Изобретение относится к новым производным индола, к способам их получения, к фармацевтическим композициям их содержащим и к их использованию в медицине.
В частности оно относится к производным индола, которые являются сильными и специфическими антагонистами возбуждающих аминокислот.
Патент США N 4960786 раскрывает, что определенные известные 2-карбоксильные производные индола являются антагонистами возбуждающих аминокислот EP-A 0396124, также говорится о том, что определенные 2-карбоксильные производные индола обладают терапевтической эффективностью при лечении нарушений ЦНС (CNS), возникающих по причине нейротоксических повреждений или нейродегенеративных заболеваний. В EP-0394905 A2 раскрыты производные 2-карбоксииндола, обладающие аналогичной заявленным соединениям активностью и содержащие их фармкомпозиции.
В настоящее время обнаружена новая группа 2-карбоксииндол производных, которые обладают очень мощной и специфической антагонистической активностью в отношении нечувствительного к стрихнину участка (сайта) связывания глицина, локализованного на NMDA рецептором комплексе.
Таким образом, данное изобретение предлагает соединение формулы (I)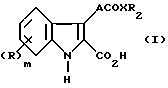
или соль, или метаболически лабильный сложный эфир, где R представляет группу, выбранную из: галоген, алкил, алкокси, амино, алкиламино, диалкиламино, гидрокси, трифторметил, трифторметокси, нитро, циано, SO2R1 или COR1, в которой R1 представляет гидрокси, метокси или амино;
m - нуль или целое число 1 или 2.
A представляет этинилгруппу или необязательно замещенный этинил, или циклопропильную группу;
X представляет -O- или NH;
R2 представляет арилгруппу и, когда X является атомом кислорода R2, может также представлять атом водорода или алкил группу.
Соединения, представленные формулой (I), могут существовать более чем в одной изомерной форме. Таким образом, когда группа A в соединениях формулы (I) представляет необязательно замещенный этенил или необязательно замещенную циклопропилгруппу, там могут существовать цис- и транс-изомеры и изобретение включает все такие изомеры и их смеси.
При использовании в медицине соли соединений формулы (I) должны быть физиологически приемлемыми для этого. Однако другие соли могут быть использованы для приготовления соединений формулы (I) или физиологически приемлемых их солей. Поэтому, если не оговорено особо, ссылки на соли включают как физиологически приемлемые соли, так и физиологически неприемлемые соли соединений формулы (I).
Подходящие физиологически приемлемые соли соединений изобретения включают основные соли присоединения и там, где они уместны, соли присоединения кислот.
Подходящие физиологически приемлемые основные соли присоединения соединений формулы (I) включают щелочной металл или соли щелочного металла, такие как соли натрия, калия, кальция, магния и аммония, образованные с аминокислотами (например лизин или аргинин) и органическими основными (например, прокаин, фенилбензиламин, этаноламин, диэтаноламин и N-метил глюкозамин).
Следует принять во внимание, что соединение формулы (I) может быть получено in vivo (в живом организме) путем метаболизма подходящего пролекарства. Такими пролекарствами могут быть, например, физиологически приемлемые метаболитически лабильные эфиры соединений общей формулы (I). Эти эфиры могут быть получены путем этерификации, например любых карбоксильных кислотных групп в исходном соединении общей формулы (I) с предварительной защитой, где уместно, любых других реакционноспособных групп, присутствующих в молекуле, и с последующим снятием защиты, если требуется. Примеры таких метаболитически неустойчивых эфиров включают C1-4 алкильные эфиры, например, метиловый или этиловый эфиры, замещенные или незамещенные аминоалкиловые эфиры (например, аминоэтиловый, 2-(N, N-диэтиламино) этиловый, или 2-(4-морфолино) этиловый эфиры) или ацилоксиалкильные эфиры, такие как ацилоксиметил ил 1-ацилоксиэтил, например: пивалоилоксиметил, 1-пивалоилокстиэтил, ацетоксиметил, 1-ацетоксиэтил, 1-метокси-1-метил-этилкарбонилоксиэтил, 1-бензилоксиэтил, изопропоксикарбонилоксиметил, 1-изопропоксикарбонилоксиэтил, циклогексилкарбонилоксиметил, 1-циклогексилкарбонилоксиэтиловый эфир, циклогексилоксикарбонилоксиметил, 1-циклогексилоксикарбонилоксиэтил, 1-(4-тетрагидропиранилоксикарбонилоксиэтил) или 1-(4-тетрагидропиранилкарбонилоксиэтил).
Предпочитаемые метаболитически лабильные эфиры соединений формулы (I) включают C1-4 алкиловые эфиры, более конкретно метиловый или этиловый, аминоалкиловые эфиры более конкретно 2-(4'-морфолино) этил, или ациклосиалкиловые эфиры, например ацетоксиметил, пивалоксиметил, 1-циклогексилоксикарбонилоксиэтил или 1-(4-тетрагидропиранилоксикарбонилокси)этил.
Соединения формулы (I) и соли и метаболитически лабильные эфиры могут образовывать сольваты, например гидраты, и изобретение включает такие сольваты.
В соединениях формулы (I) группа R может быть в любом из четырех возможных положений на конденсированном бензольном кольце и, когда m равно 2, две R группы могут быть одинаковыми или различными.
Термин алкил в том понимании как он используется здесь как группа или часть группы относится к алкильной группе с линейной или разветвленной цепью содержащей от 1 до 4 углеродных атомов. Примеры таких групп включают этил пропил, изопролпил, н-бутил, изобутил, вторичный бутил или третичный бутил.
Термин галоген относится к атому фтора, хлора или брома. Термин необязательно замещенный этенил означает этенилгруппу необязательно замещенную 1 или 2 алкильными группами, например метилгруппы, и включает как цис-, так и транс-конформации. Примеры таких групп включают этенил, 1 - метилэтенил, 2-метилэтенил и/или 1,2-диметилэтенил.
Термин необязательно замещенный циклопропил означает циклопропилгруппу необязательно замещенную 1, 2 или 3 алкилгруппами, например метилгруппы.
Для группы R2 термин арил означает необязательно замещенную фенильную группу, или 5 или 6 членную гетероарильную группу, в которой 5-членная гетероарильная группа содержит 1 или 2 гетероатома, выбранных из кислорода, серы или азота, и 6 членная гетероарильная группа содержит 1 или 2 атома азота. Примеры подходящих гетероарильных групп включают фуранил, тиенил, имидазолил, тиазолил, оксазолил, пиридинил и пиримидинил.
Термин замещенный фенил относится к фенильной группе, содержащей до трех заместителей выбранных из : галоген, C1-4 алкил, C1-4 алкокси, амино, алкиламино, диалкиламино, трифторметил, трифторметокси, гидрокси, циано, нитро, амино, SO2R1 или COR1, когда присутствует более одного заместителя, то заместители могут быть те же самые или различные.
Предпочтительным классом соединений формулы (I) являются те соединения, где R представляет хлор, m равно 1 или 2 и R находится в 4 и/или 6 позиции, и особенно m равно 2.
Когда A представляет необязательно замещенную этенилгруппу он предпочтительно находится в E конформации.
Когда группа R2 представляет замещенную фенильную группу, предпочтительно используется фенильная составляющая, замещенная одной или более группами, выбранными из : алкокси, алкил, амино, алкиламин, диалкиламин, фтор, хлоро, гидрокси, нитро, трифторометил или COR1, где R1 представляет собой гидрокси или метокси.
Предпочтительным классом соединений формулы (I) являются те соединения, где R2 представляет фенил или фенил, замещенный одной или двумя группами, выбранными из : фтор, трифторометил, алкил, например метил или изопропил, гидрокси, алкокси или нитро или особенно R2 - фенил.
Кроме того, предпочтительным классом соединений формулы (I) являются соединения, где X представляет NH.
Предпочтительной группой соединений формулы (I) являются те соединения, в которых R - хлор и m равно 1 или более предпочтительно 2, X является NH или O и R2 - необязательно замещенная фенильная группа. Внутри этой группы наиболее предпочтительные соединения включают те, в которых X является NH.
Кроме того, предпочтительной группой соединений формулы (I) являются те соединения, в которых A представляет необязательно замещенную циклопропилгруппу или в частном случае этинилгруппу или замещенную этинилгруппу, такую как 1 - метилэтинил. Внутри этой группы особенно предпочтительные соединения включают те соединения, в которых X является NH, а R2 представляет необязательно замещенный фенил, и главным образом фенил.
Для соединений формулы (I), где A представляет незамещенную этенилгруппу, предпочтительные соединения данного изобретения включают те соединения, в которых X является NH, R2 представляет необязательно замещенный фенил и группа A находится в транс (E) конфигурации. Внутри этой предпочтительной группе соединений, особенно предпочтительные соединения включают те соединения, в которых R2 представляет фенильную группу замещенную одной или двумя группами, выбранными из: фтор, трифторометил, метил, изопропил, гидрокси, алкокси например метокси или этокси, или нитро.
Особенно предпочтительным соединением изобретения, в частности благодаря его мощному и селективному воздействию на стрихнин-нечувствительный сайт связывания глицина, в сочетании с очень выгодной биологической доступностью является (E) -3-[2-(фенилкарбамоил)этенил] -4,6, дихлороиндол-2-карбоновая кислота, физиологически приемлемые соли, метаболитически лабильные эфиры. Предпочтительные соли этого соединения включают соль калия и особенно соль натрия. Предпочтительно метаболические лабильные эфиры этого соединения включают этиловый и 2-(4-мерфолино) этиловый эфиры.
Кроме того, наиболее предпочтительные соединения включают:
3-[2-(фенилкарбамоил)этинил] -4,6-дихлороиндол-2-карбоновая кислота, ее физиологически приемлемые соли и метаболически лабильыне эфиры;
3-[2-(фенилкарбамоил)пропенил]-4,6-дихлороиндол-2-карбоновая кислота, ее физиологически приемлемые соли и метаболитически лабильные эфиры;
Другие наиболее предпочтительные соединения включают:
(E)-3-[2-(4-этоксифенилкарбамоил)этенил] -4,6-дихлороиндол-2- карбоновая кислота;
(E)-3-[2-(2-гидрокси-5-нитрофенилкарбамоил)этенил)] -4,6- дихлороиндол-2-карбоновая кислота;
(E)-3-[2-(2-метил-4-метоксифенилкарбамоил)этенил)] -4,6-дихлороиндол- 2-карбоновая кислота;
(E)-3-[2-(2-изопропилфенилкарбамоил)этенил)] -4,6-дихлороиндол-2- карбоновая кислота;
(E)-3-[2-(2,4-дифторофенилкарбамоил)этенил)] -4,6-дихлороиндол-2- карбоновая кислота;
(E)-3-[2-(3,4-диметоксифенилкарбамоил)этенил)] -4,6-дихлороиндол-2- карбоновая кислота;
и их физиологически приемлемые соли, например соли натрия или калия, и метаболитически лабильные эфиры.
Соединения формулы (I) и/или физиологически приемлемые их соли являются антагонистами возбуждающих аминокислот. Более конкретно, они являются мощными антагонистами в отношении стрихнин - нечувствительного сайта связывания глицина с NMDA рецепторным комплексом. Как таковые они являются мощными антагонистами NMDA рецепторного комплекса. Кроме того, соединения изобретения обнаруживают благоприятный профиль активности, включающий хорошую биологическую доступность. Поэтому эти соединения являются полезными в лечении или профилактике нейротоксических повреждений или нейродегенеративных заболеваний. Так, соединения являются полезными для лечения нейротоксических травм, за которыми следуют церебральный приступ, тромбоэмболитический приступ, геморрагический приступ, церебральная ишемия, церебральный вазоспазм, типогликемия, анаэзия, гипоксия, аноксия, остановка сердца при перинатальной асфиксии. Соединения являются полезными в лечении хронических нейродегенеративных заболеваний, таких как болезнь Huntingdon'a, Альцгеймера - пресенильное слабоумие, боковой амиотрофический склероз, разновидность Глутаровой ацидемии, деменция множественного инфаркта, состояние эпилепсии, травмы при контузиях (например, повреждение спинного мозга), вирусные инфекции, вызывающие нейродегенерацию (например, СПИД, энцефалопатия), синдром Дауна, эпилепсия, шизофрения, депрессия, страх, боль, нейрогенный мочевой пузырь, нарушения раздраженного мочевого пузыря, лекарственная зависимость, включающие аутизм симптомы от алкоголя, кокаина, наркотиков, никотина, бензодиазепина.
Мощное и селективное воздействие соединения изобретения на стрихнин-нечувствительный сайт связывания глицина, находящийся на NMDA рецепторном комплексе, легко может быть определено использованием обычных методик испытания.
Так, способность связываться с участком стрихнин-нечувствительного связывания глицина было определено использованием методики Kishimoto et al., J. Neurochem. 1981, 37, 1015 - 1024. Селективность воздействия соединений изобретения на стрихнин-нечувствительный сайт связывания глицина была подтверждена изучениями на других известных ионотропных возбуждающих аминокислотных рецепторах. Так, найдено, что соединение изобретения обнаруживает небольшое сродство или отсутствие его к рецептору кокаиновой кислоты (кокаинат) L-амино-3-гидрокси-5-метил-4-изоксазол-пропионовой кислоты (АМРА) рецептору или к сайту связывания NMDA.
Использованием методики Chiamulera C. et al., Psychopharmacology (1990) 102, 551-552 найдено также, что соединения изобретения ингибируют NMDA индуцируемые конвульсии (cyдороги) у мышей.
Нейрозащитное действие соединений изобретения также продемонстрировано на препарате окклюзии средней церебральной артерии на мышах использованием методики, описанной Chiamulera C. et al., European Journal of Pharmacology, 216 (1992), 335-336.
Соединение было активным, когда вводилось в предишемическом либо постишемическом состоянии.
Следовательно, изобретение предусматривает применение соединения формулы (I) и/или физиологически приемлемой соли или метаболитически лабильного эфира для употребления в терапии и, в частности, использования в качестве лекарственного препарата для противодействия влияниям возбуждающих аминокислот на NMDA рецепторный комплекс.
Изобретение также предусматривает использование соединения формулы (I) и/или физиологически приемлемой соли или метаболически лабильного эфира для производства лекарственного препарата для противодействия влияниям возбуждающих аминокислот на NMDA рецепторный комплекс.
Согласно следующему аспекту изобретение также предусматривает способ противодействия влияниям возбуждающих амино кислот на NMDA рецепторный комплекс, включающий введение нуждающемуся в этом пациенту антагонистического количества соединения формулы (I) и/или физиологически приемлемой соли или метаболически лабильного эфира.
Специалистам в данной области техники будет понятен факт, что приведенная здесь рекомендация по лечению распространяется как на профилактику, так и на лечение установленных заболеваний или симптомов.
Кроме того, следует обратить внимание на то, что количество соединения изобретения, требующееся для применения в лечении, будет изменяться с характером предполагаемого способа введения лекарства, возрастом и состоянием пациента и будет в конечном счете на усмотрении врача.
В общем однако дозы, применяемые для лечения взрослого человека находятся обычно в интервале от 2 до 800 мг на день в зависимости от способа введения.
Так, ежедневная доза для парентерального введения обычно находится в интервале 20-100 мг, предпочтительно 60-80 мг на день. Для орального введения ежедневная доза будет обычно в пределах 200-800 мг, например 400-600 мг в день. Желаемая доза обычно может быть представлена единичной дозой или разделенными дозами, вводимыми с соответствующими интервалами, например как две, три, четыре или более поддоз в день.
Хотя возможно, что для использования в терапии соединение данного изобретения может назначаться в виде сырого химического вещества, предпочтительно давать активный ингредиент в виде фармацевтического состава.
Изобретение, таким образом, предлагает к тому же фармацевтический состав включающий соединение формулы (I) или его фармацевтически приемлемую соль, или метаболически лабильный эфир вместе с одним или более фармацевтически приемлемым носителей, и, необязательно, с другими терапевтическими и/или профилактическими ингредиентами. Носитель (ли) должен быть "приемлемым" в том смысле, что является совместимым с другими ингредиентами и не разрушительным по отношению к реципиенту.
Композиции изобретения включают композиции, составленные главным образом в форме для орального, трансбуккального, парентерального, путем ингаляции или инсуффляции, имплантатного или ректального введения. Предпочтительно парентеральное введение.
Таблетки и капсулы для орального введения могут содержать обычные наполнители, такие как связывающие агенты, например сироп, сок акации, желатин, сорбитол, трагакант, растительный клей крахмала или поливинилпирролидон; наполнители, например: лактоза, сахар, микрокристаллическая целлюлоза, кукурузный крахмал, фосфат кальция или сорбитол; смазывающие вещества, например стеарат магния, стеариновая кислота, тальк, полиэтилен гликоль или двуокись кремния, дезинтеграторы, например картофельный крахмал или крахмальный гликолят натрия, или увлажняющие агенты, такие как лаурил сульфат натрия. Таблетки могут быть покрыты оболочками согласно хорошо известным в технике методикам. Оральные жидкие препараты могут быть в форме, например, водных или масляных суспензий, эмульсионных растворов, сиропов или эликсиров, или могут быть представлены в виде сухого продукта для составленная перед использованием с водой или другим подходящим растворителем.
Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, например сорбитол сироп, метил целлюлоза, глюкоза/ сахарный сироп, желатин, гидроксиэтилцеллюлоза, карбоксиметил целлюлоза, гель стеарата алюминия или гидрированные съедобные жиры; эмульгирующие агенты, например лецитин, сорбитан моноолеат или сок акации; неводные растворители, которые могут включать пригодные в пищу масла, например миндальное масло, фракционированное кокосовое масло, масличные эфиры, пропилен гликоль или этиловый спирт; и консерванты, например метил или пропил п-гидроксибензоаты или аскорбиновая кислота. Композиции могут быть составлены в виде суппозиториев, например содержащих обычные суппозиторные основы, такие как масло какао или другие глицериды.
Для трансбуккального введения композиции можно изготовлять в форме таблеток или лепешек составленных обычным способом.
Согласно изобретению композиция может быть составлена для парентерального введения путем инъекции или непрерывного вливания. Составы для инъекции могут быть представлены в форме единичной дозы в ампулах или в контейнерах с множественной дозой с добавлением консервантом. Композиции могут принимать такие формы, как суспензия, растворы или эмульсии в масляных или водных растворителях, и могут содержать формующие агенты, такие как суспендирующий, стабилизирующий и/или диспергирующий агенты. Или иначе активный ингредиент может быть в форме порошка для составления с подходящим растворителем, например стерильная, пироген-свободная вода, перед использованием.
Для введения путем ингаляции соединения согласно изобретению обычно поставляют в форме упаковок под давлением, обеспечивающих аэрозольное распыление, с использованием подходящего пропеллента, такого как дихлородифторометан, трихлорофторометан, дихлоротетрафтороэтан, двуокись углерода или другой подходящий пропеллент, такой как дихлородифторометан, трихлорофторометан, дихлоротетрафтороэтан, двуокись углерода или другой пригодный газ, или из пульверизатора. В случае аэрозоля под давлением для отпуска дозирующего количества единица дозирования может определяться по выделяемому объему.
Напротив, для введения путем ингаляции или инсуффляции соединения согласно изобретению могут быть взяты в форме сухой порошкообразной композиции, например порошкообразная смесь соединения и подходящего носителя, такого как лактоза или крахмал. Порошкообразная композиция может быть представлена в форме единичной дозы, например капсулы или гильзы, покрытых, например, желатиновой или пузырьковой оболочками, из которых порошок может вводиться с помощью ингалятора или инсуффлятора.
Композиция согласно изобретению может также быть составлена как препарат для депонирования. Такие составы длительного действия могут вводиться путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Таким образом, соединения изобретения могут быть составлены с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или использоваться в виде умеренно растворимых производных, например в виде умеренно растворимой соли.
Согласно изобретению композиции могут содержать между 0,1-99% активного ингредиента, целесообразно 30-95% для таблеток и капсул и 3-50% для жидких препаратов.
Соединения общей формулы (I) и их соли могут быть приготовлены обычными методами описанными в общих чертах в дальнейшем. В следующем далее описании группы R, R1 и R2 являются такими, как определено для соединения формулы (I), если не оговорено особо.
Соединения формулы (I), в которых A представляет необязательно замещенную этенилгруппу могут быть приготовлены из соединения (II), в котором R, m и n имеют приведенные выше значения, R3 представляет карбоксилзащитную группу и R4 является атомом водорода или C1-4 алкилгруппой.
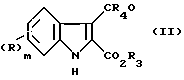
путем взаимодействия с соответствующим илидом фосфор способным превращать группу CR4O в группу ACOXR2, где X и R2 имеют те же значения, что определены выше для формулы (I) с последующим, где это необходимо или желательно, удалением карбоксил защитной группы.
Подходящие карбоксилзащитные группы включают аллил, алкил, трихлороалкил, триалкилсилалкил или арилметилгруппы такие как бензил, нитробензил или тритил. В одном воплощении данного способа реакция может проводиться использованием илида фосфора формулы (III)
(R5)3P = CHCOXR2 (III),
где R5 представляет алкил- или фенилгруппу, и X и R3 имеют указанные выше значения.
Реакция проводится в апротонном растворителе, таком как ацетонитрил, или простом эфире таком как 1,4-диоксан, и предпочтительно, нагреванием, например, до 40-12oC.
В другом воплощении способа реакция проводится использованием фосфорилида формулы (IV).
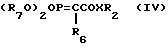
где R6 представляет водород или C1-4 алкил.
R7 представляет C1-4 алкил;
X и R3 имеют определенные выше значения.
Реакцию проводят в апротонном растворителе, таком как тетрагидрофуран, и необязательно при нагревании.
Соединения формулы (I), где A представляет необязательно замещенную циклопропилгруппу, могут быть приготовлены обработкой олефина (V)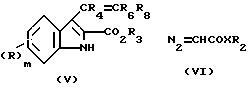
где R, R3, R4, R6 имеют определенные выше значения и R8 является атомом водорода или C1-4 алкилгруппой, диазопроизводным (VI), где группы X и R2 - такие как определено выше, с последующим, где это необходимо или желательно, удалением карбоксилзащитной группы.
Реакция проводится в растворителе, таком как 1,2-диметоксиэтан, и в присутствии родий (II) катализатора, такого как ацетат или пивалат родия.
Соединения формулы (I), где A представляет этинилгруппу, могут быть приготовлены взаимодействием алкина формулы (VII)
где R, m, X и R2 имеют значения, определенные в формуле (I),
R9 представляет группу (CH3)3 SiCH2CH2OCH2-
с хлористоводородной кислотой в этаноле, с последующим взаимодействием с подходящим основанием, таким как гидроокись лития.
Соединения формулы (I) или их защищенные производные могут быть превращены в другие соединения изобретения.
Таким образом, соединения формулы (I), в которых A представляет незамещенную этенилгруппу с цис-конфигурацией могут быть приготовлены из соответствующего соединения формулы (I), где A представляет этинил, путем восстановления с использованием водорода и палладия на носителе карбонат кальция/окись свинца в качестве катализатора.
Соединения формулы (I), в которых A представляет необязательно замещенную циклопропилгруппу, могут быть приготовлены взаимодействием соединения формулы (I), где A является необязательно замещенной этенилгруппой, или его защищенного производного, например простого эфира с диазометаном в присутствии ацетата палладия, с последующим, где необходимо или желательно, удалением любой защитной группы.
Реакция проводится в апротонном растворителе, например дихлорометан и/или диэтиловый эфир, и предпочтительно при температуре в интервале 0-20oC.
В любой из приведенных выше реакций карбоксилзащитная группа R3 может быть удалена по обычным методикам известным для удаления таких групп. Так, группа R3 может быть удалена гидролизом с использованием гидроокиси щелочного металла например гидроокиси лития в растворителе таком как этанол, с последующим добавлением там, где это желательно или необходимо, подходящей кислоты, например хлористоводородной кислоты, для получения соответствующей свободной карбоновой кислоты.
Физиологически приемлемые соли соединения формулы (I) могут быть приготовлены обработкой кислоты соответствующим основанием, например гидроокисью щелочного или щелочноземельного металла, в соответствующем растворителе, таком как алканол например метанол.
Метаболически лабильные эфиры соединений формулы (I) могут быть приготовлены путем этерификации карбоксильной кислотной группы или соли или путем трансэтерификации использованием обычных методик. Так, например, ацилоксиалкил эфиры могут быть приготовлены взаимодействием свободной карбоновой кислоты или соли с соответствующим ацилоксиалкил галогенидом в подходящем растворителе, таком как диметилформамид. Для этерификации свободной карбоксильной группы эта реакция предпочтительно проводится в присутствии галогенида четвертичного аммония такого как тетрабутиламмоний хлорид или бензилтриэтиламмоний хлорид.
Аминоалкиловые эфиры могут быть приготовлены путем трансэтерификации соответствующего алкилового эфира, например метилового или этилового эфира, путем взаимодействия с соответствующим аминоалканолом при повышенной температуре, например 5-150oC.
Соединения формулы (II), в которых R3 представляет карбоксил защитную группу, R4 - водород, могут быть приготовлены обработкой соответствующего индола (VIII)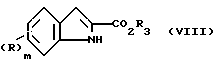
где R и m - также, как определено для формулы (I) N-метилформанилидом и оксихлоридом фосфора в растворителе, таком как 1,2-дихлороэтан.
Соединения формулы (II), в которых R3 является карбоксил защитной группой, R4 представляет алкил и n равно нулю, могут быть приготовлены обработкой индола (VIII) амидом (CH3)2NCOR4 и фосфор оксихлоридом в подходящем растворителе.
Соединения формулы (V) могут быть приготовлены обработкой соответствующего соединения формулы (II) реагентом, способным ввести группу CR4 = CR6R8.
Так, взаимодействие соединения формулы (II) с трифенилфосфин производным Ph3P+CH2R6Br- в присутствии подходящего основания, такого как бутил литий, в апротонном растворителе даст соответствующее соединение формулы (V), в котором R8 является водородом.
Соединения формулы (V), в котором R4 представляет водород и R6 и R8 независимо представляют C1-4 алкил, могут быть приготовлены обработкой соединения формулы (II), где R4 представляет водород, дважды замещенный R6R8-C-P+Ph3 в подходящем растворителе таком как N,N-диметилформамид. Предпочтительно дважды замещенный приготавливают на месте путем обработки триметилсилил производного (CH3)3SiCR6R8P+Ph3Y-, где Y является анионом, фторидом цезия. Триметилсилил производное может быть приготовлено методом Bestmann and Bomhard, Angew. Chem, Int. Ed. Eng. 21 (1982) N 7 pages 545-546.
Соединения формулы (V),где R4, R6 и R8 каждый представляет алкилгруппу могут быть приготовлены из соответствующего соединения формулы (II) путем взаимодействия с фенилсульфонатом (PhSO2CHR6R8). Реакция может быть проведена с использованием обычной реакционной методики, описанной Julia and Paris Tetrahedron Letters N 49, 4833-4836 1973.
Соединения формулы (VII) могут быть приготовлены взаимодействием бромозамещенной кислоты (IX)
с соответствующим изоцианатом R2NCСО или хлороформиатом R2OCOCl в присутствии подходящего основания, такого как т-бутил лития, и в апротонном растворителе, таком как тетрагидрофуран, с последующим взаимодействием сырого реакционного продукта с триметилсилилдиаземетаном ((CH3)3SiCHN2). Бромозамещенная (IX) кислота может быть приготовлена из индола (II), где R4 представляет водород по следующей реакционной последовательности.
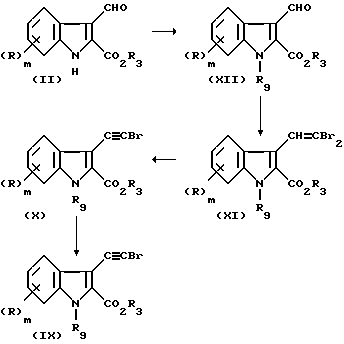
Соединения формулы (IX) могут быть приготовлены щелочным гидролизом соответствующего эфира (X). Эфир (X) может быть приготовлен из соответствующего дибромофена (XI) путем взаимодействия с подходящим основанием, таким как литий бис-триметилсилиламид, в растворителе, таком как простой эфир, например тетрагидрофуран. Дибромоэтен (XI) может быть приготовлен из соответствующего альдегида (XII) взаимодействием с трифенилфосфином и четырехбромистым углеродом в растворителе, таком как дихлорметан.
N-защищенный индол (XII) может быть приготовлен из индола (II, R4=H) взаимодействием с триметилсилилэтоксиметилхлоридом в присутствии основания, такого как натрий бис-триметилсилиламид, в полярном апротонном растворителе, таком как диметилформамид.
Индолы формулы (VIII) либо являются известными соединениями, либо могут быть приготовлены по методикам, аналогичным тем, что описаны для известных соединений.
Для того, чтобы изобретение было более детально понято, только в качестве иллюстрации приводятся следующие примеры.
Для промежуточных соединений и примеров, если не оговорено особо, температуры плавления (m.p.) определены на приборе для определения температуры плавления Gallenkamp и не корректировались. Все температуры, относящиеся к oC и к спектру, измерены на приборе FI-IR. Спектр ПМР (1H-ЯМР) записан при 300 МГц, химические сдвиги представлены в ppm (м.д.) нижней области (d) относительно Me4Si, используемого в качестве внутреннего стандарта и интерпретируются как синглеты (S), дублеты (d), двойные дублеты (dd), триплеты (t), квартеры (g) или мультиплеты (m). Колоночную хроматографию осуществляли на силикагеле (Merck AG Darmstadt, Germany). В тексте использованы следующие обозначения: EA = этилацетат, CH = циклогексан, CM = дихлорметан, ДВИ = 1,8 диазабицикло [5.4.0] ундек-7-ен. DMF = NM - диметилформамид THF = тетрагидрофуран, LiOH. H2O = моногидрат гидроокиси лития. TlC относится к тонкослойной хроматографии на пластинах из силикагеля. Раствор сушили над безводным сульфатом натрия.
Промежуточный продукт 1
Этил 4,6-дихлороиндол-2-карбоксилат
К раствору этил пирувата (2,05 мл) в абсолютном этаноле (38 мл) медленно при энергичном перемешивании добавили концентрированную серную кислоту (0,5 мл). Полученную смесь перемешивали при 23oC в течение 10 минут, затем добавили порциями 3,5-дихлорфенилгидразин гидрохлорида (4 г). Смесь нагревали до температуры кипения с обратным холодильником в течение 4 часов, охладили до 23oC, вылили в холодную воду (500 мл) и экстрагировали диэтиловым эфиром (3 • 300 мл). Органические соли отделили и высушили. Растворитель упарили при пониженном давлении для получения этилового эфира 2-(3,5-дихлорфенилгидразон) пропионовой кислоты в виде желтого твердого продукта (5 г, tlс DCM, Rf = 0,79, 0,47) в E и Z изомерной смеси. Твердый продукт добавили при перемешивании к полифосфорной кислоте (20 г) и смесь нагревали до 45oC в течение 20 минут для получения бурого продукта, который кристаллизовали из 95% этанола (300 мл) для получения названного в заглавии соединения в виде желтовато-бурого твердого продукта (3,3 г m.p. 180oC; Tlc DCM, Rf = 0,54) IR (CDCl3) Vmax(cm-1) 3440 (NH), 1772-1709 (C=0).
Промежуточный продукт 2
Этил 3-формил-4,6-дихлороиндол-2-карбоксилат
Раствор N-метил форманилида (5,19 г) и фосфор оксихлорида (5,53 г) перемешивали при 23oC в течение 15 минут. Добавили 1,2 - дихлорэтан (60 мл) и промежуточный продукт 1 (6 г) и образовавшуюся суспензию перемешивали при 80oC в течение 6 часов. Реакционную смесь вылили в 50% водный раствор ацетата натрия (300 мл) для получения путем фильтрации названного в заглавии соединения в виде желтого твердого продукта (4,1 г tlc EA/CH; 4/6; Rf = 0,4).
Промежуточный продукт 3
Этил-3-формил-1-(2-триметилсилил-этоксиметил)-4,6-дихлороиндол- 2-карбоксиловая кислота
К охлажденному раствору промежуточного продукта (2) (700 мг) в сухом DMF (20 мл) при 0oC добавили литий бис-триметилсилиламид (3,7 мл), 1M раствор) в THF.
Смесь перемешивали в течение 15 минут при 0oC, затем добавили три-метилсилилэтоксиметил хлорид (0,817 г). Спустя один час полученную смесь вылили в воду (25 мл) и экстрагировали этилацетатом (3 • 20 мл). Объединенные органические слои высушили и сконцентрировали при пониженном давлении. Остаток очистили хроматографией на силикагеле с целью получения названного в заглавии соединения (950 мг) в виде палево-желтого твердого продукта.
Rf = 0,3 EA/CH; 1.9
Промежуточный продукт 4
Этил 3-(2,2-дибромовинил)-1-(триметилсилил-этокси-метил)-4,6- дихлороиндо-2-карбоксилат
Промежуточный продукт 3 (300 мг) растворили в сухом дихлорметане (7 мл) и раствор охладили до -15oC в бане лед/соль. Затем добавили трифенилфосфин (1,14 г) и четырехбромистый углерод (719 мл) и образовавшийся раствор перемешивали в течение 1,5 часа, пока температура постепенно повышалась до 0oC. Затем добавили насыщенный NH4Cl (20 мл), разделили две фазы и водную фазу экстрагировали дважды дихлорметаном. Объединенную органическую фазу высушили, сконцентрировали и полученный остаток пропустили через слой силикагеля (CH/EA : 9/1) для получения названного в заглавии соединения (390 мг) в виде желтого масла. Rf= 0,62 CH/EA : 9/1
Промежуточный продукт 5
Этил-3-бромоэтинил-1-(2-триметилсилилэтоксиметил)-4,6- дихлориндол-2-карбоксилат
Промежуточный продукт 4 растворили в сухом (50 мл) и раствор охладили до 0oC на бане лед/вода. Из шприца медленно добавили литий бис-триметилсилиламид (7,6 мл, 1,0 M раствор в THF), смесь перемешивали при 0oC 30 минут и затем остановили реакцию при смешении с холодным насыщенным NH4Cl (20 мл). Добавили этилацетат, две фазы разделили и органический слой промыли 1н. соляной кислотой, высушили и сконцентрировали для полного высушивания. Сырой продукт очистили колоночной хроматографией до получения названного в заглавии соединения (2,9 г) в виде желтого масла.
Rf = 0,35 CH/EA : 95/5.
Промежуточный продукт 6
3-Бромоэтинил-1-(2-триметилсилилэтоксиметил)-4,6-дихлороиндол-2- карбоновая кислота
Промежуточный продукт 5(2,9 г) растворили в 95% этанола (40 мл), затем добавили LiOH • H2O и раствор перемешивали в течение ночи при 80oC. Реакционную смесь сконцентрировали затем досуха и образовавшийся остаток промыли 1н. HCl. После фильтрации полученный твердый продукт промыли водой и высушили над P2O5 для получения названного в заглавии соединения (2,6 г) в виде белого твердого продукта.
ИК (Нуйол) Vmax (см-1) 1676 (C = 0), 1600 (C=C)
1H-NMR (ДМСО) 14,00 (S), 7,90 (d), 7,38 (d), 5,92 (S), 3,41 (t), 0,76 (t), -0,13 (S).
Промежуточный продукт 7
Метил-3-фенилкарбамоилэтинил-1-(2-триметилсилилэтокси- метил)-4,6-дихлороиндол-2-карбоксилат
Промежуточный продукт 6 (454 г) растворили в сухом THF (15 мл) и раствор охладили до -78oC. Медленно добавили раствор т-бутил-лития (1,3 мл, 1,7M в гексане) и реакционную смесь перемешивали 2 часа. Затем добавили фенилизоцианат (0,12 мл) и смесь постепенно нагрели до комнатной температуры и перемешивали 3 часа. Реакцию остановили добавлением холодного насыщенного NH4Cl и экстрагировали этилацетатом. Объединенные органические фазы промыли 1н. HCl, водой и раствором соли, высушили и сконцентрировали досуха. Сырой продукт солюбилизовали затем в дихлорметане (8 мл) и метаноле (2 мл) и обработали при комнатной температуре Me3SiCHN2 (1,2 мл, 1,0 M раствор в гексане). После перемешивания в течение 30 мин раствор сконцентрировали досуха и сырой продукт очистили мгновенной хроматографией (CH/EA : 85/5) для получения названного в заглавии соединения (230 мг) в виде твердого желтого продукта.
Промежуточный продукт 8
(E)-Этил 3-[2-(фенилкарбамоил)этенил]-1-(2-триметил силилэтоксиметил)-4,6-дихлороиндол-2-карбоксилат
К охлажденному (0oC) раствору (E)-этил-3-2-(фенилкарбамоил) этенил-4,6-дихлороиндол-2-карбоксилата (300 мг) в сухом DMF (25 мл) добавили по каплям раствор натрий бис-(триметилсилил) амида (1M; 0,0814 мл). Образовавшуюся смесь перемешивали при комнатной температуре в течение 30 минут и затем охладили до 0oC. Добавляли триметилсилилэтоксиметилхлорид (185 мг), и реакционную смесь перемешивали в течение одного часа при комнатной температуре.
Образовавшийся раствор вылили в H2 O (20 мл) и экстрагировали диэтиловым эфиром (15 млх3). Органические слои высушили, сконцентрировали при пониженном давлении и продукт выделили хроматографией на силикагеле (CH/EA : 83/15) получив названное в заглавии соединение (311 мг)
Rf = 0,35 CH/EA : 85/15
Промежуточный продукт 9
Этил-3-[(2-фенилкарбамоил)-пропенил] -1-(2-триметилсилилэтоксиметил)-4,6 -дихлороиндол-2-карбоксилат
P, P-Диэтил-2-фосфоно-пропананилид (644 мг) растворили в безводном DMF (10 мл) и образовавшийся раствор охладили до 0oC и обработали Lin(Me3Si)2 (2,3 мл 1,0 M раствора в THF) в течение 1,5 часов. К этой смеси добавили промежуточный продукт 3 (784 мг), отдельно растворенный в сухом DMF (8 мл) и перемешивание продолжали в течение всей ночи. Реакцию остановили, вылив реакционную смесь в 50 мл охлажденного насыщенного NH4Cl:
водную фазу экстрагировали затем этилацетатом и органический слой промыли 1 н. соляной кислотой, водой и физиологическим раствором, высушили, отфильтровали и сконцентрировали. Окончательная очистка колоночной хроматографией дала названное в заглавии соединение (660 мг) в виде не совсем белого твердого продукта. Rf = 0,35 CH/EA 8,5/1,5.
Пример 1A.
(E) Этил-3-[2-(фенилкарбамоил)этенил]-4,6-дихлориндол-2-карбоксилат
DBU (319 мг) добавили к перемешиваемой суспензии фенилкарбамоилметилтрифенилфосфонийбромида (1 г) в ацетонитриле (100 мл) в атмосфере азота. Перемешивание продолжали при 0oC в течение 15 минут, затем добавили промежуточный продукт 2 (680 мг) и смесь нагревали до кипения с обратным холодильником в течение 6 часов. После разбавления дихлорметаном (15 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (380 мг EA/CH : 3/7, Rf = 0,5), в виде белого твердого продукта.
ИК (Нуйол) Vmax(см-1) 3305 - 3288 (NH), 1678 - 1662 (C = O), 1627 - 1601 (C = C)1 H -
NMR(DMSO) 12,61 (S), 10,20 (S), 8,27 (d), 7,73 (d), 7,52 (d), 7,36 - 7,30 (m), 7,06 (m), 6,77 (d), 4,39 (q), 1,36 (t).
Пример 1B
(E) Этил 3-[2-(4-трифторметилфенилкарбамоил)этенил]-4,6-дихлороиндол-2-карбоксилат
К перемешиваемой суспензии 4-(трифторметил) фенил - карбамоилметилтрифенилфосфоний хлорида (0,99 г) в ацетонитриле (10 мл) при 0oC в атмосфере азота, добавили ДВИ (0,3 г) Перемешивание продолжали при 0oC в течение 25 минут, затем добавили промежуточный продукт 2 (0,56 г) и смесь нагревали до кипения с обратным холодильником в течение 8 ч. После разбавления дихлорметаном (20 мл), образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (0,6 г).
EA/CH : 4/6 Rf = 0,49) в виде белого твердого продукта.
ИК (Нуйол) Vmax cv-1 33100 (NH), 1676 (C = O), 1632, 1612 (C=C)
Пример 1C
(E) Этил 3-[2-(2-2-изопропилфенил)карбамоилэтенил]-4,6-дихлороиндол-2-карбоксилат
К перемешиваемой суспензии (2-изопропилфенил) карбамоилфенилметилтрифенилфосфоний хлорида (0,83 г) в ацетонитриле (10 мл) при 0oC в атмосфере азота добавили ДВИ. Перемешивание продолжали при 0oC в течение 20 минут, затем добавили промежуточный продукт 2 (0,5 г) и смесь нагревали до кипения с обратным холодильником в течение 4 ч. После разбавления дихлорметаном (20 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (340 мг, tlc EA/CH : 4/6 Rf = 0,53) в виде белого твердого продукта. ИК (нуйол) Vmax (см-1) 3304 (NH), 1676, 1659 (C=O).
Пример 1D
(E) Этил 3-[2-(2-нитрофенилкарбамоил)этенил] -4,6-дихлороиндол-2-карбоксилат
К перемешиваемой суспензии (2 - нитрофенил) карбамоилметилтрифенилфосфоний хлорида (0,75 г) в ацетоне (10 мл) при 0oC в атмосфере азота, добавили ДВИ (238 мг). Перемешивание продолжали при 0oC в течение 20 минут, затем добавили промежуточный продукт 2 (0,45 г) и смесь нагревали до кипения с обратным холодильником в течение 4 м. После разбавления дихлорметаном (20 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (420 мг, tlc EA/CH : 4/6 Rf = 0,55) в виде желтого твердого продукта.
ИК (нуйол) Vmax (cm-1) 3348 - 3308 (NH), 1672 (C = O), 1607 - 1590 (C= C), 1556 - 1346 (NO2).
Пример 1E
(E) Этил 3-[2-(2-метил-4-метоксифениламинокарбонил)этенил]-4,6-дихлороиндол-2 -карбоксилат
К суспензии (2-метил-4-метоксифенил)аминокарбонилметилтрифенилфосфоний хлорида (0,998 г) в ацетонитриле (15 мл) при 0oC в атмосфере азота, добавили DBU (0,32 г). Перемешивание продолжали при 0oC в течение 25 минут, затем добавили промежуточный продукт 2 (0,6 г) и смесь нагревали до кипения с обратным холодильником в течение 3 часов. После разбавления дихлорметаном (20 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (0,57 г tlc EA/CH : 4 : 6 Rf = 0,34) в виде не совсем белого твердого продукта.
ИК (нуйол) Vmax (cm-1) 3302 - 3246 (NH), 1678 - 1659 (C = O), 1624 (C= C).
Пример 1F
(E) Этил 3-[2-(2-гидроксифениламинокарбонил)этенил] -4,6-дихлороиндол e-2-карбоксилат
К cуспензии (2-гидроксифенил) аминокарбонилметилтрифенилфосфоний хлорида (0,94 г) в ацетонитриле (15 мл) при 0oC в атмосфере азота добавили ДВИ (0,32 г). Перемешивание продолжали при 0oC в течение 25 минут, затем добавили промежуточное соединение 2 (0,6 г) и смесь перемешивали в течение 24 ч при комнатной температуре. Суспензию упарили досуха и остаток очистили мгновенной хроматографией (EA/CH : 3/7, затем 4/6), получив названное в заглавии соединение (0,37 г tlc EA/CH: 4/6 Rf = 0,39) в виде твердого продукта бежевого цвета.
ИК (нуйол) Vmax (cm-1) 3317 - 3290 (NH), 1678 - 1655 (C = O), 1618 (C= C).
Пример 1G
(E) Этил 3-[2-(3,4-диметоксифениламинокарбонил)этенил]-4,6-дихлороиндол -2-карбоксилат
К cуспензии (3,4-диметоксифенил) аминокарбонилметилтрифенилфосфоний хлорида (0,69 г) в ацетонитриле (10 мл) при 0oC в атмосфере азота добавили ДВИ (0,21 г). Перемешивание продолжали при 0oC в течение 25 минут, затем добавили промежуточное соединение 2 (0,4 г) и смесь перемешивали в течение ночи при комнатной температуре, затем нагревали до кипения с обратным холодильником в течение 3 часов. После разбавления дихлорметаном (20 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (0,457 г tlc EA/CH: 4/6 Rf = 0,20) в виде желтого твердого продукта.
ИК (нуйол) Vmax (cm-1) 3317 - 3254 (NH), 1678 (C = O), 1620 - 1600 (C= C).
Пример 1H
(E) Этил 3-[2-(4-этоксифениламинокарбонил)этенил]-4,6-дихлороиндол -2-карбоксилат
К cуспензии (4-этоксифенил) аминокарбонилметилтрифенилфосфоний хлорида (0,67 г) в ацетонитриле (10 мл) при 0oC в атмосфере азота, добавили ДВИ (0,21 г) ДВИ. Перемешивание продолжали при 0oC в течение 25 минут, затем добавили промежуточный продукт 2 (0,6 г) и смесь нагревали до кипения с обратным холодильником в течение 28 часов. После разбавления дихлорметаном (20 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (0,265 г tlc EA/CH : 4/6 Rf = 0,41) в виде светло-желтого твердого продукта.
ИК (Нуйол) (см-1) 3321-3260 (NH), 1676 (C=O), 1622 (C=C)
Пример 1I
(E) Этил 3-[2-(2,4-дифторофениламинокарбонил)этенил]-4,6- дихлориндол-2-карбоксилат
К суспензии (2,4-дифторофенил)аминокарбонилметилтрифенилфосфоний хлорида (0,655 г) в ацетонитриле (10 мл) при 0oC в атмосфере азота, добавили ДВИ (0,21 г). Перемешивание продолжали при 0oC в течение 25 минут, затем нагревали до кипения с обратным холодильником в течение 26 часов. После разбавления дихлорметаном (20 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (0,42 г tlc EA/CH : 4/6 Rf = 0,54) в виде светло-желтого твердого продукта.
ИК (Нуйол) Vmax (см-1) 3298 (NH), 1678-1661 (C=O), 1624 (C=C)
Пример 1J
(E) Этил 3-[2-(2-фторо-5-нитрофениламинокарбонил)этенил]-4,6- дихлороиндол-2-карбоксилат
К суспензии (2-фторо-4-нитрофенил)аминокарбонилметилтрифенилфосфоний хлорида (0,52 г) в ацетонитриле (10 мл) при 0oC в атмосфере азота, добавили DBU (0,16 г). Перемешивание продолжали при 0oC в течение 25 минут, затем добавили промежуточный продукт 2 (0,3 г) и смесь нагревали до кипения с обратным холодильником в течение 18 часов. После разбавления дихлорметаном (20 мл) образовавшийся осадок собрали фильтрацией, получив названное в заглавии соединение (0,34 г tlc EA/CH : 4/6 Rf = 0,41) в виде твердого продукта бежевого цвета.
ИК (Нуйол) nmax (см-1) 3300 (NH), 1680-1666 (C=O), 1545-1377 (NO2)
Пример 2A
(E) 3-[2-(фенилкарбамоил)этенил]-4,6-дихлороиндол-2-карбоновая кислота
К раствору примера 1A (250 мг) в этаноле (2,5 мл) добавили при 23oC гидроокись лития (104 мг). Реакционную смесь перемешивали при 50oC в течение 6 часов, затем растворитель упарили и остаток растворили в воде (5 мл). Водный слой подкислили 1 н. соляной кислотой до выпадения белого твердого осадка. Последний собрали путем фильтрации и высушили, получив названное в заглавии соединение в виде белого твердого продукта (230 мг).
ИК (Нуйол) Vmax (см-1) 3402-3281-3192 (OH, NH), 1661 (C=O), 1607-1579 (C=C).
1H-NMR (DMSO) 12,4 (s), 10,1 (s), 8,50 (d), 7,74 (d), 7,48 (s), 7,27 (t), 7,16 (s), 7,11 (d), 6,99 (t)
Исходя из той же самой общей методики, были приготовлены следующие соединения:
Пример 2B
(E) 3-[2-(Трифторометилфенилкарбамоил)этенил] -4,6-дихлороиндол- 2-карбоновая кислота
Исходя из примера 1B (585 мг), получили названное в заглавии соединение в виде светло-бурого твердого продукта (520 мг).
ИК (Нуйол) Vmax (см-1) 3430-3000 (NH, OH), 1700-1678 (C=O), 1636-1614 (C=C).
1H-NMR (DMSO) 14-13,5 (s), 12,55 (s), 10,54 (s), 8,37 (d), 7,91 (d), 7,67 (d), 7,48 (d), 7,30 (d), 6,86 (d).
Пример 2C
(E) 3-[2-(2-Изопропилфенилкарбамоил)этенил]-4,6-дихлороиндол- 2-карбоновая кислота, дилитиевая соль
Исходя из примера 1C (317 мг), получено названное в заглавии соединение в виде светло-бурого твердого продукта (288 мг).
ИК (Нуйол) (ДМСО) (см-1) 3661 (NH, OH), 1610 (C=O).
1H-ЯМР (ДМСО) 12,1 (s), 9,39 (s), 8,57 (d), 7,57 (s), 7,38-7,28 (m), 7,28-7,10 (m), 3,25 (m), 1,15 (d).
Пример 2D
(E) 3-[2-Нитрофенилкарбамоил)этенил]-4,6-дихлороиндол-2- карбоновая кислота
Исходя из примера 1D (440 мг), получено названное в заглавии соединение в виде белого твердого продукта (290 мг)
ИК (Нуйол) Vmax (см-1) 3234 (NH, OH), 1684-1636 (C=O), 1639 (C=C).
1H-ЯМР (ДМСО) 12,2 (s), 10,51 (s) 8,59 (d), 7,95 (dd), 7,81 (dd), 7,69 (m), 7,48 (d), 7,38-7,28 (m), 7,20 (d)
Пример 2E
(E) 3-[2-(2-Метил-4-метоксифениламинокарбонил)этенил] -4,6- дихлороиндол-2-карбоновая кислота
Исходя из примера 1E (0,54 г), получено названное в заглавии соединение в виде желтого твердого продукта (0,39 г).
ИК (Нуйол) Vmax (см-1) 3279 (NH, OH), 1703-1661 (C=O), 1630 (C=C).
1H-ЯМР (ДМСО) 12,41 (s), 9,39 (s), 8,26 (d), 7,48 (d), 7,36 (d), 7,27 (d), 6,90 (d), 6,80 (d), 6,75 (dd), 3,73 (s), 2,19 (s).
Пример 2F
(E) 3-[2-(2-Гидроксифениламинокарбонил)этенил]-4,6-дихлороиндол- 2-карбоновая кислота
Исходя из примера 1F (0,34 г), получено названное в заглавии соединение в виде желто-бурого твердого продукта (0,33 г).
ИК (Нуйол) Vmax (см-1) 3150 (NH, OH), 1736-1656 (C=O), 1630 (C=C).
1H-ЯМР (ДМСО) 12,56 (s), 9,97 (s), 9,76 (s), 8,24 (s), 7,8 (d), 7,49 (d), 7,30 (d), 6,96 (d), 6,96 (td), 6,88 (dd), 6,79 (td).
Пример 2G
(E) 3-[2-(3,4-Диметоксифениламинокарбонил)этенил]-4,6- дихлороиндол-2-карбоновая кислота
Исходя из примера 1G (0,41 г), получено названное в заглавии соединение в виде светло-желтого твердого продукта (0,38 г).
ИК (Нуйол) Vmax (см-1) 3420-2381 (NH), 1690-1680 (C=O), 1620-1607 (C=C).
1H-ЯМР (ДМСО) 13,8-13,6 (s), 12,53 (s), 10,08 (s), 8,23 (d), 7,47 (m), 7,29 (d), 7,20 (dd), 6,89 (d), 6,74 (d), 3,37 (s), 3,70 (s).
Пример 2H
(E) 3-[2-(4-Этоксифениламинокарбонил)этенил] -4,6-дихлороиндол- 2-карбоновая кислота IV
Исходя из примера 1H (0,25 г), получено названное в заглавии соединение в виде светло-желтого твердого продукта (0,22 г).
ИК (Нуйол) nmax (см-1) 3248 (NH, OH), 1663 (C=O), 1632-1610 (C=C)
1H-ЯМР (ДМСО) 13,7 (s), 12,50 (s), 10,04 (s), 8,22 (d), 7,61 (d), 7,47 (d), 7,29 (d), 6,86 (d), 6,74 (d), 3,97 (q), 1,29 (t).
Пример 2I
(E) 3-[2-(2,4-Дифторофениламинокарбонил)этенил] -4,6-дихлоиндол- 2-карбоновая кислота
Исходя из примера 1I (0,41 г), получено названное в заглавии соединение в виде светло-желтого твердого продукта (0,37 г)
ИК (нуйол) nmax (cm-1) 3431-3233 (NH, OH),
1707 - 1678 (C=O), 1612 (O=C).
1H-ЯМР (ДМСО) 14,0 - 13,6 (S), 12,54 (S), 9,99 (S), 8,29 (d), 7,97 (m), 7,48 (d), 7,30 (m), 7,29 (d), 7,07 (m), 6,90 (d)
Пример 3
Метил-3-[2-(фенилкарбамоил)этинил]-4,6-дихлороиндол-2- карбоксилат
Промежуточный продукт 7 растворили в этаноле 95% (18 мл), затем добавили по каплям HCl (18 мл, 5 н.) и раствор нагревали до кипения с обратным холодильником в течение 3 часов. После добавления этил ацетата (50 мл) две фазы разделили и органический слой промыли водой (2 x 40 мл), высушили и очистили хроматографически. Полученный белый твердый продукт (140 мг) растворили в THF (4 мл), воде (2 мл) и перемешивали при комнатной температуре в течение 10 минут. После охлаждения до 8oC, добавили LiOH • H2O (42 мг), полученную смесь перемешивали в течение 1 часа, затем вылили в раствор 0,01н. HCl и экстрагировали этил ацетатом. Объединенные органические слои высушили и сконцентрировали при пониженном давлении, получив остаток, который растирали в эфире и получили названное в заглавии соединение (100 мг) в виде белого твердого вещества.
Rf = 0,18 CE/EA: 70/30
ИК (нуйол) Vmax (см-1) 3273 (NH), 2220 (C=C),
1686 (C=C), 1636 (C=O).
1H-ЯМР (ДМСО) 13,5 (s), 10,71 (s), 7,68 (m), 7,52 (m), 7,40 (d), 7,35 (m), 7,11 (m), 3,96 (s)
Пример 4
3-[2-Фенилкарбамоил)этинил]-4,6-дихлороиндо-2- карбоновая кислота
Смесь примера 3 (100 мг), тетрагидрофурана (4 мл), воды (2 мл) и LiOH • H2O (39 мг) перемешивали при 45oCC в течение 12 часов. Затем ее вылили в воду (15 мл) и HCl (0,05 М, 5 мл) добавили по каплям при перемешивании. Полученный осадок собрали фильтрацией для получения названного в заглавии соединения в виде желтого твердого продукта (63 мг) t.m. = 207oC.
ИК (нуйол) Vmax (см-1) 3169 (NH-OH), 2240 (C=C), 1745 (C=O), 1661 (C=O).
1H-ЯМР (ДМСО) 13,05 (s), 14,0 (s), 10,88 (s), 10,7 (s), 7,67 (d), 7,51 (d), 7,35 (d), 7,33 (m), 7,10 (m)
Пример 5
(D. L. )-Транс-этил-3-[2-фенилкарбамоил)циклопропил]-4,6- дихлороиндол-2-карбоксилат
(a) (D.L.)-Транс-этил-3-[2-(2-фенилкарбамоил)циклопропил]-1- (2-триметилсилилэтоксиметил)-4,6-дихлороиндол-2-карбоксилат
К смеси промежуточного продукта 8 (0,1 г) и ацетата палладия (П) (4 мг) в дихлорметане (10 мл) в атмосфере азота при 0oC, добавили при перемешивании раствор диазометана в диэтиловом эфире (8 мл, 0,125 М). Получили черный твердый продукт при бурном выделении газа. Реакционную смесь перемешивали в течение 15 часов при комнатной температуре, затем растворитель и некоторую часть оставшегося диазометана упарили при пониженном давлении в токе азота. К образовавшемуся остатку добавили дихлорметан, профильтровали через целит и упарили при пониженном давлении.
Очистка мгновенной хроматографией дала смесь исходного материала, названного в заглавии соединения в соотношении 0,3 : 1 (86 мг) в виде бледно-желтого твердого вещества.
b) (D.L.)-Транс-этил-3-[2-(2-фенилкарбамоил)циклопропил]-4,6- дихлороиндол-2-карбоксилат
HCl (2 мл 5 М) добавили к продукту примера 5a (66 мг) в этиловом спирте (2 мл; 95%) и перемешивали при нагревании до температуры кипения в течение 2 часов. После охлаждения смесь вылили в холодную воду (50 мл) и экстрагировали этил ацетатом (3 x 100 мл). Органические соли объединили, высушили и растворитель упарили при пониженном давлении, получив названное в заглавии соединение (tic CH/EA = 9/4; Rf = 0,32).
ИК (нуйол) Vmax (см-1) 3312 (NH), 1672 (C=O), 1648 (C=O), 1599 (C=C) 1535 (C=C).
1H-ЯМР (CDCl3) 12,1 (s), 10,2 (s), 7,60 (d), 7,40 (d), 7,28 (t), 7,01 (m), 4,40 (m), 4,25 (m), 2,55 (m), 1,98 (m), 1,49 (m), 1,27 (t), 1,22 (m)
Пример 6
(D. L. )-Транс-3-[2-фенилкарбамоил)циклопропил]-4,6- дихлороиндол-2-карбоновая кислота
Исходя из Примера 5 b (40 мг) и LiOH и используя общую методику Примера 2a, получили названное в заглавии соединение в виде белого твердого продукта (23 мг)
ИК (нуйол) Vmax (см-1) 3271 (NH), 1663 - 1653 (C=O), 1599 (C=C).
1H-ЯМР (DMSO) 13,4 (s), 11,98 (s), 10,11 (s), 7,60 (s), 7,37 (d), 7,27 (t), 7,17 (d), 7,00 (t), 1,97 (m), 1,50 (m), 1,47 (m), 1,2 (m)
Пример 7
Этил-3-[(2-фенилкарбамоил)-пропенил]-4,6-дихлороиндол-2- карбоксилат
Промежуточный продукт 9 (660 мг) растворили в 95% EtOH (6 мл) и обработали при температуре кипения 5н. соляной кислотой (6 мл) в течение ночи. Затем раствор был поглощен этил ацетатом, промыт 1н соляной кислотой, водой и физиологическим раствором, высушен, отфильтрован и сконцентрирован. Очистка колоночной хроматографией дала названное в заглавии соединение (-20 мл) в виде твердого белого продукта.
Rf = 0,30 CH/EA 7,5/2,5
1H-ЯМР (ДМСО) 12,48 (s, 1H), 9,70 (s, 1H), 7,80 - 7,72 (m, 3H), 7,48 (d, 1H), 7,33 (t, 2H), 7,26 (d, 1H), 7,08 (m, 1H), 4,32 (q, 2H), 1,79 (d, 3H), 1,30 (t, 3H) ppm,
ИК (нуйол) (Vmax = см-1) 3317 - 3288 (str NH), 1678 (str CO)
Пример 8
3-[(2-фенилкарбамоил)пропенил]-4,6-дихлороиндол-2-карбоновая кислота
Пример 7 (210 мг) растворили в 95% EtOH (6 мл) и обработали LiOH • H2O (32 мг) при 30oC за 1,5 дня и затем в течение 2,5 дней при комнатной температуре. Затем раствор сконцентрировали досуха и обработали в течение 2 часов 1н. соляной кислотой. Образовавшийся белый осадок отфильтровали, высушили в высоком вакууме и затем перекристаллизовали из диэтилового эфира, получив названное в заглавии соединение (135 мг) в виде белого твердого продукта.
1H-ЯМР 13,5 (s, 1H), 12,37 (s, 1H), 9,70 (s, 1H), 7,76 (d, 2H), 7,75 (s, 1H), 7,45 (d, 1H), 7,31 (t, 2H), 7,23 (d, 1H), 7,06 (t, 1H), 1,78 (d, 3H) ppm:
ИК (нуйол) (Vmax = см-1) 3209 (str, NH), 1664 (str, CO)
Пример 9
(E) 3-[2-(фенилкарбамоил)этенил] -4,6-дихлороиндол-2-карбоновой кислоты натриевая соль
Метанол добавили по каплям к суспензии (E) 3-[2- (фенилкарбамоил)этенил] -4,6-дихлороиндол-2-карбоновой кислоты 200 мг в 0,5 М гидроокиси натрия (1,01 мл) до получения прозрачного раствора. После перемешивания в течение 15 минут, раствор упарили досуха и остаток высушили при 50oC в течение 12 часов для получения названного в заглавии соединения в виде белого твердого продукта (150 мг).
ИК (нуйол) Vmax (см-1) 3404-3126 (NH), 1624 (C=O), 1600 (C=C)
1H-ЯМР (ДМСО) 11,9 (S), 10,06 (S), 8,59 (d), 7,75 (d), 7,44 (d), 7,27 (t), 7,21 (d), 7,10 (d), 6,98 (t).
Пример 10
(E)-2-[-2-(N, N-диэтиламино)этил] -3-[2-(фениламинокарбонил)- этенил]-4,6-дихлороиндол-2-карбоксилат
(E) Этил-3-[2-(фенилкарбамоил)этенил] -4,6-дихлороиндол-2- карбоксилат (0,3 г) и N,N-диэтилэтаноламин (1,3 г) перемешивали в течение 20 минут перед добавлением карбоната натрия (0,073 г) и смесь нагревали при 70oC в течение 24 часов. Раствор сконцентрировали при пониженном давлении и остаток оставили стоять в течение ночи до образования белого осадка. Фильтрация и кристаллизация из этил ацетата дали названное в заглавии соединение (0,13 г Rf= 0,65 = ДСМ/MeOH 8,2) в виде белого твердого продукта.
ИК (нуйол) Vmax (см-1) 3300 (NH), 1676 (C=O), 1624 (C=C).
1H-ЯМР (ДМСО) 12,52 (S), 10,18 (S), 8,22 (d), 7,70 (d), 7,50 (d), 7,32 (d), 7,31 (t), 7,04 (t), 6,73 (d), 4,36 (t), 2,75 (t), 2,49 (q), 0,90 (t).
Пример 11
(E) 2-[4-(-2N-морфолино)этил] -3-[2-(фениламинокарбонил)-этенил]- 4,6-дихлороиндол-2-карбоксилат
Смесь (E) Этил-3-[2-(фенилкарбамоил)этенил] -4,6- дихлороиндол-2-карбоксилата (400 мг), 4-(2-гидроксиэтил)-морфолина (7 мл) и п-толуолсульфоновой кислоты (15 мг) перемешивали при 130oC в течение 120 часов. Смесь разбавили водой и экстрагировали этил ацетатом (3х100 мл). Органические экстракты высушили, сконцентрировали и собрали осадок, получив названное в заглавии соединение в виде белого твердого продукта.
1H-ЯМР (ДМСО) 10,21 (S), 8,28 (d), 7,75 (d), 7,56 - 7,35 (d,d), 7,35 (t), 7,08 (t), 6,74 (d), 4,46 (t), 3,54 (m), 2,43 (m), 2,70 (t).
Пример 12
(a) (Е)-2-(Т-Бутилкарбонилоксиметил)-3-[2-(фениламинокарбонил)этенил]-4,6- дихлороиндол-2-карбоксилат
Пример 2. (200 мг) растворили в DMF (4 мл) и добавили тетрабутиламмоний хлорид (168 мг). После перемешивания в течение 0,5 ч, добавили по каплям хлорометилпивалат (118 мг) и реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Смесь разбавили водой и экстрагировали этил ацетатом (2х100 мл). Органический слой промыли водой, высушили и упарили для получения сырого продукта, который очистили мгновенной хроматографией для получения названного в заглавии соединения в виде желтого твердого продукта (190 мг) toCпл 205oC.
ИК (нуйол) Vmax (см-1) 3383-3308 (H), 1747 (C=O), 1688 (C=O), 1634 - 1603 (C=C).
1H-ЯМР (ДМСО) 12,75 (S), 10,22 (S), 8,22 (d), 7,73 (d), 7,54 (d), 7,36 (d), 7,33 (t), 7,07 (t), 6,79 (d), 6,02 (S), 1,15 (S).
Используя ту же самую общую методику, приготовили следующее соединение:
(b) (E)-2-[1-(Тетрагидро-4-пиран-4-4-илоксикарбонилокси) этил] 3-[2-(фениламинокарбонил)этенил]-4,6-дихлороиндол-2- карбоксилат
Из примера 2 (200 мг) в сухом DMF (11 мл), бензилтриэтиламмоний хлорида (178 мг) и 1-(тетрагидро-4-H-пиран-4-илоксикарбонилокси) этил хлорида (244 мг), после 4 дней перемешивания при комнатной температуре получено названное в заглавии соединение в виде желтого твердого продукта (209 мг) toCпл = 209oCC
ИК (нуйол) Vmax (см-1) 3300 (NH), 1749 (C=O), 1730 (C=O).
1H-ЯМР (ДМСО) 12,73 (S), 10,22 (S), 8,21 (d), 7,72 (d), 7,53 (d), 7,34 (d), 7,32 (t), 7,05 (t), 6,90 (q), 6,76 (d), 4,76 (m), 3,72 (m), 1,87 - 1,53 (m), 1,61 (d).
(c) (E)-2-[1-(Циклогексилоксикарбонилокси(этил]3-[2- (фениламинокарбонил)этенил]-4,6-дихлороиндол-2-карбоксилат
Из примера 2 (300 мг) в сухом DMF (8 мл), бензилтриэтиламмоний хлорида (178 мг) и 1-(циклогексилоксикарбонилокси) этил хлорида (242 мг), после 0,5 часа перемешивания при комнатной температуре, получили названное в заглавии соединение в виде желтого твердого продукта (170 мг) toCпл = 125oC
ИК (нуйол) Vmax (см-1) 3300 (NH), 1730 (C=O),
1H-ЯМР (ДМСО) 12,71 (SO, 10, 21 (S), 8,21 (d), 7,71 (d), 7,51 (d), 7,36 - 7,26 (m), 7,05 (t), 6,85 (q), 6,76 (d), 4,54 (m) 1,79 (m), 1,51 - 1,1 (m), 1,6 (d).
(d) (E)-2-[(Метоксикарбонилметил)3-2-(фениламинокарбонил) -этенил] -4,6-дихлороиндол-2-карбоксилат
Из примера 2 (200 мг) в сухом DMF (4 мл), тетрабутиламмоний хлорида (168 мг) и метил хлорацетата (85 мг), после 48 часов перемешивания при комнатной температуре, получено названное в заглавии соединение в виде не совсем белого твердого продукта (210 мг).
toCпл = 241 - 242oC
ИК (нуйол) Vmax (см-1) 3348 (NH), 1749 (C=O), 1672 (C=O), 1634 - 1610 (C=C).
1H-ЯМР (ДМСО) 12,8 (S), 10,21 (S), 8,28 (d), 7,72 (d), 7,54 (d), 7,38 - 7,28 (m), 7,06 (t), 6,48 (d), 7,54 (d), 7,38 - 7,28 (m), 7,06 (t), 6,48 (d), 5,02 (S), 3,73 (S).
Фармацевтические примеры А.Капсулы/Таблетки
Активный ингредиент - 200,0 мг
Крахмал 1500 - 32,5 мг
Микрокристаллическая целлюлоза - 60,0 мг
Кроскармелоза натрия - 6,0 мг
Стеарат магния - 1,5 мг
Активный ингредиент смешивают с другими наполнителями. Смесь может быть использована для наполнения желатиновых капсул или прессования с целью получения таблеток при использовании соответствующих перфораторов.
Таблетки могут быть покрыты оболочками при использовании обычных методик и покрытий.
В.Таблетка
Активный ингредиент - 200,0 мг
Лактоза - 100,0 мг
Микрокристаллическая целлюлоза - 28,5 мг
Повидон - 25,0 мг
Кроскармелоза натрия - 6,0 мг
Стеарат магния - 1,5 мг
Активный ингредиент смешивают с лактозой, микрокристаллической целлюлозой и частью кроскармелозы натрия.
Смесь гранулируют с повидоном после диспергирования в подходящем растворителе (например вода). Гранулы, после высушивания и измельчения, смешиваются с оставшимися наполнителями. Смесь может быть прессована с использованием соответствующих перфораторов и таблетки могут быть покрыты оболочками при использовании обычных методик и покрытий.
С. Составы для инъекций
Активный ингредиент - 0,1 - 7,00 мг/мл
Фосфат натрия - 1,0 - 50,00 мг/мл
с NaOH до заданного pH (интервал 3-10) вода для инъекций q.s. до объема - 1 мл
Состав может быть упакован в склянку (ампулы) с резиновой пробкой (флаконы, шприцы) и пластик/металл изолирующим слоем (только флаконы).
Сухой порошок для составления с подходящим носителем
Активный ингредиент: - 0,1 - 100,00 мг
Маннитол qS в количестве до - 0,02 - 5,00 мг
упакованные в стеклянные флаконы или шприцы, с резиновой пробкой (и только флаконы) пластиково-металлическим изолирующим слоем.
Е.Гильзы для ингаляций мг/на гильзу
Активный ингредиент (микронизированный) - 5,00
Лактоза до - 25,00
Активный ингредиент предварительно микронизируется в мощной размалывающей в пыль мельнице до размера мельчайших частиц для смешения с используемой для составления таблеток лактозой обычного качества в смесителе высокой мощности. Порошкообразная смесь заполняется в соответствующую емкость для единичной дозы в виде пластыря или капсулы для использования подходящим путем:ингаляции или инсуффляции.
Активность соединения изобретения в отношении нарушенного стрихнином места связывания глицина определили использованием методики Kishimoto H.et al T. Neurochem 1981, 37, 1015 - 1024.
Значения pKi, полученные с представленными соединениями изобретения, даны в таблице 1.
Способность соединений изобретения ингибировать NМДА вовлеченные судороги у мышей определили использованием методики Chiamulera C. et al. Psychopharmacology 1990, 102, 551 - 552. В этом испытании способность соединения ингибировать распространенные эпилептические припадки, вызванные интрацеребровентрикулярной инъекцией NMDA у мышей, изучалась при ряде уровней доз. Из этих результатов была рассчитана доза, требуемая для защиты 50 % животных от конвульсивного действия NMDA. Эта доза, выраженная мг/кг, называется ED50 значением.
Изложенные результаты, полученные для соединений изобретения вводимых внутривенно или пероральным путем, приведены в таблице 2.
Соединения изобретения в основном не токсичны при терапевтическом использовании полых доз. Так, например, соединение Примера 9 не вызывает неблагоприятных побочных эффектов при введении крысам или мышам в дозах 3 - 30 мг/кг внутривенно или 30 - 300 мг/кг перорально.
Изобретение относится к производному индолу общей формулы (I)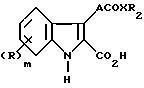
или его физиологически приемлемой соли, или метаболически лабильному сложному эфиру, где R представляет хлор в положениях 4 и 6 индольного кольца, R2 представляет фенил, возможно замещенный одной или двумя группами, выбранными из фтора, трифторметила, низших алкила и алкокси, гидрокси и нитрогрупп, Х представляет NH. Способы получения указанных производных включают взаимодействие соответствующего индола, включающего карбоксизащитную группу и группу CR4O, где R4 водород, или С1-С4-алкил, с илидом фосфора формулы
(R5)3P=CH-COXR2 или (R7O)2 OP=CR6-COXR2,
где R5 - алкил или фенил; R6 - водород или С1-С4-алкил; R7 - C4-C4алкил, а X и R2 указаны выше. Полученные производные индола в качестве активного ингредиента входят в состав фармацевтической композиции, обладающей свойством антагониста аминокислот, возбуждающих NMDA рецепторный комплекс. 4 с. и 9 з.п. ф-лы, 2 табл.
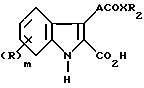
или его физиологически приемлемая соль, или метаболически лабильный сложный эфир,
где R представляет хлор в положениях 4 и 6 индольного цикла,
А представляет этинил, 1-метилэтенил, этенил или циклопропил группу;
Х представляет NH;
R2 представляет фенил, возможно замещенный одной или двумя группами, выбранными из фтора, CF3, низших алкила и алкокси, гидрокси и нитрогрупп.
(Е)-3-[2-(4-этоксифенилкарбамоил)этенил] -4,6-дихлориндол-2-карбоновую кислоту,
(Е)-3-[2-(2-гидрокси-5-нитрофенилкарбамоил)этенил] -4,6-дихлориндол-2-карбоновую кислоту,
(Е)-3-[2-(2-метил-4-метоксифенилкарбамоил)этенил] -4,6-дихлориндол-2-карбоновую кислоту,
(Е)-3-[2-(2-изопропилфенилкарбамоил)этенил]-4,6-дихлориндол-2-карбоновую кислоту,
(Е)-3-[2-(2,4-дифторфенилкарбамоил)этенил] -4,6-дихлориндол-2-карбоновую кислоту,
(Е)-3-[2-(3,4-диметоксифенилкарбамоил)этенил] -4,6-дихлориндол-2-карбоновую кислоту,
или их физиологически приемлемые соли или метаболически лабильные эфиры.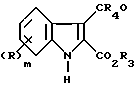
в котором R и m имеют значения, определенные в п.1;
R4 - водород или С1-С4 алкил и
R3 - карбоксизащитная группа,
с илидом фосфора формулы III
(R5)3P=CH-COXR2,
где R5 представляет алкил или фенил;
Х и R2 имеют значения, определенные в п.1,
или с илидом фосфора формулы IV
(R7O)2OP=CR6 -COXR2,
где R6 - водород или С1-С4 алкил;
R7 - С1-С4 алкил;
Х и R2 определены в п.1.
| ЭЛЕКТРОПРИВОД | 0 |
|
SU394905A1 |
| US 4960786 A, 1990 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЬ1Х ИНДОЛА | 0 |
|
SU262906A1 |
| Шланговое соединение | 0 |
|
SU88A1 |
