Настоящее изобретение связано с новой группой соединений, которые полезны в лечении нестабильности мочевого пузыря у млекопитающих, таких как человек. Более конкретно, настоящее изобретение относится к группе 4,6,7,8-тетрагидро-5(1H)-хинолонов, их применению в лечении недержания мочи у млекопитающих (включая человека), способам их получения и фармацевтическим композициям, их содержащим.
Существующие методы лечения недержания мочи в большинстве случаев неудовлетворительны, основываются на лекарственных препаратах, которые первоначально разрабатывались для других показаний. Одна группа таких лекарственных препаратов включает блокаторы кальциевых каналов, такие как нифедипин, которые первоначально были разработаны и главным образом используются как сердечно-сосудистые препараты.
Нифедипин принадлежит к структурному классу соединений, известных как дигидропиридины. Этот структурный класс был широко исследован, и структурные требования для блокирования кальциевой активности в настоящее время достаточно хорошо установлены. Так, как описано в главе 14.1 Руководства по лекарственной химии, Comprehensive Medical Chemistry, том 3, под редакцией John C. Emmett, опубликованного Pergamon Press в 1990, соединения обладают 1,4-дигидропиридиновым циклом, имеющим оптимально арильную группу в 4-положении и сложноэфирные группы в 3- и 5-положениях. Удаление эфирных групп или замена их ацетильной или циано группами связаны со снижением активности. Обычно соединения имеют метильные группы в 2- и 6-положениях. Geinshteins et al. , Khim. Geterotsikl. Soedin. (6), 1118-20, 1967 раскрывают соединения 3-циано-4-фенил-2,7, 7-триметил-4,6,7,8-тетрагидро-5(1H)-хинолон и 3-этаноил-4-фенил- 2,7,7-триметил-4,6,7,8-тетрагидро-5(1H)-хинолон. 15(1), 39-42, 1981 раскрывает исследование воздействия некоторых 4,6,7,8-тетрагидро- 5(1H)-хинолонов и имеющих сложноэфирную или циано группы в 3-положении на сердечно-сосудистую систему и на гладкую мышцу кишечника. Сообщается, что 3-циано-4-фенил-2,7,7- триметил-4,6,7,8-тетрагидро-5(1H)-хинолон проявляет гипотензивные свойства и способен к блокированию спазмогенного воздействия как ацетилхолина, так и хлорида бария на гладкую мышцу кишечника.
DE 2003148 раскрывает группу 1,4-дигидропиридин производных, включая некоторые 4,6,7,8-тетрагидро-5(1H)-хинолоны, которые содержат сложноэфирную или кето группу в 3-положении и которые, как упоминается, проявляют широкий и многогранный фармакологический спектр воздействия. Основные упомянутые воздействия, проявляемые соединениями, включают сильные мышечные спазмолитические эффекты, которые становятся очевидными в гладкой мускулатуре желудочно-кишечного тракта, мочевого тракта и респираторной системы. Установлено, что другие основные воздействия проявляются в действии на сердце (сердечно-успокаивающий эффект) и в снижении кровяного воздействия животных с нормальным и гипертоническим кровяным давлением, так что они могут быть использованы в качестве гипотензивных средств.
S. M. Tain et al., Indian Journal of Chemistry, том 30B, ноябрь, 1991, стр. 1037 - 1040 раскрывает синтез и фармакологический скрининг некоторых 9-(замещенный фенил)-1,8-(2H, 5H)-акридиндионов. Было обнаружено, что соединения обладают различными степенями гипотензивной противовоспалительной и противоимплантационной активности.
Известно, что ткань мочевого пузыря является возбудимой и что недержание мочи может быть вызвано неконтролируемыми или нестабильными сокращениями мочевого пузыря. Была найдена группа соединений, которые оказались неожиданно способными к расслаблению гладкой мышцы мочевого пузыря, обеспечивая, таким образом, предотвращение или уменьшение интенсивности неконтролируемых или нестабильных сокращений мочевого пузыря. Следовательно, соединения могут быть полезными для лечения недержания позывов, включающего, например, детруссорную нестабильность, которая может быть следствием цистита, уретрита, опухолей, камней, дивертикула или непроходимости оттоков; и детруссорную гиперрефлексию, которая может быть следствием удара, слабоумия, болезни Паркинсона, супрасакрального повреждения спинного мозга или супрасакрального заболевания спинного мозга. Было найдено, что некоторые соединения обладают дополнительным неожиданным свойством, состоящим в том, что они способны к селективному воздействию на мочевой пузырь, без существенного воздействия в то же время на сердечно-сосудистую систему, что показано измерениями частоты сердечных сокращений и кровяного давления. Таким образом, эти соединения особенно могут быть полезными при лечении недержания мочи у пациентов, таких, например, как пожилые люди, для которых сердечно-сосудистые воздействия, особенно такие, как гипотензивные воздействия, являются особенно нежелательными.
Было также неожиданно найдено, что соединения согласно изобретению служат для открытия калиевых каналов. Известно, что, выполняя действие по раскрытию калиевых каналов, соединения, открывающие калиевые каналы, могут функционировать таким образом, что расслабляют гладкую мышцу. Хотя не ставится задача теоретического ограничения, соответственно предполагается, что соединения настоящего изобретения действуют путем открытия калиевых каналов в клетках мочевого пузыря и тем самым расслабляют гладко-мышечную ткань мочевого пузыря, препятствуя, таким образом, неконтролируемым сокращениям или уменьшая интенсивность неконтролируемых сокращений мочевого пузыря, которые могут вызывать недержание мочи. Nurse D.A., Kestorick T.M. and Mundy A.R., British Journal of Urology, (1991), 68, 27 - 31 раскрывают, что хромакалим, который хорошо известен как вещество, открывающее калиевые каналы, как было установлено ими в первичных клинических испытаниях, является эффективным для лечения недержания мочи.
В соответствии с настоящим изобретением, было найдено, что структурные требования относительно активности 4,6,7,8-тетрагидро-5(1H)-хинолонов отличны от соответствующих требований, предъявляемых к дигидропиридиновым кальциевым блокаторам. Таким образом, придается большое значение тому, что настоящее изобретение основано на открытии нового фармакологического класса дигидропиридинов, являющихся миорелаксантами мочевого пузыря.
Настоящее изобретение дает соединение формулы I (формула вынесена вместе с другими формулами, обозначенными римскими цифрами, на страницы, следующие за примерами) или его фармацевтически приемлемую соль,
где:
R2 представляет водород, (1-6C)алкил или (1-4C)фторалкил; и R3 представляет водород, циано, (1-6C)алкил, (1-6C)фторалкил или этаноил; или
R2 и R3, взятые вместе, образуют 1,4-бутандиил;
R4 представляет 2- или 3-тиенил или фурил, замещенный в 4- и/или 5-положении(иях) радикалом или радикалами независимо выбранными из группы (a), состоящей из нитро, циано, гало, (1-4C)алкила, (1-4C) алкилсульфонила и 2-тиенила при условии что 3-тиенил или фурил группа могут быть замещенными только в 5-положении; или
R4 обозначает 2-пиридил, который замещен в 5- положении и/или либо в 4-положении, либо в 6-положении одним из членов вышеприведенной группы (a); или
R4 обозначает 3-пиридил, который замещен в 6-положении одним из членов вышеприведенной группы (a); или
R4 представляет собой 4-пиридил, который замещен в 2-положении одним из членов вышеприведенной группы (a); или
R4 представляет группу формулы II, в которой:
R7 является водородом; и
R8 и R9 независимо выбираются из водорода, гидрокси, (1-4C)алкокси, нитро, циано, (1-4C)фторалкила, (1-4C)фторалкокси, гало, (1-4C)алкила, (1-4C)алканоила, фенила и (1-4C)алкилсульфонила; или
R8 и R9, взятые вместе, представляют (1-3C)алкилендиокси; и
R10 и R11 каждый независимо представляют водород или (1-4C)алкил, исключая 3-циано-4-фенил-2,7,7-триметил- 4,6,7,8-тетрагидро-5(1H)-хинолон и 3-этаноил-4-фенил-2,7,7-триметил- 6,7,8-тeтрагидро-5(1H)-хинолин.
В данном описании термины "алкил" и "алкокси" включают как линейные, так и разветвленные цепные радикалы, но для того чтобы было понятно, термин, отнесенный к конкретным радикалам, таким как "пропил" или "пропокси", охватывает только линейный цепной ("нормальный") радикал, разветвленные цепные изомеры, такие как "изопропил" или "изопропокси", упоминаются особо.
Термин "гало" включает фторо, хлоро, бромо, и йодо, если не оговорено особо.
Конкретные примеры 2- или 3-тиенил или фурил заместителей в 4- и/или 5-положениях включают 4-бром-2-тиенил, 5-бром-2-тиенил, 5-метилсульфонил-2-тиенил, 5-метил-2-тиенил, 5-(2-тиенил)-2-тиенил, 4-нитро-2-тиенил, 5-нитро-2-тиенил, 4-циано-2-тиенил, и 5-нитро-3-тиенил.
Конкретные значения (1-6C)алкила включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, тpeт-бутил, пентил, 3-метилбутил, 1-этилпропил, гексил или 4-мeтилпeнтил.
Конкретные значения (1-4C)алкила включают метил, этил, пропил, изопропил, бутил, втор-бутил, и трет-бутил.
Конкретные значения (1-4C)фторалкила включают трифторметил и пентафторэтил.
Конкретные значения (1-4C) алкокси включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, и трет-бутокси.
Конкретные значения (1-4C)фторалкокси включают трифторметокси и пентафторэтокси.
Конкретным значением (1-4C)алканоила является этаноил.
Конкретное значение (1-4C)алкилсульфонила включает метиленсульфонил.
Конкретными значениями для (1-3C)алкилендиокси являются метилендиокси и этилендиокси.
Конкретные значения R8 включают водород, гидрокси, метокси, нитро, циано, трифторметил, трифторметокси, метил, этил, изопропил и гало и этаноил.
Конкретные значения R9 включают водород, гидрокси, метокси, нитро, циано, трифторметил, трифторметокси, метил, этил, изопропил и гало.
Конкретными значениями R2 являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил и трифторметил.
Конкретными значениями для R3 являются водород, пиано, этаноил, или вместе с R2 1,4-бутандиил.
Было найдено, что соединения, в которых R3 является водородом или R2 и R3, взятые вместе, образуют 1,4-бутандиил, проявляют, в частности, хорошую активность и селективность в отношении мочевого пузыря и являются, таким образом, предпочтительными.
Было найдено, что соединения, в которых R2 является трифторметилом, проявляют, в частности, хорошую активность, селективность в отношении мочевого пузыря и стабильность, и являются, таким образом, особенно предпочтительными.
R4 является предпочтительно группой формулы II.
Конкретными значениями R4 являются фенил, 3-метоксифенил, 3-нитрофенил, 3-цианофенил, 3-трифторметилфенил, 3-трифторметил-4-цианофенил, 4-трифторметилфенил, 3-трифторметоксифенил, 3-фторфенил, 3-хлорфенил, 3-хлор-4-фторфенил, 3-бромфенил, 4-фторфенил, 4-хлорфенил, 3-бром-4- фторфенил, 3,4-дихлорфенил, 4-метилфенил, 3,4-метилендиоксифенил и 4-нитро-2-тиенил. Наиболее предпочтительным R4 является 3-нитрофенил или 3-цианофенил.
Предпочтительными значениями для R10 и R11 являются водород и метил. Предпочтительно R10 и R11 оба представляют водород или оба представляют метил.
Особенно предпочтительной группой соединений формулы I являются те, в которых либо
R2 обозначает метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил или трифторометил; и
R3 обозначает водороду либо
R2 и R3, взятые вместе, образуют 1,4-бутaндиил; и
R4 обозначает 3-нитрофенил или 3-цианофенил; и
R10 и R11 оба являются водородом.
Особенно предпочтительными соединениями являются 2- трифторметил-4-(3-нитрофенил)-4,6,7,8-тетрагидро-5(1H)-хинолон и 2- трифторметил-4-(3-цианофенил)-4,6,7,8-тетрагидро-5(1H)-хинолон. Было найдено, что оба эти соединения проявляют удивительно высокую активность in vitro и неожиданно высокую селективность в отношении мочевого пузыря in vivo (в живом организме). Было также найдено, что они проявляют химическую стабильность, превосходящую стабильность соответствующих соединений формулы I, в которых R2 является метилом.
Специалистами в соответствующей технике будет оценено, что соединения формулы I содержат асимметрически замещенный углерод и соответственно могут существовать и быть выделены в оптически-активной и рацемической формах. Некоторые соединения могут проявлять полиморфизм. Понятно, что настоящее изобретение включает любую рацемическую, оптически-активную, полиморфическую или стереоизомерную форму, или их смеси, чья форма обладает свойствами, полезными в лечении недержания мочи, специалисту в соответствующей технике хорошо известно, как приготовить оптически-активные формы (например, разделением рацемической формы, синтезом из оптически-активных исходных материалов, хиральным синтезом, или хроматографическим разделением, использующим хиральную неподвижную фазу) и как определить в лечении недержания мочи стандартными методиками, описанными здесь далее.
Соединение формулы I может быть получено способом, включающим способ, известный в химической технике для получения структурно аналогичных соединений. Такие способы для производства 4,6,7,8-тетрагидро-5(1H)-хинолонов формулы I, как определено выше, даны в качестве дальнейшей особенности изобретения и иллюстрируются следующими методиками, в которых значения основных радикалов такие, как приведено выше, если не оговорено особо. Такой способ может быть эффективным в основном:
(a) для соединения формулы I, в которой R3 обозначает водород, при использовании декарбоксилирования соответствующей карбоновой кислоты формулы III, в которой R12 и R13 вместе представляют связь, или декарбоксилирования и дегидратации карбоновой кислоты формулы III, в которой R12 обозначает водород, а R13 обозначает гидрокси;
b) при использовании взаимодействия ненасыщенного кетона формулы IV с соответствующим 1,3-циклогександионом и аммиаком или солью аммония;
c) для соединения формулы I, в которой R2 и R3, взятые вместе, образуют 1,4-бутандиил, при взаимодействии акридиндиона формулы VI с восстанавливающим агентом;
d) для соединения формулы I, в которой R2 и R3, взятые вместе, образуют 1,4-бутандиил, при взаимодействии кетона формулы VII с соответствующим 1,3-циклогександионом и аммиаком или солью аммония;
е) при взаимодействии ендиона (дикетона с одной двойной связью) формулы III с енамином (амином с двойной связью) формулы XIII, в которой обозначает имино группу в присутствии аммиака или соли аммония;
f) для соединения формулы I, в которой R3 обозначает этаноил или циано при взаимодействии соединения формулы XV, в которой R3a обозначает этаноил или циано с соответствующим 1,3-циклогександионом и соответствующим альдегидом формулы R4CHO.
Декарбоксилирование, описанное в (a), удобно выполнять при повышенной температуре, например, в интервале от 50 до 250oC, предпочтительно в интервале от 90 до 120oC. Подходящие для реакции растворители включают спирты, например, метанол или этанол; диметилсульфоксид; ароматические углеводороды, такие как толуол, и простые эфиры, такие как, например, 1,2-диметоксиэтан или диглим. Взаимодействие удобно осуществлять в присутствии кислоты, например, концентрированной серной кислоты или п-толуолсульфоновой кислоты.
Реакцию (b) удобно выполнять при температуре, например, в интервале от 0 до 150oC, предпочтительно при повышенной температуре, например в интервале от 50 до 120oC. Подходящие для реакции растворители включают спирты, например метанол или этанол; диметилсульфоксид; простые эфиры, такие как 1,2-диметоксиэтан или диглим; и карбоновые кислоты, например уксусная кислота. Подходящей солью аммония является, например ацетат аммония.
Реакцию (c) удобно проводить при температуре, например, в интервале от 20 до 80oC, предпочтительно при температуре в интервале от 50 до 80oC. Подходящие растворители включают спирты, например, этанол, пиридин и их смеси. Подходящие восстанавливающие агенты включают, например, боргидрид натрия.
Реакцию (d) удобно выполнять при температуре, например, в интервале от 0 до 150oC, предпочтительно при повышенной температуре, например в интервале от 50 до 120oC. Подходящие растворители для взаимодействия включают спирты, например метанол или этанол; диметилсульфоксид; простые эфиры, такие как 1,2-диметоксиэтан или диглим; и карбоновые кислоты, например уксусная кислота. Подходящей солью аммония является, к примеру, ацетат аммония.
В реакции (е), Z может быть, к примеру, диалкиламиногруппой, такой как диметилимино, или циклической имино группой, такой как пирролидино или пиперидино.
Енамин - исходный продукт формулы XIII удобно получать на месте по реакции соответствующего кетона формулы R2COCH2R3 с соответствующим амином.
Реакцию удобно проводить при температуре в интервале от 20 до 80oC. Подходящие растворители включают спирты, такие как этанол.
Реакцию (f) удобно выполнять при температуре в интервале от 0 до 150oC предпочтительно от 50 до 120oC. Подходящие растворители включают спирты, такие как этанол.
Если нет готовых коммерческих продуктов, необходимые вещества для описанных ниже способов могут быть изготовлены по методикам, выбранным из стандартных органических химических методик, методикам, являющимся аналогами синтезов известных структурно подобных соединений, или по методикам, аналогичным вышеописанным методикам или методикам, описанным в примерах.
Промежуточный продукт формулы III может быть приготовлен взаимодействием ацетоуксусного эфира формулы X, в котором OR2 является спиртовым остатком, таким, например, как 2-цианоэтокси или этокси с альдегидом формулы R4CHO и соответствующим 1,3-циклогександионом, что дает сложный эфир формулы VIII, как показано в схеме 1, приведенной после формул. Гидролиз сложного эфира, например, обработкой водной гидроокисью натрия в 1,2-диметоксиэтане дает кислоту формулы III. В некоторых случаях может быть получен сложный эфир формулы VIIIa вместо сложного эфира формулы VIII, и он может быть гидролизован, что дает соединение формулы IIIa. Такая методика иллюстрируется далее в примере 15.
Промежуточный продукт формулы IV может быть приготовлен взаимодействием соответствующего бензальдегида формулы R4CHO с кетоном формулы XIV. Реакцию удобно проводить в присутствии основания, например гидроокиси натрия.
Промежуточный акридиндион формулы VI может быть приготовлен, как показано в схеме II, приведенной после формул, взаимодействием соответствующего альдегида формулы R4CHO или ацеталя, или его полуацеталя с аммиаком или солью аммония (такой, как ацетат аммония) и соответствующего 1,3-циклогександиона. Реакцию выполняют при температуре в интервале от 0 до 150oC, предпочтительно при повышенной температуре, например в интервале от 50 до 120oC. Подходящие для реакции растворители включают спирты, например метанол или этанол; диметилсульфоксид; простые эфиры, такие как 1,2-диметоксиэтан или диглим; и карбоновые кислоты, например уксусную кислоту.
Как показано в cхеме II, промежуточный акридиндион формулы VI может быть получен взаимодействием соединения формулы IX с альдегидом формулы R4CHO или его ацеталем, или полуацеталем, или его реакционноспособным производным. Реакция выполняется при температуре в интервале от 0 до 150oC, предпочтительно при повышенной температуре, например в интервале от 50 до 120oC. Подходящие для реакции растворители включают спирты, например метанол или этанол; диметилсульфоксид; простые эфиры, такие как 1,2-диметокси этан или диглим; и карбоновые кислоты, например уксусную кислоту.
Промежуточный продукт формулы VII может быть приготовлен конденсацией 1-морфолиноциклогекcена и альдегида формулы R4CHO с последующим гидролизом как показано в cхеме III, приведенной после формул.
Промежуточный продукт формулы XII может быть приготовлен взаимодействием альдегида формулы R4CHO с соответствующим 1,3- циклогександионом. Реакция наиболее эффективна при температуре в интервале от 0 до 80oC, предпочтительно от 40 до 50oC. Подходящие для такой реакции растворители включают спирты, такие как этанол.
Фармацевтически приемлемые соли могут получены использованием стандартных методик, хорошо известных в соответствующей технике эксперимента, например взаимодействием соединения формулы 1 c подходящей кислотой или основанием, дающими физиологически приемлемый противоион.
При использовании для лечения недержания мочи соединение формулы I обычно вводится в виде соответствующей фармацевтической композиции, которая включает соединение формулы I, определенное ранее с фармацевтически приемлемым растворителем или носителем, композиция является адаптированной для конкретного выбранного способа введения. Такие композиции являются предметом изобретения.
Поэтому согласно иному аспекту изобретение обеспечивает фармацевтические композиции, которые включают соединение формулы I или его фармацевтические приемлемую соль, как определено выше, и фармацевтически приемлемый растворитель или носитель.
Композиции могут быть получены применением oбычных методик и эксципиентов и связывающих веществ и могут быть представлены в разнообразных дозированных формах. Например, они могут быть в форме таблеток, капсул, растворов или суспензий для перорального введения; в форме суппозиториев для ректального введения; в форме стерильных растворов или суспензий для введения путем внутривенной, интравентрикулярной, подкожной или внутримышечной инъекции или вливанием; или в форме пластыря для транcдермального применения.
Далее изобретение дает способ лечения недержания мочи, включающий введение млекопитающему, нуждающемуся в такой обработке, эффективного количества соединения формулы I, как определено выше, или его фармацевтически приемлемой соли.
Обработка, использующая соединение согласно изобретению, может быть лечебной или терапевтической, производимой введением соединения в начальной стадии или при развитом недержании мочи у пациента. Обработка может также быть профилактической или перспективной, производимой введением соединения в ожидании возможного развития недержания мочи, например у пациентов, которые в прошлом страдали недержанием.
Согласно дальнейшему аспекту изобретение дает применение соединения формулы I, определенной выше, в производстве лекарственного препарата для лечения недержания мочи. Поскольку соединения согласно изобретению действуют таким образом, что открывают калиевые каналы клеток, они могут также быть полезными в качестве терапевтических средств при лечении других состояний или болезней, при которых действие терапевтического средства, которое открывает калиевые каналы, является желательным или известно, что оно обеспечивает улучшение. Такие состояния или болезни включают гипертонию, астму, заболевание периферических сосудов, недостаточность правых отделов сердца, застойную сердечную недостаточность, ангину, ишемическую болезнь сердца, заболевание сосудов головного мозга, глаукому, почечную колику, нарушения, связанные с почечными камнями, синдром раздраженной толстой кишки (слизистый колит), форму мужской гнездовой плешивости, преждевременные роды, и пептическую язву.
Доза соединения формулы I, которая вводится, будет обязательно варьироваться в соответствии с принципами, хорошо известными в соответствующей технике с учетом способа введения, тяжести состояния недержания, размеров и возраста пациента. В основном соединение формулы I вводится теплокровному животному (такому как человек) таким образом, что эффективная доза является общепринятой, обычно суточная доза выше 0,005, например в интервале приблизительно от 0.01 до 10 мг/кг веса тела. Предпочтительно в этом дозовом интервале соединение вводится перорально.
Специалистам в соответствующей технике должно быть понятно, что соединение формулы I может вводиться совместно с другими терапевтическими или профилактическими средствами и/или лекарственными препаратами, которые не являются с ним лекарственно несовместимыми. Не было найдено, чтобы соединения, находящиеся в рамках объема данного изобретения и приложенных пунктов, оказывали какие-либо неблагоприятные побочные действия на животных, используемых в лабораторных испытаниях при многократном превышении эффективной дозы.
Действие соединений формулы 1 в качестве миорелаксантов, полезных как терапевтические средства для лечения недержания мочи, проявляемое в открытии калиевых каналов и гиперполяризации мембранного потенциала в детруссорной гладкой мышце может быть показано использованием соответственно моделированных испытаний in vitro (в лабораторном сосуде), таких как описаны далее. Было найдено, что соединения согласно изобретению являются в этих испытаниях активными при 30 мкМ (микромолей) или менее. При испытании найдено, что для соединений, приведенных здесь в качестве примеров, характерно давать IC50 порядка 30 мкM или ниже.
Например, соединение, описанное в примере 9, дает при испытании IC50, равное 8.11 мкM. "IC50" - хорошо известный термин и означает концентрацию испытуемого соединения, которая вызывает 50%-ное снижение in vitro сокращения ткани мочевого пузыря, описанное в следующем испытании.
Мужских особей морской свинки-альбиноса Hartley (450 - 500 г) умерщвляют с помощью двуокиси углерода, вызывающей асфиксию, и быстро cпускают кровь. Вскрывают нижнюю часть брюшной полости и отделяют мочевой пузырь. Мочевой пузырь очищают от окружающей соединительной ткани и жировой ткани, и часть, расположенную выше отверстий мочеточников, удаляют и промывают в буферном растворе Krebs-Henseleit'L следующего состава (в мМ): N2Cl 118.0, KCl 4.7, MSO4 1.2, KH2PO4 1.2, CaCl2 2.5, NaHCO3 25.0 и d-глюкоза 11.1. Раствор нагревают до 37oC и насыщают смесью 95% O2 и 5% CO2. При энергичном барботировании, раствор должен иметь значение pH, близкое к 7.4.
Купол промытого мочевого пузыря обрезают и отбрасывают, оставшийся мочевой пузырь помещают на марлю в чашку Петри, содержащую буферный раствор. Производят средневентральный продольный разрез ножницами, чтобы открыть мочевой пузырь. Из купола нарезают полоски и край основания отбрасывают. Оставшийся детруссорный средний отдел разрезают на две горизонтальные полоски приблизительно шириной 2.0 мм. Эти две полоски затем делят пополам по среднедорсальному отделу, получая четыре полоски одинакового размера. Таким образом, каждая полоска содержит как дорсальную, так и вентральную области мочевого пузыря.
Два конца каждой отдельной подоски привязывают к стеклянному стержню, служащему основой, и преобразователю силового смещения (модель Grass'L FR03) соответственно с помощью 4 - 0 черной плетеной шелковой шовной нити. Преобразователь подключают к полиграфу (модель Grass'L 7E), который калибруют при 5 м/см, и калибровку проверяют на линейность грузами 5 и 0.5 г. Аналогичные электрические выходящие сигналы полиграфа приобретают вид пиков благодаря преобразующей сигналы системе модулирующего устройства Micro 5000, использующей Biowindon файлы от Acquisition Software, которая работает c операционной системой Microsoft OS/2 на IBM-совместимой PC. Детруссорные полоски на стеклянном стержне укрепляются в 20 мл ванны для тканей и уравновешиваются под предварительной нагрузкой 2 г. Во время следующего от 45 до 60 мин периода уравновешивания ткани промывают свежим буферным раствором с 15-минутным интервалом c нагрузкой, приведенной, если необходимо, перед промыванием, к 2 г. После периода уравновешивания применяется начальная доза 15 мМ KCl (общая концентрация в ванне). Через 10 мин ткань промывают и промывают вдвое больше при 15-мин интервалах с нагрузкой, приведенной к 2 г, перед каждым промыванием.
Когда ткань после последнего промывания релаксируется до устойчивого состояния, снова применяют 15 мМ KCl. Когда миогенная активность ткани достигает устойчивого состояния, c помощью системы, использующей Biowindows файлы от Acquisition, получают основные (нулевые) данные путем 5-мин усреднения миогенных данных, полученных при 32 Гц. Как только нулевая линия получена, экспериментальные соединения вводят определенными дозами кумулятивным способом в половинных инкрементах структурных единиц. Время контакта с каждой дозой составляет 10 мин, причем последнее с 5 мин, являeтся периодом времени, которое требуется для получения ответных дозовых характеристик. Если 30 мМ испытуемого соединения не уничтожают детруссорную механическую активность, тогда вводится 30 мМ хромакалима, вещества, считающегося способным открывать калиевые каналы, чтобы получить максимальный отзыв. Действие соединения при каждой дозе выражается в % максимально ингибирующего отклика, который далее нормализуют по отношению к соответствующему эффекту носителя соединения в качестве контроля. Нормализованный отклик используют затем для получения lC50 релаксирующей активности соединения путем применения методики приведения нелинейной итерактивной кривой Marquardt'L стандартной функции доза-отклик. Способность соединений согласно изобретению открывать калиевые каналы в детруссорной гладкой мышце может быть дополнительно продемонстрирована вторым испытанием in vitro.
Это второе испытание in vitro аналогично описанному выше в отношении подготовки ткани и получения данных. Однако необходимо отметить следующие исключения. В этом втором испытании сокращение детруссорных полос на начальном периоде и после периода уравновешивания осуществляют с помощью 80 мМ KCl вместо 15 мМ KCl (общая концентрация в ванне). Постоянное напряжение в ткани после этой высокой KCl-стимуляции является очевидным, так как чувствительные к напряжению кальциевые каналы делаются открытыми, что разрешает приток кальция в клетки и развитие тонизирующего напряжения. Это напряжение полностью устраняется 300 мкМ папаверина, который поэтому используется для создания максимального отклика в данном испытании. Обычные блокаторы кальциевых каналов, такие как нифедипин, нимодипин, израдипин и верапамил, способны уменьшать напряжение и снижать миогенную активность детруссорных полосок морской свинки в обоих испытаниях путем воздействия их блокирующей активности на кальциевые каналы. Однако всевышеупомянутые блокаторы кальциевых каналов более сильно действуют во втором испытании, когда используют 80 мкМ KCl, чем в первом испытании, где используют 15 мкМ KCl. Напротив, хотя хромакалим, вещество предположительно открывающее калиевые каналы, обладает сильнодействующей релаксантной активностью в первом испытании с IC50 в интервале от 0.6 до 0.9 мкМ, он проявляет незначительную релаксантную активность во втором испытании при таких высоких концентрациях, как 30 мкМ. Таким образом, показатели более высокой релаксантной активности в первом испытании по сравнению со вторым для соединений согласно изобретению указывают на то, что соединения действуют как вещества, открывающие калиевые каналы.
Способность соединений согласно изобретению воздействовать в качестве веществ, открывающих калиевые каналы, на ткань мочевого пузыря может быть дополнительно продемонстрирована путем стандартного испытания, которое измеряет воздействие испытуемого соединения на скорость истечения рубидия из ткани.
Специалистами в соответствующей технике будет дополнительно оценен тот факт, что эффективность соединений согласно изобретению может быть продемонстрирована стандартными испытаниями in vivo. Ниже приведено описание такого стандартного испытания. Самцы крыс Wistar, весящие 450 - 550 г, подвергнуты анастезии 20 мг/кг i.p. (внутрибрюшинно) Нембутала и 80 мг/кг i.p. Кетамина. Трахел катетеризируется для препятствия обструкции дыхательных путей. Температурa тела поддерживается с помощью грелки-подушки. Артериальное кровяное давление и частота сердечных сокращений могут быть измерены датчиком давления, соединенным с полиэтиленовой трубкой (РЕ50), которая может быть вставлена в правую сонную артерию. Правая яремная вена катетеризируется для введения лекарства. Производят срединное чревосечение мочевого пузыря и выпускают мочу, применяя слабое надавливание рукой. Катетер (РЕ50) вводят через верхушку cвода мочевого пузыря приблизительно на 3 - 4 мм в его полость и привязывают шовной нитью (4 - 0 шелк) для предотвращения утечки. Катетер мочевого пузыря соединяют с датчиком давления для измерения давления мочевого пузыря. Мочевой пузырь помещают затем обратно в брюшную полость и надрез закрывают швом за исключением того места, где катетер выходит из полости. Мочевому пузырю дают уравновеситься в течение приблизительно 15 мин. После периода уравновешивания крысам проводят вливание солевого раствора непосредственно в мочевой пузырь при скорости 0.05 мл/мин за чистое время эксперимента. Затем регистрируют с помощью приборов давление мочевого пузыря для исходных сокращений мочевого пузыря. После того, как начинаются сокращения, животному дают стабилизировать свой паттерн сокращений приблизительно от 30 до 45 мин перед введением лекарства. Испытуемые соединения вводятся i. v. (внутривенно). Эффективность испытуемого соединения измеряется в сравнении с известным в литературе лекарством хромакалим (Smithkline-Beecham), которое вводится i.v. в дозовом интервале от 0.05 до 0.5 мг/кг.
Приведенное выше испытание in vivo дает возможность оценки как кровяного давления, так и цистометрической активности испытуемых соединений. Кровяное давление измеряют немедленно после введения лекарства и спустя 5, 15 и 30 мин. Позывы на мочеиспускание вызывают медленным непрерывным введением раствора соли непосредственно в мочевой пузырь. Для каждого соединения приведено среднее превращение (в секундах от контроля) в длительности интервала между сокращениями (время между сокращениями) через приблизительно 20-минутный период.
Далее приведено описание испытания in vivo, которое является добавочным к вышеописанным испытаниям и которое может быть использовано для выяснения, является ли испытуемое соединение активным, и обладает ли испытуемое соединение селективностью в отношении мочевого пузыря без существенного сердечно-сосудистого воздействия, когда оно вводится перорально в дозированной форме. Соединение, описанное в примере 1, является активным и селективным в этом испытании, когда оно вводится перорально в дозированной форме при 3 мг/кг веса тела.
Самцов крыс wistar (400 - 500 г) анестезировали 50 мг/кг Нембутала, i.p. У каждой крысы, брюшная область и передняя и задняя части шеи были выбриты и на кожу нанесены повидов-иод. Для катетеризации сонной артерии левую сонную артерию подвергли воздействию через небольшой вентральный цервикальный надрез. Область воздействия была промыта сильной струей 2% лидоксинового HCl раствора, чтобы ослабить сосуд. Катетер, наполненный 0.9% солевым раствором, ввели приблизительно на 2,4 см в артерию, так чтобы его наконечник находился в дуге аорты. Дистальный конец катетера был завернут наружу в задней части шеи, наполнен гепарином (1000 единиц/мл) и теплоизолирован. Для катетеризации мочевого пузыря произвели срединное чревосечение мочевого пузыря. Троакар пропустили через абдоминальную мышцу на расстояние около 1 см от верхнего края сечения и затем провели подкожной туннелизацией так, чтобы выходил наружу в задней части шеи. Заполненный солевым раствором катетер провели через троaкар. Небольшое отверстие в своде мочевого пузыря создали термокаутером Асси-Тетр. Катетер ввели в мочевой пузырь и закрепили с помощью 4 - 0 шелковой лигатуры. Катетер заполнили солевым раствором и отметили раскрытое состояние сосуда. Наружный конец катетера теплоизолировали, чтобы предупредить утечку мочи. На абдоминальную мышцу и кожу наложили швы. Оба катетера были пропущены через иглу из нержавеющей стали с фиксирующим крючком (Instech), которую затем пришили к подкожной мышце в точке выворачивания наружу. На кожу наложили шов через крючок. Животным дали выздороветь после анестезии.
Через 24 - 48 ч после оперативного вмешательства каждую крысу поместили в камеру для метаболизма и подсоединили с помощью фиксирующего крючка к Instech пружинному ограничителю и поворотной системе, чтобы защитить катетеры от повреждения и позволить животному свободно двигаться в камере. Каротидный катетер подсоединили к Gould P23xL датчику давления для измерения давления крови. Катетер мочевого пузыря подсоединили к насосу для вливания раствора соли и к датчику давления посредством РЕ 50 труб и 4-ходового запорного крана. Весы с верхней загрузкой со сборным стаканчиком поместили под камерой для измерения выделенной мочи.
Крыс взвесили, ввели перорально стимулирующую дозу (введена дозирующей иглой, но не препятствующей вытеканию), и начали и продолжали во время эксперимента введение раствора соли. Изменения кровяного давления, частоты сердечного сокращения, внутрипузырного давления и выделения мочи регистрировали либо на полиграфе Grass'a, либо Gould TA4000 регистрирующей системе. Животным дали уравновеситься до тех пор, пока позывы на мочеиспускание стали устойчивыми (приблизительно 45 - 90 мин). С этого момента отметили нулевой уровень каждого экспериментального параметра и крысам ввели с помощью перорального желудочного зонда соответствующую дозу соединения (в 75% PEG 400-солевом носителе) в таких концентрациях, что объем составлял 1 мг/кг веса тела. Воздействие соединений на экспериментальные параметры продолжалось в течение 5 ч после введения.
Экспериментальные результаты для интервалов между сокращениями и для частоты сердечных сокращений выражены в виде ±S.E.M. (Стандартная ошибка измерения) % изменения от нулевого уровня для каждого животного, служащего его индивидуальным контролем. МАР выражается как ±мм Hg изменения от нулевого уровня.
Соединения согласно изобретению являются активными в одном или более вышеописанных испытаний.
Далее соединение иллюстрируется следующими неограничивающими примерами в которых, если не оговорено особо:
i) температура дана в градусах Цельсия (oC); операции проводились при комнатной температуре или температуре окружающей среды, т.е. при температуре в интервале 18 - 25oC;
ii) органические растворители высушивали над безводным сульфатом магния; упаривание растворителя проводили, используя роторный испаритель при пониженном давлении (600 - 4000 Па, 4.5 - 30 мм Hg) с температурой ванны при 60oC;
iii) хроматография означает "мгновенную хроматографию; обращенно-фазовая хроматография означает мгновенную хроматографию на носителе с октадецилсилановым (ODS) слоем, имеющeм диаметр частиц 32 - 74 мкм, известном как "PReP - 40 - ODS" (Ard 731740-100 from Bodman Chemicals, Aston, PA, USA). Тонкослойная хроматография (TLC) выполнена на силикагелевых пластинах;
(iv) обычно за ходом реакции следили с помощью и реакционного времени, данных только для иллюстрации;
v) температуры плавления не корректировались, и (dec) означает разложение; температуры плавления даны в том виде, как получены для продуктов, приготовленных по описанной методике; полимoрфизм может приводить к выделению продуктов с различными температурами плавления в разных приготовлениях;
vi) конечные продукты имели удовлетворительный протонный ядерный магнитный резонансный спектр (NMR); (ПМР).
vii) выходы даны только для иллюстрации и не являются необходимыми для тех выходов, которые могут быть получены при тщательном выполнении способа; приготовление повторяли, если требовалось большое количество продукта;
viii) когда приведены, ЯМР данные выражены в форме дельта значений для основных используемых для характеристики протонов, приведенных в миллионных долях (ppm) относительно тетраметилсилана (TMC) как внутреннего стандарта, определенные при 300 МГц использованием пердейтеротиметил силоксида (DMCO-d6) в качестве растворителя; использованы обычные обозначения форм сигналов:
q - квартет,
s - синглет,
m - мультиплет,
d - дублет,
t - триплет,
dd - двойной дублет
постоянная связи (J) дана в Гц; AГ означает ароматический протон, когда делается такая интерпретация спектра;
(ix) химические символы имеют их обычное значение; использовали единицы и символы системы СИ;
(x) пониженное давление приведено как абсолютное давление в Па (P2); повышенное давление приведено как давление манометра в барах;
(xi) приведены объемные соотношения растворителей в объем:объем (об./об. ) терминах; и
(xii) масс-спектр (MC) снят c электронной энергией 70 электрон-вольт в модах электронного соударения (E1), иcпользующих датчик непосредственного обнаружения; где указанная ионизация являлась действием химической ионизации (C1) или бомбардировки быстрыми атомами (FAB); значения для m/z приведены; обычно указаны только ионы, которые означают исходную массу.
Пример 1. 2-Метил-4-(3-нитрофенил)-4,6,7,8-тетригидро- 5(1H)-хинолон.
2-Метил-4-(3-нитрофенил)-5-оксо-1,4,5,6,7,6- гексагидро-3-хиноликарбоновую кислоту (4.66 г) суспендировали в этоноле (200 мл) с концентрированной серной кислотой (0.5 мл). Смесь нагревали до кипения с обратным холодильником в течение 5 ч, за это время все твердые продукты перешли в раствор. Растворитель упарили, и остаток распределили между 2 н водной гидроокисью натрия и этилацетатом. Водную порцию экстрагировали этилацетатом. Объединенные экстракты промыли (вода, физиологический раствор), высушили, и упарили, получив остаток, который очистили хроматографией, элюируя этилацетатом с целью получения названного в заглавии соединения в виде бледно-желтого твердого продукта (1.6 г); Т.пл. 183 - 185oC: 284 (М); 250 МГц ЯМР: 1.76 (s, 3, CH), 187 (m, 2, CH), 2.16 (m, 2, CH), 2.46 (m, 2, CH2), 4.58 (d, 1, J = 4,9 CH), 4.64 (d, 1, J = 4,9, CH), 7.53 (t, 1, 7.6, АГ), 7.62 (d, 1, J = 7.6, АГ), 7.96 (s, 1, АГ), 7.98 (m, 1, АГ), 8.80 (s, 1, NH). Анализ для C16H16N2O3:
Рассчитано,%: C 67,59; H 5.67; N 9.35
Найдено,%: C 67,44; H 5,75; N 9.56
Исходную 2-метил-4-(3-нитрофенил)-5-оксо-1,4,5,6,7,8- гексагидро-3-хинолинкарбоновую кислоту приготовили как описано далее.
а. 2-Цианоэтил 2-метил-4-(3-нитрофенил)-5-оксо-1,4,5,6,7,8- гексагидро-3-хинолинкарбоксилат. 2-Цианоэтил ацетоацетат (4.27 г), 3-нитробензальдегид (4.24 г), 1,3-циклогександион (3.09 г) и ацетат аммония (4.26 г) смешали в этаноле (240 мл) и нагревали при температуре кипения 18 ч. Растворитель упарили и остаток хроматографировали, элюируя этилацетатом, что дало хинолинкарбоксилат в виде желтого твердого продукта (7.42 г); МС: 381(М); 250 МГц ЯМР: 1.75-2.00 (m, 2, CH2), 2.22 (m, 2, CH2), 2.35 (s, 3, CH3), 2.81 (m, 2, CH2), 3.34 (m, 2, CH2), 4.12 (t, 2, J = 5.9, CH2), 5.01 (s, 1, CH), 7.51 (m, 1, АГ), 7.84 (dd, 1; J = 7,7, 1.1 АГ): 7.97 (m, 1, АГ) 7.99 (s, 1, АГ), 9.43 (s, 1, NH).
b. 2-Метил-4-(3- нитрофенил)-5-оксо-1,4,5,6,7,8-гексагидро-3-хинолинкарбоновая кислота. К суспензии 2-цианоэтид-2-метид-4-(3-нитрофенил)-5-оксо- 1,4,5,6,7,8-гексагидро-3-хинолинкарбоксилата (0.47 г) в этиленгликoль диметиловом эфире (2 мл) добавили 1 н гидроокись натрия (4 мл). Смесь перемешивали 20 ч, за это время весь твердый продукт перешел в раствор. Смесь разбавили водой, промыли этил ацетатом, и подкислили добавлением концентрированной соляной кислоты. Образовавшийся осадок собрали вакуумной фильтрацией и высушили, получив кислоту в виде желтого твердого вещества (0.31 г): Т.пл. 253 - 255oC; MC: 328(М); ЯМР: 1.63-2.00 (m, 2, CH), 2.20 (m, 2, CH), 2.31 (s, 3, CH3), 2.49 (m, 2, CH2), 5.01 (s, 1, CH), 7.49-7.61 (m, 2, АГ), 7.97 (m, 2, АГ), 9.24 (s, 1, NH). Анализ для C17H16N2O5:
Рассчитано,%: C 62.19; H 4.91; N 8.53
Найдено,%: C 62.16; H 5.02; N 8.33
Примеры 2 - 4.
За исключением особо оговоренных случаев, следующие 4-арил-2-метил-4,6,7,8-тетрагидро-5(1H)хинолоны формулы I, в которой R4 обозначает 4-арил радикал, приготовлены из соответствующих 4-арил-8-метил-5-оксо-1,4,5,6,7,8-гексагидро-3- хинолинкарбоновых кислот формулы III с использованием методик, аналогичных описанной в примере I, с теми изменениями, что указаны.
Пример 2. R4 = 3-цианофенил; продукт примера 2.b (1.10 г) суcпендировали в диэтилен гликоле (10 мл) и нагревали при 100oC в течение 25 мин. Реакционную смесь очистили хроматографически, элюируя этилацетатом, что дало названное в заглавии соединение в виде бледно-желтого твердого вещества (0.52 г, 56); Т.пл. 163 - 165oC; MC: 264(М); ЯМР: 1.74 (s, 3, CH3), 1.86 (m, 2, CH2), 2.16 (m, 2, CH2), 2.44 (m, 2, CH), 4.47 (d, 1, J = 4,8, CH), 4.60 (d, 1, J = 4,2, CH), 7.51 (m, 4, АГ), 8.55 (s, 1н, NH). Анализ для C17H16N2O•0,15H2O:
Рассчитано,%: C 76,47; H 6.14; N 10.49
Найдено,%: C 76.36; H 6.17; N 10.45
Пример 3. R4 = 3-бром-4-фторфенил. Хроматография дала твердый продукт, который растерли с диэтиловым эфиром, профильтровали и высушили, получив названное в заглавии соединение в виде желтого твердого продукта (1.52 г); Т.пл. 177 - 179oC; MC: 336 (М); 400 МГц ЯМР: 1.74 (s, 3, CH3), 1.75-1.90 (m, 2, CH2), 2.14 (m, 2, CH2), 2.43 (m, 2, CH2), 4.41 (d, 1, J = 4.7, CH), 4.58 (d, 1, J = 4,8, CH), 7.18 (m, 2, АГ), 7.37 (dd, 1, J = 6.9, 2.0, АГ), 8.52 (s, 1, NH). Анализ для C16H15BrFNO:
Рассчитано,%: C 57.16; H 4.50; N 4.17
Найдено,%: C 57.11; H 4.61; N 4.07
Пример 4. R4 = 4-фторофенил; реакционную смесь упарили и остаток растворили в водном бикарбонате натрия и экстрагировали этилацетатом. Объединенные органические экстракты промыли (вода, физиологический раствор) и упарили. Остаток хроматографировали, элюируя этилацетатом, получив масло, которое затвердело при стоянии. Твердый продукт растерли с диэтиловым эфиром, содержащим несколько капель этилацетата. Фильтрация и высушивание дали названное в заглавии соединение в виде бледно-желтого твердого продукта (0.17 г); Т. пл. 170 - 173oC; MC: 257 (М); 250 МГц ЯМР: 1.72 (s, 3, CH3), 1.83 (m, 2, CH2), 2,42 (m, 2, CH2), 4.38 (d, 1, J = 4,8, CH), 4.59 (d, 1, J = 4.9, CH), 7.04 (M, 2, АГ), 7.14 (m, 2, АГ), 8.45 (S, 1, NH). Анализ для C16H16FNO:
Рассчитано,%: C 74.69; H 6.27; N 5.44
Найдено,%: C 74.45; H 6.32; N 5.34
Исходные 4-арил-2-метил-5-оксо-1,4,5,6,7,8-гексалидро-3- хинолинкарбоновые кислоты для примеров 2 и 3 приготовлены как описано далее.
За исключением особо оговоренных случаев, следующие 4-арил-2- метил-5-оксо-1,4,5,6,7,8-гексагидро-3-хинолинкарбоксилаты формулы VIII, в которой R2 обозначает 2-цианоэтил и R4 обозначает 4-арил радикал, приготовлены из соответствующих производных бензальдегида формулы R4CHO использованием методик, аналогичных описанной в примере 1а, элюируя указанным хроматографическим растворителем.
2а. R4 = 3-цианофенил; хроматографический растворитель: этилацетат; получен в виде бледно-желтого твердого вещества; MC: 361(М); 250 МГц ЯМР: 1.70-2.00 (m, 2, CH2), 2.23 (m, 2, CH2), 2.35 (s, 3, CH3), 2.52 (m, 2, CH2), 2.82 (m, 2, CH), 4.13 (t, 2, J = 5,7, CH), 4.94 (s, 1, CH), 7.44 (m, 1, АГ), 7.54 (m, 3, АГ), 9.37 (s, 1, NH).
3a. R4 = 3-бром-4-фторфенил; хроматографический растворитель: этилацетат; получен в виде желтого твердого продукта (77%), MC; 433 (М); ЯМР: 1.67-2.00 (m, 2, CH2), 2.20 (m, 2, CH2), 2.32 (m, 3, CH3), 2.50 (s, 2, CH2), 2,82 (m, 2, CH2), 4.13 (t, 2, J = 5,9, CH2), 4.87 (s, 1, CH), 7.18 (m, 2, АГ), 7.38 (d, 1, J = 6,1, АГ), 9.33 (s, 1, NH).
4a. R4 = 4-фторфенил; хроматографический растворитель: этилацетат; получен в виде бледно-желтого вещества (86%); MC, 354 (М) 250 МГц ЯМР: 1,63-2.00 (m, 2, CH2), 2.18 (m, 2, CH2), 2.29 (s, 3, CH3), 2.50 (m, 2, CH2) 2.81 (m, 2, CH2) 4.11 (t, 2, J = 5.4, CH2), 4.87 (s, 1, CH), 6.97 (t, 2, J = 8,8, АГ), 7.18 (m, 2, АГ), 9.25 (s, 1, NH).
Если не оговорено особо, следующие 4-арил-2-метил-5-оксо-1,4,5,6,7,8-гексагидро-3-хинолинкарбоновые кислоты формулы III, в которой R4 обозначает 4-арил радикал, приготовлены гидролизом соответствующих 2-цианоэтил сложных эфиров формулы VIII, описанной выше, использованием методик аналогичных описанной в примере 1b.
2b. R4 = 3-цианоэтил; реакционную смесь вылили в ледяную воду и подкислили 1 н соляной кислотой. Образовавшийся осадок собрали вакуумной фильтрацией и высушили, получив названное в заглавии соединение в виде бледно-желтого твердого продукта (95%); МC: 308 (М); ЯМР: 1.62-1.97 (m, 2, CH2), 2.19 (m, 2, CH2), 2.30 (s, 3, CH3), 2.49 (m ,2, CH, 4.92 (s, 1, CH), 7.47 (m, 3, АГ), 7.56 (m, 1, АГ), 9.17 (s, 1, NH), 11.80 (уширен, s, 1, CO2H).
3b. R4 = 3-бром-4-фторфения; реакционную смесь подкислили 6 и соляной кислотой. Образовавшийся осадок собрали вакуумной фильтрацией и высушили, получив названное в заглавии соединение в виде не совсем белого твердого вещества (69%); MC: 380 (М); 250 МГц ЯМР: 1.65-200 (m, 2, CH2), 2.21 (m 2, CH2), 2.30 (s, 3, CH3), 2.51 (m , 2, CH2), 4.88 (s, 1, CH), 7.20 (m, 1, АГ), 7.36 (d, 1, J = 6.8, АГ), 9.17 (s, 1, NH), 11.81 (уширен, s, 1, CO2H).
4b. R4 = 4-фторфенил, получен в виде белого твердого вещества (94%); Т. пл. 235 - 237oC (разл.); MC: 301 (М) 250 МГц ЯМР: 1.63-2.00 (m, 2, CH2), 2.19 (m, 2, CH2), 2.28 (s, 3, CH3), 2.47 (m, 2, CH2), 4.88 (s, 1, CH), 7.00 (m, 2, АГ), 7.15 (m, 2, АГ), 9.09 (s, 1, NH), 11.73 (уширен. s, 1, CO2H). Анализ для C17H16FNO3:
Рассчитано,%: C 67.76; H 5.35; N 4.65
Найдено,%: C 67.63; H 5.35; N 4.61
Пример 5. 2-Метил-4-фенил-4,6,7,8-тетрагидро-5(1H)-хинолон.
Транc-4-фенил-3-бутен-2-он (1.63 г), 1,3-циклогександион (1.30 г), и ацетат аммония (1.82 г) смешали в этоноле (100 мл) и нагревали до температуры кипения с обратным холодильником в течение 5 ч. Растворитель упарили и остаток хроматографировали, элюируя смесью этилацетат:гексан (2:1). Порцию образовавшегося твердого продукта растерли с горячим этилацетатом, профильтровали, и высушили, получив тетрагидрохинолон в виде бледно-желтого твердого вещества (0.53 г); Т.пл. 227 - 229oC; 239 (М); 250 МГц ЯМР: 1.72 (s, 3, CH3), 1.73-1.95 (m, 2, CH2), 2.14 (m, 2, CH2), 2.43 (m, 2, CH2), 4.37 (d, 1, J = 4,8, CH), 4.60 (d, 1, J = 5,0, CH), 7.15 (m, 5, АГ), 8.42 (s, 1, NH). Анализ для C16H17NO:
Рассчитано,%: C 80,30; H 7.16; N 5,85
Найдено,%: C 80.16; H 7.18; N 5.76
Пример 6. 2-Метил-4-(3,4-метилендиоксифенил)-4,6,7,8- тетрагидро-5(1H)-хинолон.
Используя методику аналогичную описанной в Примере 5, но заменяя 4-(3,4-Метилендиоксифенил)-3-бутен-2-он на транс-4-фенил-3-бутен-2он, получили названное в заглавии соединение. Хроматография при элюировании этилацетатом дала желтый твердый продукт. Вторичная хроматография при элюировании ацетонитрил:дихлорметаном (20:80) дала твердое вещество, которое при растирании с диэтиловым эфиром дало тетрагидрохинолон (13%) в виде бледно-желтого твердого продукта; Т.пл. 217 - 220oC; MC: 283 (М), 250 МГц ЯМР: 1.72(s, 3, CH3), 1.84 (m, 2, CH2), 2.11 (m, 2, CH2), 2.42 (m, 2, CH2), 4.30 (d, 1, J = 4,7, CH), 4.57 (d, 1, J = 4.2, CH), 5.91 (s, 2, CH2), 6.58 (m, 1, АГ), 6.65 (d, 1, J = 1,3, АГ), 6.73 (d, 1, J = 8,0, АГ), 8.42 (s, 1, NH). Анализ для C17H17NO:
Рассчитано,%: C 71.62; H 6.08; N 4.91
Найдено,%: C 71,59; H 6,16; N 5.25
Пример 7. 4-(4-Хлорфенил)-2-метил-4,6,7, в-тетрагидро-5(1H)-хинолон.
4-(4-Хлорфенил)-3-бутен-2-он (2.83 г), 1.3-циклогександион, (1.81 г), и ацетат аммония (2.60 г) смешали в этаноле (125 мл) и нагревали при температуре кипения с обратным холодильником в течение 5.5 ч. Растворитель упарили, и остаток растворили в воде и экстрагировали этилацетатом. Объединенные органические экстракты промыли (вода, физиологический раствор), высушили, и упарили, что дало остаток, который хроматографировали, элюируя смесью гексан-этилацетат (1:1), получив названное в заглавии соединение в виде бледно-желтого твердого вещества (1.46 г); Т.пл. 184 - 187oC; MC: 273 (М); 1.73 (s, 3, CH3), 1.75-1.98 (m, 2, CH2), 2.15 (m, 2, CH2), 2.43 (t, 2, J = 6.0, CH2), 4.39 (d, 1, J = 4.7, OH), 4.58 (d, 1, J = 4,8, CH), 7.14 (d, 2, J = 8,4, АГ), 7.26 (d, 2, J = 8.4, АГ), 8.48 (s, 1, NH). Анализ для C16H16ClNO:
Рассчитано,%: C 70.20; H 5.89; N 5.12
Найдено,%: C 70.21; H 5.67; N 5.02
Пример 8. 2-Метил-4-(3-трифторметилфенил)-4,6,7,8-тетрагидро- 5(1H)-хинолон.
Смесь 4-(3-трифторметилфенил)-3-бутен-2-она (4.9 г), 1,3-циклогександиона (2.60 г), ацетата аммония (2.65 г) и 75 мл этанола нагревали при температуре кипения с обратным холодильником в течение 8 ч и затем охладили до комнатной температуры. Растворитель упарили и остаток распределили между водой и этилацетатом. Органический слой высушили, отфильтровали, и упарили, получив названное в заглавии соединение (2.8 г) в виде не совсем белого твердого продукта; Т.пл. 184 - 185oC; ЯМР: 1.74 (s, 3, CH3), 1.87-1.98 (m, 2, CH2), 2.13-2.19 (m, 2, CH2), 2.42-2.49 (m, 2, CH2), 4.50 (d, 1, J=4.9 CH), 4.63 (d, 1, J=4.9, CH), 7.45(s, 4, АГ), 8.53 (s, 1, NH); (Cl, CH4), MC: m/z=308(M+1). Анализ для C17H16F3NO:
Рассчитано,%: C 66.44; H 5.25; N 4.56
Найдено,%: C 65.90; H 5.33; N 4.43
Пример 9. 2-Метил-4-(4-трифторметилфенил)-4,6,7,8-тетрагидро-5(1H)-хинолон.
Смесь 4-(4-трифторметилфенил)-3-бутен-2-она (4.9 г), 1,3-циклогександиона (2.68 г), ацетата аммония (2.65 г) и 76 мл этанола нагревали при температуре кипения с обратным холодильником в течение 8 ч, затем охладили до комнатной температуры. Растворитель упарили и остаток распределили между водой и этилацетатом. Органический слой высушили, отфильтровали и упарили, получив желтое масло. Хроматография с использованием смеси гексан:этилацетат (1: 1) в качестве элюента, и перекристаллизация из смеси толуол:гексан дали названное в заглавии соединение в виде желтого твердого продукта (4.0 г); Т. пл. 116 - 118oC; ЯМР: 1.73 (s, 3, CH3), 1.87-1.98 (m, 2, CH2), 2.14-2.16 (m, 2, CH2), 2.45-2.49 (m, 2, CH2), 4.49 (d, 1, J=4,9, CH), 4.60(d, 1, J=4,9 CH), 7.35 (d, 2, J=8,0, Аг), 7.57 (d, 2, J=8,0, Аг), 8.54 (s, 1, NH), (CI, CH4) MC: m/z 308 (М+1). Анализ для C17H16F3NO:
Рассчитано,%: C 66,44; H 5.25; N 4.56
Найдено,%: C 66.72; H 5.34; N 4.41
Пример 10. 4-(3-Хлорфенил)-2-метил-4,6,7,8-тетрагидро-5(1H)хинолон.
Смесь 4-(4-хлорфенил)-3-бутен-2-она (5.0 г), 1,3- циклогександиона (3.24 г), ацетата аммония (3.20 г) и 90 мл этанола нагревали при температуре кипения с обратным холодильником в течение ночи и затем охладили до комнатной температуры. Растворитель упарили и остаток распределили между водой и этил ацетатом. Органический слой высушили, отфильтровали, и упарили, получив желтый твердый продукт. Перекристаллизация из этанола дала названное в заглавии соединение в виде желтого твердого вещества (3.0 г); T.пл. 201 - 203oC; ЯМР: 1.73 (s, 3, CH3), 1.87-1.98 (m, 2, CH2), 2.13-2.17 (m, 2, CH2), 2.43-2.49 (m, 2, CH2), 4.40 (d, 1, J=4,9, CH) 4.60 (d, 1, J=4.9, CH), 7.09-7.25 (m, 4, АГ), 8.50 (s, 1, NH); (Cl, CH4) MC: m/z = 274 (M+E). Анализ для C16H16ClNO:
Рассчитано,%: C 69.51; H 5.94; N 5.07
Найдено,%: C 69.47; H 6.03; N 4.91
Пример 11. 2-Meтил-4-(4-мeтилфeнил)-4,6,7,8-тетрагидpo-5(1H)-хинолон.
Смесь 4-(4-метилфенил)-3-бутен-2-она (5.0 г), 1.3-циклогександиона (3.66 г), ацетата аммония (3.61 г) и 100 мл этанола нагревали при температуре кипения с обратным холодильником в течение ночи, и затем охладили до комнатной температуры. Растворитель упарили и остаток распределили между водой и этилацетатом. Органический слой высушили, отфильтровали и упарили, получив янтарное масло. Хроматография с использованием смеси гексан:этилацетат (1:1) в качестве элюента, и растирание с диэтиловым эфиром дало названное в заглавии соединение в виде не совсем белого твердого вещества (2.0 г); Т.пл. 196 - 198oC; ЯМР: 1.73 (s, 3, CH3), 1.87-1.98 (m, 2, CH2), 2.14-2.16 (m, 2, CH2), 2.22 (s, 3, CH3), 2.43-2.49 (m, 2, CH2), 4.34 (d, 1, CH), 4.57 (d, 1, CH), 7.01 (s, 4, АГ), 8.40 (s, 1, NH), (Cl, CH4) MC: m/z=254(M+1). Анализ для C17H19NO:
Рассчитано,%: C 80.60; H 7.56; N 5.53
Найдено,%: C 80.24; H 7.68; N 5.36
Пример 12. 9-(3-Нитрофенил)-3,4,5,6,7,8,9,10-октагидpo-1(2H)-акридинон.
Смесь 2-(3-нитрофенилметилен)циклогексанона (4.69 г), 1,3-циклогександиона (2.28 г), ацетата аммония (2.35 г) и 90 мл этанола перемешивали при температуре кипения в атмосфере азота в течение 48 ч. Смесь охладили в ледяной бане и оранжевые кристаллы собрали фильтрацией. Продукт хроматографировали с дихлорметаном, этиловым эфиром:дихлорметаном (2:98) и этиловым эфиром: дихлорметаном (10: 90) в качестве элюентов. Растворитель упарили и остаток перекристаллизовали из смеси толуол/гексан, получив названное в заглавии соединение в виде желтых кристаллов (0.50 г); Т. пл. 218 - 221oC: ЯМР (CDCl3): 1.57-1.59 (m, 4, CH2) 1.74-1.80 (m, 2, CH2) 1.88-2.01 (m, 2, CH2) 2.14 (уширен. δ 2, CH2) 2.28-2.36 (m, 2, CH2) 2.42-2.46 (m, 2, CH2) 4.50 ( δ , 1, CH) 5.40 (s, 1, NH) 7.38 (t , 1, J=7,8, АГ) 7.71 (d, 1, J=7,6, АГ) 7.98 (q, 1, J=7,3, 1.4, Аг) 8.09 (d, 1, J=1,9, Аг); MC (Cl, CH4) : m/z=325(M+l). Анализ для C19H20N2O3•0.6H20:
Рассчитано,%: C 68.08; H 6.37; N 8.36
Найдено,%: C 68.27; H 6.28; N 8.01
Исходный 2-(3-нитрофенилметилен)циклогексанон приготовили как описано ниже.
1-Морфолино-1-циклогексан (13.20 г) добавили по каплям к перемешиваемому раствору 3-нитробензадвдегида (10.0 г) и 65 мл сухого толуола, в атмосфере азота. После перемешивания при комнатной температуре в течение 5 дней смесь обработали 75 мл 5 н HCl, перемешали дополнительно 15 мин и разделили слои. Водную фазу экстрагировали тремя дополнительными 75 мл порциями толуола, толуольные экстракты высушили (MgSO4), отфильтровали и растворитель упарили. Образовавшееся желтое масло, которое превратилось в зеленоватое при хранении в течение ночи, хроматографировали, используя в качестве элюента метиленхлорид, получив 2-( α -гидрокси-3-нитробензил)циклогексанен (6.35 г) в виде желтого масла; MC: (Cl,CH4): m/z в 250(М+1). Желтое масло растворили в диэтиловом эфире (50 мл) и обработали 50 мл концентрированной соляной кислоты. После перемешивания в течение ночи слои разделили и водную фазу экстрагировали диэтиловым эфиром. Объединенные экстракты высушили (MgSO4), отфильтровали и упарили получив неочищенный 2-(3- нитрофенил)метиленциклогексанон в виде низкоплавких оранжевых кристаллов (4.69 г), который использовали без дальнейшей очистки. ЯМР (CDCl3): 1.79-1.89 (m, 2, CH2), 1.93-2.01 (m, 2, CH2), 2.56-2.60 (m, 2, CH2), 2.82-2.87 (m, 2, CH2), 7.47 (s,1, олефиновый), 7.58 (t, 1, J=7.9, Аг), 7.69 (d, 1, J=7,7, Аг), 8.19 (d, 1, J=8,9, Аг), 8.24 (s, 1, Аг); MC (CI, CH4: m/z=232(M+1).
Пример 13. 9-(3-Цианофенил)-3,4,5,6,7,8,9,10-октагидpo-1(2H)-акридинеон.
Смесь 9-(3-цианофенил)-3,4,5,6,7,9,10- гексагидро-1,8-(2H,5H)-акридиндиона (3,18 г), боргидрида натрия (2.50 г) и 50 мл этанола перемешивали при 70oC в течение ночи. Смесь обработали приблизительно 300 мл воды, перемешивали несколько часов и твердый продукт собрали фильтрацией с отсосом, суспендировали на воронке с небольшим количеством этанола и отсосали досуха на воронке. Твердый продукт объединили с продуктом из предыдущего опыта, и объединенный материал хроматографировали, используя смесь метанол:дихлорметан (2.5: 97,5) в качестве элюента. Растворитель упарили и остаток растворили в 50 мл метанола и приблизительно 20 мл метиленхлорида. Раствор концентрировали на паровой бане до тех пор, пока не начали образовываться кристаллы, обработали равным объемом этилацетата и охладили за ночь. Названное в заглавии соединение получили в виде светло-желтых кристаллов (1.16 г); Т.пл. 246 - 249oC; ЯМР: 1.47 (уширен, s, 2, CH2), 1.65-1.87 (m, 6, CH2), 2.11 (m, 4, CH2), 2.39-2.51 (s, 2, CH2), 4.25 (m, 1, CH), 7.41-7.57 (m, 4, АГ), 8.45 (s, 1, NH); (Cl, CH4), MC: m/z 305(M+1). Анализ для C20H20N2O:
Рассчитано,%: C 78.92; H 6.62; N 9.20
Найдено,%: C 78.64; H 6.65; N 9,08
Промежуточный 9-(3-цианофенил)-3,4,6,7,9,10-гексагидро-1,8- (2H,5H)-акридинон может быть приготовлен по следующей методике.
Перемешиваемую смесь 3-цианобензальдегида (1.48 г), 1,3- циклогександиона (2.53 г) и ацетата аммония (1.24 г) в этаноле (20 мл) нагревали до кипения с обратным холодильником в течение 18 ч. Смесь вылили в воду, и желтый твердый продукт собрали и высушили при пониженном давлении получив названный в заглавии акридинон (3,22 г); Т.пл. 285 - 288oC; ЯМР: 1.80-1.93 (m, 4), 2.19-2.22 (m, 4), 2.50-2.54 (m, 4), 4.91 (s, 1) 7.37-7.42 (m, 1) 7.48-7.54 (m,3) 9.55 (s, 1); MC: m/z=319(M+1). Анализ для C20H18N2O2:
Рассчитано,%: C 75,44; H 5.71; N 8.80
Найдено,%: C 75.27; H 5.71; N 8.80
Найдено,%: C 75.27; H 5.66; N 8.77
Пример 14. 2-Изoбутил-4- фeнил-4,6,7,8-тeтpaгидpo-5(1H)-хинолон. 5-метил-1-фенил-1-гексан-3-он (5.05 г), 1,3- циклогександиона (3.31 г) и ацетата аммония (4.91 г) объединили в 200 мл этанола и нагревали до температуры кипения с обратным холодильником в течение 7 ч. Смесь упарили и остаток очистили хроматографически, используя этилацетат в качестве элюента, что дало названное соединение в виде белого твердого продукта (1.48 г); Т.пл. 182 - 184oC: MC: 281 (М); ЯМР: 0.79 (d, 3, J=5,5, CH3); 0.85 (d, 3, J=5,5 CH3), 1.73-1.93 (m, 5, CH2, CH2, CH), 2.13 (m, 2, CH2), 2.42 (m, 2, CH2), 4.38 (d, 1, J= 4,9, CH), 4.58 (d, 1, J=5.0, CH), 7.03-7.23 (m, 5, АГ), 8.30 (s, 1, NH). Анализ для C19H23NO:
Рассчитано,%: C 81.80; H 8.24; N 4.98
Найдено,%: C 80,91; H 8.21; N 5.00
Пример 15. 2-Трифторметил-4-(3-нитрофенил)-4,6,7,8-тетрагидро-5(1H)хинолон.
Суспензию 3-карбокси-2-трифторметил-2- гидрокси-4-(3-нитрофенил)-4,6,7,8-тетрагидро-5(1H)-хинолона (3.17 г) в толуоле (150 мл) обработали п-толуолсульфоновой кислоты моногидратом (0.32 г), смесь энергично кипятили с насадкой Дина-Старка в течение 4 ч и затем распределили между водой и этилацетатом. Органическую фазу промыли (вода и физиологическим раствором) и упарили. Остаток очистили хроматографически (гексан/этилацетат, 1:1 и смесью метиленхлорид/ацетонитрил 95: 5), что дало названное в заглавии соединение (0.83 г) в виде бледно-желтого твердого продукта; Т.пл. 215 - 216oC, ЯМР: 1.77-2.00 (m, 2, CH2), 2.16-2.36 (m, 2, CH2), 2.50-2.71 (m, 2, CH2), 4.79 (d, 1, J=4.1, CH), 5.67 (d, 1, J=5.4, CH), 7.57-7.67 (m, 2, Аг), 8.04 (m, 2, Аг), 9.48 (s, 1, NH), MC: m/z=338(M). Анализ для C16H13F3N2O3:
Рассчитано,%: C 56.81; H 3.87; N 8.28
Найдено,%: C 56.74; H 4.02; N 8.28
Исходный 3-карбокси-2- трифторметил-2-гидрокси-4-(3-нитрофенил)-4,6,7,8-тетрагидро-5(1H)- хинолон получили как приведено ниже:
Перемешиваемую смесь этил 4,4,4-трифторацетоацетата (6.00 мл), 1,3-циклогександиона (4.65 г), 3-нитробензальдегида (6.31 г), и ацетата аммония (6.57 г) в этоноле (350 мл) нагревали до кипения с обратным холодильником в течение 4,5 ч. После удаления растворителя оранжевый остаток обработали диэтиловым эфиром, и образовавшийся осадок собрали фильтрацией. Твердый продукт растерли с горячим диэтиловым эфиром, собрали, и очистили хроматографией (этилацетат), что дало 3-карбоэтокси-2-трифторметил-2-гидрокси-4-(3- нитрофенил)-4,6,7,8-тетрагидро-5(1H)-хинолон (8.47 г) в виде белого твердого продукта. ЯМР: 0.85 (t, 3, J=7,1, CH3), 1.87 (m, 2, CH2), 2.07 (m, 2, CH2), 2.27-2.44 (m, 1, CH2), 2.57-2.72 (m, 1, CH2), 2.75 (d, 1, J=11,9, CH), 3.83 (m, 2, CH2), 4.07 (d, 1, J=11,4, CH), 7.30 (s, 1, OH), 7.50 (m, 1, Аг); 7.57 (m, 1, Аг), 7.87 (m, 1, Аг), 8.01 (m, 1, Аг), 8.16 (s, 1, NH); MC: m/z= 428(М).
Суспензию 3-карбоэтокси-2-трифторметил-2-гидрокси-4-(3-нитрофенил)- 2,3,4,5,7,8-гексагидро-5(1H)-хинолона (7.02 г) в этаноле (40 мл) и воде (40 мл) обработали моногидратом гидроокиси лития (1.44 г). Смесь нагревали при 90oC, в течение 30 мин, разбавили водой, подкислили 1 н соляной кислотой, и экстрагировали этилацетатом. Объединенные органические экстракты промыли (вода и физиологический раствор), высушили, отфильтровали и десорбировали, получив коричневое масло, которое в дальнейшем очистили хроматографически (этилацетат и этилацетат/метанол, 4: 1). 3-Карбокси-2-трифтормeтил-2-гидрокси-4-(3- нитрофенил)-4,6,7,8-тетрагидро-5(1H)-хинолон (2.07 г) получили в виде белого твердого продукта. ЯМР: 1.85 (m, 2, CH2), 2.10 (m, 2, CH),. 2.33-2.60 (m, 3, CH2, CH), 4.21 (d, 1, J=8.7, CH) 7.47 (m, 2, Аг), 7.81 (s, 2, OH или Nн, АГ), 7.96 (m, 1H, АГ); MC: m/z=400(M).
Пример 16. 2,7,7-Триметил-4-(3-нитрофенил)-4,6,7,8-тетрагидро-5(1H)хинолон.
4-(3-Нитрофенил)-3-бутен-2-он (0.87 г), 5,5-диметил-1,3- циклогександион (0.66 г) и ацетат аммония (0.82 г) смешали в этаноле (30 мл) и нагревали до кипения с обратным холодильником в течение 6.5 ч. После удаления растворителя и хроматографии (гексан/этилацетат, 1:1) названное в заглавии соединение было получено (1.02 г) в виде желтого твердого продукта; Т.пл. 162 - 164oC: ЯМР: 0.94, (s, 3, CH3), 1.01 (s, 3, CH3), 1.76 (s, 3, CH3), 1.9-2.18 (m, 2, CH2), 2.25-2.43 (m, 2, CH2), 4.55 (d, 1, J=4.7, CH), 4.64 (d, 1, J=4.8 CH), 7.54 (m, 1, Аг), 7.63 (m, 1, Аг), 7.96 (s, 1, Аг), 7.97 (m, 1, Аг), 8.53 (s, 1, NH), MC: m/z=312 (M). Анализ для C18H20N2O3:
Рассчитано,%: C 69,21; H 6.45; N 8.97
Найдено,%: C 69.07; H 6.56; N 8.85
Исходный 4-(3-Нитрофенил)-3-бутен-2-он приготовили как описано ниже:
2 н Гидроокись натрия (2.4 мл) добавили до каплям при 5-10oC к предварительно охлажденному раствору 3-нитробензальдегида (10.04 г) в ацетоне (50 мл). Смесь нагрели до комнатной температуры, перемешивали 20 мин, разбавили водой, подкислили 2 н соляной кислотой и экстрагировали этилацетатом. Объединенные органические экстракты промыли (вода и физиологический раствор) и высушили. Хроматография образовавшегося оранжевого масла (метиленхлорид) дала названное в заглавии соединение (2.73 г) в виде бледно-желтого твердого продукта. ЯМР: 2.37 (s, 3, CH3), 7.01 (d, 1, J=16,0, CH), 7.73 (m, 1, Аг), 7.78 (d, 1, J=16.2, CH), 8.20 (d, 1, J=7,9, Аг), 8.26 (m, 1, Аг), 8.55 (s, 1, Аг), MC: m/z= 191(М).
Пример 17. 4-(3-Xлорфенил)-2,7,7-триметил-4,6,7,8-тетрагидро-5(1H)хинолон.
Смесь 4-(3-хлорфенил)бут-3-ен-она (5.0 г), 5,5-диметил-1,3-циклогександиона (3.88 г), ацетата аммония (3.20 г) и этанола (90 мл) нагревали при температуре кипения с обратным холодильником в течение 9 ч. Реакционную смесь обработали, как описано в примере 8, и перекристаллизовали из смеси этилацетат/гексан получив названное в заглавии соединения (4.7 г) в виде светло-желтого твердого продукта; Т.пл. 183 - 185oC; ЯМР 0.94 (s, 3, CH3), 1.00 (s, 3, CH3), 1.74 (s, 3, CH3), 1.95 (d, 1, J=16, CH)), 2.11 (d, 1, J= 16, CH), 2.25 (d, 1, J=16.7, CH), 2.35 (d, 1, J=16,7, CH), 4.38 (d, 1, J= 4,6, CH), 4.59 (d, 16, J=4,6, CH), 7.09-7.28 (m, 4, Аг), 8.44 (s, 1, NH); (Cl, CH4) MC; m/z=302 (М+1). Анализ для C18H20ClNO:
Рассчитано,%: C 71.63; H 6.68; N 4.64
Найдено,%: C 71.60; H 6.69; N 4.54
Пример 18.
2-Этил-4-(3-нитрофенил)-4,6,7,8-тетрагидро-5(1H)-хинолон.
Смесь 1-(3-нитрофенил)-1-пентен-3-она (2.42 г), 1,3-циклогександиона (1.36 г), ацетата аммония (2.00 г) и этанола (70 мл) нагревали при температуре кипения 7.5 ч. Удаление растворителя, хроматография (этилацетат/гексан, 2: 1 и метиленхлорид/ацетонитрил, 9:1), растирание с горячим диэтиловым эфиром и перекристаллизация из этилацетата дали названное в заглавии соединение (1.29 г) в виде желтого твердого продукта; Т.пл. 182 - 184oC; ЯМР: 1.03 (t, 3, J=7,4, CH3), 1.70-1.95 (m, 2, CH2), 2.07 (q, 2, J=7,5, CH2), 2.17 (m, 2, CH2), 2.48 (m, 2, CH), 4.59 (d, 1, J=4,8, CH), 4.66 (dd, 1, J=4.8, 0.9, CH), 7.53 (m, 1, Аг), 7.64 (m, 1, Аг), 7.98 (m, 2, Аг), 8.55 (s, 1, NH), MC: m/z= 298 (M). Анализ для C17H18N2O3:
Рассчитано,%: C 68.44; H 6.08; N 9.39
Найдено,%: C 68.43; H 6.09; N 9.39
Исходный 1-(3-Нитрофенил)-1-пентен-3-он приготовили следующим образом:
2-Бутанон (3.90 мл) добавили к смеси пирролидина (3.70 мл) и ледяной уксусной кислоты (2.50 мл) при 5oC. Убрали охлаждающую баню и к смеси добавили по каплям раствор 3-нитробензальдегида (6.70 г) в толуоле (30 мл) и диэтиловый эфир (10 мл). Перемешивание продолжали при комнатной температуре в течение 48 ч. Реакционную смесь распределили между собой и этилацетатом и подкислили 2 н соляной кислотой, таким образом, чтобы водная часть оставалась кислой. Органическую часть промыли (вода и физиологический раствор), высушили, растворитель отогнали и образовавшийся коричневый твердый продукт хроматографировали (гексан/этилацетат 2: 1). Растирание со смесью гексан/этилацетат дало названное в заглавии соединение (2.45 г) в виде желтого твердого продукта: ЯМР: 1.04 (t, 3, J=7,3, CH3), 2.75 (q, 2, J=7,3, CH2), 7.10 (d, 1, J=16.3, CH), 7.72 (m, 1, Аг), 7.74 (d, 1, J=16.3, CH), 8.20 (m, 1, Аг), 8.25 (m, 1, Аг), 8.56 (m, 1, Аг), MC: m/z 205 (M).
Пример 19. 2-Изопропил-4-(3-нитрофенил) -4,6,7,8- тетрагидро-5(1H)хинолон.
4-Метил-1-(3-нитрофенил)-1-пентен-3-он (2.90 г), 1,3-циклогександион (1.50 г) и ацетат аммония (2 г) смешали в этаноле (100 мл) и нагревали при температуре кипения с обратным холодильником в течение 7 ч. После удаления растворителя, хроматографии (гексан/этилацетат, 1:1) и растирания с диэтиловым эфиром получили названное в заглавии соединение в виде желтого твердого вещества; Т.пл. 195 - 196oC; ЯМР: 1.06 (d, 3, J=6.9, CH3), 1.07 (d, 3, J= 6,9, CH3), 1.67-1.95 (m, 2, CH2), 2.07-2.22 (m, 2, CH2), 2.33 (m, 1, CH), 2.49 (m, 2, CH2), 4.60 (d, 1, J=5.O, CH), 4.67 (d, 1, J=4,9, CH), 7.53 (m, 1, АГ), 7.64 (m, 1, АГ), 7.97 (m, 2, АГ), 8.42 (s, 1, NH); MC: m/z=312 (М). Анализ для C18H20N2O3:
Рассчитано,%: C 69.21; H 6.45; N 8.97
Найдено,%: C 69.03; H 6.50: N 8.60
Исходный 4-метил-1-(3-нитрофенил)-1-пентен-3-он получили следующим образом:
3-Метил-2-бутанон (5.00 мл) добавили к смеси пирролидина (3.40 мл) и ледяной уксусной кислоты (2.30 мл) при 5oC. Охлаждающую баню убрали и к смеси добавили по каплям раствор 3-нитробензальдегида (6.16 г) в толуоле (30 мл) и диэтиловый эфир (10 мл). После 48 ч при комнатной температуре смесь вылили в 2 н соляную кислоту и экстрагировали этилацетатом. Органический слой промыли (вода и физиологический раствор), высушили и удалили растворитель. Очистка хроматографией (гексан/этилацетат, 3: 1) дала 1-гидрокси-4-метил-1-(3-нитрофенил) пентан-3-он (3.95 г) в виде желтого твердого продукта. ЯМР: 0.96 (d, 3, CH3), 1.00 (d, 3, CH3), 2.62 (m, 1, CH), 2.73-2.97 (m, 2, CH2), 5.14 (m, 1, CH), 5.66 (d, 1, J=4,7, OH), 7.62 (m, 1, Аг), 7.81 (d, 1, J=7,7, АГ), 8.11 (m, 1, АГ), 8.23 (m, 1, АГ): (C1, CH4) MC: m/z=238(М+1).
Смесь 1-гидрокси-4-метил-1-(3-нитрофенил) дентан-3-она 3.32 г), моногидрата п-толуолсульфоновой кислоты (0.03 г) и толуола (50 мл) нагревали до кипения с обратным холодильником в течение 1 ч в условиях Дина-Старка. Смесь разбавили этилацетатом, промыли (насыщенный бикарбонат натрия, вода, физиологический раствор) и удалили растворитель. Хроматография (гексан/этилацетат, 3: 1) дала 4-метил-1-(3-нитрофенил)-1-пентен-3-он (2.98 г) в виде желтого твердого вещества. ЯМР: 1.11 (d, 6, J=6.8, CH3), 3.02 (m, 1, CH), 7.27 (d, 1, J=16.2, CH), 7.73 (m, 2, CH, АГ), 8.24 (m, 2, АГ), 8.61 (m, 1, АГ), MC: m/z=219 (M).
Пример 20. 2-(т-Бутил)-4- (3-нитрофенил)-4,6,7,8-тетра-гидро-5-(1H)-хинолон.
т-Бутил-3-нитростирилкетон (1.81 г), 1.3-циклогександион (0.82 г) и ацетат аммония (1.25 г) смешали в этаноле (50 мл) и нагревали до кипения с обратным холодильником в течение 7,5 ч. Последующим удалением растворителя и хроматогрфией (гексан/этилацетат; 1:1 и метиленхлорид/ацетонитрил 9:1) получили названное в заглавии соединение (0.54 г) в виде белого твердого продукта; Т. пл. 180 - 181,5oC; ЯМР: 1.12 (s, 9, CH2), 1.68-1.95 (m, 2, CH2), 2.06-2.27 (m, 2, CH2), 2.42-2.67 (m, 2, CH2), 4.61 (d, 1, J=5.2, CH), 4.72 (dd, 1, J=5.2, 1.8; CH); 7.53 (m, 1, АГ), 7.64 (m, 1, АГ), 7.64 (m, 1, АГ), 7.97 (m, 2, АГ), 8.10 (s, 1,NH), MC: m/z= 326(M). Анализ для C19H22N2O3:
Рассчитано,%: C 69.92; H 6.70; N 8.58
Найдено,%: C 69.96; H 6.80; N 8.60
Пример 21. 4-(3,4-Дихлорфенил)-2-метил-4,6,7,8- тетрагидро-5(1H)-хинолон.
Смесь 1-(3,4-дихлорфенил)бут-1-ен-3- она (5.0 г), 1,3-циклогександиона (2.71 г), ацетата аммония (2.70 г) и этанола (75 мл) нагревали до кипения с обратным холодильником в течение 10 ч. Реакцию проводили как описано в примере 8 и перекристаллизация из смеси этилацетат-гексан дала названное в заглавии соединение (2.6 г) в виде не совсем белого твердого продукта; Т.пл. 199 - 201oC; ЯМР: 1.74 (s, 3, CH3), 1.84-1.88 (m, 2, CH2), 2.14-8.19, (m, 2, CH2), 2.41-2.46 (m, 2, CH2), 4.42 (d, 1, J=4.7, CH), 4.59 (d, 1, J=4.7, CH), 7.12 (dd, 1, J= 8.3, 2.0, Аг); 7.31 (d, 1, J=2,0, Аг), 7.48 (d, 1, J=8.3, Аг), 8.55 (s, 1, NH); (Cl, CH4) MC: m/z=308(M+1). Анализ для C16H15Cl2NO:
Рассчитано,%: C 62,35; H 4.91; N 4,54
Найдено,%: C 62.03; H 5.09; N 4.44
Пример 22. 4-(3-Метоксифенил)-3-метил-4,6,7,8-тетра-гидро-5(1H)-хинолон.
Раствор 1-(9-метоксифенил)бут-1-он-3-она (2.07 г), 1,3- циклогександиона (1.37 г) и ацетата аммония (1.39 г) в этаноле (20 мл) перемешивали при температуре кипения в течение 10 ч. Реакцию выполняли как описано в примере 8 и очищали хроматографией (10% об./об. диэтиловый эфир в метиленхлориде), что дало названное в заглавии соединение (1.14 г) в виде не совсем белого твердого продукта. Перекристаллизация из смеси этанол/гексан дала аналитически чистый продукт; Т.пл. 164 - 167oC; ПМР: 1.71 (s, 3, CH3), 1.32-1.88 (m, 2, CH2), 2.13-2.17 (m, 2, CH2), 2.40-2.44 (m, 2, CH2), 3.67 (s, 3, CH3), 4.35 (d, 1, J=4,8, CH), 4.60 (d, 1, J=4,6, CH), 6.64-6.66 (m, 2, АГ), 6.71 (d, 1, J=7,7, АГ) 7.12 (dd, 1, J=7,7, 3,8, АГ), 8.41 (s, 1, NH), (C1, CH4), MC: m/z=270(М+1). Анализ для C17H19NO2•0,5H2O:
Рассчитано,%: C 73.36; H 7.24; N 5.03
Найдено,%: C 73.49; H 6.98; N 4.93
Пример 23. 3-Ацетил-2-метил-4-(3-нитрофенил)-4,6,7,8-тетрагидро-5(IH)-хинолон.
4-Амино-пентен-2-он (2.00 г), 1,3-циклогександион (2.30) г, и 3-нитробензальдегид (3,08 г) смешали в этаноле (180 мл) и нагревали до кипения с обратным холодильником в течение 5 ч. При охлаждении до температуры окружающей среды образовывался желтый твердый продукт. Твердый продукт очистили растиранием с горячим этил ацетатом/этанолом, что дало названное в заглавии соединение (2.65 г) в виде желтого твердого вещества; Т.пл. 250oC; ПМР: 1.67-1.97 (m, 2, CH2), 2.13 (s, 3, CH3), 2.22 (m, 2, CH2), 2.35 (s, 3, CH3), 2.47 (m, 2, CH2), 5.11 (s, 1, CH), 7.52 (m, 1, АГ), 7,60 (d, 1, J=7,7, АГ), 7.97 (m, 2, Аг), 9.32 (s, 1, NH); MC: m/z=326(M). Анализ для C18H18N2O4:
Рассчитано,%: C 66.25; H 5,56; N 8.58
Найдено,%: C 65.99; H 5,61; N 8.40
Пример 24. 2-(Изобутил)-4-(3-нитрофенил)-4,6,7,8- тетрагидро-5(1H)хинолон.
Раствор 5-метил-1-(3-нитрофенил)-1- гексен-3-она (2.52 г), 1,3-циклогександиона (1.22 г) и ацетата аммония (1.88 г) в этаноле (65 мл) перемешивали при температуре кипения в течение 7.5 ч. После удаления растворителя остаток хроматографировали (этилацетат/гексан 2:1), что дало названное в заглавии соединение (1.51 г) в виде желтого твердого вещества, Т.пл. 158 - 160oC; ПМР: 0.81 (d, 3, J=5.8, CH3), 0.86 (d, 3, J=5,8, CH3), 1.77-1.97 (m, 5, CH2, CH), 2.10-2.23 (m, 2, CH2), 2.46 (m, 2, CH2), 4.59 (d, 1, J=4,8, CH), 4.64 (d, 1, J=5.8, CH), 7.53 (m, 1, Аг), 7.65 (m, 1, Аг), 7.97 (m, 2, Аг), 8.49 (s, 1, NH), MC: m/z=326(M). Анализ для C19H22NO3:
Рассчитано,%: C 69,92; H 6.79; N 8,58
Найдено,%: C 70.03; H 6.81; N 8.54
Исходный 5-метил-1-(3-нитрофенил)-1-гекоен-3-он получили следующим образом:
4-Метил-2-пентанон (5.00 мл) добавили к смеси пирролидина (3.30 мл) и ледяной уксусной кислоты (2.30 мл) при 5oC. Охлаждающую баню убрали, и к смеси добавили по каплям раствор 3-нитробензальдегида (6.12 г) в толуоле (25 мл) и диэтиловом эфире (10 мл). После 48 ч при комнатной температуре реакционную смесь разбавили водой, подкислили 2 н соляной кислотой и экстрагировали этилацетатом. Объединенные органические слои промыли (вода и физиологический раствор), высушили и образовавшийся маслянистый остаток очистили хроматографией (гексан/этилацетат, 3:1), что дало названное в заглавии соединение (5.42 г) в виде бледно-желтого твердого продукта. ПМР: 0.92 (d, 3, J= 6.6, CH3), 0.93 (d, 3, J=6,6, CH3), 2.14 (m, 1, CH), 2.60 (d, 2, J=7,0, CH), 7.09 (d, 1, J=16.4, CH), 7.72 (m, 1, Аг), 7.75 (d, 1, J=16,0, CH), 8.20 (d, 1, J=7,8, АГ), 8.25 (m, 1, Аг), 8.57 (s, 1, Аг); MC; m/z=233(M).
Пример 25. 9-(3-Нитрофенил)-3,4,5,6,7,8,9,10-октагидро- 1(2H)-акридинон.
К смеси 9-(3-нитрофенил)-3,4,6,7,9,10-гексагидро-1,8(2H, 5H)- акридиндиона (3.00 г) в этаноле (30 мл) добавили боргидрид натрия (2.21 г) в этаноле (20 мл). Смесь нагревали при температура кипения с обратным холодильником в течение 16 ч, охладили и добавили дополнительное количество боргидрида натрия (1.11 г). После дополнительного часа при температуре кипения реакционную смесь профильтровали в горячем состоянии и фильтрат разбавили водой. Этанол удалили и водную порцию экстрагировали этилацетатом (2х150 мл). Объединенные органические слои высушили, сконцентрировали до коричневого масла и хроматографировали (5 - 10% диэтиловый эфир/метиленхлорид), что дало названный в заглавии акридинон в виде желтого твердого продукта (1.12 г); Т.пл. 213 - 220oC; ПМР: 1.46-1.52 (m, 3, CH2), 1.62-1.98 (m, 5) 2.05-2.19 (m, 4, CH2), 2.40-2.46 (m, 2, CH2), 4.35 (s, 1, CH), 7.49-7.55 (m, 1, Аг), 7.61-7.65 (m, 1, Аг), 7.95-7.99 (m, 2, Аг), 8.52 (s, 1, NH); (Cl, CH), MC: m/z= 325(M+1). Анализ для C19H20N2O3:
Рассчитано,%: C 70.35; H 6.21; N 8.64
Найдено,%: C 70.06; H 6.25; N 8,34.
Получение необходимого акридинон исходного материала описано в патентной заявке Германии, опубликованной под номером E 2003148.
Пример 26. 9-(3-Метоксифенил)-3,4,5,6,7,8,9,10-октагидро-1(2H)-акридинон.
К смеси 9-(3-метоксифенил)-3,4,6,7,9,10-гексагидро- 1,8-(2H, 5H)-акридиндиона (5.75 г) в этаноле (150 мл) добавили боргидрид натрия (4.04 г). Смесь нагревали при температуре кипения с обратным холодильником в течение 16 ч, охладили и добавили дополнительное количество борогидрида натрия (2,69 г). Смесь нагревали до температуры кипения в течение 8 ч, охладили и добавили дополнительно борогидрид натрия (3.50 г). Смесь нагревали при температуре кипения 16 ч, вылили в вoду и отфильтровали, получив не совсем белый твердый продукт. Перекристаллизация из этанола дала названный в заглавии акридинон в виде белого твердого продукта (5.36 г); Т.пл. 240 - 242oC; ПМР: 1.46-1.52 (m, 3, CH2), 1.64-1.87 (m, 5, CH2), 2.08-2.13 (m, 4, CH2), 2.37-2.43 (m, 2, CH2), 3.68 (s, 3, CH3), 4.12 (s, 1, CH), 6.63-6.74 (m, 3, АГ), 7.10 (t, 1, J= 7,76, АГ), 8.34 (s, 1, NH); (C1, CH4) MC: m/z=310(M+1). Анализ для C20H23N2:
Рассчитано,%: C 75.44; H 7.59; N 4.40
Найдено,%: C 75.20; H 7.30; N 4.37
Необходимый акридинон исходный материал может быть приготовлен согласно методике, описанной в S.M. Jain et. al., Indian Journal of Chemistry, v. 30 B, Nov. 1991, p. 1037-1040.
Пример 27. 2-(3-Трифторметоксифeнил)-3,4,5,6,7,8,9,10- октагидро-1 (2H)-акридинон.
К смеси 9-(3-трифторметоксифенил)- 3,4,6,7,9,10-гексагидро-1,3(2H,5H)-акридинона (2.70 г), этанола (25 мл) и диметилформамида (15 мл) добавили боргидрид натрия (1.62 г). Смесь нагревали при температуре кипения с обратным холодильником в течение 16 ч, охладили и добавили дополнительно (1.62 г) боргидрида натрия. Смесь нагревали до кипения 2 ч, охладили и добавили дополнительное количество боргидрида натрия (2.00 г). Смесь нагревали при температуре кипения 2 ч, вылили в воду и отфильтровали получив не совсем белый твердый продукт. Очистка хроматографией (5% этилацетат/метиленхлорид) дала названный в заглавии акридинон в виде белого твердого продукта (1.41 г); Т. пл. 214 - 216oC. ПМР: 1.46-1.52 (m, 3, CH2), 1.65-1.88 (m, 5, CH2), 2.08-2.14 (m, 4, CH2), 2.38-2.43 (m, 2, CH2), 4.23 (s, 1, CH), 7.06 (m, 2, Аг), 7.18 (d, 1, J=7.73, Аг) , 7.33 (m, 1, Аг) 3.44 (s, 1, NH); (Cl, CH4) MC: m/z=364(M+1). Анализ для C20H20NO2F3•0,2H2O:
Рассчитано,%: C 65.46; H 5.60; N 3.82
Найдено,%: C 65.42; H 5.53; N 3.62
Необходимый акридинон исходный продукт приготовили согласно методике, описанной в примере 28 ниже, но используя 3-трифторметоксибензальдегид вместо 3-трифторметилбензальдегида; Т.пл. 273 - 275oC.
Найдено для C20H18FNO3,%: C 63.55; H 4.71; N 3.67.
Пример 28. 9-(3-Трифторметилфенил)-3,4,5,6,7,8,9,10-октагидро- 1(2H)-акридинон.
Смесь 9-(3-трифторметилфенил)-3,4,6,7,9.10- гексагидро-1,8(2H, 5H)-акридиндиона (2.0 г), борогидрида натрия (2.1 г) и этанола (50 мл) нагревали при 70oC в течение ночи и охладили до комнатной температуры. Смесь распределили между водой и этилацетатом; органический слой высушили, отфильтровали и сконцентрировали. Хроматография (элюат: метиленхлорид/метанол; 98/2) и перекристаллизация из смеси этанол/гексан дали названное в заглавии соединение (1.0 г) в виде желтого твердого продукта; Т.пл. 250 - 251oC; ПМР: 1.47-1.52 (m, 3, CH2, 1.66-1.90 (m, 5, CH2), 2.10-2.15 (m, 4, CH2), 2.40-2.45 (m, 2, CH2), 4.28 (s, 1, CH), 7.45 (s, 4, Аг), 8.44 (s, 1, NH); (Cl, CH4) MC: m/z =348(М+1). Анализ для C20H20F3NO:
Рассчитано,%: C 69.15; H 5.80; N 4.03
Найдено,%: C 69.01; H 5.83; N 3.98
Названное в заглавии соединение разделили на два его энантиомера с помощью жидкостной хроматографии высокого давления, используя 250 х 250 мм колонку, заполненную основанным на двуокиси кремния насадочном материале с совомукоидной неподвижной фазой (Ultron ES-OVM), элюируя смесью 30% ацетонитрил: 70% 0.013 и KH2PO4, доведенной до pH 5 с помощью 1.0 М KOH, с объемной скоростью потока 15.6 мл/мин. Энантиомеры анализированы спектрометрически при длине волны 250 нм.
Необходимый акридинон исходный продукт может быть приготовлен следующим образом:
Перемешиваемую смесь 3- трифторметилбензальдегида (3.48 г, 1.3-циклогександиона (4.49 г), ацетата аммония (2.31 г) и этанола (40 мл) нагревали до кипения с обратным холодильником в течение 18 ч. Смесь охладили и желтые игольчатые кристаллы собрали, промыли водой и высушили при пониженном давлении, что дало названное в заглавии соединение (6.38 г); Т.пл.> 300oC; ПМР: 1.7-2.0 (m, 4), 2.19-2.25 (m, 4), 2.50-2.56 (m, 4), 4.98 (s, 1), 7.41 (s, 3), 7.48 (s, 1), 9.48 (s, 1); MC: m/z =362(М+1).
Найдено для C20H18F3NO2,%: C 66.44; H 5.00; N 3.80.
Пример 29. 9-(3-Фторфенил)-3,4,5,6,7,8,9,10-октагидpo-1(2H)- акридинон.
Смесь 9-(3-фторфенил)-3,4,6,7,9,10- гексагидро-1,8-(2H, 5H)-акридинона (5.0 г), боргидрида натрия (6.1 г), этанола (140 мл) и пиридина (50 мл) нагревали при 70oC в течение ночи и охладили до комнатной температуры. Растворитель удалили и образовавшийся желтый твердый продукт собрали и хорошо промыли водой. Хроматография (этилацетат/гексан; 80/20) дала названное в заглавии соединение (2.1 г) в виде желтого твердого продукта; Т.пл. 249 - 251oC; ПМР: 1.45-1.55 (m, 3, CH2), 1.66-1.90 (m, 5, CH2); 2.10-2.15 (m, 4, CH2), 2.39-2.45 (m, 2, CH2), 4.19 (s, 1, CH), 6.87-6.92 (m, 2, Аг), 7.00 (d, 1, J=7,8, АГ) 7.20-7.27 (m, 1, Аг), 8.41 (s, 1, NH); (Cl, C5H4) МC: m/z=298 (M+1). Анализ для C19H20FNO:
Рассчитано,%: C 76.74; H 6.78; N 4.71
Найдено,%: C 76.32; H 6.71; N 4.62
Необходимый акридинон исходный продукт приготовили согласно методике примера 28, но используя 3-фторбензальдегид вместо 3-трифторметилбензальдегида. Т.пл. > 350oC.
Найдено для C19H18NFO2,%: C 73.30; H 5.87; N 4.37
Пример 30. 9-Фенил-3,4,5,6,7,8,9,10-октагидро-1(2H)-акридинон.
Смесь 9-фенил-3,4,6,7,9,10-гексагидро-1,8(2H, 5H)-акридинона (5.0 г), боргидрида натрия (6.5 г), этанола (150 мл) и пиридина (50 мл) нагревали при 70oC в течение ночи и охладили до комнатной температуры. Растворитель удалили, остаток распределили между водой и этилацетатом, органический слой высушили и растворитель удалили. Хроматография (метиленхлорид/метанол; 98/2) дала названное в заглавии соединение (3.0 г) в виде белого твердого продукта. Перекристаллизация из смеси этанол/гексан обеспечила аналитически чистое названное в заглавии соединение; Т.пл. 252 - 254oC, ПМР: 1.41-1.51 (m, 3, CH2), 1.60-1.88 (m, 5, CH2), 2.03-8.13 (m, 4, CH2), 2.38-2.44 (m, 2, CH2), 4.14 (s, 1, CH), 7.04-7.21 (m, 5, Аг), 8.34 (s, 1, NH); (Cl, CH4) MC: m/z= 280(М+1). Анализ для C19H21NO•0,2H2O:
Рассчитано,%: C 80.64; H 7.62; N 4.95
Найдено,%: C 80.84; H 7.62; N 4.80
Необходимый акридинон исходный продукт может быть приготовлен согласно методике описанной в примере 28, но с использованием бензальдегида вместо 3-трифторметилбензальдегида.
Пример 31. 9-(3-Бромфенил)-3,4,5,6,7,8,9,10-октагидро-1 (2H)-акридинон
К перемешиваемой при 70oC смеси 9-(3-бромфенил)-3,4,6,7,9,10-гексагидро-1,8(2H, 5H)-акридиндиона (5.0 г, 13,4), этанола (120 мл) и пиридина (40 мл) добавили боргидрид натрия (7.5 г) в три порции за 6 ч. Растворитель удалили: образовавшийся желтый твердый продукт хорошо промыли водой и высушили на воздухе, что дало названное в заглавии соединение (4.8 г). Перекристаллизация из смеси этанол/гексан обеспечила аналитически чистый желтый твердый продукт; Т. пл. 258 - 260oC, ПМР: 1.41-1.51 (m, 3, CH2), 1.60-1.88 (m, 5, CH2), 2.09-2.15 (m, 4, CH2), 2.39-2.45 (m, 2, CH2), 4.17 (s, 1, CH), 7.16-7.21 (m, 2, Аг), 7.26-7.29 (m, 2, Аг), 8.44 (s, 1, NH); (C1, CH4) MC: m/z=360(M+1). Анализ для C19H20BrNO:
Рассчитано,%: C 63.69; H 5.59; N 3.91
Найдено,%: C 63.73; H 5.69; N, 3.94
Необходимый акридинон исходный продукт может быть приготовлен как описано в примере 28, но с использованием 3-бромбензальдегида вместо 3-трифторметилбензальдегида; Т.пл. 336 - 339oC.
Найдено для C19H18BrNO2,%: C 61.12; H 4.94; N 3.64
Пример 32. 9-(3-Хлорфенил)-3,4,5,6,7,8,9,10-октагидро-1(2H)- акридинон.
К перемешиваемой при 70oC смеси 9-(3-хлорфенил)-3,4, 6,7,9,10-гексагидро-1,8(2H, 5H)-акридиндиона (5.0 г), этанола (120 мл) и пиридина (40 мл) добавили боргидрид натрия (7.5 г) в две порции за 6 ч. Растворитель удалили; образовавшийся твердый желтый продукт хорошо промыли водой и высушили при пониженном давлении, получив названное в заглавии соединение (5.0 г) в виде желтого твердого продукта. Перекристаллизация из этанола обеспечила аналитически чистый продукт; Т. пл. 249 - 250oC; ПМР: 1.41-1.51 (m, 3, CH2), 1.60-1.88 (m, 5, CH2), 2.09-2.15 (m, 4, CH2), 2.39-2.45 (m, 2, CH2), 4.17 (s, 1, CH), 7.09-7.15 (m, 3, Аг), 7.21-7.26 (m, 1, Аг), 8.43 (s, 1, NH), (Cl, CH4) MC: m/z=314(М+1). Анализ для C19H20ClNO:
Рассчитано,%: C 72.72; H 6.37; N 4.46
Найдено,%: C 72,55; H 6.48; N 4.49
Необходимый акридиндион исходный материал может быть приготовлен как описано в примере 28, но с использованием 3-хлорбензальдегида вместо 3-трифторметилбензальдегида; Т.пл. >300oC.
Найдено для C19H18ClNO2,%: C 69.34; H 5.57; N 4.22
Пример 33. 9-(4-Хлорфенил)-3,4,5,6,7,8.9,10-октагидро-1 (2H)-акридинон.
К перемешиваемой при 70oC смеси 9-(4-хлорфенил)-3,4,6, 7,9,10-гексагидро-1,8(2H, 5H)-акридиндиона (6.0 г), этанола (160 мл) и пиридина (50 мл) добавили боргидрид натрия (7.5, 198,4 ммоль) в две порции за 6 ч. Растворитель удалили; образовавшийся желтый твердый продукт хорошо промыли водой и высушили при пониженном давлении. Хроматография (метиленхлорид/этилацетат; 85/15) и перекристаллизация из этанола дали названное в заглавии соединение (1,0 г) в виде белого твердого продукта; Т.пл. 253 - 254oC; ПМР: 1.41-1.51 (m, 3, CH2), 1.60-1.88 (m, 5, CH2), 2.07-2.14 (m, 4, CH2), 2.38-2.41 (m, 2, CH2), 4.16 (s, 1, CH), 7.16 (d, 2, J=8.4, Аг); 7.25 (d, 2, J=8,4, Аг)), 8.39 (s, 1, NH); (C1, CH4) MC: m/z=314(М+1). Анализ для C19H20ClNO:
Pассчитано,%: C 72.72; H 6.37; N 4.46
Найдено,%: C 72.35; H 6.60; N 4.29
Необходимый исходный акридинон продукт может быть получен как описано в примере 28, но c использованием 4-хлорбензальдегида вместо 3-трифторметилбензальдегида; Т.пл. > 300oC.
Найдено для C19H18N2O5•0.5H2O,%: C 62.74; H 5.26; N7.53
Пример 34. 9-(3-Бром-4-фторфенил)-3,4,5,6,7,8,9,10-октагидро- 1(2H)-акридинон.
Смесь 9-(3-бром-4-фторфенил)-3,4,6,7,9,10-гексанидро- 1,8(2H,5H)-акридинона (1.8 г), боргидрида натрия (1.74 г), этанола (40 мл) и пиридина (14 мл) нагревали при 70oC 3,5 ч. Растворитель удалили, остаток распределили между водой и этил ацетатом, органический слой высушили и растворитель удалили. Хроматография (метиленхлорид/этилацетат: 7/3) дала названное в заглавии соединение (1.1 г) в виде желтого твердого продукта; Т.пл. 254 - 257oC; ПМР: 1.41-1.51 (m, 3, CH2), 1.60-1.88 (m, 5, CH2), 2.03- 2.13 (m, 4, CH2), 2.38-2.44 (m, 2, CH2), 4.18 (s, 1, CH), 7.13-7.24 (m, 2, Аг), 7.38 (dd, 1, J= 6.9, 2.0, Аг) 8.45 (s, 1, NH); (Cl, CH4) MC: m/z = 378 (M+1). Анализ для C19H19FBr:
Рассчитано,%: C,60.65; H 5.09; N 3.72
Найдено,%: C 60.68; H 5.09; N 3.65
Необходимый акридинон исходный материал может быть получен как описано в примере 28, но с использованием 3-бром-4-фторбензальдегида вместо 3- трифторметилбензальдегида; Т.пл. 308 - 310oC.
Найдено для C19H17BrFNO2,%: C 58.31; H 4.42; N 3.58
Пример 35. 4-(3-Бромфенил)-2-метил-4,6,7,8-тетрагидро-5(1H)хинолон.
Смесь 3-бромбензальдегидa (2.0 г) 1,3-циклогександиона (1.27 г) и этанола (30 мл) нагревали до кипения в течение 3 ч. Добавили ацетон (0.75 г), ацетат аммония (1.25 г) и раствор пирролидин ацетата (1.89 г) в 4 мл этанола и продолжали нагревание при температуре кипения в течение ночи. Смесь охладили, отфильтровали и фильтрат сконцентрировали при пониженном давлении. Хроматография (элюат:метиленхлорид/метанол; 98/2) и растирание с диэтиловым эфиром дали названное в заглавии соединение (0.28 г) в виде желтого твердого продукта; Т. пл. 187 - 191oC, ПМР: 1.73 (s, 3, CH3), 1.80-1.90 (m, 2, CH2), 2.10-2.18 (m, 2, CH2), 2.40-2.45 (m, 2, CH2), 4.39 (d, 1, J=4.7, CH), 4.59 (d, 1, J=4.7, CH), 7.12-7.28 (m, 4, Аг), 8.50 (s, 1, NH), (Cl, CH4) MC: m/z =320 (M+1). Анализ для C16H16BrNO:
Рассчитано,%: C 60.39; H 5.07; N 4.40
Найдено,%: C 60.19; H 5.27; N 4.10
Пример 36. 4-(3-Трифторметоксифенил)-2-метил-4,6,7,8-тетрагидро-5(1H)-хинолон.
Раствор 3-(трифторметокси)бензальдегида (10.00 г) и 1,3- циклогександиона (5.90 г) в этаноле (50 мл) перемешивали при температуре кипения в течение 3 ч. К охлажденной смеси добавили ацетат аммония (6.09 г) и ацетон (3.67 г), и впоследствии раствор уксусной кислоты (3.79 г) и пирролидина (4.49 г) в этаноле (15 мл). Смесь перемешивали при кипении в течение ночи, вылили в воду и экстрагировали этилацетатом (2 х 200 мл). Объединенные органические слои высушили и сконцентрировали до масла, которое очистили хроматографией (0 - 40% об./об. этилацетат и метиленхлорид), что дало названное в заглавии соединение (2.36 г) в виде не совсем белого твердого продукта; Т.пл. 144 - 146oC; ПМР: 1.74 (s, 3, CH3), 1.78-1.92 (m, 2, CH2), 2.14-2.19 (m, 2, CH2), 2.42-2.47 (m, 2, CH2), 4.46 (d, 1, J=4.8, CH), 4.63 (d, 1, J=4.9, CH), 7.01-7.05 (m, 2, Аг), 7.17 (d, 1, J=7.7), 7.36 (dd, 1, J=7.7, 3.2, Аг); 6.52 (s, 1, NH); (Cl, CH4) MC: m/z = 324 (М+1). Анализ для C17H16N2F3:
Pассчитано,%: C 63.15; H 4.99; N 4.33
Найдено,%: C 62.88; H 5,05; N 4.33
Пример 37. 4-(3-Фторфенил)-2-метил-4,6,7,8-тетрагидро-5(1H)хинолон.
Смесь 3-фторбензальдегида (7.0 г), 1,3-циклогександиона (6.61 г) и этанола (150 мл) нагревали при 70oC в течение ночи и затем охладили до комнатной температуры. Добавили ацетон (3.93 г), ацетат аммония (6.52 г) и раствор пирролидин ацетата (9.83 г) в этаноле (10 мл) и нагревали при 70oC в течение 3 ч, а затем охладили до комнатной температуры. Растворитель удалили и остаток распределили между водой и этилацетатом. Органический слой высушили, отфильтровали, сконцентрировали при пониженном давлении, что дало янтарное масло. Хроматография (метиленхлорид/этилацетат; 9.0/1.0) и перекристаллизация из этанола дали названное в заглавии соединение (0.12 г) в виде белого твердого продукта; Т.пл. 216 - 218oC; ПМР: 1.73 (s, 3, CH3), 1.78-1.90 (m, 2, CH2), 2.14-2.19 (m, 2, CH2), 2.41-2.46 (m, 2, CH2), 4.42 (d, 1, J= 4.8, CH), 4.61 (d, 1, J=4,8, CH), 6.86-7.00 (m, 3, Аг), 7.25 (m, 1, Аг), 8.49 (s, 1, NH); (Cl, CH4) MC: m/z =258 (М+1). Анализ для C16H16FNO:
Pассчитано,%: C 74.69; H 6.27; N 5.44
Найдено,%: C 74.39; H 6.30; N 5.35
Пример 38. 3-Циано-2-метил-4-(3-нитрофенил)-4,6,7,8- тетрагидро-5(1H)-хинолон.
3-Аминокротонитрил (0.91 г), 1.3-циклогександион (1.25 г), и 3- нитробензальдегид (1.70 г) растворили в этаноле (100 мл) и нагревали при температуре кипения с обратным холодильником в течение 7 ч. Удаление растворителя и хроматография (этилацетат) дали названное в заглавии соединение (2.40 г) в виде желтого твердого продукта; Т.пл. 214 - 216oC. ПМР: 1.73-2.00 (m, 2, CH2); 2.08 (s, 3, CH3), 2.13- 2.31 (m, 2, CH2), 2.41-2.63 (m, 2, CH2), 4.66 (s, 1, CH), 7.61 (m, 1, Аг), 7.68 (dd 1, J=7,7, 1.1 Аг), 7.98 (s, 1, Аг), 8.07 (m, 1, Аг), 9.66 (s, 1, NH); MC: m/z =309 (М). Анализ для C17H15N3O3:
Рассчитано,%: C 66.01; H 4.89; N 13.58
Найдено,%: C 65.78; H 4.99; N 13.45
Пример 39. 2-Трифторметил-4-(3-цианофенил)-4,6,7,8-тетрагидро-5(1H)-хинолон.
Суспензию 3-карбокси-2-трифторметил-2-гидрокси-4-(3-цианофенил)- 4,6,7,8-тетрагидро-5(1H)-хинолон (2.25 г) в толуоле (100 мл) обработали моногидратом п-толуол сульфоновой кислоты (0.33 г). Смесь энергично кипятили в условиях Дина-Старка в течение 1.5 ч и затем распределили между водой и этилацетатом. Органическую часть промыли (вода и физиологический раствор) и упарили. Остаток очистили хроматографией (гексан/этилацетат, 1:1), что дало названное в заглавии соединение (1.04 г) в виде белого твердого продукта; Т. пл. 182 - 183oC; ПМР: 1.80-2.00 (m , 2, CH2), 2.13-2.30 (m, 2, CH2), 2.50-2.67 (m, 2, CH2), 4.68 (d, 1, CH, J=5.2), 5.61 (d, 1, CH, J=5.3), 7.51 (m, 2, Аг), 7.60 (s, 1, Аг), 7.64 (m, 1, Аг), 9.42 (s, 1, NH); MC: m/z = 318 (M). Анализ для C17H13F3N2O:
Рассчитано,%: C 64.15; H 4.12; N 8.80
Найдено,%: C 64.15; H 4.10; N 8.63
Исходный 3-карбокси-2-трифторметил-2- гидрокси-4-(3-цианофенил)-4,6,7,8-тетрагидро-5(1H)-хинолон приготовили как описано далее:
Смесь этил 4,4,4-трифторацетоацетата (5.0 мл), 1,3-циклогександиона (3.87 г), 3-цианобензальдегида (4.61 г), и ацетата аммония (5.63 г) в этаноле (250 мл) нагревали при температуре кипения с обратным холодильником в течение 5 ч. Образовавшийся осадок собрали фильтрацией (3.39 г). Фильтрат сконцентрировали и очистили хроматографией (гексан/этилацетат), что дало продукт присоединения (5.77 г). Продукт 3-карбоэтокси-2-трифторметил-2-гидрокси-4-(3-цианофенил)-4,6,7,8-тетрагидро-5(1H)- хинолон получили в виде белого твердого вещества; МC: m/z = 408 (М); ПМР: 0.86 (t, 3, CH3, J=7.0), 1.08-1.97 (m, 2, CH2), 2.00-2.13 (m, 2, CH2), 2.27-2.40 (m, 1, CH2), 2.53-2.67 (m, 1, CH2), 2.74 (d, 1, CH, J=12,2), 3.77-3.80 (m, 2, CH2), 3.98 (d, CH, J=11.7), 7.24 (s, 1, OH) 7.33-7.47 (m, 2, Аг), 7.50 (s, 1, Аг), 7.59 (m, 1, Аг), 8.10 (s, 1, NH).
Суспензию 3-карбоэтокси-2-трифторметил-2-гидрокси-4- (3-цианофенил)-4,6,7,8-тетрагидро-5(1H)-хинолона (3.39 г) в этаноле (20 мл) и воде (20 мл) обработали моногидратом гидроокиси лития (0.75 г). Смесь нагревали при 90oC в течение 1.5 ч, разбавили водой, подкислили 1 М соляной кислотой и экстрагировали этилацетатом. Объединенные органические экстракты промыли (вода и физиологический раствор), высушили, отфильтровали, и упарили, получив пеку, которую очистили хроматографией (этилацетат и этилацетат/метанол, 3:1), получив 3-карбокси-2-трифторметил-2-гидрокси-4-(3-цианофенил)-4,6,7,8-5(1H)-хинолон (2.67 г) в виде бледно-желтой пены; MC: m/z=381 (m).
Пример 40. 2-Метил-4-(3-хлор-4-фторфенил)-4,6,7,8- тетрагидро-5(1H)-хинолон.
Смесь 3-хлор-4-фторбензальдегида (2.7 г) 1,3- циклогександиона (1.99 г) и 47 мл этанола нагревали при 50oC в течение ночи и затем охладили до комнатной температуры. Добавили ацетон (1.18 г), ацетат аммония (1.97 г) и предварительно смешанный раствор пирролидин ацетата (20.4 ммоль) в 5 мл этанола и нагревали при 70oC в течение 3 ч и затем охладили до комнатной температуры. Растворитель упарили при пониженном давлении и остаток распределили между водой и этилацетатом. Органический слой высушили над сульфатом магния, отфильтровали, и сконцентрировали при пониженном давлении. Хроматография (элюат: диэтиловый эфир/гексан; 9.0/1.0) на силикагеле дала названное в заглавии соединение 0.140 г в виде желтого твердого продукта; Т.пл. 164 - 167oC. ПМР (300 МГц, d6 - ДMCO): 1.74 (s, 3H, CH3), 1.81-1.88 (m, 2H, CH2) 2.15-2.19 (m, 2H, CH2), 2.41-2.48 (m, 2H, CH2), 4.41 (d, 1H, J=4.8 Гц CH), 4.60 (d, 1H, J=4.8 (Гц, CH), 7.10-7.28 (m, 2H, ароматическ.), 8.54 (s, 1H, NH); MC:(Cl, CH4): 292 (М+1): Анализ для C16H15ClFNO:
Рассчитано,%: C 65.87; H 5.18; N 4.80
Найдено,%: C 65.67; H 5.50; N 4.46
Пример 41. 9-(3-Трифторметил-4-цианофенил)-3,4,5,6,7,8,9,10- октагидро-1-(2H)-акридинон.
Смесь 9-(3-трифтор-4-цианометилфенил)-3,4,6,7,9,10- гексагидро-1,8-(2H, 5H)-акридиндиона (0.75 г) и боргидрида натрия (0.73 г) и 17 мл этанола и 6 мл пиридина нагревали при 70oC в течение 2 ч и затем охладили до комнатной температуры. Растворитель удалили при пониженном давлении и остаток распределили между водой и этилацетатом. Органические слои высушили над сульфатом магния, отфильтровали и сконцентрировали при пониженном давлении. Хроматография (элюат: метиленхлорид/этилацетат; 9.0/1.0) и растирание со смесью диэтиловый эфир/гексан дали названное в заглавии соединение (11 г) в виде желтого твердого продукта; Т. пл. 210 - 212oC; ПМР (300 МГц, d6-ДMCO); 1.47-1.89 (m, 8H, CH2), 2.05-8.16 (m, 4H, CH2), 2.40-2.45 (m, 2H, CH2), 4.41 (s, 1H, CH), 7.63 (d, 1H, J=8.0 Гц, ароматич.), 7.74 (s,1H, ароматич.) 8.04 (d, 1H, J= 8.0 Гц, ароматич.) 8.59 (s, 1H, NH); MC (Cl, CH4): 373 (M+1). Анализ для C21H19F3N2O:
Рассчитано,%: C 67.73; H 5.14; N 7.52
Найдено,%: C 67.76; H 5.31; N 7.46
Необходимый акриндиндион исходный продукт может быть приготовлен как описано в примере 28, но c использованием 4-циано-3-трифторметилбензальдегида; Т.пл. 289 - 291oC.
Найдено для C21H17F3N2O2,%: C 65.08; H 4.40; N 7.22
Пример 42. 9-(3-Хлор-4-фтор)-3,4,5,6,7,8,9,10-октагидро-1-(2H)-акридинон.
Смесь 9-(3-хлор-4-фтор)-3,4,6,7,9,10-гексагидро-1.8-(2H, 5H)-акридиндиона (1.5 г) и боргидрида натрия (1.64 г) и 38 мл этанола пиридина нагревали при 70oC в течение 3.5 ч и затем охладили до комнатной температуры. Растворитель удалили при пониженном давлении и остаток распределили между водой и этил ацетатом. Органический слой высушили над сульфатом магния, отфильтровали и сконцентрировали при пониженном давлении. Хроматография (элюат: метиленхлорид/этилацетат; 8.0/2.0) на силикагеле дала названное в заглавии соединение (0.75 г) в виде желтого твердого продукта; Т.пл. 236 - 239oC. ПMP (300 МГц, d6 - ДMCO): 1.41-1.51 (m, 3H, CH2) 1.60-1.88 (m, 5H, CH2), 2.09-2.15 (m, 4H, CH2) 2.38-2.44 (m, 2H, CE2), 4.19 (s, 1H, CH), 7.10-7.16 (m, 1H, ароматич.) 7.21-7.27 (m, 2H, ароматич.) 8.45 (s, 1H, NH), MC (Cl, CH4): 332 (M+1); Анализ для C19H19ClNO•0.1 H2O:
Рассчитано,%: C 68.40; H 5.80; N 4.20
Найдено,%: C 68.30; H 5.84; N 4.05
Необходимый акридиндион исходный материал может быть приготовлен как описано в примере 28, но с использованием 3-хлор-4-фторбензальдегида; Т.пл. 325 - 328oC.
Найдено для C19H17ClFNO2,%: C 65.78; H 4.88; N 3.98.
Пример 43. Далее приведены иллюстрированные примеры фармацевтических дозированных форм содержащих соединение формулы I, например как иллюстрировано в любом из приведенных ранее примеров (далее здесь упоминается как "соединение X") для терапевтического или профилактического применения для человека:
(а) Таблетка - мг/таблетка
Соединение X (пример 39) - 50.0
Маннитол (фармакопея США) - 223.75
Натрий кроскармелоза - 6.0
Кукурузный крахмал - 15.0
Гидроксипропилметилцеллюлоза - 2.25
Стеарат магния - 3.0
(b) Капсула
Соединение X (примера 39) - 10.0
Маннитол - 488.5
Натрий кроскармелоза - 15.0
Стеарат магния - 1.5
Приведенные выше составы могут быть получены обычными способами, хорошо известными в фармацевтической технике. Таблетки могут быть покрыты энтеросолюбильной оболочной обычными способами, например, может быть использовано покрытие из ацетатфталата целлюлозы.
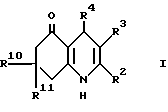
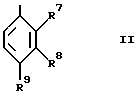
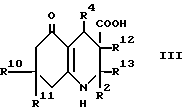
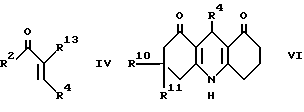
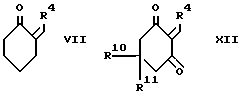
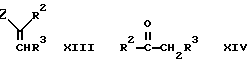
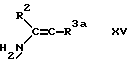
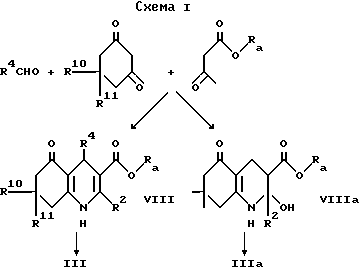
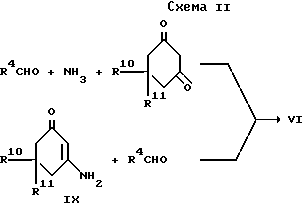
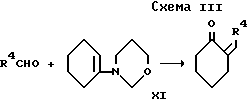 р
р
Изобретение относится к хинолоновым и акридиноновым соединениям формулы I или их фармацевтически приемлемым солям, где R2 - H, (C1-C6)алкил (C1-C4)фторалкил; R3 - H, CN, (С1-C6)алкил, (C1-C6)фторалкил, этаноил или R2 и R3 образуют 1,4-бутандиил; R4 означает группу формулы II; R7 - H, R8 - H, (C1-C4)алкокси, NO2, CN, (C1-C4)фторалкил, галоген, (C1-C4)алкил; R9 - H, галоген, (C1-C4)алкил или (C1-C4)фторалкил или R8 и R9, взятые вместе, образуют (C1-C3)алкилендиокси; R10 и R11 - H, (C1-C4)алкил. Предложено также соединение формулы III, которое подвергают декарбоксилированию или дегидратированию для получения соединения I, где R3 - H. Предложены также другие способы получения соединений I. На основе этих соединений может быть приготовлена фармацевтическая композиция, обладающая релаксирующим действием в отношении гладкой мышцы мочевого пузыря. 8 с. и 10 з.п. ф-лы.
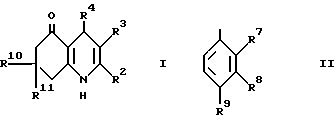
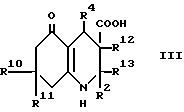
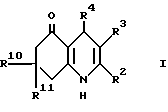
или их фармацевтически приемлемые соли,
где R2 представляет водород, (1-6С)алкил или (1-4С) фторалкил;
R3 представляет водород, циано, (1-6С) алкил, (1-6С) фторалкил или этаноил
R2 и R3, взятые вместе, образуют 1,4-бутандиил;
R4 представляет группу формулы II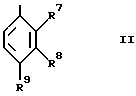
в которой R7 представляет водород;
R8 представляет водород, (1-4С) алкокси, нитро, циано, (1-4С) фторалкил, галоген, (1-4С) алкил;
R9 представляет водород, галоген, (1-4С) алкил или (1-4С) фторалкил
или R8 и R9, взятые вместе, представляют (1-3С) алкилендиокси;
R10 и R11 представляют каждый независимо водород или (1-4С) алкил, за исключением 3-циано-4-фенил-2,7,7-триметил-4,5,7,8-тетрагидро-5(1H)-хинолона и 3-этаноил-4-фенил-2,7,7-триметил-4,6,7,8-тетрагидро-5(1H)-хинолона.
R3 представляет водород, циано или этаноил или
R2 и R3, взятые вместе, образуют 1,4-бутандиил.
R3 представляет водород или
R2 и R3, взятые вместе, образуют 1,4-бутандиил.
R3 представляет водород или
R2 и R3, взятые вместе, образуют 1,4-бутандиил; R4 представляет 3-нитрофенил или 3-цианофенил;
R10 и R11 оба представляют водород.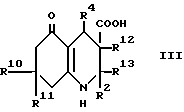
в которой R12 и R13 вместе представляют связь,
подвергают декарбоксилированию, или карбоновую кислоту формулы III, в которой R12 представляет водород и R13 представляет гидроксигруппу, подвергают декарбоксилированию и дегидратированию с последующим, в случае необходимости, переводом полученного соединения в фармацевтически приемлемую соль реакцией с подходящей кислотой или основанием, дающими физиологически приемлемый противоион.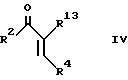
в которой R2, R4 и R13 имеют значения, определенные в п. 1,
подвергают реакции с соответствующим 1,3-циклогександионом и аммиаком или аммониевой солью с последующим, в случае необходимости, переводом полученного соединения в фармацевтически приемлемую соль реакцией с подходящей кислотой или основанием, дающими физиологически приемлемый противоион.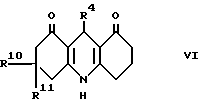
в которой R4, R10 и R11 имеют значения, определенные в п. 1,
или его фармацевтически приемлемую соль подвергают реакции с восстанавливающим агентом с последующим, в случае необходимости, переводом полученного соединения в фармацевтически приемлемую соль реакцией с подходящей кислотой или основанием.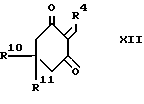
в которой R2, R3, R4, R10 и R11 имеют значения, определенные в п. 1,
подвергают реакции с енамином формулы XIII
в которой Z представляет имино-группу.
в присутствии аммиака или аммониевой соли с последующим, в случае необходимости, переводом полученного соединения в фармацевтически приемлемую соль реакцией с подходящей кислотой или основанием, дающими физиологически приемлемый противоион.
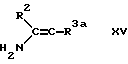
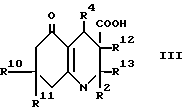
Приоритет по пунктам:
29.10.92 - по п. 1, где R10 и R11 означает водород;
20.04.93 - по пп. 12-18.
| US 4645758 A, 24.02.87 | |||
| Смешанные ангидриды хинолинкарбоновой кислоты и борной кислоты как промежуточные продукты для получения производных пиперазинил-3-хинолинкарбоновой кислоты, обладающих антибактериальной активностью | 1989 |
|
SU1838302A3 |
| 10-Метил-5-кетодекагидрохинолин или его соль,или его оптические изомеры,как промежуточные продукты для синтеза физиологически активных веществ,и способ их получения | 1977 |
|
SU602499A1 |
| Способ электролитического покрытия латунью | 1956 |
|
SU110496A1 |
| Уплотнитель мычки в вытяжном приборе прядильных и т п машин | 1961 |
|
SU142920A1 |
| US 3910917 A, 07.10.75. | |||

