Изобретение относится к способу получення новых производтгых 1,2,4-триазола, которые могут найти применение В сельском хозяйстве, Известен способ алкклирования триазо- лов галоиднь ми алкилами, содержащими активирующие галоидзаместители, в среде инертного органического растворителя при температуре от О до 150°С 1д. Целью изобретения является получение новых 1фоизоодных 1,2(4 триазола, обладающих фунгицидной активностью. Поставленная цель достигается описываемым способом получения производных 1,2,4 триазола общей формулы I NО - C-Yгде R - фенил, незамещенный или заме- щ.еиный галоидом, алкилом с атомами углерода, галоидалкилом с 1-4 атомами углерода, нитрогруппой, аминогруппой, фенилом, метоксиг{5уппой; R - водород, алкил с 1-4 атомами углерода, фенил; Ц - алкил с 1-4 атомами углерода, циклоалкил с &-6 атомами углерода, фе1Й1Л, галоидфенил, -- -СО-,-е-, -c(oH)f-, II :NOH или их солей. Способ заключается в том, что соединение общей формулы II Hal R-O-C-Y-R где R , R , R и Y имеют указанные выще значения и На означает галоген.
подвергают взаимодействию с 1,2, а30лом общей формулы ИI
jf-ll lj,.N.
I
н
при 20-150°С, лучше 80-120°С, в среде полярного растворителя в присутствии связывающего кислоту агента,с последующим выделением нелевого продукта в виде основания или соли.аИ означает галоген, предпочтительно хлор или бром.
Для образования соли соединений Ij2, 4 триазола формулы 1 используются следующие кислоты:
галогепБодородлью, например хлористои бромйстоводородные кислоты, в особен- НОСТ11 хлорпстг,)водородпая, фосфорная кислота, моно- и бнфункдиоиальвая карбо1и вая кислота и оксикарбоиовые кислоты, например уксусная, малеиповая, янтарная, фумаровая, винная, лимонная, салишышьая, сорбиновая, молочная, 1;5 Нафталипдисуль- фоновая кислоты.
В качестве растворителей пспольоуют полярные органические растворители такие как нитрилы, например ацетонитрил, днметилсульфоксид, формалиды, например диметилформамнд, кетоиы такие как ацетон, эфи- ры, например диэт шовый эфир и тетрагидро фураи, нитроалканы, например нитрометац, и несимметричные хлоруглеводороды, такие как хлористый метилен и хлорофо зм,
В качестве свяа тощего кислоту вещест ва используют избыток 1,2,4 триазола. Для этой же цели- используют органически основания, такие как третишн е алкиламины или аралкиламины, нанриктер трп.этлл™ амин или диметилбензилампн.
Реакцию проводят в широком интервале температур от 20 до 150 С, предио.чтительно при 8О-120°С.
На 1 моль соединения II бе™ рут предпочтительно, 1 моль Ij2,4триазола и 1 моль связующего кислоту вещества. Для выделения соед))1ий формулы 1 ., растворитель выпаривают в вакууме и остаток растворяют в оргаии™ ческом растворителе. Затем следует экотракдия водой для удаления образовавше-гося 1,2,4 триазолипгидрохлорица и упаривание раствора воздуха. Из остатка полу чают осиоы1ние после перекристал; И.заш-1И, соль нолучают обработкой оснований, сосп ветствующей кислотой обычными методами
Полученные П1юдлагаемьи4 способом .соединения формулы 1 6i.iTb обычными методами переведены в фушщиональ
ные производные (кетогруппы) и/иля в их соли.
Пример 1,
О
N
.,0- Н-СО-Ц Нз)з
11,2 г (0,033 моль) 1-брогл-1-(2, 4 -дихлорфенокси)-3,Зг-диметилбута№-2она и 9,9 г (О,13 моль) 1,2,4-триазола
растворяют в 80 мл ацетонитрила и нагревают в течение 48 ч с обратны 1 холодильником. Затем растворитель отгоняют в вакууме, остаток поглощают в 150 мл воды и водный раствор три раза извлекают путем встряхивания с метиленхлоридом в К5 личестве каждый раз по 40 мл. Органическую базу два раза промывают водой в количестве 150 мл каждый раз, сущат над суль(1)атом натрия, растворитель упаривают. Пол5ченное в остатке масло перекристаллизовывают из небольщого количества эфира, причем сначала получают 1 г продукта с т. пл. который не был идентифицирован как побочный .кт, а затем 7,6 г (70% от теоретического) ,2,4-триазолил-(1 ) -1--(2,
дих.лор)фенокси1-3,3-диметилбутан4-о,
.с, т. пл. . Исходное соединение
Бг J
f у н-со-(1,1енз);
получают следующим образом,
К полученному из 32,6 г (0,2 моль) 2,4--дихлорфенола и 4,6 г (О,2 моль) натрия в 130 мл абсолютного спирта, 2,4дихлорфеноляту натрия прикапывают 35,8 г (0,2 моль) Xt -.бромпииаколона в 5О мл эфира уксусной кислоты и в течение ночи нагревают до кипения. Образовавшийся бромид натрия отфильтровывают в горячем состоянии, фильтрат упаривают в вакууме и твердый остаток перекристаллизовывают из небольшого количества лигроина.
Получают 38 г (73% от теоретического) 1(2,4 -дихлорфенокси)3,3-димети№бутан-2-она с т. пл. 65С.
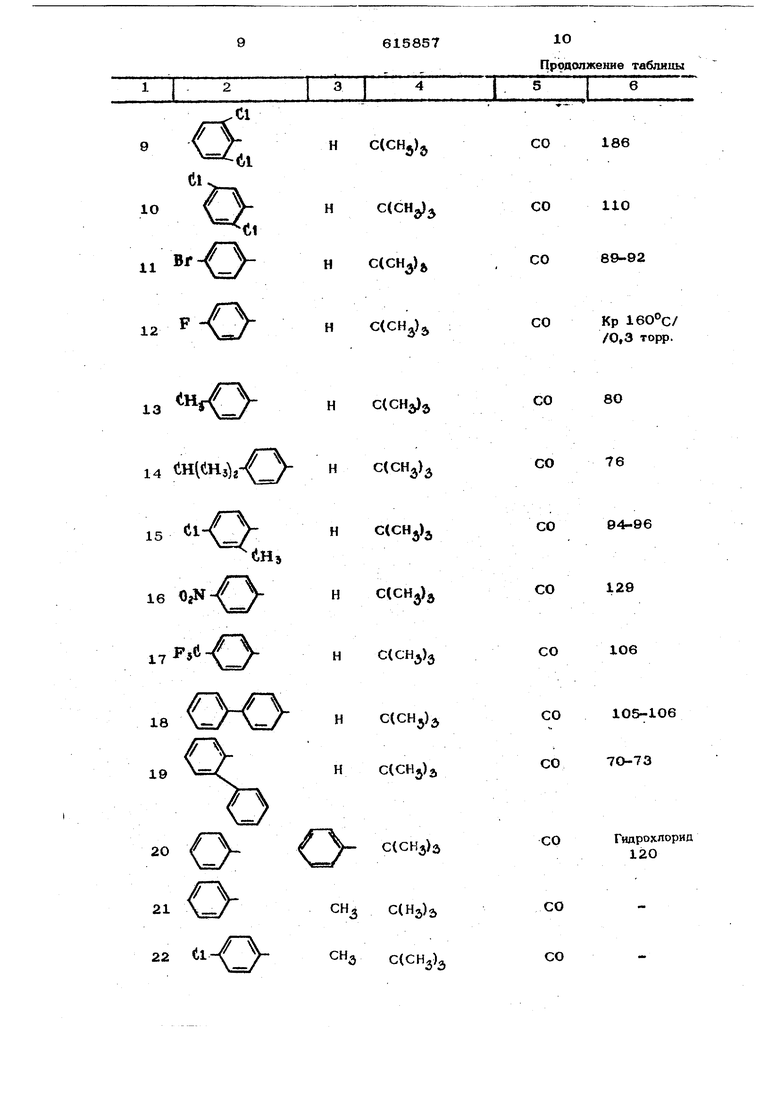
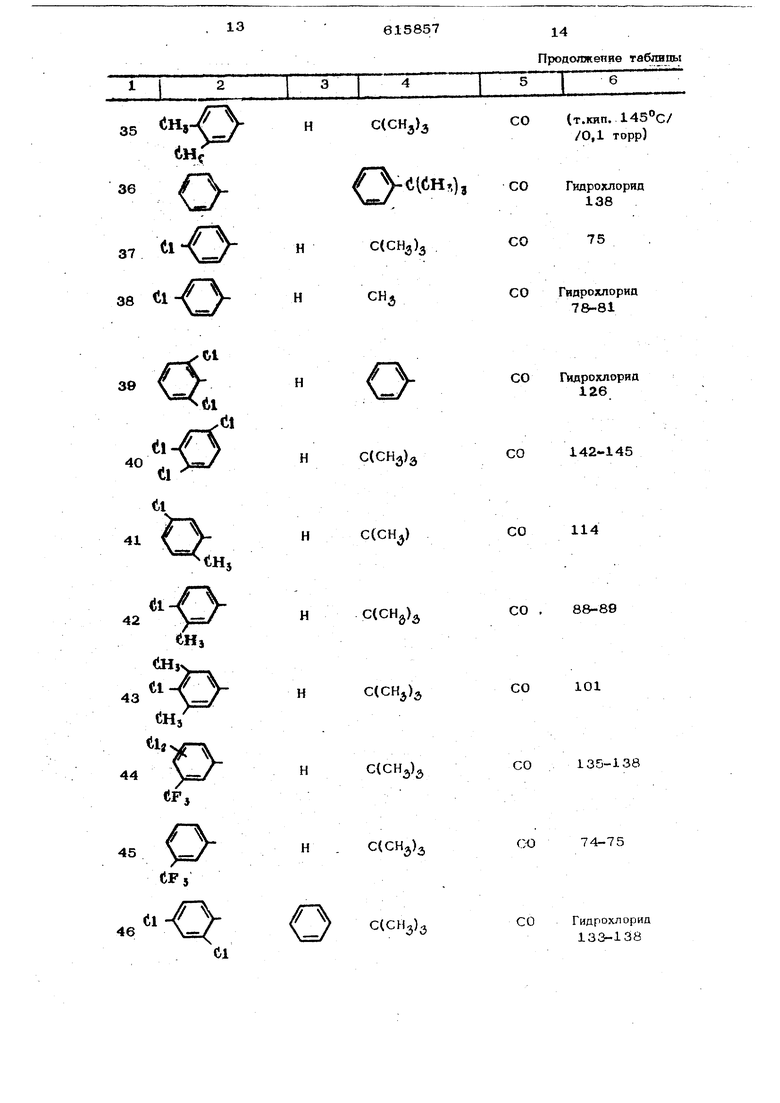
К 26,1 г (0,1 моль) 1-(2,4-дихло{ фенокси)-3,3-ДИметш1бутан-2-она добавляют 6 мл (0,11 моль) брома и смесь н&Гревают в течение 1 ч с обратным холодильником до . Полученный маслянистый остаток растворяют в петролейном эфире, причем он кристаллизуется; твердый остаток отфильтровывают и хорошо промывают. Получают ЗО г (89% от теоретического) 1-бром-1-(2,4-йихлорфенокси)-Э -аиметилбутан-2-она ст. пл. . Гидрохлорид U о- CH-d/o-CCdHjjj -Hcii получают следующим образом. ,2,4-триазолил-{1 )( 2 ,4-дихлор)феноксиЗ-3,3-диметилбутан-2он суспендируют в безводном эфире н добавляют эфирную соляную кислоту. При этом наступает постепенно растворение. Эфир отгоняют в вакуу.е. Оставшийся ос;таток перекристаллизавывают из изонропанола. Полученный гидрохлорид ,2,4-триазолил-{1 ) -1-(2 ,4 - дихлорфенокси)-3-диметилбутан-2-она имеет т, пл. 153 С П р и м е р 2, 18,O г (0,05 моль) 60 -бром-60-(2,б-дихлорфенокси)-ацетофенона и 15 г (0,22 моль) 1,2,4 триазола растворяло т в 12Омл ацетонитрила и нагревают с обратным холодильником в течение 48 ч. После отгонки в вакууме растворителя остаток растворяют в 4ОО мл воды. Этот водный раствор описанным уже методом экстрагир тот метиленхлоридом и органическую фазу, промывают два раза водой в количестве 10О мл каждый раз, затем высушивают над сульфатом натрия и, в вакууме, отгоняют растворитель. Полученный маслянисть й остаток кристаллизуется при нагревании с простым эфиром. После перекристаллизации из этиленхлорида получают 7 г (40% от теоретического) д) ,2,4 триазолил-( 1 )-(2,б-пихлор)-фенокси1 здетофенона с т. Ш1, 166°С. Применяемый в качестве нсход1 ого W-бром-(л)-(2 .6 --дихлор)-феноfvCнадетофонол получают обычными методами - кон- денсаш1ей 2,6-д}1хлорфенола c{) -хлорацетофеноном и бромированием образовавшегс « ся CJ -(2 ,6 дихлор))еноксиацетофенона. Полученный таким образом исходный гфодукт имеет т. пл. 58°С. Пример 3. NО- (l/H-liO-li CiHs) (СНз)/ 30 г (0,12 моль) 1-бром-1- 4- р8Ти-бутил)-фенокси-3,3-диметилбутан-2-1жа и 24 г(О,35моль) 1,2,4-трназола растворяют в 24О мл ацетонитрила и в течение 24 ч нагревают с обратным холодильником. Затем в вакууме отгоняют растворитель, в остаток добавляют ледяную воду и три раза экстрагируют метиленхлорчдом (каждый раз в количестве 4О мл). После отделения органической фазы ее два раза промывают водой в количестве 20О мл каждый раз, сушат над сульфатом натрия и отгоняют растворитель в вакууме. Остаток пе эекристаллизовьшают из лигроина. Получают 26 г ( 69%- от теоретического) ,2,4-триазолил-1 )1-1-|(4 -трет,-бутил) фенокси-3,3 диметил6утан- -2-она с т. пл. И5°С. Применяемый в качестве исходного мг териала 1-бром-1- 4 -трет.-бутил)-фенокси -3,3-диметилиутан-2-он (т. пл. ) получают посредством ксжденса- цни .-бутилфенола с с, - бромпина- колоном-(2) и последующего бромировп- ни я. Пример 4. 4V -1,(Нз)з 19,о Г (О,О5 моль) 1-бром-i-фенил- -1-( 4 - хлорфеноксн)-3,3-димeтилбyтaH- -2-она растворяют в 12О мл ацетонитрила, затем добавляют 12 г (0,175 моль) 1,2,4-трназола и раствор нагревают с обратным холодильником в течение 12 ч. После упаривания растворителя в вакууме добавляют 2ОО мл ледяной воды. В закл очение четыре раза экстрагируют каждый раз 5О мл метиленхлорида, отделяют органическую фазу и трижды промыва ют 5О мл воды. Ее высушивают и растворитель упаривают в вакууме. Маслянистый остаток перекристаллизовывают из лигроина. Получают 5,3 г (29% от теоретическ го) 1-фенил-1(4 -хлорфенокси)1-11,2 4-триаэолип( 1 ),3 метилбутан-2-он с т. пл. 13О°С. Исходное соединение Вг О о-Сн-(1,-е((1нз), нолучают следующим образом. Иа 38 г (0,3 моль) бензилхлорида и 7,3 г (0,3 моль) магния в 300 ми без водного эфира получают реактив Гриньяра К нему при температуре кипения прикапывают 21 г (0,25 моль) пивалонйгрила в 1ОО мл безводного эфира и в течяние 3 ч выдерживают при температуре кипеЕ1ия с обратным холодильником. После охлаждения реакционную смесь выливают в 1,5 л ледяной воды, эфирную фазу отделяют и выбрасывают, а водную фазу размешивают в течение 2 ч на водяной ванне. При этом смесь принимает постепенно маслянистую консистенцию. Масло многократно экстрагируют с 250м метиленхлорида, органическую фазу промывают водой, высушивают и упаривают в вакууме. Получают 40,5 г (92% от теоретического) 1-фенил-3,3-диметилбутан-2 она с т,кип, Kpj 86-88 С. 17,6 г (ОД моль) )енил-3,3-диметилбутан-2-она растворяют в 100 мл четы ре ххлорис того эфира при перемешивании, К раствору и с обратным холодильником прикапывают 5 мл (0,1 моль) и в течение 1 ч нагревают по кипении, После охлаждения и упаривания растворителя получают количественный выход (25,4 г) 1-бром-1-фенил-3,8-диметилбутан-2-она с т, пл. Зв-42 С, Раствор из f25,4 г (0,1 моль) 1-бром-1-фенил-3,3-диметилбутан-2-она в 5О мл эфира уксусной кислоты при кипении прикапывают в раствор из 12,85 г (ОД моль) 4-хлорфенила и 2,3 г (ОД моль) натрия в 1ОО мл этанола. После кипячения с обратным холодильником в течение 12ч отфильтровывают в горячем состоянии от выделившегося бромида натрия. Фильтрат упаривают в вакууме, и оставшийся твердый остаток перекристаллизовывают из лигроина. Получают 20,2 г (67% от теоретического) 1-бром-1-фенил-1-(4 -хлорфенокси)-д -3,3-диметилбутан-2 она с т.пл.ЮЗ С. Аналогично примерам 1-4 получают приведенные в следующей таблице соединения.




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ борьбы с грибками | 1973 |
|
SU648045A3 |
| Способ получения производных 1,2,4--триазола | 1975 |
|
SU556727A3 |
| АЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОТИВОГРИБКОВАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ, СПОСОБ ИНГИБИРОВАНИЯ РАЗВИТИЯ И/ИЛИ РАЗРАСТАНИЯ ГРИБОВ У ТЕПЛОКРОВНЫХ ЖИВОТНЫХ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2156764C2 |
| Способ получения производных имидазола или их солей | 1975 |
|
SU552027A3 |
| Способ получения производных 1,4-диазепина | 1980 |
|
SU1056904A3 |
| Способ получения производных 1,2,4-триазола или их солей | 1975 |
|
SU577988A3 |
| Способ получения производных триазолбензодиазепинов | 1972 |
|
SU444371A1 |
| Замещенные 4-(азол-1-илметил)-1-фенил-5,5-диалкилспиро-[2.5]октан-4-олы, способ их получения (варианты), фунгицидная и рострегуляторная композиции на их основе | 2016 |
|
RU2648240C1 |
| Способ получения производных диазепина | 1972 |
|
SU472505A3 |
| Способ получения производных диазепина | 1973 |
|
SU482045A3 |
Н С( СН J 65-70 101-104
9
22 U1
СНд С(СН.)
10
615857
Продолжение таблицы
СО
ЗЭ
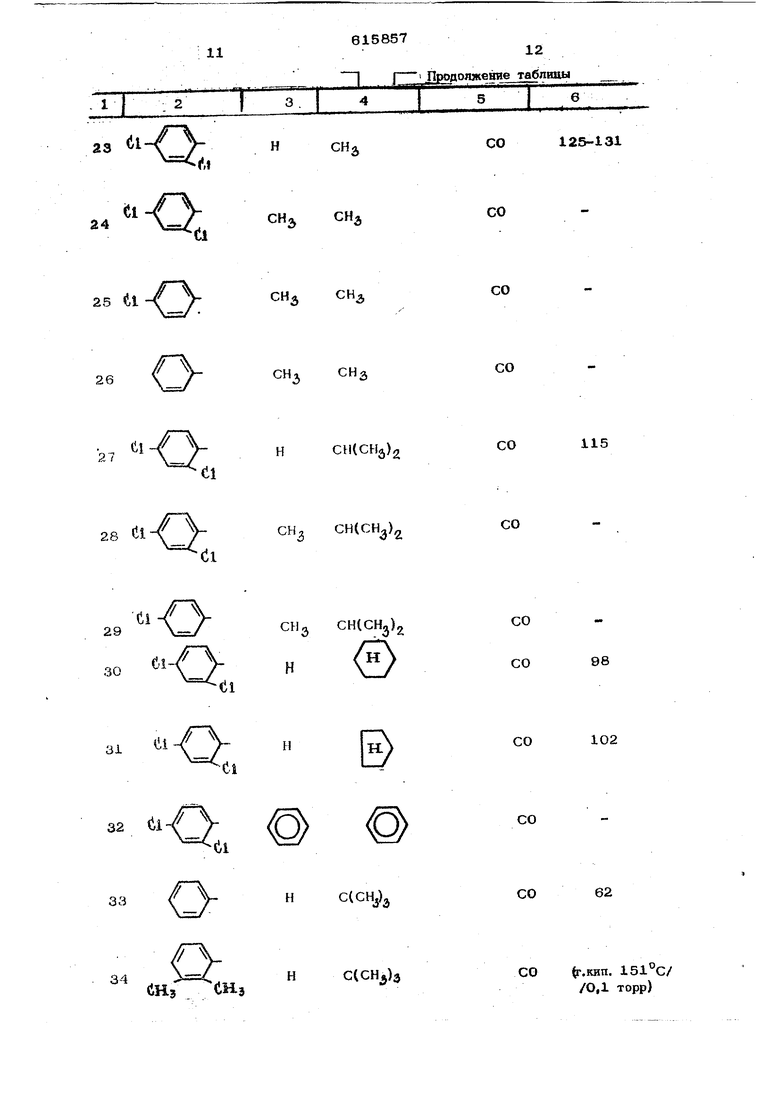
11
TIZI
ei
23
НСН
Л|
tl
СНаCHi
24
ei
25 ul
CHiСН
26
НСИ(СН)2
27
dl
28 «il- -СН СН(СН)
Ci
Ci
CHg CHICH)
H
ei
ti
31
615857
12 дояжсние таблицы
6
со125-131
СО
СО
СО
СО115
СО
CO
CO98
в
CO1O2
32 i
ei
CO
C(CH)
H
33
ен,,
CO62
CO tf.KHn. 151°C/ /0,1 Topp)
Продолжение таботпы
Формула ис)обретения1
N
-а
,N
Ж
О
,
в которой TJ - фенил, незамещенный ил замвщв 1ный галоидом, алкилом, с 1-4 атомами углерода, галоидалкилом с 1-4 атомами углерода, нигрогруппой, аминогруппой, метоксигруппой, фенилом;
R - водород, алкил с 1-4 атомами углерода, фенил{
- алкил с 1-4 атомами углерода, циклоалкил 5-6 атомами углерода, фенил, галоидфенил;
Y- -ео-,-с-, -c oHij-, в
NOH
или их солей, отли-чающийся тем, что соединение общей формулы И Hdt
R-O-U-Y-R
к
.
.t
где, 1 , R . У имеют указанные выше значения и HotR -означает галоген, подвергают взаимодействию с 1,2,4-тоиазопом общей формулы 1И
V
н
при 20-150С в среде полярного растворителя в присутствии связывающего кислоту агента с последующим выделением целевого продукта в виде основания или соли.
Источники информации, принятые, во Внимание при экспертизе;
