Изобретение относится к новым производным аминокислоты с ценными биологическими свойствами и к фармацевтической композиции на их основе.
Известны производные аминокислоты, например, 2-[3(S)- [[N-(бензилокси-карбонил)-L-аспарагинил] амино-2-(R)-гидрокси- 4-фенилбутил]-N-фенилбутил]-N-трет.бутил-декагидро(4aS, 8aS)- изохинолин-3(S)-карбоксамид, которые могут представлять собой активное вещество фармацевтической композиции, обладающей антивирусной активностью, в частности ингибирующей протеазу HIV активностью (см. например, EP 0432695 A2, МПК C 07 D 217/26, A 61 K 31/47, 1991 г.).
Задачей изобретения является расширение ассортимента производных аминокислоты с биологической активностью, в частности обладающей антивирусной активностью, преимущественно ингибирующей протеазу HIV активностью.
Данная задача решается предлагаемыми производными пипеколиновой кислоты общей формулы (I)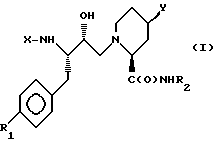
где X - группы R3OC(O), где R3 означает низший алкил или бензил, R4C(O), где R4 означает фенил, замещенный низшим алкилом и/или галоидом, R5OCH2C(O), где R5 означает фенил, незамещенный или одно-, дву- или трехкратно замещенный низшим алкилом,  где R8 означает водород, низший алкил, карбамилметил. (N-метил)карбамилметил или гидроксиметил, а A означает группу R9-C(O), где R9 означает низший алкилокси, бензилокси, нафталенил или хинолинил, или R6NR7C(O), где R6 означает пиридинилметил, а R7 - низший алкил,
где R8 означает водород, низший алкил, карбамилметил. (N-метил)карбамилметил или гидроксиметил, а A означает группу R9-C(O), где R9 означает низший алкилокси, бензилокси, нафталенил или хинолинил, или R6NR7C(O), где R6 означает пиридинилметил, а R7 - низший алкил,
R1 - водород или галоид,
R2 - низший алкил,
Y - циклогексил, фенил, бензил, или группа W(CH2)nZ, где W означает оксо, тио или сульфонил, Z - фенил, незамещенный или монозамещенный галоидом, пиридинил или пиримидинил, незамещенный или замещенный низшим алкилом, n - 0 или 1,
и их солями, в частности терапевтически приемлемыми кислотно-аддитивными солями.
Предпочтительными являются соединения формулы (I) где X означает группу 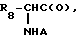 где R8 означает низший алкил, карбамилметил, или гидроксиметил, а A означает группу R9C(O), где R9 означает бензилокси, 2-нафталенил или 2-хинолинил, R1 - водород или фтор, R2 - 1,1-диметилэтил, Y - фенил, бензил, фенокси, 2-пиридинилокси, 2-пиримидинилокси, (2,6-диметил-4-пиримидинил) окси, 2-пиридинилметокси, 3-пиридинилметокси, 4-пиридинилметокси, фенилтио, фенилсульфонил, 2-пиридинилтио, 3-пиридинилтио, 4-пиридинилтио, 2-пиримидинилтио, (4,6-диметил-2-пиримидинил)тио, (2-пиридинилметил)тио, (3-пиридинилметил)тио или (4-пиридинилметил)тио и их терапевтически приемлемые кислотно-аддитивные соли.
где R8 означает низший алкил, карбамилметил, или гидроксиметил, а A означает группу R9C(O), где R9 означает бензилокси, 2-нафталенил или 2-хинолинил, R1 - водород или фтор, R2 - 1,1-диметилэтил, Y - фенил, бензил, фенокси, 2-пиридинилокси, 2-пиримидинилокси, (2,6-диметил-4-пиримидинил) окси, 2-пиридинилметокси, 3-пиридинилметокси, 4-пиридинилметокси, фенилтио, фенилсульфонил, 2-пиридинилтио, 3-пиридинилтио, 4-пиридинилтио, 2-пиримидинилтио, (4,6-диметил-2-пиримидинил)тио, (2-пиридинилметил)тио, (3-пиридинилметил)тио или (4-пиридинилметил)тио и их терапевтически приемлемые кислотно-аддитивные соли.
В частности предпочитаются соединения формулы (I), где X означает (2-метилфенокси)ацетил, (2,6-диметилфенокси)ацетил или (2,4,6-триметилфенокси)ацетил, R1 - водород, R2 - 1,1- диметилэтил, Y - фенил, бензил, фенокси, 2-пиримидинилокси, 2-пиридинилметокси, 3-пиридинилметокси, 4-пиридинилметокси, фенилтио, фенилсульфонил, 2-пиридинилтио, 3-пиридинилтио, 4-пиридинилтио, 2-пиримидинилтио, (4,6-диметил-2-пиримидинил)тио, (2-пиридинилметил)-тио, (3-пиридинилметил)тио или 4-(пиридинилметил)тио, и их терапевтически приемлемые кислотно-аддитивные соли.
Благодаря вышеуказанной биологической активности соединений формулы (I) другим объектом изобретения является фармацевтическая композиция, обладающая ингибирующей протеазу HIV активностью, содержащая по меньшей мере один фармацевтически приемлемый носитель и по меньшей мере одно производное пипеколиновой кислоты вышеприведенной общей формулы (I) или его терапевтически приемлемую кислотно-аддитивную соль в качестве активного вещества в эффективном количестве.
Производные пипеколиновой кислоты вышеприведенной общей формулы (I) можно получать следующими известными способами.
(а) Эпоксид формулы (II)
где X и R1 имеют вышеуказанные значения,
подвергают взаимодействию с пиперидинкарбоксамидом формулы (III)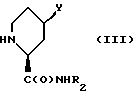
где R2 и Y имеют вышеуказанные значения.
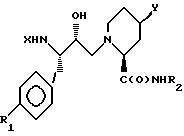
(б) Соединение формулы (IV)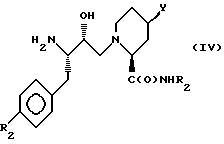
где R1, R2 и Y имеют вышеуказанные значения,
подвергают взаимодействию с реакционноспособным производным карбоновой кислоты формулы X-OH, где X означает группу R5OCH2C(O), где R5 имеет вышеуказанные значения.
(в) Соединение вышеприведенной формулы (IV), где R1, R2 и Y имеют вышеуказанные значения, подвергают взаимодействию с соответствующей α- аминокислотой.
Получаемое согласно реакциям (а) - (в) соединение формулы (I) можно переводить в соответствующую терапевтически приемлемую кислотно-аддитивную соль.
Следует отметить, что соединение общей формулы (I), где X означает обычно используемую N-защитную группу, такую, как, например, трет.-бутилоксикарбонил, бензилоксикарбонил, 9-флуоренил-метоксикарбонил или п-метоксибензилоксикарбонил, целесообразно получают согласно реакциям (а) и (в). Получаемое при этом соединение представляет собой промежуточный продукт для получения по реакции (б) соединений общей формулы (I), где X имеет значение, отличное от обычно применяемой N-защитной группы. Поэтому данное промежуточное соединение общей формулы (I) подвергают снятию защитной группы и получаемый при этом N-концевой свободный амин применяется в качестве исходного соединения общей формулы (IV) или (V).
Вышеуказанную реакцию (а) можно проводить в среде инертного растворителя, например, этанола, тетрагидрофурана или диметилсульфоксида, при температуре 20 - 110oC. Время реакции зависит от температуры и природы реагентов формул (II) и (III). Она обычно составляет 2 - 24 часов.
Подходящими для осуществления реакции (б) реакционноспособными производными карбоновой кислоты формулы X - OH являются агенты ацилирования, способные к отдаче подходящего ацила X - CO. Примерами являются соответствующие галоидангидриды, предпочтительно хлор- или бромангидриды, активные сложные эфиры, ангидриды или смешанные ангидриды. Реакцию осуществляют известными приемами и в известных условиях. Так, например, реакцию обычно осуществляют в среде инертного растворителя, такого, как, например, тетрагидрофуран, диметилформамид или хлористый метилен, при температуре 0 - 50oC в течение 15 минут до 24 часов.
Для осуществления реакции (в) применяют известные агенты сочетания, которые содействуют дегидратационному сочетанию свободной карбоксильной группы одного реагента со свободной аминогруппой другого реагента. В качестве примеров подходящих агентов сочетания можно назвать 1,1'-карбониддиимидазол, N, N'-дициклогексилкарбодиимид, 1-окси-бензотриазол в сочетании с N,N'-дициклогексилкарбодиимидом или N-этил-N' -[(3,3'-диметиламино)пропил]карбодиимидом. В качестве агента сочетания целесообразно применять легкодоступный гексафторфосфат (бензотриазол-1-илокси)трис-(диметиламино)фосфония, при необходимости в сочетании с 1-оксибензотриазолом. Другим целесообразным агентом сочетания является легкодоступный фторборат 2-(1H-бензотриазол-1-ил)-N,N, N'N'-тетраметилурония.
Реакцию сочетания осуществляют в среде инертного растворителя, например, хлористого метилена, ацетонитрила или диметилформамида. Для поддержания реакционной смеси на pH примерно 8 добавляют избыток органического амина, например, диизопропилэтиламина или N-метилморфолина. Реакцию обычно осуществляют при температуре от -20oC примерно до +30oC в течение 15 минут до 8 часов.
Эпоксид формулы (II), применяемый для осуществления реакции (а), либо известен, либо может получаться известными методами.
Другие исходные соединения для осуществления вышеприведенных способов, например, пиперидинкарбоксамиды общей формулы (III) и соединения формулы (IV), являются новыми. Соединения общей формулы (IV) можно получать указанным выше образом. Пиперидинкарбоксамиды формулы (III) можно получать путем взаимодействия подходящего 4-замещенного пиперидина с реагентом, позволяющим ведение карбоксамидной функции в положение 2 пиперидина. Подходящий способ получения пиперидинкарбоксамида общей формулы (III) иллюстрируется соответствующими примерами, представленными ниже.
Как уже указывалось выше, соединения общей формулы (I) могут получаться в виде терапевтически приемлемой кислотно-аддитивной соли. Примерами являются соли с органическими кислотами, такими, как, например, уксусная, молочная, янтарная, бензойная, салициловая, метансульфоновая и п-толуолсульфоновая кислоты, а также с полимерными кислотами, такими, как, например, дубильная кислота или карбоксиметилцеллюлоза, а также соли с органическими кислотами, такими, как, например, галоидводородные кислоты, например, хлористоводородная кислота, серная кислота или фосфорная кислота. В случае необходимости определенную кислотно-аддитивную соль можно переводить в другую кислотно-аддитивную соль, такую, как, например, нетоксичная фармацевтически приемлемая соль, путем обработки подходящим ионитом известными приемами.
Для борьбы с инфекциями, вызываемыми вирусом HIV, соединения общей формулы (I) или их терапевтически приемлемые соли можно применять орально, локально или парентерально. При этом соединения применяются в сочетании по меньшей мере с одним фармацевтически приемлемым носителем, количество которого определяется растворимостью и химической природой активного вещества, методом аппликации и требованиями биологической практики. Для оральной аппликации соединение формулы (I) или его терапевтически приемлемую соль применяют в виде дозировочных единиц, таких, как, например, капсулы или таблетки, которые содержат определенное количество активного вещества, например, 5 - 150 мг, а также фармацевтически приемлемый носитель. Для местной аппликации соединение общей формулы (I) или его соли применяют в виде препаратов, содержащих 0,01 вес.%, предпочтительно 0,05 - 1 вес.% активного вещества, остаток - фармацевтически приемлемый носитель. Такими препаратами являются, например, кремы, лосьоны, подъязычные таблетки или же предпочтительно трансдермальные или ротовые препараты для парентеральной аппликации. Соединение общей формулы (I) или его соли применяют в сочетании с фармацевтически приемлемым носителем путем внутривенной, подкожной или внутримышечной инъекции. Если впрыскивать, то соединение предпочтительно применяют в виде раствора в стерильной водной среде, которая может также содержать другие растворенные вещества, такие, как, например, буферные вещества или консерванты, а также достаточное количество фармацевтически приемлемых солей или глюкозы с тем, чтобы раствор был изотоническим.
Подходящие вспомогательные вещества или носители для приготовления вышеупомянутых препаратов можно найти в стандартной фармацевтической литературе, например, в "Remington's Pharmaceutical "Sciences", 18-ое издание, издательство Mack Publishing Company, г. Истон, штат Пенсильвания, США, 1990 г.
Доза активного вещества зависит от вида аппликации и его природы. Кроме того, она также зависит от общего состояния пациента. В начале процесса лечения обычно дают дозы активного вещества, которые существенно меньше оптимальной дозы. Затем дозы постепенно увеличивают до достижения оптимального действия в каждом конкретном случае. Новые соединения в общем дают в концентрациях, которые обеспечивают антивирусное действие без отрицательных или вредных побочных эффектов.
В случае оральной аппликации новые соединения и их терапевтически приемлемые соли дают в количестве 0,5 - 15 мг/кг веса тела в сутки, предпочтительно 0,5 - 5 мг/кг/сутки. В случае системных аппликаций новые соединения формулы (I) дают в дозах от 1 - 100 мкг/кг/ веса тела в сутки.
Хотя вышеуказанные препараты представляют собой эффективное и относительно безопасное средство для лечения инфекций, вызываемых вирусом HIV, возможность совместной аппликации этих препаратов с другими антивирусными средствами или агентами для получения дополнительных положительных результатов не исключена. Такими другими антивирусными средствами или активными веществами являются растворимый протеин CD4, зидовудин, диданозин, зальцитабин, фосфоноформиат тринатрия, рибаварин, ацикловир или антивирусные интерфероны, например, α- интерферон или интерлейкин-2.
Нижеследующие примеры далее поясняют данное изобретение. Если ничего другого не указано, то %-ные данные или соотношения по составу растворов относятся к объему. Для проведения спектров ЯМР применялся спектометр фирмы Брукер мощностью 200 МГц, причем химические сдвиги, (δ) указаны в млн.долях.
Пример 1.
Получение 1-(трет.-бутилоксикарбонил)-4-(фенилтио)пиперидина
Раствор 3,0 г (14,9 ммоль) 1-(трет.-бутилоксикарбонил)-4- пиперидинола в 30 мл тетрагидрофурана охлаждают до температуры 0oC, после чего последовательно добавляют 3,2 мл (1,5 эквивалента) триэтиламина и 1,26 мл (1,1 эквивалента) метилсульфонилхлорида. Реакционную смесь перемешивают при температуре 0oC в течение двух часов, после чего добавляют 30 мл диэтилового эфира и 20 мл воды и получаемую смесь перемешивают при температуре 0oC в течение дополнительных 30 минут. Реакционную смесь разбавляют 200 мл диэтилового эфира, органический слой последовательно промывают водой, 10%-ной водной лимонной кислотой, два раза насыщенным водным раствором бикарбоната натрия и солевым раствором. В результате сушки органического слоя над сульфатом магния и сгущения под пониженным давлением получают 4,0 г (96%) метилсульфоната 1-(трет. -бутилоксикарбонил)-4- пиперидинола в качестве желтоватого твердого вещества.
1H ЯМР (CDCl3) δ 4,90 (м, 1H), 3,72 (ддд, J = 4,3, 6,5, 13,5 Гц, 2H), 3,32 (ддд, J = 4,3, 8,1, 13,5 Гц, 2H), 3,05 (с, 3H), 1,47 (с, 9H).
Получаемый таким образом метилсульфонат далее используют без дальнейшей очистки. 1,84 мл (17,9 ммоль) тиофенола медленно добавляют к суспензии 334 мг (14,3 моль) гидрида натрия в 8 мл диметилформамида при температуре 0oC. Смесь перемешивают в течение 20 минут, после чего добавляют раствор 2,0 г (7,17 ммоль) метилсульфоната в 6 мл диметилформамида и получаемую смесь перемешивают при комнатной температуре (20 - 22oC) в течение 18 часов. Смесь разбавляют диэтиловым эфиром, органическую фазу последовательно промывают 1 м. водным раствором гидроокиси натрия (три раза) и солевым раствором, сушат над сульфатом магния и сгущают досуха под пониженным давлением. В результате очистки остатка флеш-хроматографией на двуокиси кремния с применением в качестве элюента смесей этилацетата и гексана в соотношениях 1 : 9 и 1 : 6 получают 1,82 г (86%) вышеуказанного целевого продукта в качестве масла, которое затвердевает при стоянии.
1H ЯМР (CDCl3) δ 7,48 - 7,2 (2м, 2H + 3H), 3,97 (м, 2H), 3,22 (м, 1H), 2,80 (ддд, J = 3,8, 10,5, 13,5 Гц, 2H), 1,47 (с, 9H).
Масс-спектр (бомбардировка быстрыми атомами), m/z: 294 (М + H)+.
Пример 2.
Получение d, 1-цис-N-трет. -бутил-1-(трет. -бутилоксикарбонил) -4-(фенилтио)пиперидин-2-карбоксамида
Раствор 3,57 г (12,2 ммоль) целевого соединения примера 1 в 60 мл диэтилового эфира охлаждают до температуры 78oC. К охлажденному раствору добавляют 4,6 мл (2,5 эквивалента) N,N,N',N'-тетраметилендиамина с последующей порционной добавкой 12,0 мл (1,3 эквивалента) 1,3 м. втор.- бутиллития в циклогексане. Получаемую смесь перемешивают в течение 3,5 часа при температуре - 78oC, после чего быстро добавляют 2,1 мл (1,5 эквивалента) трет.-бутилизоцианата и реакционную смесь перемешивают в течение 40 минут при температуре - 78oC. К реакционной смеси добавляют 10%-ную водную лимонную кислоту, после чего ей дают нагреваться до комнатной температуры. Органическую фазу отделяют и водную фазу экстрагируют диэтиловым эфиром. Объединенные органические фазы промывают насыщенным водным раствором бикарбоната натрия и солевым раствором, сушат над сульфатом магния и упаривают под пониженным давлением. Остаток очищают путем флеш-хроматографии на двуокиси кремния с применением в качестве элюента смесей гексана и этилацетата в соотношениях 6 : 1 и 4 : 1, в результате чего получают 4,34 г (90%) целевого соединения в качестве бесцветного масла, которое затвердевает при стоянии.
1H ЯМР (CDCl3) δ 7,42 (м, 2H), 7,28 (м, 3H), 5,85 (широк, с, 1H), 4,43 (дд, J = 4,0, 7,0 Гц, 1H), 3,92 (ддд, J = 3,5, 5,0, 13,5 Гц, 1H), 3,49 (м, 1H), 3,32 (ддд, J = 4,0, 11,5, 13,5 Гц, 1H), 1,48 (с, 9H), 1,39 (с, 9H).
Масс-спектр (бомбардировка быстрыми атомами), m/z: 393 (М + H)+.
Пример 3.
Получение d, 1-цис-N-трет. -бутил-1- (трет.-бутилоксикарбонил)-4-(2-пиридинилокси)пиперидин-2- карбоксамида
Раствор 5,2 г (25,9 ммоль) 1-(трет.-бутилоксикарбонил)-4- пиперидинола, 4,07 г (1,05 эквивалента) трет.-бутилдиметилсилилхлорида и 2,7 г (1,5 эквивалента) имидазола в 20 мл диметилформамида перемешивают в течение 16 часов. Разбавляют диэтиловым эфиром, получаемый раствор последовательно промывают два раза водой, 10%-ной водной лимонной кислотой, насыщенным водным раствором бикарбоната натрия и солевым раствором. Органический слой сушат над сульфатом магния и сгущают досуха. Остаток очищают высокопроизводительной жидкостной хроматографией, осуществляемой в двух колонках на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношении 19 : 1 (используют аппарат WA TERS LC-500 фирмы Millipore Corporation, г. Мильфорт, штат Массачьюсетс, США), в результате чего получают 7,54 г (92%) 1-(трет.- бутилоксикарбонил)-4-(трет.-бутилдиметилсилокси)пиперидина.
1H ЯМР (CDCl3) δ 3,87 (м, 1H), 3,61 (ддд, J = 3,5, 7,5, 13,0 Гц, 2H), 3,24 (ddd, J = 3,7, 8,0, 13,0 Гц, 2H), 1,48 (с, 9H), 0,88 (с, 9H), 0,07 (с, 6H).
Далее работают аналогично примеру 2, в результате чего получают d,1-цис-N-трет. -бутил-1-(трет. -бутилоксикарбонил)- 4-(трет.-бутилдиметилсилокси)пиперидин-2-карбоксамид.
1H ЯМР (CDCl3) δ 5,70 (с, 1H), 4,47 (дд, J = 2,7, 8,0 Гц, 1H), 4,07 (м, 1H), 3,83 (м, 1H), 3,22 (ддд, J = 5,4, 10,5, 13,5 Гц), 1,48 (с, 9H), 1,35 (с, 9H), 0,88 (с, 9H), 0,1 и 0,08 (2с, 6H).
К раствору 700 мг (1,69 ммоль) полученного карбоксамида в 10 мл тетрагидрофурана добавляют раствор 2,15 мл (1,25 эквивалента) 1 м. фторида тетрабутиламмония в тетрагидрофуране. Реакционную смесь перемешивают при комнатной температуре в течение 30 минут, после чего разбавляют диэтиловым эфиром, получаемую смесь промывают два раза водой и раз солевым раствором, органический слой сушат над сульфатом магния и сгущают досуха под пониженным давлением. Остаток очищают флеш-хроматографией на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношении 1 : 1, в результате чего получают 386 мг (76%) d,1-цис-N-трет.-бутил-1- (трет.-бутилоксикарбонил)-4-оксипиперидин-2-карбоксамида в качестве белого твердого вещества.
Масс-спектр (бомбардировка быстрыми атомами), m/z: 301 (М + H)+.
К охлажденному до температуры 0oC раствору 220 мг (0,73 ммоль) вышеуказанного карбоксамида, 244 мг (2,0 эквивалента) 4- нитробензойной кислоты и 288 мг (1,5 эквивалента) трифенилфосфина в 13 мл смеси бензола и тетрагидрофурана в соотношении 5 : 1 добавляют 173 мкл (1,5 эквивалента) диэтилазодикарбоксилата. Получаемую реакционную смесь перемешивают при температуре 0oC в течение 30 минут и затем при комнатной температуре в течение трех часов. Растворитель удаляют под пониженным давлением, остаток очищают флеш-хроматографией на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношении 4 : 1, в результате чего получают 280 мг d, 1-цис-N-транс-N-трет. -бутил-1- бутилоксикарбонил)-4-(4-нитробензоилокси)-2-карбоксамида, содержащего примерно 25% примеси. Этот карбоксамид далее используют без дальнейшей очистки.
Смесь 404 мг (0,9 ммоль) вышеуказанного карбоксамида и 28 мг (0,2 эквивалента) карбоната калия в 9 мл метанола перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют под пониженным давлением, остаток растворяют в хлороформе, получаемый раствор промывают водой, сушат над сульфатом магния и сгущают досуха под пониженным давлением. Остаток очищают флеш-хроматографией на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношениях 1 : 1 и 1 : 2, в результате чего получают 194 мг (71%) d, 1-транс-трет.-бутил-1- (N-трет.-бутилоксикарбонил)-4-оксипиперидин-2-карбоксамида.
Раствор 145 мг (0,48 ммоль) полученного карбоксамида, 68 мг (1,5 эквивалента) 2-оксипиридина и 187 мг (1,5 эквивалента) трифенилфосфина в 12 мл смеси бензола и тетрагидрофурана в соотношении 5 : 1 охлаждают до температуры 0oC, после чего добавляют 114 мкл (1,5 эквивалента) диэтилазодикарбоксилата. Реакционную смесь перемешивают в течение 90 минут при температуре 0oC и затем при комнатной температуре в течение 30 минут. Растворитель удаляют под пониженным давлением и остаток очищают путем флеш-хроматографии на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношении 2 : 1. При этом получают 70 мг (38%) вышеуказанного целевого продукта.
1H ЯМР (CDCl3) δ 8,12, 7,43, 6,85 и 6,62 (4 м, 4H), 5,72 (с, 1H), 5,39 (м, 1H), 4,63 (м, 1H), 4,05 (м, 1H), 3,29 (м, 1H), 1,48 (с, 9H), 1,36 (с, 9H).
Пример 4.
Получение (d, 1-цис-М-трет.-бутил-1- (трет.-бутилоксикарбонил)-4-фенилсульфонил)пиперидин-2-карбоксамида
Смесь 1,68 г (4,28 ммоль) целевого продукта примера 2 и 2,2 г (12,83 ммоль) 3-хлорпероксибензойной кислоты в 20 мл хлористого метилена перемешивают при комнатной температуре в течение 18 часов, после чего к реакционной смеси добавляют 10%-ный водный раствор сульфита натрия и разбавляют этилацетатом. Органический слой отделяют, последовательно промывают насыщенным водным раствором бикарбоната натрия, водой и солевым раствором, сушат над сульфатом магния и сгущают под пониженным давлением. Получаемый твердый остаток перемешивают вместе со смесью 18 мл гексана и 12 мл этилацетата и фильтруют. Получают 1,57 г (86%) целевого продукта в качестве белого твердого вещества.
1H ЯМР (CDCl3) δ 7,90 (м, 2н), 7,75 - 7,55 (м, 3H), 5,95 (с, 1H), 4,07 (дд, J = 8,0, 9,5 Гц, 1H), 3,88 (дт, J = 5,4, 13,5 Гц, 1H), 3,32 - 3,05 (м, 2H), 1,45 (с, 9H), 1,35 (с, 9H).
Масс-спектр (бомбардировка быстрыми атомами), m/z: 425 (М + H)+.
Если повторять данный пример с той лишь разницей, что применяют 1-мольный эквивалент 3-хлорпероксибензойной кислоты, то получают d,1-цис-N-трет. -бутил-1-(трет. -бутилоксикарбонил)-4- (фенилсульфинил)пиперидин-2-карбоксамид.
Пример 5.
Получение N-трет. -бутил-1-[3(S)-трет.-бутилоксикарбониламино)-2(R)-окси-4- фенилбутил]-4(R)-(фенилтио)пиперидин-2(S)-карбоксамида
(а) Раствор 3,04 г (7,76 ммоль) целевого продукта примера 2 в 6 н. растворе соляной кислоты в диоксане перемешивают при комнатной температуре в течение 20 минут, после чего сгущают досуха под пониженным давлением. Получают d, 1-цис-N-трет.- бутил-4-(фенилтио)пиперидин-2-карбоксамид, к которому добавляют 50 мл этилацетата и 20 мл водной гидроокиси натрия и получаемую реакционную смесь перемешивают при комнатной температуре в течение 15 минут. Органический слой отделяют, промывают небольшим количеством воды и солевым раствором, сушат над сульфатом магния и сгущают досуха под пониженным давлением. Получаемое масло сушат в высоком вакууме в течение примерно 45 минут. Получаемое масло смешивают с 2,45 г (9,36 ммоль) 3 (S)- (трет.-бутилоксикарбониламино)-1,2(R)-эпокси-4-фенилбутана и 40 мл абсолютного этанола. Получаемую смесь нагревают с обратным холодильником в течение 18 часов, после чего добавляют дополнительное количество эпоксида (600 мг) и дополнительно нагревают с обратным холодильником в течение 4 часов. Реакционную смесь сгущают досуха под пониженным давлением и сырой продукт очищают путем высокопроизводительной жидкостной хроматографии в двух колонках на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношении 6 : 4 (хроматографию осуществляют в вышеуказанном аппарате WATERS LC- 500). При этом получают 1,46 г (34%) целевого изомера в качестве белой пены.
Масс-спектр (бомбардировка быстрыми атомами), m/z: 556 (М + H)+.
Аналогично примеру 5 можно получать другие соединения формулы (I), в которой X, R1, R2 и Y имеют вышеуказанные значения. Так, например, если вместо 3(S)- (трет. -бутилоксикарбониламино)-1,2(R)-эпокси-4-фенилбутана применять эквивалентное количество 3(S)-(трет.-бутилоксикарбониламино)- 1,2(R)эпокси-4-(4-фторфенил)бутана, то получают N-трет. -бутил-1- {3(S)-{ (бензилоксикарбонил)амино} -2-(R)-окси-4-(4- фторфенил)бутил} -4-(R)-(фенилтио)-пиперидин-2(S)-карбоксамид.
Масс-спектр (бомбардировка быстрыми атомами), m/z: 608 (М + H)+].
Кроме того, можно получать еще соединения формулы (I), сведенные в таблице I. В этой таблице также указаны исходное эпокси-соединение формулы (II) и исходное пиперидинкарбоксамид формулы (III), которые применяют в количествах, эквивалентных соответствующим исходным соединениям, применяемым в примере 5.
В таблице I используют следующие условные сокращения:
Бок = бензилоксикарбонил;
БуТ = трет.-бутил;
БТок = трет.-бутилоксикарбонил.
Приведенные в скобках для характеристики целевых продуктов цифры представляют собой результаты масс-спектра в условиях бомбардировки быстрыми атомами.
Пример 6.
A: Получение N-трет.-бутил-1-{3(S)-{{N-(трет.- бутилоксикарбонил)валил} амино}-2(R)-окси-4-фенилбутил}-4(R)- (фенилтио)пиперидин-2(S)-карбоксамида.
Раствор 1,14 г (2,04 ммоль) целевого продукта примера 5 в 10 мл 6 н. соляной кислоты в диоксане перемешивают при комнатной температуре в течение 20 минут, после чего растворитель удаляют под пониженным давлением, белый твердый остаток перемешивают вместе с диэтиловым эфиром, фильтруют и сушат. Получают 1,06 г (98%) соответствующего амина без защитной группы в виде гидрохлорида. 341 мг (0,645 ммоль) этого гидрохлорида растворяют в 3,5 мл хлористого метилена и к получаемому раствору добавляют 225 мкл (1,29 ммоль) диизопропилэтиламина, 145 мг (0,667 ммоль) защищенной трет.-бутилоксикарбонилом - аминоизовалериановой кислоты и 342 мг (0,774 ммоль) гексафторфосфата (бензотриазол-1-илокси)-трис- (диметиламино)-фосфония. Реакционную смесь перемешивают при комнатной температуре в течение 3,5 часа. При этом ее поддерживают по величине pH 8 путем постоянного контроля и добавления требуемого количества диизопропилэтиламина. Затем реакционную смесь разбавляют этилацетатом, последовательно промывают насыщенным водным раствором бикарбоната натрия (2 раза), водой и солевым раствором. Органический слой сушат над сульфатом магния и сгущают под пониженным давлением. В результате очистки остатка путем флеш-хроматографии на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношении 1 : 1 получают 338 мг (80%) вышеуказанного целевого продукта в качестве белого твердого вещества.
Масс-спектр (бомбардировка быстрыми атомами), m/z: 655,3 (М + H)+.
Б: Получение N-трет. -бутил-1-{ 3(S)-{3(S)-{{N-(трет.- бутилоксикарбонил)аспарагинил} амино} -2(R)-окси-4-фенилбутил}- 4(R)-(фенилтио)пиперидин-2(S)-карбоксамида.
1,97 г (14,57 ммоль) 1-оксибензотриазола добавляют к охлажденному до температуры 0oC раствору 16,08 ммоль N,N'- дициклогексилкарбодиимида в 6,7 мл хлористого метилена и 45 мл тетрагидрофурана. Реакционную смесь перемешивают в течение 15 минут, после чего добавляют 3,30 г (14,57 ммоль) защищенной трет. -бутилоксикарбонилом аспарагиновой кислоты и раствор 3,30 г (7,24 ммоль) соответствующего незащищенного амина целевого продукта примера 5 в 40 мл диметилформамида (примечание: незащищенный амин получают аналогично первому абзацу варианта A данного примера с последующим переводом гидрохлорида в свободное основание). Реакционной смеси дают медленно нагреваться до комнатной температуры, после чего перемешивают в течение 18 часов. Затем смесь разбавляют этилацетатом и водой, органическую фазу отделяют, промывают насыщенным водным раствором бикарбоната натрия, водой и солевым раствором, сушат над сульфатом магния и сгущают досуха под пониженным давлением. В результате очистки твердого остатка путем флеш-хроматографии на двуокиси кремния с применением в качестве элюента смеси хлороформа и метанола в соотношении 97,5 : 2,5 получают 3,56 г (73%) вышеуказанного целевого продукта в качестве белого твердого вещества.
Масс-спектр (бомбардировка быстрыми атомами), m/z: 670 (М + H)+.
Аналогично примеру 6 можно получать другие соединения формулы (I). Так, например, если повторять вариант A данного примера с той лишь разницей, что вместо целевого соединения примера 5 применяют эквивалентное количество также упомянутого в примере 5 N-трет.-бутил-1-{3(S)-{ (бензилоксикарбонил)амино}-2- (R)-окси-4-(4-фторфенил)бутил}-(R)-(фенилтио)пиперидин-2-(S)- карбоксамида, то получают N-трет.-бутил-1-{3(S)-{ { N- (бензилоксикарбонил)валил} амино)-2(R)-окси-4-(4- фторфенил)бутил} -4(R)-(фенилтио)пиперидин-2(S)-карбоксамид.
Масс-спектр, m/z: 707 (М+H)+}.
Аналогично можно получать соединения, сведенные в таблице II. В этой таблице также указаны исходные соединения, то есть соответствующий целевой продукт таблицы I, и защищенную аминокислоту формулы PG-AA-OH, где PG означает защищающий α- аминогруппу остаток, а AA - указанный аминокислотный остаток. Исходные соединения применяют в количествах, эквивалентных количествам соответствующих исходных соединений данного примера. Приведенные в скобках цифры для характеристики целевых продуктов представляют собой результаты масс-спектра (М + H)+.
Приведенные в таблице II условные сокращения имеют следующие значения:
Бок = бензилоксикарбонил;
БТок=трет.-бутилоксикарбонил;
Asn = аспарагин;
Ile = изолейцин;
Val = валин.
Пример 7.
Получение N-тpeт. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ {N-(2- хинолинилкарбонил)-валил}амино}бутил}-4-(R)-(фенилтио)пиперидин- 2(S)-карбоксамида.
Раствор 167 мг (0,255 ммоль) полученного согласно варианту A примера 6 целевого продукта в 2,0 мл 6 н. соляной кислоты в диоксане перемешивают при комнатной температуре в течение 20 минут, после чего растворитель удаляют под пониженным давлением. Получаемый твердый остаток сушат в высоком вакууме в течение 20 минут с получением соответствующего незащищенного амина в виде гидрохлорида, который растворяют в 2 мл хлористого метилена и к получаемому раствору соли добавляют 89 мкл (0,510 ммоль) диизопропилэтиламина, 48,6 мг (0,280 ммоль) 2-хинолинкарбоновой кислоты и 135 мг (0,306 ммоль) гексафторфосфата (бензотриазол-1-ил-окси)трис-(диметиламино)-фосфония. Реакционную смесь перемешивают при комнатной температуре в течение 3,5 часа. При этом pH смеси поддерживают при 8 путем постоянного контроля и добавления требуемого количества диизопропилэтиламина. Затем реакционную смесь разбавляют этилацетатом и последовательно промывают два раза насыщенным водным раствором бикарбоната натрия, два раза водой и солевым раствором. Органический слой сушат над сульфатом магния и сгущают досуха под пониженным давлением. Получаемое бесцветное масло очищают путем флеш-хроматографии на двуокиси кремния с применением в качестве элюента смеси гексана и этилацетата в соотношении 2 : 3. Получают 161 мг (89%) целевого продукта в качестве белого твердого вещества
Масс-спектр (бомбардировка быстрыми атомами), m/z: 710 (М + H)+.
Аналогично примеру 7 или же варианту Б примера 6 можно получать еще соединения формулы (I), сведенные в таблице III.
Пример 8.
Получение N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{{N- {(2-пиридинилметокси)-карбонил} изолейцил} амино}бутил}-4(R)- фенилпиперидин-2(S)-карбоксамида.
N-тpeт. -бутил-1-{ 3(S)-aминo-2(R)-oкcи-4- фeнилбутил}-4(R)-фeнилпипepидин-2(S)-карбоксамид (получаемый гидрированием 0,605 мг (0,108 ммоль) N-трет. -бутил-1-{ 3-(S)- (бензиоксикарбониламино)-2-(R)-окси-4-фенилбутил}-4(R)- фенилпиперидин-2(S)-карбоксиамида (см. целевой продукт N 1 таблицы I) в среде метанола на палладиевом катализаторе (5% Pd/C) при атмосферном давлении в течение двух часов) растворяют в 1,6 мл диметилформамида. К получаемому раствору добавляют 586 мг (0,228 ммоль) литиевой соли N-{(2-пиридинилметокси)карбонил}изолейцина, 32 мг (0,237 ммоль) 1- оксибензотриазола и 45,4 мг (0,237 ммоль) N-этил-N'-{3- (диметиламино)пропил}карбодиимида. Реакционную смесь перемешивают при комнатной температуре в течение 18 часов, после чего ее разбавляют диэтиловым эфиром, последовательно промывают водой, два раза насыщенным водным раствором бикарбоната натрия и солевым раствором, сушат над сульфатом магния и упаривают досуха. Получают желтое масло, которое очищают флеш-хроматографии на двуокиси кремния с применением в качестве элюента смесей хлороформа и метанола в соотношениях 97,5 : 2,5 и 95 : 5, в результате чего получают 58,7 мг целевого продукта в качестве белого твердого вещества.
Масс-спектр (бомбардировка быстрыми атомами), m/z: 672 (М + H)+.
Пример 9.
Получение N-трет. -бутил-1-{ 2(R)-окси-3(S)-{ N-{{N- метил-N-(2-пиридинилметил)-амино} карбонил} -валил} -4- фенилбутил}-4(R)-фенилтио)пиперидин-2(S)-карбоксамида.
9,41 мл (17,89 ммоль) 1,9 м. фосгена в толуоле добавляют к 1,0 г (5,96 ммоль) H-Val-OCH3 • HCl (Val = валин). Реакционную смесь нагревают с обратным холодильником в течение 2 часов с применением конденсатора на сухом льду, охлаждают до комнатной температуры, при интенсивном перемешивании подают азот в течение 90 минут и затем сгущают досуха. К остатку добавляют 5 мл толуола и получаемый раствор сгущают досуха с получением сложного метилового эфира (S)-2-изоцианато-3-метилбутановой кислоты, который сушат в высоком вакууме в течение 5 минут.
1H ЯМР (CDCl3) δ 3,95 - 3,94 (д, J = 3,82 Гц, 1H), 3,81 (с, 3H), 2,35 - 2,22 (м, 1H), 1,04 - 1,02 (д,02 (д, J = 6,8 Гц, 3H), 0,91 - 0,89 (д, J = 6,74 Гц, 3H).
471 мг (3,00 ммоль) указанного сложного метилового эфира растворяют 5 мл толуола и к полученному раствору добавляют 366 мг (3,00 ммоль) N-метил-N-(2-пиридинилметил)амина. Получаемую смесь перемешивают в атмосфере азота при температуре 90oC в течение 16 часов, после чего растворитель упаривают и остаток очищают путем флеш-хроматографии на двуокиси кремния с применением в качестве элюента смеси этилацетата и метанола в соотношении 24 : 1. При этом получают 616 мг (73%) сложного метилового эфира N-{{N-метил-N- (2-пиридинилметил)-амино}карбонил}валина в качестве оранжевого масла.
1H ЯМР (CDCl3) δ 8,58 - 8,55 (д, 1H), 7,72 - 7,65 (т, 1H), 7,29 - 7,19 (м, 2H), 6,20 - 6,05 (широк., с, 1H), 4,55 (с, 2H), 4,45 - 4,40 (м, 1H), 3,71 (с, 3H), 3,04, (с, 3H), 2,21 - 2,12 (м, 1H), 1,0 - 0,92 (дд, 6H).
К интенсивно перемешиваемому раствору 400 мг (1,43 моль) вышеуказанного сложного метилового эфира в 4 мл диоксана и 1 мл воды добавляют 1,72 мл (1,72 ммоль) 1 н. гидроокиси лития при комнатной температуре в течение 3 часов. Реакционную смесь перемешивают при комнатной температуре в течение 18 часов, после чего упаривают досуха, поучаемый остаток переводят в порошок и сушат над полупятиокисью фосфора в высоком вакууме. Получают 390 мг (100%) литиевой соли N-{{N-метил-N-(2- пиридинилмети)амино}карбонил}валина, которую аналогично примеру 8 подвергают взаимодействию с N-трет.-бутил-1-{3(S)-амино-2- (R)окси-4-фенилбутил-{ -4(R)-(фенилтио)пиперидин-2(S)- карбоксамидом (получаемым гидрированием целевого продукта N 1 таблицы II). Получают вышеуказанный целевой продукт следующей характеристики: масс-спектр (бомбардировка быстрыми атомами), m/z: 703 (М + H)+.
Пример 10.
Получение N-трет. -бутил-1-{3(S)-{{(2,6- диметилфенокси)ацетил}амино}-2(R)-окси-4-фенилбуnил} -4(R)-{ (3- пиридинилметил)тио} пиперидин-2(S)-карбоксамида.
Целевой продукт N 14 таблицы I переводят в соответствующий первичный амин, то есть N-трет.-бутил-1-(3(S)- амино-2(R)-окси-4-фенилбутил)-4(R)-{(3-пиридинилметил)тио} пиперидин-2 -карбоксамид, за счет снятия защитной трет. -бутилоксикарбонильной группы. Первичный амин выделяют в виде трисгидрохлорида. 154 мг (0,27 ммоль) этого трисгидрохлорида, 55,1 мг (0,31 ммоль) (2,6-диметилфенокси)-уксусной кислоты и 147 мг (0,33 ммоль) гексафторфосфата (бензотриазол-1-илокси)трис- (диметиламино)-фосфония размешивают в 4 мл безводного диметилформамида. Добавляют 185 мкл (1,06 ммоль) диизопропилэтиламина, после чего смесь перемешивают при комнатной температуре в течение 10 минут. Добавляют еще 95 мкл (0,55 ммоль) диизопропилэтиламина и смесь перемешивают при той же температуре в течение 18 часов. Затем реакционную смесь разбавляют 25 мл этилацетата, последовательно промывают насыщенным водным раствором бикарбоната натрия, водой и солевым раствором, сушат над сульфатом магния и сгущают досуха. В результате очистки флеш-хроматографией на двуокиси кремния с применением в качестве элюента смесей этанола и этилацетата при градиенте 0,1% - 5% этанола получают 107 мг (64%) целевого продукта в качестве желтоватого твердого вещества.
Масс-спектр (бомбардировка быстрыми атомами), m/z: 633 (М+H)+.
Способность соединений формулы (I) и их терапевтически приемлемых солей к торможению протеазы HIV и защите клеток иллюстрируется следующими примерами.
Пример 11.
Опыт по определению торможения рекомбинантной протеазы HIV
Энзим
Протеаза HIV экспрессировалась в Е. coli [конструкт pBRT1 prt+, см W.G. Farmerie и др., Science, 236, стр. 305 (1987)] нижеописанным образом. Применяемые в данном опыте растворы являются водными, если ничего другого не указано.
1. Ферментация
Клетками Е.coli, содержащими плазмиду pBRTI prt+, инокулировалась состоящая из бульона Luria-Bertani культура, включающая 100 мкг/мл ампицилина. Культура инкубировалась в колбах при температуре 37oC в течение 17 часов при размешивании. Содержащийся в колбах емкостью 2 л стерильный бульон М9, дополненный 100 мкг/мл ампицилина, инокулировался инкубировавшей культурой, взятой в количестве 1% от объема бульона. Общий объем в каждой колбе составляет 500 мл. Колбы инкубировались при температуре 37oC при размешивании до достижения концентрации клеток, соответствующей оптической плотности 0,6 при λ = 540 мкм (без разбавления). Данный результат достигался обычно через 3 - 4 часа. Затем в колбы добавлялись 5 ммоль изопропилгиогалактозида, после чего инкубация продолжалась до концентрации клеток, соответствующей оптической плотности 0,2 при 16-кратном разведении. Затем в колбы подавался 1 ммоль фенилметилсульфонилфторида, после чего колбы быстро охлаждались до температуры 4oC. Бактериальные клетки отделялись путем центрифугирования при температуре 4oC, после чего подавались на хранение при температуре - 70oC.
2. Получение энзима, требуемого для осуществления опыта качества
Все нижеописанные приемы осуществлялись при температуре 4oC, если ничего другого не указано. Клетки размораживались и смешивались с буфером А [50 ммоль трис(оксиметил)аминоэтана в виде гидрохлорида (трис-HCl, pH 7,4); 0,6 ммоль этилендиаминтетрауксусной кислоты; 0,375 моль хлористого натрия, 0,2% неионогенного поверхностно-активного вещества (торговый продукт Нонидет Р - 40 фирмы БДХ Кэмикальз Лтд., Пуль, GB); 1 ммоль фенилметилсульфонилфторида] в массовом соотношении 1 : 9. Затем добавлялась диатомовая земля (торговый продукт Celite 545, фирмы John Manville, Ломпок, штат Калифорния, США) в количестве 2 частей на 1 часть влажных клеток. Получаемая суспензия гомогенизировалась с высокой скоростью (примерно 20000 об/мин) с применением промышленного смесителя Waring (импульсы: 8 х 15 сек). Клетки и диатомовая земля собирались путем центрифугирования и получаемый центрифугат экстрагировался буфером A, взятым в количестве 4,5 части на часть влажных веществ, при гомогенизации в вышеуказанных условиях. Надосадочные жидкости обеих стадий гомогенизации объединялись и растворимый белок осаждался твердым сульфатом аммония, взятым в количестве, обеспечивающем 75%-ное насыщение. Смесь размешивалась в течение 60 минут и остаток отделялся путем центрифугирования. Получаемый центрифугат суспендировался в буфере Б [50 ммоль трис-HCl, pH 8; 30 ммоль хлористого натрия; 1 нмоль DL-дитиотреитола; 1 ммоль этилендиаминтетрауксусной кислоты; 1 моль фенилметилсульфонилфторида и 10% глицерина], после чего подвергался диализу в отношении того же буфера в течение 18 часов.
Аликвот диализованного экстракта, включающего 150 мг белка, подавался на анионнообменную колонку марки Sephadex А25 фирмы Фармация, Упсала, Швеция, в которой слой неподвижной фазы имел длину 70 см, и диаметр - 2,5 см. Элюация проводилась изократически буфером Б при линейной скорости 10 см/час. Проявляющие активность протеазы HIV фракции объединялись и растворимый белок осаждался насыщенным водным раствором сульфата аммония, взятым в количестве, обеспечивающем 85%-ное насыщение сульфатом аммония. Осадок отделялся путем центрифугирования и получаемый центрифугат растворялся в буфере В [50 ммоль 2-(4- морфолино)этансульфокислоты, pH 5,5; 150 ммоль хлористого натрия; 1 ммоль D, L-дитиотреитола; 1 ммоль этилендиаминотетрауксусной кислоты; 10% глицерина] , после чего проводился диализ с применением того же буфера В течение 18 часов с последующим замораживанием при температуре - 70oC. Все сырые экстракты очищались путем хроматографии в описанных выше условиях, которой подвергались аликвоты, содержащие 150 мг белка. Препараты от всех групп объединялись, подразделялись на аликвоты объемом 34 мкл и хранились при температуре - 70oC. В результате ферментации культуры объемом 20 л обычно получают 300 мг целевого белка, имеющего удельную активность протеазы HIV, равную 18,2 ммоль расщепляемого субстрата/мин/мг.
Перед применением аликвоты разбавлялись вышеуказанным буфером до объема, составляющего 1/38 первоначального объема. Получаемый при этом препарат представляет собой рабочий раствор энзима.
Субстрат
В качестве субстрата использовался VSFNFPQITL-NH2 с молярной массой 1164 (см. Krausslich и др. Proc. Natl. Acad. Sci. USA, 86, стр. 807, 1989). Субстрат переводился в 10-ммолярный раствор в диметилсульфоксиде, который хранился при температуре 4oC. Перед применением раствор разбавлялся буфером, взятым в количестве, обеспечивающем 400-мкмолярный раствор, который представляет собой рабочий раствор субстрата.
Буфер
100 ммоль 2-(4-морфолино)этансульфокислоты, 300 ммоль хлористого калия и 5 ммоль этилендиаминотетрауксусной кислоты растворялись в 90 мл дистиллированной воды и раствор доводился до pH 5,5 концентрированной водной гидроокисью натрия. Получаемый раствор разбавлялся водой до 100 мл.
Осуществление опыта
20 мкл рабочего раствора субстрата смешивались с 10 мкл раствора исследуемого соединения в 10% диметилсульфоксида и 10 мкл рабочего раствора энзима. Полученная смесь инкубировалась при температуре 37oC в течение 30 минут, после чего реакция прекращалась путем добавления 200 мкл 2%-ной водной трифторуксусной кислоты. Субстрат и продукты реакции, то есть VSFNF и PQITL-NH2, выделялись путем высокопроизводительной жидкостной хроматографии, для чего 100 мкл реакционной смеси подавались на колонку марки 3 х 3 CRC8 (фирмы Перкин Эльмер Инк. , Норфок, штат Коннектикат, США). Хроматография осуществлялась со скоростью 4 мл/мин с соблюдением следующего градиента:
0,0 - 0,5 мин, 70% А/30% Б;
0,5 - 3,0 мин, 67% А/33% Б;
3,0 - 5,0 мин, 20% А/80% Б;
5,0 - 6,5 мин, 70% А/30% Б.
При этом А представляет собой 3 ммоль додецилсульфата натрия и 0,05% фосфорной кислоты в воде, а Б - 0,05%-ную фосфорную кислоту в ацетонитриле. Элюация проводилась при 210 нм. В качестве контроля применялась вышеупомянутая смесь, но без добавки исследуемого соединения.
Определение активности исследуемого соединения
Продукты расщепления и оставшийся исходный субстрат количественно определялись либо с помощью высоты пика, либо с помощью интеграции соответствующих пиков высокопроизводительной жидкостной хроматографии. Конверсия субстрата определялась по следующему уравнению:
Торможение энзима, проявляемое исследуемым соединением, определялось следующим образом: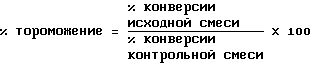
Концентрация исследуемого соединения, которая вызывала 50%-ное торможение активности протеазы HIV, то есть КТ50, определялась следующим образом. %-ное торможение энзима определялась с учетом по меньшей мере трех различных концентраций исследуемого соединения. Затем КТ50 определялась графическим путем за счет составления диаграммы процентного торможения энзима и концентрации исследуемого соединения.
Результаты данного опыта сведены в нижепредставленной таблице IV, в которой указаны исследуемое соединение и его КТ50 в нмоль.
Пример 12.
Определение антивирусной активности новых соединений формулы (I)
В данном опыте применялись трансформированные клетки HTLV-I. Эти клетки применялись из-за быстроты, с которой ретровирусы HIV воспроизводятся в таких клетках. Исследуемое соединение растворялось в диметилсульфоксиде до концентрации 5 мг/мл. Полученный раствор может хранятся при температуре 4oC до применения. Перед применением раствор четырехкратно разбавлялся средой RPMI 1640. После разведения раствор должен применяться в течение четырех часов. 50 мкл раствора подавались на плоскодонную микротитровую пластинку, имеющую 96 углублений. Кроме того, на соответствующие контрольные пластинки подавались 50 мкл среды RPMI. Во все углубления подавались 5 • 104 клеток C 8166 в 50 мкл содержащей буфер HEPES (=N-2-оксиэтилпиперазин- N'-2-этансульфокислоту) среды RPMI 1640 (pH 7,2), включающей 10% инактивированной термообработкой эмбриональной телячьей сыворотки и 12,5 мкл/мл гентамицина. Среда указанного состава далее обозначается как полная среда. Раствор H9/HTLV-IIIB, (который хранился в среде жидкого азота в качестве надосадочной жидкости культуры в 50% эмбриональной телячьей сыворотки) в 100 мкл полной среды добавлялся во все углубления. Титр инфекционности вирусного препарата определялся за счет серийного разведения в клетках C 8166. Вирусные препараты устойчивы в течение 6 - 12 месяцев, если хранят их при температуре - 193oC. Микротитровые пластинки размещались на 72 часа в инкубационной камере при температуре 37oC в присутствии 5% двуокиси углерода. Затем пластинки вынимались из камеры и центры синцитиев рассчитывались в каждом углублении путем низкомощной фазово-контрастной световой микроскопии. Каждое скопление клеток, которое показывало малейшее образование синцитиев, рассматривалось как центр синцитиев. Контрольные углубления должны были иметь 25 - 75 центров синцитиев на углубление. %-ное торможение образования синцитаев определялось согласно следующему уравнению:
Концентрация исследуемого соединения, которая обеспечивает 50%-ное торможение образования синцитиев, то есть доза ЭД50, определялась путем известного приема по серийному разведению вышеуказанного раствора соответствующего исследуемого соединения и составления диаграммы обнаруженного процентного торможения образования синцитиев при различных концентрациях соответствующего исследуемого соединения.
Результаты данного опыта сведены в следующей таблице IV, в которой указаны исследуемые соединения и их доза ЭД50 в нмоль.
В следующих таблицах V - VII сведены дальнейшие соединения общей формулы (I), охарактеризованные соответствующими данными масс-спектра в условиях бомбардировки быстрыми атомами (МС/ББА). Для этих соединений также приведены данные по КТ50 и ЭД50.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ МОДУЛЯЦИИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПОЛУЧЕНИЯ ЭТИХ СОЕДИНЕНИЙ | 2000 |
|
RU2255937C2 |
| АНТАГОНИСТЫ 3-АМИНОАЛКИЛ-1,4-ДИАЗЕПАН-2-ОН МЕЛАНОКОРТИНА-5 РЕЦЕПТОРА | 2009 |
|
RU2530017C2 |
| СПОСОБЫ МОДУЛИРОВАНИЯ АКТИВНОСТИ МС5 РЕЦЕПТОРА И ЛЕЧЕНИЕ СОСТОЯНИЙ, ОТНОСЯЩИХСЯ К ДАННОМУ РЕЦЕПТОРУ | 2009 |
|
RU2555343C9 |
| ПРОИЗВОДНОЕ ИНДАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2817737C2 |
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТ И ИХ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1990 |
|
RU2071470C1 |
| ПРОИЗВОДНОЕ СУЛЬФОНИЛБЕНЗАМИДА И ЕГО КОНЬЮГАТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2839468C1 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| ПРОИЗВОДНОЕ ФЕНИЛПРОПАНАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2738207C2 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |



















Производные пипеколиновой кислоты общей формулы I, где Х - группы R3ОСО, R4СО, R5ОСН2СО, R8-СНNНА-СО; R3 -низший алкил или бензил, R4 - фенил, замещенный низшим алкилом или галоидом; R5 - фенил, возможно одно или двукратно замещенный низшим алкилом; R8 - Н, низший алкил, карбамилметил, (N-метил)карбамил или гидроксиметил; А -группа R9-СО; R9 - низший алкилокси, бензилокси, нафталенил или хинолинил или R6NR7СО; R6 - пиридинилметил; R2, R7 -низший алкил; R1 - Н, галоид; Y - циклогексил, фенил, бензил, W(СНR2)2Z, W - оксо, тио или сульфонил, Z - фенил, возможно монозамешенный галоидом, пиридинил или пиримидинил, n = 0 или 1, и их соли, обладают антивирусной активностью, преимущественно ингибирующей протеазу активностью. 2 с. и 3 з. п.ф-лы, 7 табл.
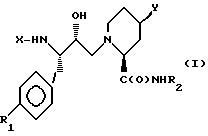
где Х - группы R3ОС(О), где R3 означает низший алкил или бензил, R4С(О), где R4 означает фенил, замещенный низшим алкилом и/или галоидом, R5ОСН2С(О), где R5 означает фенил, незамещенный или одно-, дву- или трехкратно замещенный низшим алкилом,  где R8 означает водород, низший алкил, карбамилметил, (N-метил)карбамилметил или гидроксиметил, а А означает группу R9-С(О), где R9 означает низший алкилокси, бензилокси, нафталенил или хинолинил, или R6NR7С(О), где R6 означает пиридинилметил, а R7 - низший алкил;
где R8 означает водород, низший алкил, карбамилметил, (N-метил)карбамилметил или гидроксиметил, а А означает группу R9-С(О), где R9 означает низший алкилокси, бензилокси, нафталенил или хинолинил, или R6NR7С(О), где R6 означает пиридинилметил, а R7 - низший алкил;
R1 - водород или галоид;
R2 - низший алкил;
Y - циклогексил, фенил, бензил, или группа W(СН2)nZ, где W означает оксо, тио или сульфонил, Z - фенил, незамещенный или монозамещенный галоидом, пиридинил или пиримидинил, незамещенный или замещенный низшим алкилом; n = 0 или 1,
и их соли. где R8 означает низший алкил, карбамилметил, или гидроксиметил, а А означает группу R9С(О), где R9 означает бензилокси, 2-нафталенил или 2-хинолинил, R1 - водород или фтор, R2 - 1,1-диметилэтил, Y - фенил, бензил, фенокси, 2-пиридинилокси, 2-пиримидинилокси, (2,6-диметил-4-пиримидинил)окси, 2-пиридинилметокси, 3-пиридинилметокси, 4-пиридинилметокси, фенилтио, фенилсульфонил, 2-пиридинилтио, 3-пиридинилтио, 4-пиридинилтио, 2-пиримидинилтио, (4,6-диметил-2-пиримидинил)тио, (2-пиридинилметил)тио, (3-пиридинилметил)тио или (4-пиридинилметил)тио и их терапевтически приемлемые кислотно-аддитивные соли.
где R8 означает низший алкил, карбамилметил, или гидроксиметил, а А означает группу R9С(О), где R9 означает бензилокси, 2-нафталенил или 2-хинолинил, R1 - водород или фтор, R2 - 1,1-диметилэтил, Y - фенил, бензил, фенокси, 2-пиридинилокси, 2-пиримидинилокси, (2,6-диметил-4-пиримидинил)окси, 2-пиридинилметокси, 3-пиридинилметокси, 4-пиридинилметокси, фенилтио, фенилсульфонил, 2-пиридинилтио, 3-пиридинилтио, 4-пиридинилтио, 2-пиримидинилтио, (4,6-диметил-2-пиримидинил)тио, (2-пиридинилметил)тио, (3-пиридинилметил)тио или (4-пиридинилметил)тио и их терапевтически приемлемые кислотно-аддитивные соли.
N-трет. -бутил-1-{ 3(S)-(бензилоксикарбониламино)-2-(R)-окси-4-(4-фторфенил)бутил}-4(R)-(фенилтио)пиперидин-2(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-(бензилоксикарбониламино)-2-(R)-окси-4-фенилбутил} -4(R)-фенилпиперидин-2(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-(бензилоксикарбониламино)-2-(R)-окси-4-фенилбутил} -4(R)-бензилпиперидин-2(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-(бензилоксикарбониламино)-2-(R)-окси-4-фенилбутил} -4(R)-(фенилсульфонил)-пиперидин-2(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-(бензилоксикарбониламино)-2-(R)-окси-4-фенилбутил} -4(R)-(фенилтио)пиперидин-2-(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-(бензилоксикарбониламино)-2-(R)-окси-4-фенилбутил} -4(R)-феноксипиперидин-2-(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-(бензилоксикарбониламино)-2-(R)-окси-4-фенилбутил} -4(R)-циклогексилпиперидин-2-(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-бензилоксикарбонил)-валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-фенилтио)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{ { N-бензилоксикарбонил)-аспарагинил}амино}-2-(R)-окси-4-фенилбутил}4-(R)-фенилтио)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-бензилоксикарбонил)-валил}амино}-2(R)-окси-4-фенилбутил}4(R)-фенилпиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{3(S)-{{N-бензилоксикарбонил)-изолейцил}амино}-2(R)-окси-4-фенилбутил}-4(R)-фенилпиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{ { N-бензилоксикарбонил)-аспарагинил}амино}-2-(R)-окси-4-фенилбутил}-4(R)-фенилпиперидин-2-(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-бензилоксикарбонил)-валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-бензилпиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-бензилоксикарбонил)-валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(фенилсульфонил)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{ { N-бензилоксикарбонил)-аспарагинил}амино}-2-(R)-окси-4-фенилбутил}-4(R)-(фенилсульфонил)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-бензилоксикарбонил)-валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-феноксипиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{ { N-бензилоксикарбонил)-аспарагинил}амино}-2-(R)-окси-4-фенилбутил}-4(R)-феноксипиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-бензилоксикарбонил)-валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(2-пиридинилокси)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-бензилоксикарбонил)-валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-циклогексилпиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинкарбонил)-валил} -амино}бутил}-4(R)-фенилтио)пиперидин-2-(S)-карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ {N-(2-хинолинкарбонил)-аспарагинил}амино}бутил}-4(R)-феноксипиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ {N-(2-хинолинкарбонил)-аспарагинил}амино}бутил}-4(R)-фенилсульфонил)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ {N-(2-хинолинкарбонил)-аспарагинил}амино}бутил}-4(R)-фенилтио)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{{N-(2-нафталенилкарбонил)-валил}амино}бутил}-4(R)-фенилтио)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{2(R)-окси-3(S)-{{N-(2-нафталенилкарбонил)-аспарагинил} -амино}-4-фенилбутил}-4(R)-фенилтио)пиперидин-2-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{ {N-(2-бензилоксикарбонил)-валил}амино}2(R)-окси-4-(4-фторфенил)бутил}-4(R)-фенилтио)пиперидин-2(S)карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ {N-(2-пиридинилметокси)-карбонил}изолейцил}амино}бутил}-4(R)-фенилпиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{3(S)-(бензилоксикарбониламино)-2(R)-окси-4-фенилбутил} -4(R)-(2-пиридинилтио)-пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{3(S)-(бензилоксикарбониламино)-2(R)-окси-4-фенилбутил} -4(R)-(4-пиридинилтио)-пиперидин-2(R)-карбоксамид,
N-трет. -бутил-1-{3(S)-(бензилоксикарбониламино)-2(R)-окси-4-фенилбутил} -4(R) -(2-пиримидинилтио)-пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{3(S)-(бензилоксикарбониламино)-2(R)-окси-4-фенилбутил} -4(R)-(4,6-диметил-2-пиримидинилтио)-пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{3(S)-(бензилоксикарбониламино)-2(R)-окси-4-фенилбутил} -4(R)-(бензилтио)-пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{3(S)-{{N-бензилоксикарбонил)валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(2-пиридинилтио)-пиперидин-2(S)- карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-(бензилоксикарбонил)валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)-пиперидин-2(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-{{N-(бензилоксикарбониламино)валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(2-пиримидинилтио)-пиперидин-2(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-{{N-(бензилоксикарбониламино)валил}амино}-2(R)-окси-4-фенилбутил} -4(R)-(4,6-диметил-2-пиримидинилтио)-пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{N-(бензилоксикарбонил)валил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(бензилтио)-пиперидин-2(S)- карбоксамид,
N-трет. -бутил-1-{2(R)-окси-3(S)-{N-{{N-метил-N-(2-пиридинилметил)амино] карбонил}валил}-4-фенилбутил}-4(R)-(фенилтио)-пиперидин-2(S)-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(2-пиридинил)тио}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(4-пиридинилметилтио}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(2-пиридинилметокси)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-{(4,6-диметил-2-пиримидинил)тио)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(4-пиридинилтио)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(2-пиридинилтио)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-феноксипиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(3-пиридинилметил)тио}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-{(2-пиридинилметил)тио}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(2-пиримидинилокси)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино} бутил} -4(R)-{ (4,6-диметил-2-пиримидинил)-окси}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-{(4-метил-2-пиримидинил)окси}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино} бутил} -4(R)-{ (2,6-диметил-4-пиримидинил)-окси}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(фенилсульфонил-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R){(4-фторфенил)-окси}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(4-пиридинилметокси)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-{(2-пиридинилметил)сульфонил-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-{(3-пиридинилметил)сульфонил-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-{(4-пиридинилметил)сульфонил-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(2-пиридинилсульфонил)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(4-пиридинилсульфонил)-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино} бутил} -4(R)-{(2,6-диметил-4-пиримидинил)-тио}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-{(4-метил-2-пиримидинил)-тио}-пиперидин-2-карбоксамид,
N-трет.-бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-валил} -амино}бутил}-4(R)-(3-пиридинилметокси)-пиперидин-2-карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ { N-(2-хинолинилкарбонил)-трет-бутилглицил}амино}бутил}-4(R)-(фенилтио)-пиперидин-2-карбоксамид,
N-трет-бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ {N-(2-хинолинилкарбонил)-аспарагинил} амино} бутил} -4(R)-{(4,6-диметил-2-пиримидинил)-тио}-пиперидин-2-карбоксамид,
N-трет-бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ {N-(2-хинолинилкарбонил)-аспарагинил}амино}бутил}-4(R)-(2-пиримидинилтио)-пиперидин-2-карбоксамид,
N-трет-бутил-1-{ 2(R)-окси-4-фенил-3(S)-{ { N-(2-хинолинилкарбонил)- N4-метил)-аспарагинил}амино}бутил}-4(R)-фенокси-пиперидин-2-карбоксамид,
N-трет. -бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-трет. -бутилглицил} амино} бутил}-4(R)-{(3-пиридинилметил)-тио}пиперидин-2-карбоксамид,
N-трет. -бутил-1-{ 2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-треонил}-амино}бутил}-4(R)-(фенилсульфонил)-пиперидин-2-карбоксамид,
N-трет. -бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-трет. -бутилглицил}амино}бутил}-4(R)-(4-пиридилсульфонил)-пиперидин-2-карбоксамид,
N-трет. -бутил-1-{2(R)-окси-4-фенил-3(S)-{{N-(2-хинолинилкарбонил)-трет. -бутилглицил} амино} бутил} -4(R)-(2-пиридинилсульфонил)-пиперидин-2-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{(2,6-диметилфенокси)ацетил}-амино}-2(R)-окси-4-фенилбутил}-4(R)-{(3-пиридинилметил)тио}пиперидин-2-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{{(2,4,6-триметилфенокси)-ацетил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{(феноксиацетил)амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)-пиперидин-2-(S)-карбоксамид,
N-трет. -бутил-1-{ 2(S)-{{(2,6-диметилфенокси)ацетил}-амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)-пиперидин-2(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{ {2-метилфенокси)ацетил}-амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)пиперидин-2-(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-{{(2,4-дихлорфенил)карбонил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)-пиперидин-2-(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-{{(2,5-дихлорфенил)карбонил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)пиперидин-2-(S)-карбоксамид,
N-трет.-бутил-1-{3(S)-{{(2,6-дифторфенил)карбонил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)-пиперидин-2-(S)-карбоксамид,
N-трет. -бутил-1-{ 3(S)-{ {(5-фтор-2-метилфенил)карбонил}амино}-2(R)-окси-4-фенилбутил}-4(R)-(4-пиридинилтио)пиперидин-2-(S)-карбоксамид.
где Х - группы R3ОС(О), где R3 означает низший алкил или бензил, R4С(О), где R4 означает фенил, замещенный низшим алкилом и/или галоидом, R5ОСН2С(О), где R5 означает фенил, незамещенный или одно-, дву- или трехкратно замещенный низшим алкилом,  где R8 означает водород, низший алкил, карбамилметил, (N-метил)карбамилметил или гидроксиметил, а А означает группу R9-С(О), где R9 означает низший алкилокси, бензилокси, нафталенил или хинолинил, или R6NR7С(О), где R6 означает пиридинилметил, а R7 - низший алкил;
где R8 означает водород, низший алкил, карбамилметил, (N-метил)карбамилметил или гидроксиметил, а А означает группу R9-С(О), где R9 означает низший алкилокси, бензилокси, нафталенил или хинолинил, или R6NR7С(О), где R6 означает пиридинилметил, а R7 - низший алкил;
R1 - водород или галоид;
R2 - низший алкил;
Y - циклогексил, фенил, бензил, или группа W(СН2)nZ, где W означает оксо, тио или сульфонил, Z - фенил, незамещенный или монозамещенный галоидом, пиридинил или пиримидинил, незамещенный или замещенный низшим алкилом, n = 0 или 1,
или его терапевтически приемлемую кислотно-аддитивную соль в эффективном количестве.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| СПОСОБ ПОЛУЧЕНИЯ МИКРОКАПСУЛ | 0 |
|
SU346842A1 |
| Одноцепная линия электропередачи | 1974 |
|
SU498680A1 |
| ЗАМОК ДЛЯ РАЗРЕЗНЫХ УПРУГИХ УПЛОТНИТЕЛЬНЫХ КОЛЕЦ | 0 |
|
SU361341A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, ч.2, с.380. | |||
