксид кремния, оксид кремния - оксид алюминия и т.д. характеризуются тем, что они имеют низкую селективность в олефины вследствие их кислотной природы, вызывающей паразитические реакции, такие как изомеризация, крекинг, коксообразование и ароматизация, которые являются типичными кислотно катализируемыми реакциями.
Селективность в олефины увеличивается модификацией состава за счет добавки оксидов щелочного или щелочноземельного металла для уменьшения кислотных свойств.
Однако в литературе сообщается (Jiurnal of Physical Chemistry, том 66, 1962 г.), что добавка большого количества оксидов щелочного металла с целью улучшения селективности подвергает опасности каталитические свойства составов: сильное взаимодействие с оксидом хрома подавляет дегидрирующую активность, тогда как остаточный хром в окисленном состоянии с валентностью более чем +3 не может быть восстановлен, поскольку он стабилизирован такой большой загрузкой щелочного металла.
Наиболее близким к предлагаемому является способ получения олефиновых углеводородов дегидрированием соответствующих парафиновых углеводородов при повышенной температуре в присутствии катализатора состава (мас.%):
Cr2O3 - 12,2
K2O - 1,4
SiO2 - 2
Al2O3 - Остальное
(Патент РФ N 1366200, публ. Б.И. N 2, 15.01.88 г).
Выход олефина по данному способу является недостаточно высоким, что связано с недостаточно высокой селективностью катализатора.
Задачей, решаемой настоящим изобретением, является повышение выхода олефинового углеводорода. Предлагается способ получения олефиновых углеводородов путем дегидрирования соответствующих парафиновых углеводородов при температуре 450-800oC, давлением от 0,1 до 3 атмосфер абсолютных и объемной скорости газа от 100 до 1000 час-1 в присутствии катализатора состава (мас. %):
Cr2O3 - 6,0 - 30,0
SnO - 0,1 - 3,5
Me2O - 0,4 - 3,0
SiO2 - 0,08 - 3,0
Al2O - Остальное
где Me - щелочной металл.
После стадии дегидрирования катализатор направляют на регенерацию.
Предпочтительными являются следующие варианты осуществления нового способа:
- используют катализатор, содержащий Al2O3 в дельта, тета, дельта и тета, тета и альфа, или дельта, тета и альфа фазах;
-используют катализатор состава, мас.%:
Cr2O3 - 13 - 25
SnO - 0,2 - 2,8
MeO - 0,5 - 2,5
SiO2 - 0,08 - 3,0
Al2O3 - Остальное
где Me - щелочной металл,
в качестве щелочного металла в катализаторе используют калий;
в составе катализатора используют Al2O3 с удельной поверхностью менее 150 м2/г;
дегидрирование и регенерацию осуществляют в псевдоожиженном слое катализатора;
дегидрирование в псевдоожиженном слое катализатора осуществляют при температуре от 450 до 650oC, атмосферном или несколько большем давлении, объемной скорости газа от 100 до 1000 час-1 и времени пребывания катализатора в зоне псевдоожижения от 5 до 30 минут. При этом предпочтительной является объемная скорость от 150 до 200 час-1, а время пребывания катализатора от 10 до 15 минут;
при осуществлении дегидрирования и регенерации в псевдоожиженном слое катализатора регенерацию проводят в присутствии воздуха, или кислорода или другого газа, поддерживающего горение, при температуре выше средней температуры дегидрирования и атмосферном или несколько большем давлении, объемной скорости газа от 100 до 1000 час-1 и времени пребывания катализатора от 5 до 60 минут.
Использование в заявляемом способе дегидрирования катализатора, к которому добавлен оксид олова, значительно улучшает селективность в требуемые олефины, т.к. добавка олова резко снижает образование продуктов, образуемых в кислотно катализируемых побочных реакциях.
Процесс получения каталитической системы, применяемой в новом способе, состоит главным образом из диспергирования соединений хрома, щелочного металла и олова на носителе, состоящем из оксида алюминия и оксида кремния. Ниже приводятся методики такого диспергирования. Следует понимать, что изобретение ими не ограничивается.
Диспергирование может осуществляться пропиткой указанного носителя раствором, содержащим предшественники оксидов хрома, калия и олова, с последующей сушкой и прокаливанием, или ионной абсорбцией с последующим отделением жидкости, сушкой и прокаливанием твердого вещества. Предпочтение отдается пропитке согласно методу "начальной влажности" носителя раствором, содержащим все предшественники активных элементов.
Что касается олова, приводятся другие методики, с помощью которых его можно добавлять к каталитической системе:
- добавка олова к носителю перед диспергированием предшественников оксидов хрома и калия;
обработка твердого вещества, содержащего оксиды хрома и калия раствором, содержащим соединения олова, с помощью ионного обмена, пропитка и т.д.
- осаждение олова с помощью пароосаждения на носитель до добавки предшественников оксида хрома и оксида калия, используя летучие соединения веществ, которые осаждаются;
- осаждение олова с помощью пароосаждения на твердое вещество, содержащее: оксид алюминия, оксид хрома, оксид калия, используя летучие соединения веществ, которые осаждаются.
Среди вышеуказанных методик предпочтительными являются сопропитка носителя раствором, содержащим предшественники активных элементов: оксидов хрома, калия и олова, и пароосаждение олова.
Как неорганические, так и органические соли олова или металлоорганических производных могут использоваться в качестве предшественников оксида двухвалентного и/или четырехвалентного олова.
Неорганические или органические соли, не очень растворимые в воде, могут использоваться после контроля pH раствора, величины, которая влияет на их растворимость.
Допускается применение металлоорганических производных, растворенных в органических растворителях, в каталитической системе согласно методикам, описанным выше.
Регенерация катализатора а предлагаемом способе проводится в атмосфере воздуха и/или кислорода с увеличением температуры самой каталитической системы до соответствующих величин, например, с помощью сжигания подходящего топлива. За регенерацией должна следовать стадия восстановления катализатора с целью восстановления катализатора с целью восстановить шестивалентный хром, образованный на стадии регенерации.
Заявленный процесс может применяться для любой технологии дегидрирования: в неподвижном, псевдоожиженном или движущемся слое катализатора.
Предпочтительнее, процесс следует проводить в системе псевдоожиженного слоя в основной своей части, состоящей из реактора, где проходит реакция, и регенератора, где катализатор регенерируется за счет выжигания на его поверхности кокса, образованного на стадии реакции.
В системе реактор-регенератор катализатор в псевдоожиженном состоянии циркулирует непрерывно между реактором и регенератором, позволяя проводить процесс непрерывно и снабжая реакцию необходимым теплом за счет регенерированного катализатора, который поступает в реактор при температуре, которая выше средней реакционной температуры. В реакторе катализатор поддерживается в псевдоожиженном состоянии с помощью реакционного газа, который поступает в каталитический слой снизу через специальную систему распределения.
Прореагировавший газ выходит из реактора сверху после прохождения системы циклонов или других соответствующих систем отделения порошкообразных веществ; затем газ направляется в теплообменник для подогрева подаваемого сырья, а затем в секцию разделения, где выделяются полученные олефины, тогда как непрореагировавшие парафины могут быть возвращены на стадию синтеза, а побочные продукты отделяются и могут быть использованы в регенераторе в качестве топливного газа.
Если в производстве после установки дегидрирования есть установка этерификации, то секция разделения используется только для удаления побочных продуктов.
В реакторе катализатор в псевдоожиженном состоянии движется противотоком по отношению к газовой фазе: газ поступает в каталитический слой снизу через распределитель, который равномерно распределяет его по поверхности слоя. Катализатор выходит из реактора снизу, проходя самотеком в зону десорбции, которая является частью реактора с диаметром, меньшим или равным диаметру реакционной зоны, где газ, находящийся между частицами, перемещается и десорбируется при введении азота или метана снизу, так, что перемещенный или десорбированный газ вновь поступает в реактор, не допуская потерь реагентов или продуктов.
Катализатор, находящийся в псевдоожиженном состоянии, затем направляется пневматически в регенератор.
В реакторе в псевдоожиженном слое предпочтительно иметь следующие рабочие условия:
- поддерживание температуры на уровне от 450oC до 650oC с помощью скорости потока регенерированного катализатора, в зависимости от обрабатываемых парафинов или их смеси;
- давление атмосферное или несколько выше;
- объемная скорость от 100 до 1000 час-1 (N л газа в час и на л катализатора), наиболее предпочтительно от 150 до 200;
- время пребывания катализатора в зоне псевдоожижения от 5 до 30 минут, предпочтительно от 10 до 15 минут, а в зоне десорбции от 0,2 до 10 минут.
Решетки со свободной площадью от 10 до 90%, предпочтительно от 20 до 40%, могут быть установлены горизонтально внутри реактора на расстоянии от 20 до 200 см друг от друга.
Такие решетки устанавливаются с целью предотвратить обратное перемешивание газа и твердого вещества, так что поток газа внутри реактора выглядит как вытесняющий: таким способом достигаются максимальные конверсия парафинов и селективность в требуемые олефины.
В частности, селективность можно еще увеличить с помощью аксиального термического профиля, который устанавливается по слою катализатора при максимуме температуры в верхней части, куда поступает регенерированный катализатор, и минимуме температуры в нижней части: разница в температуре по слою предпочтительно от 15 до 65oC.
С целью оптимизации аксиального термического профиля можно также распределять регенерированный катализатор, изменяя высоту каталитического слоя. Система пневмотранспортера из реактора в регенератор состоит из транспортной линии с, по крайней мере, одной зоной, в которой катализатор имеет движение в направлении вниз и в которой поддерживаются промежуточные условия от минимальной флюидизации до минимального образования пузырьков, путем введения соответствующего количества газа на соответствующей высоте, и зоной, где катализатор имеет движение вверх до тех пор, пока он не достигнет верхней части катализаторного слоя регенератора, путем введения газа в основание, что значительно снижает плотность текучей среды.
Предпочтительно иметь регенератор аналогичных размеров, что и реактор.
Соответствующий распределитель распределяет катализатор, поступающий из реактора в регенератор, по поверхности каталитического слоя. Регенерация происходит внутри слоя за счет выжигания кокса, отложенного на катализаторе, а нагрев катализатора происходит при сжигании метана или топливного газа в присутствии воздуха или кислорода или другого топливного газа при температуре, которая выше средней температуры реактора.
До подачи регенерированного катализатора в реактор он подвергается восстановительной обработке при температурах от 650 до 680oC и в течение периода от 0,2 до 10 минут с целью удаления шестивалентного хрома, который образуется при сжигании кокса.
В регенераторе также движение газа и твердого вещества происходит противотоком: воздух подается в низ каталитического слоя, тогда как топливный газ поступает на соответствующую высоту слоя.
Газ, выходящий из регенератора, состоящий из азота и продуктов горения, можно пропускать через циклоны или другую систему, расположенную в верхней части аппарата, для отделения накопленного порошка, а затем после регенератора его можно направлять в теплообменник для подогрева воздуха, предназначенного для процесса горения.
До выпуска в атмосферу эти газы пропускают через систему фильтров или другие устройства для снижения содержания пыли до нескольких десятков миллиграмм на Nм3.
Поскольку процесс горения происходит при температуре, которая ниже 700oC, содержание оксида углерода и оксидов азота в отходящем газе такого, что не требуется дальнейшей очистки.
Рабочее давление в реакторе предпочтительнее либо атмосферное, либо несколько выше, объемная скорость от 100 до 1000 час-1, а время пребывания твердого вещества в пределах от 5 до 60 минут, наиболее предпочтительно от 20 до 40 минут.
Регенерированный катализатор передается в реактор таким же образом, как и истощенный катализатор в регенератор.
Система реактора-регенератора, задуманная таким образом, позволяет поддерживать постоянными рабочие параметры и характеристики в течение всего срока технической эксплуатации установки.
Аликвотные доли катализатора периодически выгружаются из системы и заменяются равными аликвотными долями свежего катализатора, не прерывая при этом работу установки.
Преимущества использования системы реактора с псевдоожиженным слоем - регенератора можно суммировать следующим образом:
- оптимальный температурный профиль в реакторе позволяет довести до максимума выход олефина;
- тепло передается непосредственно в реакцию с помощью регенерированного катализатора: отсутствие теплообменных поверхностей и сильного обратного перемешивания флюидизированного слоя предотвращает образование высоких температурных точек, которые снижают селективность;
- процесс в пседоожиженном слое не требует рециклов водорода, который вреден с термодинамической точки зрения, но необходим в других конфигурациях для поддержания температурного контроля;
- все прочие операции, имеющие место в данном процессе, непрерывны, и нет необходимости модифицировать рабочие параметры в течение всего срока эксплуатации установки;
- что касается проектной мощности, то установка может работать с широким диапазоном гибкости, подразумевая под этим производственную мощность:
- реакция и регенерация протекают в физически разделенных зонах, и не может быть никакого смешивания углеводородных потоков с потоками, содержащими кислород:
- процесс протекает при атмосферном давлении или несколько выше: поэтому воздух извне не может проникать в реакционную зону:
- нет специальной обработки, необходимой для снижения эмиссии газовых загрязняющих веществ.
На чертеже представлено возможное использование реакционно-регенераторной схемы, описанной ниже.
Углеводороды (1) подаются в реактор (A) через соответствующий распределитель (на чнртеже не показано), тогда как газы после реакции выводятся из линии (4) реактора после прохождения через циклоны FA.
Регенерированный катализатор (5) поступает на верх катализаторного слоя и выходит из реактора (A), проходя через десорбер (B), где он вступает в контакт с десорбирующим газом (2). Затем катализатор поступает в транспортную линию (6), по которой он направляется в регенератор (D), конкретно в верхнюю часть каталитического слоя.
В этом случае представлено (3) по одной линии ввода газа в транспортные линии. Катализаторопровод, заявленный здесь, характеризуется U-образной формой соединения между нижней и верхней частью. Катализатор спускается по регенератору (D), поступает в восстановитель (E), затем в десорбер (G), после него в транспортную линию (C), а затем направляется в реактор. Регенерирующий воздух (8), газ для горения, тот же, что и для восстановления катализатора в (E), и десорбирующий газ (10) поступают через соответствующие распределители (на чертеже не показано).
Газы после прохождения циклонов FD выходят через (7).
Представленные здесь примеры не следует рассматривать как ограничивающие данное изобретение.
Пример 1 (сравнительный)
Микрокристаллический псевдобемит с добавкой оксида кремния (1,2 мас.%) получали с размером диаметра частиц от 5 до 300 микрон методом распыления-сушки коллоидного раствора гидратированного оксида алюминия и Ludox оксида кремния.
Образец псевдобемита подвергают термообработке, состоящей из первого прокаливания при температуре 450oC в течение часа и второго при 1030oC в течение 4 часов в потоке сухого воздуха.
Полученный продукт имел удельную поверхность 100 м2/г, пористость 0,34 см3/г и состоял, главным образом, из дельта и тета переходных оксидов алюминия с небольшим количеством альфа оксида алюминия.
200 г такого оксида алюминия пропитывали, используя методику "начальной влажности", водным раствором (68 см3), содержащим 67,5 г CrO3 (99,8 мас.%) и 6,4 KOH (90 мас.%) в деионизированной воде при температуре 85oC. Пропитанный продукт оставляли при комнатной температуре на 1 час, затем высушивали при температуре 90oC в течение 15 часов. Высушенный продукт активировали в потоке сухого воздуха при температуре 750oC в течение 4 часов.
Получен катализатор, имеющий состав, мас.%:
Cr2O3 - 20
K2O - 1,87
SiO2 - 1,25
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана в интервале температур 540 - 580oC. Условия осуществления процесса описаны ниже. Результаты приведены в таблице 1.
Пример 2.
200 г микросферического оксида алюминия, полученного как описано в примере 1, пропитывали методом, описанным здесь ранее, водным раствором (68 см3), содержащим 68,3 г CrO3 (99,8 мас.%) и 6,48 г KOH (90 мас.%) и 4,13 г SnC2O4 (99,9 мас.%) в деионизированной воде при той же температуре, что и в примере 1.
Пропитанный продукт обрабатывали так же, как описано в примере 1, получая катализатор следующего состава, мас.%:
Cr2O3 - 20
K2O - 1,89
SnO - 0,9
SiO2 - 1,23
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
Пример 3.
200 г микросферического оксида алюминия, полученного как описано в примере 1, пропитывали методом, описанным здесь ранее, водным раствором (68 см3), содержащим 68,8 г CrO3 (99,8 мас.%) и 6,52 КОН (90 мас.%) и 5,61 г SnC2O4 (99,9 мас.%) в деионизированной воде при той же температуре, что и в примере 1.
Пропитанный продукт обрабатывали так же, как описано в примере 1, получая катализатор следующего состава, мас.%:
Cr2O3 - 20
K2O - 1,89
SnO - 1,4
SiO2 - 1,22
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана. Результаты представлены в таблице 1.
Пример 4.
200 г микроскопического оксида алюминия, полученного как описано в примере 1, пропитывали методом, описанным здесь ранее, водным раствором (68 см3), содержащим 67,9 г CrO3 (99,8 мас.%) и 6,44 г КОН (90 мас.%) и 1,78 г SnC2O4 (99,9 мас.%) в деионизированной воде при той же температуре, что и в примере 1.
Пропитанный продукт обрабатывали так же, как описано в примере 1, получая катализатор следующего состава, мас.%:
Cr2O3 - 20
K2O - 1,89
SnO - 0,45
SiO2 - 1,22
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
Пример 5.
200 г микросферического оксида алюминия, полученного как описано в примере 1, пропитывали методом, описанным здесь ранее, водным раствором (68 см3), содержащим 67,7 г CrO3 (99,8 мас.%) и 6,42 г КОН (90 мас.%) и 0,91 г SnC2O4 (99,9 мас.%) в деионизированной воде при той же температуре, что и в примере 1.
Пропитанный продукт обрабатывали так же, как описано в примере 1, получая катализатор следующего состава, мас.%:
Cr2O3 - 20
K2O - 1,89
SnO - 0,23
SiO2 - 1,25
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
Пример 6.
200 г микросферического оксида алюминия, полученного и пропитанного как описано в примере 1, дополнительно пропитывали по методике начальной влажности раствором метанола (44 см3), содержащим 3,99 г растворенного диметоксидибутил олова (CH3O)2(Sn(C4H9)2 в атмосфере азота.
Пропитанный продукт выдерживали при комнатной температуре в течение 1 часа, затем сушили при температуре 90oC до полного удаления метанола. Высушенный продукт окончательно прокаливали при 750oC в течение 4 часов в атмосфере сухого воздуха.
При этом получили катализатор состава, мас.%:
Cr2O3 - 20
K2O - 1,89
SnO - 0,87
SiO2 - 1,23
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
Пример 7.
200 г катализатора, полученного как в примере 6, модифицировали оловом, используя методику пароосаждения. С этой целью образец носителя загружали в кварцевый реактор, снабженный карманом для термометра и керамическим распределителем с откалиброванной пористостью с целью получения гомогенного распределения азота внизу слоя. Реактор с материалом помещали в электропечь с частичным подогревом и подавали азот (40 - 45N л/час) через пористый распределитель, что поддерживало флюидизацию материала. При достижении заданной температуры в 200oC, требуемой для осаждения олова, определяли продольный термический профиль слоя, прежде чем подавать предшественник олова.
Убедившись, что температура слоя гомогенна в пределах ± 1oC по отношению к заданной температуре, в слой вводили азот, насыщенный дибутилдиметоксиоловом (CH3O)2 (Sn(C4H9)2, со скоростью 10 - 15 N л/час и при температуре от 150 до 170oC. Насыщенный поток подавали сверху реактора через кварцевую трубку и пропускали его через каталитический слой и пористый распределитель, смешивали в нижней части перегородки с азотом. Поток, выходящий из реактора, охлаждался для выделения непрореагировавшего дибутилдиметоксиолова.
Количество олова дозировалось с помощью веса остаточного предшественника в сатураторе. Когда требуемое количество предшественника расходовалось на получение теоретической загрузки олова, работа прекращалась.
Температура каталитического слоя увеличивалась до 750oC и поддерживалась на этом уровне в течение 4 часов с целью активации материала. Активированный продукт анализировали с целью определения процентного состава, который в результате был следующим, мас.%:
Cr2O3 - 20
K2O - 1,89
SnO - 0,33
SiO2 - 1,24
Al2O3 - Остальное
Полученный катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
Пример 8 (сравнительный)
100 г образца псевдобемита, полученного как в примере 1, подвергали термообработке следующим образом: первое прокаливание при 450oC в течение 1 часа, второе прокаливание при 1000oC в течение 4 часов в потоке сухого воздуха. Прокаленный продукт имел удельную поверхность 130 м2/г, пористость 0,49 см3/г и состоял из дельта и тета переходного оксида алюминия.
150 г такого оксида алюминия пропитывали, используя метод начальной влажности, водным раствором (74 см3), содержащим 66,8 CrO3 (99,8 мас.%) и 5,36 г карбоната калия (45 мас.%./мас. КОН) и выдерживали при той же температуре, что в примере 1. Пропитанный продукт оставляли при комнатной температуре на 1 час, затем высушивали при температуре 90oC в течение 15 часов. Высушенный продукт активировали в потоке сухого воздуха при температуре 750oC в течение 4 часов.
Получили катализатор состава, мас.%:
Cr2O3 - 25
K2O - 1
SiO2 - 1,8
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования пропана при температуре от 560 до 600oC. Результаты приведены в таблице 2.
Пример 9.
150 г оксида алюминия, полученного как описано в примере 8, пропитывали по методике начальной влажности раствором метанола (74 см3), содержащим 3,75 г диметоксидибутил олово (CH3O)2(Sn(CH4H9)2.
Пропитанный продукт выдерживали в течение 1 часа, затем сушили при температуре 90oC до полного удаления метанола. Высушенный продукт окончательно прокаливали при 600oC в течение 2 часов в атмосфере сухого воздуха. Прокаленный продукт пропитывали по методу, описанному в примере 8, водным раствором (74 см3), содержащим 67,6 г CrO3 (99,8 мас.%) и 5,42 г карбоната калия (45 мас. %./мас. КОН) и выдерживали при той же температуре, что и в примере 1. Полученный катализатор имел следующий состав, мас.%:
Cr2O3 - 25
K2O - 1
SnO - 0,84
SiO2 - 1,18
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования пропана. Результаты представлены в таблице 2.
Пример 10.
150 г оксида алюминия, использованного в примере 9, пропитывали по методике начальной влажности, как в примере 9, раствором метанола (74 см3), содержащим 7,63 г диметоксидибутил олова (CH3O)2(Sn(C4H9)2. Прокаленный продукт при условиях, описанных в примере 9, пропитывали по методу, описанному в примере 8, водным раствором (74 см3), содержащим 68,4 г CrO3 (99,8 мас.%) и 5,48 г карбоната калия (45 мас.%./мас. КОН) и выдерживали при той же температуре, что и в примере 1. Полученный катализатор имел следующий состав, мас.%:
Cr2O3 - 25
K2O - 1
SnO - 1,68
SiO2 - 1,17
Al2O3 - остальное
Этот катализатор использовали в процессе дегидрирования пропана. Результаты приведены в таблице 2.
Пример 11.
150 г оксид алюминия, использованного в примере 9, пропитывали по методике начальной влажности, как в примере 9, раствором метанола (74 см3), содержащим 11,61 г диметоксидибутил олова (CH3O)2(Sn(C4H9)2. Прокаленный продукт при условиях, описанных в примере 9, пропитывали по методу, описанному в примере 8, водным раствором (74 см3), содержащим 69,2 г CrO3 (99,8 мас. %) и 5,55 г карбоната калия (45 мас.%./мас. KOH) и выдерживали при той же температуре, что и в примере 1. Полученный катализатор имел следующий состав, мас.%:
Cr2O3 - 25
K2O - 1
SnO - 2,52
SiO2 - 1,14
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования пропана. Результаты приведены в таблице 2.
Пример 12.
150 г оксид алюминия, использованного в примере 8, пропитывали водным раствором (74 см3), при температуре как в примере 1, в котором были растворены следующие продукты: 68,4 г CrO3 (99,8 мас.%) и 5,49 г карбоната калия (45 мас. %./мас. KOH) и 5,35 г SnC2O4 (99,9 мас.%./мас.). Сушку и активацию проводили согласно методике, описанной в примере 1. Полученный катализатор имел следующий состав, мас.%:
Cr2O3 - 25
K2O - 1
SnO - 1,68
SiO2 - 1,5
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования пропана. Результаты приведены в таблице 2.
Пример 13.
150 г катализатора, полученного по методике, описанной в примере 8, пропитывали раствором метанола (39 см3), содержащим 3,03 г (CH3O)2(Sn(C4H9)2, согласно методике, описанной в примере 6. Катализатор после активации аналитизировали с целью определения его состава и использовали в реакции дегидрирования пропана. Полученный катализатор имел следующий состав, мас.%:
Cr2O3 - 24,8
K2O - 0,99
SnO - 0,91
SiO2 - 1,17
Al2O3 - Остальное
Результаты приведены в таблице 2.
Пример 14.
235 г катализатора получали методом, описанным в примере 2, пропиткой 200 граммов оксида алюминия, того же, что и примере 2, водным раствором (68 см3), содержащим 37,2 г CrO3 (99,8 мас.%) и 5,87 г KOH (90 мас.%) и 3,26 г SnC2O4 (99,9 мас.%) и выдерживали при температуре 85oC. Полученный катализатор имел следующий состав, мас.%:
Cr2O3 - 12
SiO2 - 1,36
K2O - 1,89
SnO - 0,9
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
Пример 15 (сравнительный)
200 г оксида алюминия с удельной поверхностью 104 м2/г и пористостью 0,34 см3/г, полученного прокаливанием образца псевдобемита по методу, описанному в примере 1, но без оксида кремния, пропитывали водным раствором (68 см3), содержащим 68,3 г CrO3 (99,8 мас.%) и 6,48 г KOH (90 мас.%) и 4,13 г SnC2O4 (99,9%). Полученный катализатор имел следующий состав, мас.%:
Cr2O3 - 20
K2O - 1,89
SnO - 0,9
Al2O3 - Остальное
Этот катализатор использовали в процессе дегидрирования изобутана. Результаты представлены в таблице 1.
Пример 16.
Образец катализатора получили так же как в примере 2, с тем же оксидом алюминия, получили следующий состав, мас.%:
Cr2O3 - 20
K2O - 3
SnO - 0,9
SiO2 - 1,22
Al2O3 - Остальное
Катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
Пример 17.
Образец катализатора получили так же как в примере 2, с тем же оксидом алюминия. Получили следующий состав, мас.%:
Cr2O3 - 20
K2O - 0,2
SnO - 0,9
SiO2 - 1,27
Al2O3 - Остальное
Катализатор использовали в процессе дегидрирования изобутана. Результаты приведены в таблице 1.
УСЛОВИЯ ОСУЩЕСТВЛЕНИЯ ПРОЦЕССОВ ПОЛУЧЕНИЯ ОЛЕФИНОВЫХ УГЛЕВОДОРОДОВ
Катализаторы, полученные в примерах 1-17, использовались в псевдоожиженном слое в кварцевом реакторе, снабженном распределителем, так же сделанном из кварца, с откалиброванной пористостью. Расширитель помещается на верхнюю часть реактора и выполняет функции торможения потока, заставляя мелкие частицы падать обратно на слой катализатора. Каталитический цикл, имитирующий поведение промышленного реактора, состоит из реакционной фазы, при которой углеводороды подаются в течение 15 минут; фазы очистки, когда азот пропускают для освобождения катализатора от адсорбированных продуктов, в течение 10 минут; фазы регенерации, когда газ регенерации (воздух) подается в течение 30 минут (в данных экспериментах); фазы отмывки азотом, в течение, по крайней мере, 10 минут, с последующей реакционной фазой в течение 15 минут. Технические условия промышленного процесса дегидрирования в псевдоожиженном слое предполагают проведение регенерации при температурах, которые выше температуры реакции: в данном случае регенерацию и восстановление проводили при 650oC, тогда как реакцию проводили в температурных пределах от 560 до 600oC при дегидрировании пропана и от 540 до 580oC в случае дегидрирования изобутана.
Объемная скорость реагентов имеет величину 400 ± л/кат. час. При первом использовании каждый катализатор до проведения реакции дегидрирования восстанавливали согласно описанной методике.
Реагент, направляемый в реактор, дозировался по весу.
Потоки, отходящие из реактора во время реакции и фазы очистки, пропускают вначале через холодную ловушку для задерживания тяжелых продуктов, вес, углеродный и водородный % которых определяли, а затем собирали в многослойный мягкий резервуар для проб, не имеющий сродства с углеводородами. Содержимое резервуара замеряли валюметрическим насосом и анализировали газовой хроматографией.
И наконец после 10-минутной очистки азотом образец катализатора отбирается для определения количества образовавшегося кокса. Данные, полученные таким образом, вводились в персональный компьютер для расчета материального баланса, конверсии и селективности различных продуктов.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР ДЛЯ ДЕГИДРИРОВАНИЯ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ | 1999 |
|
RU2160634C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВЫХ УГЛЕВОДОРОДОВ | 1999 |
|
RU2156233C1 |
| КАТАЛИЗАТОР ДЛЯ ДЕГИДРИРОВАНИЯ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ | 2000 |
|
RU2167709C1 |
| КАТАЛИЗАТОР ДЛЯ ДЕГИДРИРОВАНИЯ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ | 2000 |
|
RU2176157C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВЫХ УГЛЕВОДОРОДОВ | 2000 |
|
RU2178398C1 |
| КАТАЛИЗАТОР ДЛЯ ДЕГИДРИРОВАНИЯ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ | 2000 |
|
RU2183988C1 |
| КАТАЛИЗАТОР ДЛЯ ДЕГИДРИРОВАНИЯ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ | 2000 |
|
RU2177827C1 |
| КАТАЛИЗАТОР ДЛЯ ДЕГИДРИРОВАНИЯ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ | 2000 |
|
RU2188073C2 |
| НОСИТЕЛЬ ДЛЯ КАТАЛИЗАТОРОВ | 2000 |
|
RU2190466C2 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА И СПОСОБ ДЕГИДРИРОВАНИЯ ЭТИЛБЕНЗОЛА ДО СТИРОЛА | 1998 |
|
RU2159151C2 |

Использование: нефтехимия. Парафиновые углеводороды дегидрируют при температуре от 450 до 800oС, давлении от 0,1 до 3 атм. абс. и объемной скорости газа от 100 до 1000 ч-1 в присутствии катализатора, имеющего состав, мас. %: (предпочтительно) Cr2O3 6,0 - 30,0 (13 - 25), Sn 0 0,1 - 3,5 (0,2 - 2,8), MeO 0,4 - 3,0 (0,5 - 2,5), SiO2 0,08 - 3,0 (0,08 - 3,0), Al2O3 остальное, где Ме - щелочной металл, предпочтительно калий. Способ позволяет повысить выход целевого продукта. 8 з.п. ф-лы, 2 табл. 1 ил.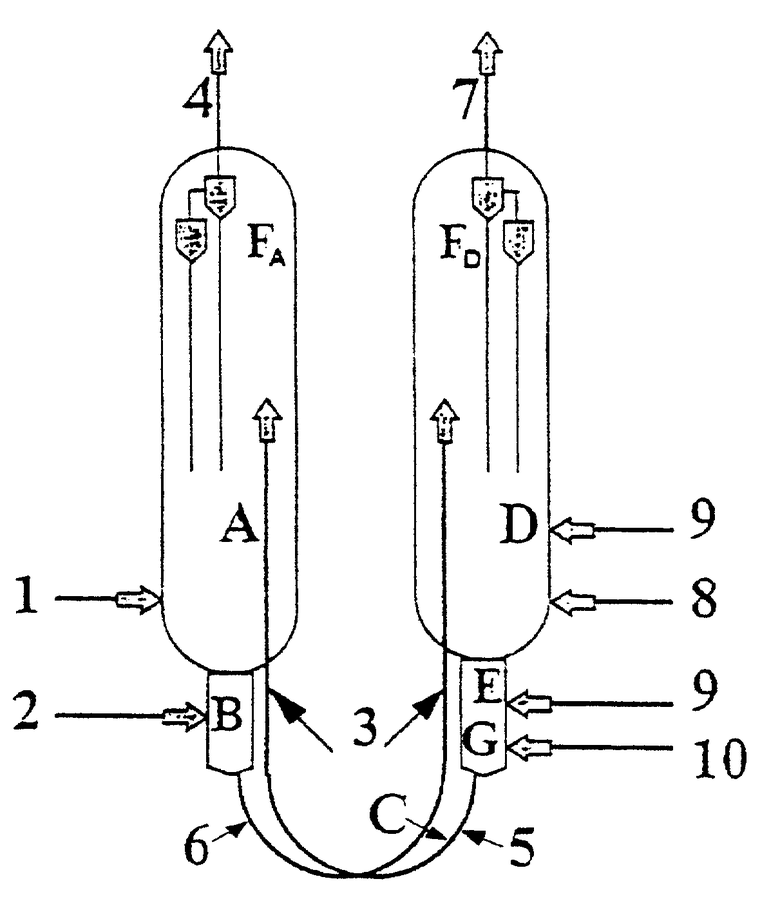
Cr2O3 - 6,0 - 30,0
SuO - 0,1 - 3,5
MeO - 0,4 - 3,0
SiO2 - 0,08 - 3,0
Al2O3 - Остальное
где Me - щелочной металл,
и дегидрирование осуществляют при температуре от 450 до 800oC, давлении от 0,1 до 3 атм.абс. и объемной скорости газа от 100 до 1000 ч-1.
Cr2O3 - 13 - 25
SuO - 0,2 - 2,8
Me2O - 0,5 - 2,5
SiO2 - 0,08 - 3,0
Al2O3 - Остальное
где Me - щелочной металл.
| SU 1366200 A1, 15.010.88 | |||
| Катализатор для дегидрирования углеводородов | 1991 |
|
SU1836140A3 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| US 4827072 A, 02.05.89. | |||
