Эта заявка является частичным продолжением U.S. Patent Application Serial (неизвестен), поданной 31 января 1995, ожидающей решения, которая является частичным продолжением U.S. Patent Application Serial 08/361811, поданной 22 декабря 1994, ожидающей решения, которая является частичным продолжением U. S. Patent Application Serial N 08/320300, поданной 11 октября 1994, ожидающей решения, которая является частичным продолжением U.S. Patent Application Serial 08/278083, поданной 20 июля 1994, ожидающей решения, которая является частичным продолжением U. S. Patent Application Serial 08/221020, поданной 1 апреля 1994, отозванной, которая является частичным продолжением U. S. Patent Application Serial 08/212164, поданной 14 марта 1994, в настоящее время отозванной.
Область изобретения
Данное изобретение касается нового белка, имеющего активность специфического стимулирования или увеличения образования тромбоцитов in vivo или усиления пролиферации и дифференциации клеток-предшественников мегакариоцитов, последовательности ДНК, кодирующей этот белок, и способа его получения.
Предпосылки изобретения
Мегакариоциты представляют собой большие богатые цитоплазмой многоядерные клетки, которые продуцируют тромбоциты и могут быть обнаружены в основном в костном мозгу. Мегакариоциты происходят из плюрипотентных гемопоэтических (кроветворных) стволовых клеток в костном мозгу. Примитивная плюрипотентная стволовая клетка дифференцируется до некоторой степени в клетки-предшественники мегакариоцитов, которые дают начало ряду поколений мегакариоцитов. Клетки-предшественники мегакариоцитов размножаются и дифференцируются в мегакариоциты. Затем мегакариоциты подвергаются полиплоидизации и цитоплазматическому созреванию и в конце концов выделяют их безъядерные цитоплазматические фрагменты, а именно, тромбоциты, в кровообращение. В среднем из одного зрелого мегакариоцита образуются 2-4 тысячи тромбоцитов. Хотя механизм образования тромбоцитов во многих отношениях пока неясен, считают, что мегакариоциты обычно локализованы на белковой поверхности эндотелия синуса в костном мозгу и производят цитоплазматические отростки, распространяющиеся в синусоид, где происходит фрагментация тромбоцитов.
Имеется много неясных моментов относительно механизма образования тромбоцитов, хотя возможно, что мегакариоциты присутствуют локально в дермальном слое оболочки венозного синуса костного мозга и что цитоплазма проходит через дермальную оболочку, образуя струноподобный выступ на внутренней стенке венозного синуса, в результате чего происходит выбрасывание тромбоцитов.
Высказывалось предположение, что имеется специфическая функция для образования и регулирования образования тромбоцитов в гемопоэзе мегакариоцитов. В здоровых людях и животных сохраняется количество эффективных тромбоцитов, хотя известно, что при введении антител к тромбоцитам здоровым животным, например, количество тромбоцитов резко уменьшается, затем их количество временно увеличивается до уровня, более высокого, чем обычный уровень, и наконец они возвращаются к нормальному уровню. Также в области клиники известно, что уменьшение в числе тромбоцитов (тромбоцитопения) или увеличение в числе тромбоцитов (тромбоцитоз) наблюдаются даже при нормальных количествах эритроцитов и лейкоцитов.
Иногда наиболее важной функцией тромбоцита является образование тромба в гемостатическом (кровоостанавливающем) механизме. Если гемостатический механизм не функционирует должным образом вследствие уменьшения числа тромбоцитов, образуется тенденция в направлении возникновения гемостаза.
Было высказано предположение о наличии специфического регуляторного механизма мегакариоцитопоэза и тромбопоэза. Тромбоциты поддерживаются в эффективных количествах в здоровых людях и нормальных животных. Однако известно, что при введении антител к тромбоцитам нормальному животному количество тромбоцитов резко уменьшается в течение короткого периода времени, после этого начинает увеличиваться и временно превышает нормальный уровень, однако в конце концов возвращается к нормальному уровню. Известно, что в клинической области, уменьшение в числе тромбоцитов (тромбоцитопения) или увеличение в их числе (тромбоцитоз) происходят даже при нормальных количествах эритроцитов и лейкоцитов. Однако до настоящего времени не было сообщений об успехе в выделении и идентификации специфического регуляторного фактора, участвующего в образовании тромбоцитов (например, подобного эритропоэтину в образовании эритроцитов).
Наиболее важной функцией тромбоцитов является образование сгустка крови в гемостатическом (кровоостанавливающем) механизме. Склонность к кровотечению имеет место, когда нормальная функция гемостатического механизма нарушена тромбоцитопенией. В области лучевой терапии и химиотерапии раковых опухолей тромбоцитопения, вызванная угнетением костного мозга, является летальным осложнением, и для предотвращения склонности к кровотечению таким больным делают трансфузию тромбоцитов. Трансфузию тромбоцитов применяют также после пересадки костного мозга или гипопластической анемии.
Тромбоциты для применения в такой трансфузии готовят тромбоцитофорезом из крови здоровых доноров крови, однако такие тромбоциты для применения в трансфузии имеют короткий срок хранения и могут быть загрязнены бактериальной инфекцией. Трансфузия тромбоцитов представляет собой возможную опасность попадания в больных опасных вирусов, таких как вирус иммунодефицита человека (Н IV) или различные вирусы гепатита, опасность образования антител, специфических для антигена главного комплекса гистосовместимости (Н L А) переливаемых тромбоцитов или возникновения реакции "трансплантат против хозяина" (GVHD), обусловленной примесью лимфоцитов в тромбоцитах, применяемых для трансфузии.
Вследствие этого будет очень полезно, если в больных тромбоцитопенией можно будет стимулировать внутреннее образование тромбоцитов и в то же время можно будет снизить их зависимость от трансфузии тромбоцитов. Кроме того, если тромбоцитопению в раковых больных можно будет корректировать или предотвращать, такие обработки могут проводиться более безопасно, интенсивность лечения может быть повышена и можно будет ожидать дальнейшего улучшения противоракового действия.
В силу этих причин проводился ряд интенсивных исследований по выделению и идентификации специфических регуляторных факторов, участвующих в регуляции образования мегакариоцитов и тромбоцитов. Согласно исследованиям in vitro, регуляторные факторы, управляющие мегакариоцитопоэзом, грубо разделены на следующие два фактора (см. Williams et al., J, Cell Physiol., vol. 110, pp. 101-104, 1982). Колониестимулирующий фактор мегакариоцитов (Мeg-CSF) является регуляторным фактором, стимулирующим пролиферацию и дифференцировку CFU-MК с образованием колоний мегакариоцитов в полутвердой культуральной среде. Другой регуляторный фактор, называемый фактором потенциирования мегакариоцитов (Meg-Pot), фактором стимуляции мегакариоцитов, фактором стимуляции тромбоцитопоэза или т.п., действует главным образом на незрелые или зрелые мегакариоциты, усиливая их дифференцировку и созревание. Мeg-Pot обнаруживают в комбинации с Мeg-CSF - активностью в некоторых случаях. Кроме того, поскольку количества тромбоцитов увеличивались при введении сыворотки или плазмы, взятой из экспериментально индуцированных животных с тромбоцитопенией, другим нормальным животным, было сделано предположение, что существует гуморальный фактор, названный тромбоэтином (ТРО), способный усиливать образование тромбоцитов in vivo.
В последние годы некоторые цитокины, гены которых были клонированы, испытывались на их способность стимулировать мегакариоцитопоэз и тромбоцитопоэз. Человеческий IL-3 стимулировал образование колоний мегакариоцитов человека (Bruno et al., Exp. Hematol., vol.16, pp.371-377, 1988) и, по меньшей мере в обезьянах, увеличивая число тромбоцитов (Donahue et al., Science, vol. 241, p. 1820, 1988). Однако, поскольку IL-3 является фактором, действующим на пролиферацию и дифференцировку всех гематопоэтических клеток, его следует отличать от специфических регуляторных факторов, регулирующих мегакариоцитопоэз и образование тромбоцитов. Человеческий IL-6 не обнаружил Мeg-CSF активности, но действовал на незрелые мегакариоциты и затем усиливал их дифференцировку в зрелые мегакариоциты (Williams et al., Exp. Hematol., vol 18, p.69, 1990). Введение in vivo IL-6 индуцировало образование тромбоцитов и усиливало созревание и вызывало сдвиг в сторону более высокой плоидности мегакариоцитов костного мозга в приматах, но также вызвало побочные действия, такие как снижение массы тела, индукция белка острой фазы (Asano et al. , Blood, vol. 78, pp. 1467-1475, 1991). IL-11 человека не обнаружил Мeg-CSF активности, но проявлял Meg-Pot активность и усиливал образование тромбоцитов в мышах (Neben et al., Blood, vol. 81, pp. 901-908, 1993). Кроме того, LIF (ингибитор миграции лимфоцитов) человека значительно увеличивал количества тромбоцитов в приматах (Мауеr et al., Blood, vol. 81, pp.3226-3233, 1993), однако его действие in vitro на мегакариоциты было слабым (Burstein et al., J, Cell. Physiol., vol. 153, pp. 305-312, 1992).
Хотя можно ожидать клинического применения этих цитокинов в качестве факторов увеличения тромбоцитов, их функции не являются специфическими для ряда поколений мегакариоцитов и они вызывают побочные действия. Поэтому требовалось нахождение увеличивающего количество тромбоцитов фактора, который является специфическим для системы мегакариоцит-тромбоцит и вызывает меньшие побочные эффекты, для клинического применения.
Meg-CSF, Meg-Pot или ТРО активность была обнаружена в сыворотке, плазме или моче больных тромбоцитопенией или животных или в культуральном супернатанте некоторых культивируемых линий клеток человека. Однако в настоящее время неизвестно, обусловлены эти активности присутствием одного фактора или комбинацией нескольких факторов и отличаются ли они от известных цитокинов.
Hoffman et al. обнаружили, что сыворотки больных с гипопластической анемией и амегакариоцитной тромбоцитопенической пурпурой содержали Meg-CSF активность, которая значительно увеличивала образование колоний мегакариоцитов человека (Hoffman et al., N. Eng. J. Med., vol. 305, pp. 533-538, 1981). После этого Mazur et аl. сообщили, что Мeg-CSF активность присутствует в сыворотке больных с апластической анемией и она отличается как от IL-3, так и от GМ-CSF (Мazur et al., Blood, vol.76, pp.290-297, 1990). Сходная Мeg-CSF активность была найдена в сыворотках раковых больных, получивших интенсивную цитотоксическую химиотерапию, и больных с пересадкой костного мозга (Mazur et al. , Exp. Hematol., vol.12, pp.624-628, 1984; de Alarcon and Schmieder, Prog. Clin. Bio. Res., vol.215, pp.335-340, 1986).
Hoffman et al. сообщили, что Meg-CSF со средней мол. массой 46000 был очищен из плазмы больных с гипомегакариоцитной тромбоцитопенией (Hoffman et al. J. Clin. Invest.,vol 75, pp. 1174-1182, 1985), но дальнейшие исследования показали, что этот материал не был такого уровня чистоты, который позволил бы точное аминокислотное секвенирование (Hoffman, Blood, vol. 74, pp. 1196-1212, 1989). Вещество, имеющее ТРО-подобную активность, которое усиливало включение 75Se-селенометионина во вновь образующиеся тромбоциты в мышах, было частично очищено из плазмы тромбоцитопенических больных или мочи больных идиопатической тромбоцитопенической пурпурой (неясного происхождения) (1ТР). Средняя мол. масса полученного из плазмы фактора была 40000 (Grossi et al., Hematologica, vol. 72, pp. 291-295, 1987; Vannucchi et al., Leukemia, vol.2, pp. 236-240, 1988).
Meg-CSF и ТРО-подобная активности были обнаружены также в пробах мочи больных с апластической анемией и тяжелой ITP (Kawakita et al., Br. J. Haematol. , vol. 48, pp. 609-615, 1981; Kawakita et al., Blood, vol. 556-560, 1983). Kawakita et al. сообщили также, что Meg-CSF активность, обнаруженная в экстракте мочи больных с апластической анемией, обнаружила среднюю мол. массу 45000, определенную гель-фильтрацией при диссоциирующих условиях (Kawakita et al. , Br. J. Haematol., vol.62, pp.715-722, 1986). Erikson-Miller et al. также сообщили об очистке Meg-CSF из подобных проб мочи, однако не дали информации о его структуре (Erikson-Miller et al., "Blood Cell Growth Factors: their present and future use in hematology and oncology" ed. by Murphy, AlphaMed Press, Dayton, Ohio, pp.204-220, 1992). Turner et al. очистили фактор стимуляции мегакариоцитов (MSF), имеющий Meg-CSF активность, из мочи больных с пересаженным костным мозгом и клонировали его ген (Turner et al., Blood, vol.78, p.1106, 279a, 1991, (abstr., Suppl.1)). Этот MSF имеет мол. массу 28000-35000. Идентичность этого фактора с Meg-CSF, обнаруженного до сих пор в пробах сыворотки и плазмы тромбоцитопенических больных, и его активность в увеличении числа тромбоцитов еще должны быть выяснены.
Вещество, имеющее ТРО-подобную активность с мол. массой 32000, было очищено из культурального супернатанта линии эмбриональных почечных клеток человека (НЕК клетки) и его биологические и биохимические свойства интенсивно исследовались, однако его структура пока неизвестна (McDonald et al., J. Lab. Clin. Hed. , vol. 106, pp. 162-174, 1985; McDonald, Int. J. Cell Cloning, vol. 7, pp. 139-155, 1989). Другие исследователи, напротив, сообщали, что основная активность в кондиционированной среде НЕК клеток, которая усиливает созревание мегакариоцитов in vitro, обусловлена известными цитокинами, а именно, IL-6 и ЕРО (withy et аl., J., Cell. Physiol., vol. 15, pp. 362-372, 1992).
Что касается полученных из животных факторов, Evatt et al. сообщали, что ТРО-подобная активность, усиливающая включение 75Se-селенометионина во вновь образующиеся тромбоциты в кроликах и мышах, была обнаружена в плазме тромоцитопенических кроликов, индуцированных инъекцией сыворотки против тромбоцитов (Evatt et al., J. Lab. Clin. Med., vol. 83, pp. 364-371, 1974). В дополнение к этому, ряд подобных исследований были опубликованы с 60-х по 70-е годы (например, Оdell et al., Proc. Soc. Biol. Med., vol. 108, pp. 428-431, 1961; Evatt and Levin, J. Clin. Invest., vol. 48, pp. 1615-1626; Harker, Am. J. Physiol., vol. 218, pp. 1376-1380, 1970; Shreiner and Levin. J. Clin, Invest, vol.49, pp.1709-1713, 1970; and Penington, Br. Med. J. vol. 1, pp.606-608, 1970). Evatt et аl., и Hill и Levin провели частичную очистку ТРО-подобной активности из плазмы тромбоцитопенических кроликов (Evatt et al., Blood, vol.54, pp.377-388, 1979; Hill and Levin, Exp. Hematol., vol.14, pp.752-759, 1986).
После этого очистка этого фактора была продолжена, была проверена активность в усилении дифференцировки и созревания мегакариоцитов in vitro, a именно, Meg-Pot активность, было обнаружено, что средняя мол. масса была 40000-46000, определенная гель-фильтрацией (Kеllеr et al., Exp. Hematol., vol. 16, pp.262-267, 1988; Hill et al., Exp. Hematol., vol.20, pp. 354-360, 1992). Поскольку IL-6 активность не обнаруживалась в плазме кроликов с тяжелой острой тромбоцитопенией, индуцированной введением антисыворотки против тромбоцитов, было высказано предположение, что эта ТРО-подобная активность обусловлена фактором, иным, чем IL-6 (Hill et аl., Вlооd, vol. 80, pp. 346-351, 1992).
Tayrien Rosenberg также очистили фактор, имеющий среднюю мол. массу 15000, из плазмы тромбоцитопенических кроликов и культурального супернатанта НЕК клеток, который стимулирует образование фактора 4 тромбоцитов в линии мегакариоцитных клеток крыс, однако не сообщили информации о его структуре (Tayrien and Rosenberg, J. Biol., vol.262, pp. 3262-3268, 1987).
Кроме того, Nakeff обнаружил Мeg-CSF активность в сыворотке тромбоцитопенических мышей, индуцированных введением антисыворотки против тромбоцитов (Nakeff, "Experimental Неmаtology Today" ed. by Baum and Ledney, Springer-Verlag, NY, pp. 111-123, 1977). С другой стороны, сыворотки из тромбоцитопенических кроликов усиливали созревание мегакариоцитов (Keller et al., Exp. hematol., vol.16, pp.262-267, 1988; Hill et al., Exp. Hematol., vol.17, pp.903-907, 1989) и стимулировали морфологическое изменение мегакариоцитов в тромбоциты (Leven and Yee, Blood, vol.69, pp. 1046-1052, 1989), но не имели детектируемой Meg-CSF активности.
Miura et al. обнаружили Meg-CSF активность в плазме крыс, ставших тромбоцитопеническими под влиянием сублетального облучения всего тела (Miura et al. , Blood, vol. 63, pp. 1060-1066, 1984), и предположили, что индукция Мeg-CSF активности in vivo связана с уменьшением мегакариоцитов, но не с уменьшением тромбоцитов, поскольку эта активность не изменялась при трансфузии тромбоцитов (Мiura et al. , Exp.Hematol vol. 16, pp. 139-144, 1988), Mazur и South обнаружили Мef-CSF активность в сыворотке облученных сублетально собак и сообщили, что этот фактор имеет среднюю мол. массу 175000 при измерении гель-фильтрацией (Мazur and South, Exp.Hematol., vol. 13, pp. 1164-1172, 1985). В дополнение к вышеизложенному, факторы, полученные из сыворотки, плазмы и мочи, сообщались другими исследователями, например, Straneva et al. , (Straneva et al., Exp. Hematol., vol.15, pp. 657-663, 1987).
Таким образом, как описано выше, различные активности, стимулирующие мегакариоцитопоэз и тромбоцитопоэз, были обнаружены в биологических пробах, полученных из тромбоцитопенических больных и животных, но выделение этих факторов, их биохимическая и биологическая идентификация и определение их свойств не были достигнуты вследствие их чрезвычайно малых количеств в природных источниках, таких как кровь и моча.
Краткое изложение существа изобретения
Целями данного изобретения являются выделение белка ТРО из природных источников и его идентификация, причем белок ТРО обладает активностью стимулирования или увеличения образования тромбоцитов in vivo и/или усиления пролиферации и дифференцировки клеток-предшественников мегакариоцитов (далее называемой "ТРО активностью"), и выделение гена, кодирующего белок ТРО, и обеспечение способа получения этого белка в гомогенном виде и в большом количестве технологией рекомбинантных ДНК. Успех в достижении этих целей приведет к замене или снижению частоты применяемой в настоящее время трансфузии тромбоцитов, и такой новый белок будет также применен для лечения и диагностики нарушений, связанных с тромбоцитами.
Таким образом, данное изобретение касается (i) очищенной и выделенной последовательности ДНК, кодирующей белок с ТРО активностью, выбранной из группы, состоящей из:
(а) последовательностей ДНК, показанных в SEQ ID 194, 195 и 196, или их комплементарных цепей, и
(b) последовательностей ДНК, гибридизующихся при строгих условиях с последовательностями ДНК п.(а), или их фрагментов, и
(с) последовательностей ДНК, которые гибридизовались бы с последовательностями ДНК пунктов (а) и (в), если бы не вырожденность генетического кода,
(ii) способа получения белка с ТРО активностью, предусматривающего стадии:
выращивание при подходящих условиях питания прокариотических или эукариотических клеток хозяина, трансформированных для трансфицированных указанной последовательностью ДНК таким образом, чтобы была возможна экспрессия данного белка, и
выделение целевого белкового продукта, полученного экспрессией указанной последовательности ДНК, и
(iii) белкового продукта, полученного экспрессией в прокариотической или эукариотической клетке хозяина указанной последовательности ДНК.
Кроме того, данное изобретение касается фармацевтической композиции, содержащей эффективное количество белка с ТРО активностью, и способа лечения связанных с тромбоцитами нарушений, в частности, тромбоцитопении, предусматривающего введение указанного белка больным с такими нарушениями.
Краткое описание чертежей
Фиг. 1 показывает Sephacryl S-200HR гель-фильтрационную хроматографию на Phenyl Sepharose 6 FF/LS F2, полученной из XRP.
Фиг.2 показывает Capcell Pak C1 хроматографию с обращенной фазой УМС-pack CN-AP ТРО-активной фракции, полученной из пробы низкомолекулярного ТРО (Sephacryl S-200HR F3) XRP.
Фиг. 3 показывает анализ в ПААГ-ДСН (SDS-PAGE) Capcell Раk C1 ТРО-активной фракции (PA) из пробы низкомолекулярного ТРО XRP.
Фиг.4 показывает пептидные карты на C18 HPLC с обращенной фазой крысиного ТРО, выделенного электрофорезом в ПААГ-ДСН (SDS-PAGE). Пептидные фрагменты были получены систематическим гидролизом с применением трех протеаз.
Фиг. 5 показывает ТРО активность, полученную из XRP в крысиной тест-системе CFU-MK.
Фиг.6 показывает конструирование экспрессирующего вектора pEF18S.
Фиг. 7 показывает ТРО активность в культуральном супернатанте COSI клеток, в которые был введен pEF18S-A2α, в крысиной тест-системе CFU-MK.
Фиг. 8 показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pEF18S-HL34, в крысиной тест-системе CFU-MK.
Фиг. 9 показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pHT1-231, в крысиной тест-системе CFU-MK.
Фиг.10а показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pHTF1 в крысиной тест-системе CFU-MK.
Фиг.10b показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pHTF1, в тест-системе M-07е.
Фиг. 11 показывает рестрикционную карту клона λНGT1 и конструкцию pHGT1 pEFHGTE.
(E:EcoR1, H: Hind 111, S; Sal1)
Фиг.12а показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pEFHGTE, в крысиной тест-системе CFU-MK.
Фиг.12b показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pEFHFTE, в тест-системе М-07е.
Фиг.13а показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pHT1-211 1, pHT1-191 1 или pHT1-171 2, в крысиной тест-системе CFU-МК.
Фиг.13b показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pHT1-163 2, в крысиной тест-системе CFU-МK.
Фиг. 14 показывает ТРО активность в культуральном супернатанте COS1 клеток, в которые был введен pHT1-211 1, pHT1-191 1, pHT1-171 2 или pHT1-163 2, в тест-системе М-07е.
Фиг. 15 является хроматограммой, полученной хроматографией с обращенной фазой (колонка Vydac), для очистки человеческого ТРО из культурального супернатанта СНО клеток, в которые был введен вектор pDEF202-hTPO-P1, экспрессировавший ТРО.
Фиг. 16 является фотографией, показывающей разделение в SDS-PAGE (ПААГ-ДСН) человеческого ТРО, очищенного из культурального супернатанта СНО клеток, в которые вводили pDEF2020 hTPO-P1 и давали экспрессироваться ТРО.
Фиг. 17 является хроматограммой, полученной хроматографией с обращенной фазой для очистки человеческого ТРО из E.coli, в которую вводили pCFM536/h 6T(1-163) для экспрессии ТРО.
Фиг. 18 является фотографией, показывающей разделение в SDS-PAGE (ПААГ-ДСН) варианта человеческого ТРО (1-163), выделенного очищенного из Е.соli, в которую вводили рСFМ536/h6Т (1-163) и позволяли экспрессироваться ТРО.
Фиг. 19 показывает рисунок элюции hТРO163 на колонке Superdex 75 рg в связи с очисткой hTРО 163 из культурального супернатанта в качестве исходного материала, полученного трансфекцией экспрессирующей человеческий ТРО плазмиды pDEF202 hTРO163 в СНО клетки. Количество белка определяли при 220 нм.
Фиг.20 показывает анализ SDS-PAGE стандартного рТРО163, элюированного из колонки Superdex 75 pg, при очистке hТРO163 из культурального супернатанта в качестве исходного материала, полученного трансфекцией экспрессирующей человеческий ТРО плазмиды pDEF202-hTPO163 в СНО клетки. Белок hTPO163 окрашивали серебром на геле.
Фиг.21 показывает структуру экспрессирующего вектора pSMT201.
Фиг. 22 показывает график, изображающий ТРО активность, определенную тестом М-07е, в культуральном супернатанте COS7 клеток, в которые вводили βGL-TPO, 3/ТРО или 09/ТРО и затем в них происходила экспрессия.
Фиг. 23 является графиком, изображающим ТРО активность, определенную тестом М-07e, в супернатанте культур COS7 клеток, в которые вводили производное с инсерцией или делецией человеческого ТРО, после чего проводили экспрессию.
Фиг.24 показывает увеличенное число тромбоцитов в мышах, которым вводили ТРО при помощи внутривенной и подкожной инъекций.
Фиг.25 показывает зависимое от дозы увеличение числа тромбоцитов в мышах после подкожной инъекции ТРО.
Фиг. 26 показывает индуцированное ТРО увеличение числа тромбоцитов после обработки мышей 5-FU для индуцирования тромбоцитопении.
Фиг. 27 показывает индуцированное ТРО увеличение числа тромбоцитов после обработки мышей гидрохлоридом нимустина для индуцирования тромбоцитопении.
Фиг. 28 показывает индуцированное ТРО увеличение числа тромбоцитов после пересадки костного мозга.
Фиг. 29 показывает индуцированное ТРО увеличение числа тромбоцитов после облучения рентгеновскими лучами мышей для индуцирования тромбоцитопении.
Фиг. 30 показывает зависимое от дозы увеличение числа тромбоцитов после введения укороченного ТРО (аминокислоты 1-163 в SEQ 1D 6).
Фиг. 31 показывает увеличение числа тромбоцитов после введения укороченного ТРО (аминокислоты 1-63 в SEQ 1D 6) после введения гидрохлорида нимустина для индуцирования тромбоцитопении.
Фиг.32 показывает, что увеличение концентраций Мр1-Х, добавленного к системе культуры человеческих мегакариоцитов, приводит к увеличивающемуся блокированию развития мегакариоцитов.
Фиг. 33 представляет ТРО активность, определенную в тесте М-07е, производных ТРО [Met-2, Lys-1, Ala1, Val3, Arg133]- TPO(1-163), [Met-2, Lys-1, Ala1, Val3, Pro148] TPO(1-163) [Met-2, Lys-1, Ala1, Val3, Arg115] TPO(1-163), экспрессированных в E.coli.
Фиг.34 описывает ТРО активность, определенную в тесте M-07e, производных ТРО [Met-2, Lys-1, Ala1, Val3, Arg129]- TPO(1-163), [Met-2, Lys-1, Ala1, Val3, Arg143] TPO(1-163), [Met-2, Lys-1, Ala1, Val3, Leu82] TPO(1-163), [Met-2, Lys-1, Ala1, Val3, Leu146] TPO(1-163) [Met-2, Lys-1, Ala1, Val3, Arg59] TPO(1-163), экспрессированных в E.coli.
Детальное описание изобретения
Данное изобретение специфически обеспечило полипептиды тромбопоэтина (ТРО), имеющие биологическую активность специфического стимулирования или увеличения образования тромбоцитов и содержащие аминокислотную последовательность 1-332 SEQ 1D 6, или их производные. Приводимые в качестве иллюстрации полипептиды включают полипептиды, которые состоят из аминокислотных последовательностей 1-163 SEQ 1D 6, 1-232 SEQ 1D 6, 1-151 SEQ 1D 6, зрелую последовательность аминокислот SEQ 1D 2, 4 и 6, в том числе полипептиды, имеющие делетированные 1-6 аминокислот NН2-конца. Дополнительные приводимые для иллюстрации полипептиды данного изобретения включают [Thr33, Thr333, Ser334, Ile335, Gly336, Tyr337, Pro338, Tyr339, Asp340, Val341, Pro342, Asp343, Tyr244, Ala345, Gly346, Val347, His348, His349, His350, His351, His352, His353, ] TPO, [Asn25, Lys231, Thr333, Ser334, Ile335, Gly336, Tyr337, Pro338, Tyr339, Asp340, Val341, Pro342, Asp343, Tyr344, Ala345, Gly346, Val347, His348, His349, His350, His351, His352, His353] TPO, [Asn25] TPO [Thr33] TPO. Другие полипептиды этого изобретения включают производные TPO полипептиды [ΔHis33] TPO(1-163), [ΔАrg117] ТРO(1-163), [ΔGly116] TPO (1-163), [His33, Thr33', Рrо34] TPO(1-163), [His33, Ala33', Pro34] TPO(1-163), His33, Gly33', Pro34, Ser38] TPO(1-163), [Gly116, Asn116', Arg177] TPO(1-163), [Gly116, Ala116', Arg177] TPO(1-163), [Gly116, Gly116', Arg117] -TPO(1-163), [Ala1, Val3, Arg129] TPO(1-163), [Ala1, Val3, Arg133] -TPO(1-163), [Ala1, Val3, Arg143] TPO(1-163), [Ala1, Val3, Leu82] -TPO(1-163), [Ala1, Val3, Leu146] TPO(1-163), [Ala1, Val3, Pro148] -TPO(1-163), [Ala1, Val3, Аrg59] ТРО(1-163) и [Ala1, Val3, Arg115] -TPO(1-163).
TPO полипептиды этого изобретения также включают такие полипептиды, которые ковалентно связаны с полимером, предпочтительно полиэтиленгликолем. Кроме того, TPO полипептиды изобретения могут включать аминокислоты [Met-2-Lys-1] , [Met-1] или [Gly-1]. Изобретение обеспечивает ДНК, которые кодируют TPO полипептид и описанные выше производные, причем эти ДНК обеспечены в виде кДНК, геномной ДНК и синтетических ДНК.
Изобретение также обеспечивает способы получения TPO полипептида, описанного выше, предусматривающего стадии экспрессии полипептида, кодируемого ДНК данного изобретения в подходящем хозяине, и выделение этого TPO полипептида. Если экспрессируемый TPO полипептид является Мet-2-Lys-1-пoлипептидом, то эти способы предусматривают стадию отщепления Мet-1-Lys-1 от выделенного TPO полипептида.
Изобретение обеспечивает также способы получения ТРО полипептидов, таких как полипептиды, слитые с глутатион-S-трансферазой (GST). ДНК, кодирующую амино-концевой GST полипептид, пептид, узнающий тромбин, и ТРО полипептид вводят в подходящего хозяина, выделяют слитый полипептид и GST-часть удаляют обработкой тромбином. Полученные ТРО полипептиды имеют [Gly-1]-структуру.
Дополнительно обеспечены прокариотические и эукариотические хозяйские клетки, трансформированные или трансфицированные последовательностью ДНК согласно изобретению таким образом, что клетки хозяина способны экспрессировать полипептид, имеющий биологическую активность стимулирования или увеличения образования тромбоцитов.
Фармацевтические композиции этого изобретения содержат эффективное количество ТРО полипептида или его производного в комбинации с фармацевтически приемлемым носителем и допустимы для применения в лечении связанных с тромбоцитами нарушений, в частности, для лечения тромбоцитопении, например, вызванной химиотерапией, лучевой терапией или трансплантацией костного мозга. Изобретение обеспечивает соответствующие способы лечения.
Наконец, данное изобретение обеспечивает антитела, специфически иммунореагирующие с ТРО полипептидами и их производными, описанными выше. Такие антитела применимы в способах выделения и определения количества ТРО полипептидов изобретения.
Согласно данному изобретению, обеспечена новая последовательность ДНК, кодирующая белок, имеющий ТРО активность (называемая далее последовательностью ДНК данного изобретения). Последовательность ДНК данного изобретения содержит последовательность ДНК, которая кодирует аминокислотную последовательность ДНК, которая кодирует аминокислотную последовательность, показанную в SEQ 1D 2, 4 или 6 прилагаемого списка последовательностей.
Также последовательностью ДНК данного изобретения называют последовательность ДНК, кодирующую частично модифицированную (заменой, делецией, инсерцией или добавлением) версию вышеупомянутой аминокислотной последовательности, показанной в SEQ 1D 2, 4 или 6, при условии, что такие модификации не нарушают ТРО активность. Т.е. последовательности ДНК, кодирующие производные ТРО, также включены в данное изобретение.
Другими словами, последовательность ДНК данного изобретения включает последовательности ДНК, кодирующие белковые молекулы, аминокислотные последовательности которых по существу являются аминокислотными последовательностями, показанными в SEQ 1D 2, 4 или 6. Фраза "аминокислотные последовательности являются по существу аминокислотными последовательностями, показанными в SEQ 1D 2, 4 или 6" в применении здесь означает, что эти аминокислотные последовательности включают последовательности, представленные SEQ 1D 2, 4 или 6, а также последовательности, представленные SEQ 1D 2, 4 или 6, которые имеют частичную модификацию, такую как замена, делеция, инсерция, добавление и т.п., при условии, что такие модификации на нарушают ТРО активность.
Далее, последовательность ДНК данного изобретения состоит по существу из последовательности ДНК, кодирующей белок с ТРО активностью.
Выражение "последовательность ДНК, кодирующая аминокислотную последовательность" включает в себя все последовательности ДНК, которые могут иметь вырожденность в нуклеотидных последовательностях.
Последовательность ДНК данного изобретения включает также следующие последовательности:
(а) последовательности ДНК, представленные SEQ 1D 7, 194, 195 и 196, или комплементарные им цепи,
(в) последовательности ДНК, гибридизующиеся при строгих условиях с последовательностями ДНК, определенными в (а), или их фрагментами, или
(с) последовательности ДНК, которые гибридизовались бы с последовательностями ДНК (а) и (в), если бы не вырожденность генетического кода.
Другими словами, последовательность ДНК данного изобретения также включает следующие последовательности:
(а) последовательность ДНК, которая интегрирована в вектор pEF18S-A2α (депозитный FERM BP-4565), включаемый в штамм DH5 E.coli., вектор pHT1-231 (депозитный FERМ ВР-4564), включенный в штамм DH5 E.coli. вектор pHTGI (депозитный FERМ ВР-4617), включенный в штамм DH5 E.coli. вектор рНGT1 (депозитный FERM ВР-4616), включенный в штамм DH5 E.coli., и кодирует аминокислотную последовательность белка, имеющего ТРО активность, или
(в) последовательности ДНК, которые гибридизуются (при строгих условиях) с последовательностями ДНК, описанными в (а), или их фрагментами, и кодируют аминокислотную последовательность белка, имеющего ТРО активность.
В применении здесь, репрезентативные "строгие" условия гибридизации представляют собой такие условия, которые применены в примерах по PCR амплификации ДНК этого изобретения при помощи вырожденных и/или уникальных олиногуклеотидных праймеров (зондов). См. также, например, главы 11 и 14 Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989) Unit 2.10 Current Protocols in Molecular Biology, Ausubel et al., eds. Current Protocols, USA, 1993).
Также последовательностью данного изобретения является последовательность ДНК, которая кодирует белок с ТРО активностью и содержит нуклеотидную последовательность, кодирующую положения 1-163 аминокислотной последовательности, представленной SEQ 1D N 6.
Такие последовательности ДНК могут содержать также сайт расщепления рестриктазой и/или дополнительную последовательность ДНК при сайте инициации, терминации или промежуточном сайте, которые облегчают конструирование легко экспрессируемых векторов. При использовании хозяина, не являющегося млекопитающим, может быть включен предпочтительный кодон для экспрессии гена в этом хозяине.
Примером последовательности ДНК данного изобретения является молекула кДНК, полученная выделением мРНК из клеток млекопитающих, в том числе человека, с последующим скринингом этой кДНК обычным путем из библиотеки кДНК, полученной известным способом. Источниками мРНК в этом случае могут быть клетки полученной из гепатоцитов крысы клеточной линии McA-R Н8994, НТС клетки, Н411-Е клетки, печень крысы, почки, мозг и тонкая кишка крысы, печень человека и т.п.
Другим примером последовательности ДНК данного изобретения является молекула геномной ДНК, которую получают скринингом ее обычным путем из геномной библиотеки, полученной известным способом из клеток млекопитающих, в том числе человека. Источниками геномной ДНК в этом случае могут быть препараты хромосомной ДНК, полученные из человека, крысы, мыши и т.п.
Последовательность ДНК, кодирующая производное TРО, может быть получена из полученной описанным образом последовательности кДНК, кодирующей белок с ТРО активностью, путем модификации этой кДНК с применением сайт-направленного мутагенеза, с частичной модификацией тем самым соответствующей аминокислотной последовательности.
После выяснения аминокислотной последовательности или последовательности ДНК белка, имеющего ТРО активность, в данном изобретении, последовательность ДНК, кодирующая частично модифицированную аминокислотную последовательность, может быть легко получена химическим синтезом.
Последовательность ДНК данного изобретения является ценным материалом для широкомасштабного получения белка, имеющего TPО активность, при помощи различных способов рекомбинантных ДНК.
Последовательность ДНК данного изобретения применима также в качестве меченого зонда для выделения гена, кодирующего родственный ТРО белок, а также кДНК и геномной ДНК, кодирующих ТРО других видов млекопитающих. Она применима также в генной терапии человека и других видов млекопитающих. Кроме того, последовательность ДНК данного изобретения применима для развития трансгенных видов мелкопитающих, которые могут использоваться в качестве эукариотического хозяина для широкомасштабного получения ТРО (Palmiter et al., Science, vol. 222, pp. 809-814, 1983).
Данное изобретение обеспечивает также вектор, в который интегрирована указанная выше последовательность ДНК, кодирующая белок, имеющий ТРО активность, хозяйские клетки, трансформированные этим вектором, и способ получения белка с ТРО активностью, предусматривающий культивирование хозяйских клеток и отделение и очистку экспрессируемого белка, имеющего ТРО активность.
Примерами клеток хозяина, применимых в этом случае, являются прокариотические клетки, такие как клетки Е.соli и т.п., и клетки эукариот, таких как дрожжи, насекомые, млекопитающие и т.п. Иллюстративными примерами клеток млекопитающих являются COS клетки, клетки яичника китайского хомячка (СЕО), С-127 клетки, клетки почек детеныша хомячка (ВНК) и т.п. Иллюстративными примерами дрожжей являются пекарские дрожжи (Saccharomyces cerevisiae), ассимилирующие метанол дрожжи (Pichia pastoris) и т.п. Иллюстративными примерами клеток насекомых являются культивируемые клетки тутового шелкопряда и т. п.
Что касается векторов для применения в трансформации этих хозяйских клеток, то для трансформации клеток Е. coli можно принять рКС30 (Shimatake H. and M. Rosenberg, Nature, 292, 128-132, 1981), pTrC99A (Amann E. et al., Gene, 69, 301-315, 1988) и т.п. Для трансформации клеток млекопитающих можно применять pSV2-neo (Southern and Berg. J. Mol. Appl. Genet., 1, 327-341, 1982), pCAGGS (Niwa et al., Gene, 108, 193-200, 1991), pcDL-SRα296 (Takebe et al. , Mol. Cell. Biol. 8, 466-472, 1988) или т.п. Для дрожжевых клеток можно применять рG-1 (Schena M. and Yamamoto K.R., Science, 241, 965-967, 1988) или т.п. Для трансформации тутового шелкопряда можно применять трансдуцирующий вектор для рекомбинантной вирусной конструкции, например, рАс373 (Luckow et al., Bio/Technology, 6, 47-55, 1988).
Если требуется, каждый из этих векторов может содержать начало репликации, маркер (маркеры) селекции, промотор и т.п., а также сайт сплайсинга РНК, сигнал полиаденилирования и т.п. в случае векторов для применения в эукариотических клетках.
Что касается начала репликации, в векторах для клеток млекопитающих можно применять последовательность, полученную, например, из SV40, аденовируса, вируса бычьей папилломы или т.п. В векторах для клеток Е.coli можно применять последовательность, полученную из ColE1, R фактор, F фактор или т. п. В векторах для дрожжевых клеток можно применять последовательность, полученную из 2 мкм ДНК, ARS1 или т.п.
Что касается промоторов для генной экспрессии, в векторах для клеток млекопитающих можно применять промоторы, полученные, например, из ретровируса, вируса полиомы, аденовируса, SV40 и т.п. Промотор, полученный из бактериофага λ, например, trp, lpp, lac или tac промотор, можно применять в векторах для клеток E.coli. ADH, РНO5, GPD, PGK или MAFα промотор можно применять в векторах для клеток пекарских дрожжей, а АОХ1 промотор или т.п. в векторах для ассимилирующих метанол дрожжей. Промотор, полученный из вируса ядерного полиэдроза, можно применять в векторах для тутового шелкопряда.
Типичными примерами селектируемых маркеров, применимых в векторах для клеток млекопитающих, являются ген устойчивости к неомицину (nео), ген тимидинкиназы (ТК), ген дигидрофолатредуктазы (DHFR), ген ксантин-гуанинфосфорибозилтрансферазы E.coli (ECOgpt) и т.п. Иллюстративными примерами селектируемых маркеров, применимых в векторах для клеток E.coli, являются ген устойчивости к канамицину, ген устойчивости к ампициллину, ген устойчивости к тетрациклину и т.п., а маркерами для дрожжевых клеток являются гены Leu2, Trp1, Ura3 и т.п.
Получение белка с ТРО активностью с использованием подходящих комбинаций хозяин-векторных систем может выполняться путем трансформации подходящих клеток хозяина рекомбинантной ДНК, полученной встраиванием гена данного изобретения в подходящий сайт указанного вектора, культивирования образующегося трансформанта с последующим отделением и очисткой целевого полипептида из полученных клеток или культуральной среды или фильтрата. Обычно применяемые средства и процедуры можно использовать для этого способа в комбинации.
При экспрессии целевого гена исходная сигнальная последовательность может быть модифицирована или заменена сигнальной последовательностью, полученной из другого белка, для того чтобы получить гомогенный N-конец экспрессируемого продукта. Гомогенизация N-конца может выполняться модификацией (заменой или добавлением) аминокислотных остатков в N-конце или вблизи него. В случае экспрессии при помощи E.coli в качестве клетки-хозяина, например, остаток лизина может быть добавлен в дополнение к остатку метионина.
Новый белок данного изобретения, имеющий ТРО активность (называемый далее "белком данного изобретения"), включает белки, каждый из которых содержит аминокислотную последовательность, показанную в SEQ 1D 2, 4 или 6. Производные ТРО, аминокислотные последовательности которых частично модифицированы (заменой, делецией, инсерцией или добавлением), также включены в данное изобретение, при условии, что ТРО активность не нарушена такой модификацией.
Другими словами, белок данного изобретения включает белковые молекулы, аминокислотные последовательности которого являются по существу аминокислотными последовательностями, показанными в SEQ 1D 2, 4 или 6.
Выражение "аминокислотные последовательности являются по существу аминокислотными последовательностями, показанными в (или представленными) SEQ 1D 2, 4 или 6", в применении здесь означает, что указанные аминокислотные последовательности включают последовательности, показанные в SEQ 1D 2, 4 или 6, а также последовательности, показанные в SEQ 2, 4 или 6, которые имеют частичную модификацию, такую как замена, делеция, инсерция, добавление и т.п., при условии, что такие модификации не нарушают ТРО активность.
Белок данного изобретения включает белок, содержащий положения 7-151 аминокислотной последовательности, показанной в SEQ 1D 6, и имеющий ТРО активность. Также белком данного изобретения является белок, который имеет ТРО активность и содержит положения 1-163 аминокислотной последовательности, представленной SEQ 1D 6.
Примеры других ТРО производных данного изобретения включают производное, стабильность и продолжительность жизни которого in vivo были улучшены модификацией аминокислот (заменой, делецией, инсерцией или добавлением), производное, в котором по меньшей мере одно потенциальное гликозилирование было изменено делецией или добавлением, производное, в котором по меньшей мере один остаток цистеина был делетирован или заменен другим аминокислотным остатком (например, остатком аланина или серина).
Предпочтительно, белок данного изобретения отличается тем, что его отделяют и очищают из клеток хозяина, трансформированных рекомбинантным вектором, содержащим молекулу кДНК, молекулу геномной ДНК или фрагмент ДНК, полученный химическим синтезом.
Когда внутриклеточную экспрессию проводят с применением бактерии, такой кaк E.coli в качестве хозяина, получают белок, в котором инициирующий остаток метионина добавлен к N-концевой стороне белковой молекулы, имеющей ТРО активность, что также включено в данное изобретение. В зависимости от применяемого хозяина полученный белок с ТРО активностью может быть гликозилированным или негликозилированным и каждый из этих случаев включен в данное изобретение.
Белок данного изобретения также включает природно встречающиеся ТРО-активные белки, очищенные и выделенные из природных источников, таких как среда культуры клеток, имеющая ТРО активность, моча, сыворотка или плазма человека.
Способ очистки ТРО из таких природных источников также включен в данное изобретение. Такой способ очистки может быть выполнен путем применения одной или комбинации обычно применяемых стадий очистки белков, таких как ионообменная хроматография, аффинная хроматография с применением лектина, аффинная хроматография с триазиновым красителем, гидрофобная хроматография, гель-хроматография, хроматография с обращенной фазой, аффинная хроматография с гепарином, хроматография с сульфатированным гелем, хроматография на гидроксиапатите, изоэлектрическое фокусирование, хроматография с применением хелатов металлов, препаративный электрофорез, гель-электрофорез с изоэлектрическим фокусированием и т.п. Процесс очистки, в котором применяют определенные способы в комбинации, с использованием физико-химических свойств ТРО, которые могут быть выведены из примеров этой заявки, также включен в данное изобретение. Кроме того, можно также использовать аффинную хроматографию с применением антител, способных узнавать ТРО. Далее, было обнаружено, что ТРО является лигандом для Мрl (de Sauvage et al., Nature, 369; 533-538 (1984); Bartley et al., Cell, 77: 1117-1124 (1994); Kaushansky et al. , Nature, 369: 565-568 (1984), при помощи которого ТРО может быть очищен с применением аффинной гель-колонки, с которой связан Мрl. Более конкретно, примером такой колонки является Мрl-Х колонка, приготовленная путем связывания смолы с внеклеточным районом Mpl (Мрl-Х), полученным способами рекомбинантных ДНК, с применением СНО клетки в качестве хозяина (Bartley et al., Supra).
Как описано здесь, полипептиды ТРО этого изобретения отличаются далее способностью связываться с Мрl рецептором и конкретно с его внеклеточным (растворимым) доменом.
В данное изобретение включен также белок, кодируемый частью ДНК, которая комплементарна кодирующей белок цепи человеческой кДНК или последовательности геномной ДНК гена ТРО, а именно, "комплементарно инвертированный белок", описанный
Tramontanо et al. (Nucl. Acids Res., vol.12, pp.5049-5059, 1984).
Также включен в данное изобретение белок данного изобретения, который помечен детектируемым маркером, например, 1251, или биотинилированием, с обеспечением реагента, применимого для детектирования и количественного определения ТРО или экспрессирующих рецептор ТРО клеток в твердых пробах, таких как ткани, и жидких пробах, таких как кровь, моча и т.п.
Биотинилированный белок данного изобретения применим в случае его связывания с иммобилизованным стрептавидином для удаления мегакариоцитов из костного мозга во время аутогенной трансплантации костного мозга. Он также применим в случае его связывания с иммобилизованным стрептавидином для концентрирования аутогенных или аллогенных мегакариоцитных клеток во время аутогенной или аллогенной трансплантации костного мозга. Конъюгат ТРО с токсином, таким как рицин, дифтерийный токсин или т.п., с радиоактивным изотопом применим в противоопухолевой терапии и в кондиционировании трансплантации костного мозга.
Данное изобретение обеспечивает также материал нуклеиновых кислот, который применим при мечении детектируемым маркером, в том числе радиоактивным маркером или нерадиоактивным маркером, таким как биотин, или при использовании в гибридизационной процедуре для детектирования положения человеческого гена ТРО и/или родственного семейства генов на хромосомной карте. Такой материал применим также для доказательства нарушений в гене ТРО человека на уровне ДНК, и он может быть использован как генетический маркер для подтверждения нарушений прилегающих генов.
Данное изобретение обеспечивает также фармацевтическую композицию, содержащую терапевтически эффективное количество белка данного изобретения вместе с полезным и эффективным разбавителем, антисептическим агентом, солюбилизирующим агентом, эмульгатором, адъювантом и/или носителем. Термин "терапевтически эффективное количество" в применении здесь означает количество, которое обеспечивает терапевтическое действие для заявленных состояний и путей введения и кондиционирования. Такую композицию применяют в форме жидкости, лиофилизированного или высушенного препарата. Она содержит разбавитель, выбранный из различных буферов (Трис-HCl, ацетатный буфер и фосфатный буфер, например), имеющих различные величины рН и ионную силу, предотвращающую поверхностное поглощение добавку, такую как альбумин или желатин, поверхностно-активное вещество, такое как Tween 20, Tween 80, Pluronic F68 или соль желчной кислоты, солюбилизирующий агент, такой как глицерин или полиэтиленгликоль, антиоксидант, такой как аскорбиновая кислота или метабисульфит натрия, антисептический агент, такой как тимеросал, бензиловый спирт или парабен, и носитель или агент тоничности, такой как лактоза или маннит. Также применима композиция, предусматривающая ковалентное связывание белка данного изобретения с полимером, таким как полиэтиленгликоль, хелатирование белка с ионами металла, включение белка в гранулярный препарат или на поверхности, состоящей из полимерного соединения, такого как полимолочная кислота, полигликолевая кислота или гидрогель, или включение этого балка в липосомы, микроэмульсии, мицеллы, однослойные или многослойные везикулы, "тени" эритроцитов или сферобластов. Такая композиция будет оказывать влияние на физические условия, растворимость, стабильность, скорость высвобождения in vivo и клиренс in vivo TPO.
Выбор композиции проводят в зависимости от физических и химических свойств применяемого ТРО-активного белка. Данное изобретение также обеспечивает гранулярную композицию, в которой гранулы покрыты полимером, таким как полоксамер или полоксамин, и TPO, который связывается с антителами для тканеспецифических рецепторов, лигандов или антигенов или с тканеспецифическими лигандами рецепторов. Другими примерами композиции данного изобретения являются композиции в форме гранул, которые имеют защитное покрытие и содержат ингибитор протеазы или усилитель проникновения, для применения в различных способах введения, таких как парентеральное, легочное, трансназальное и пероральное введение.
Фармацевтическая композиция, содержащая белок данного изобретения, может вводиться несколько раз в день, обычно в количестве 0,05 мкг - 1 мг/кг (в расчете на белок TPO) веса тела в зависимости от состояния и пола больного, способа введения и т.п.
Фармацевтическая композиция, содержащая белок данного изобретения, может вводиться в дозе 25000-500000 активного ингредиента (в единицах относительной активности, получаемых при помощи теста М-07е, который будет описан позднее) на кг веса тела один раз или несколько раз в день в течение 1-7 дней в неделю в зависимости от симптомов, пола больного и способа введения.
Авторы данной заявки подтвердили, что в отношении С-концевой стороны человеческого TPO можно отметить, что активность сохраняется, когда аминокислотные остатки до 152-й аминокислоты в аминокислотной последовательности, представленной в SEQ 1D 6, делетированы, и что касается N -концевой стороны, активность сохраняется даже в том случае, если аминокислотные остатки до 6-го остатка аминокислотной последовательности делетированы.
Таким образом, белок, имеющий ТРО активность, содержащий аминокислотную последовательность от 7-го до 151-го аминокислотного остатка SEQ 1D 6, и модифицированный (заменой, делецией, инсерцией или добавлением) в других частях, может предпочтительно использоваться в качестве эффективного ингредиента данного исследования так же, как и немодифицированный белок. Более предпочтительным производным ТРО является производное, имеющее аминокислотную последовательность 1-163 последовательности SEQ 1D 6.
Композиция, содержащая по меньшей мере один из дополнительных гематопоэтических факторов, таких как ЕРО, G-CSF, GM-CSF, M-CSL, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, LIF и SCF, в дополнение к белку данного изобретения, также включена в данное изобретение.
Белок данного изобретения применим для лечения различных тромбоцитопенических заболеваний, либо в отдельности, либо в комбинации с другими дополнительными гематопоэтическими факторами. Примерами других дополнительных гематопоэтических факторов являются ЕРО, G-CSF, GM-CSF, M-CSF, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, LIF и SCF.
Имеется много болезней, характеризующихся связанными с тромбоцитами нарушениями, таких как тромбоцитопения, обусловленная нарушениями в образовании тромбоцитов или уменьшением срока жизни тромбоцитов (вызываемым разрушением или расходованием тромбоцитов), которые могут излечиваться белком данного изобретения. Например, он может быть применен для усиления восстановления тромбоцитов в тромбоцитопеническом больном, болезнь которого вызвана врожденной гипопластической анемией с панцитопенией и другими аномалиями (синдромом Фанкони) или гипопластической анемией, вызванной химиотерапией или облучением, миелодиспластическим синдромом, острой миелоцитной лейкемией, апластическим кризом после трансплантации костного мозга. Тромбоцитопения, вызванная уменьшением срока жизни тромбоцитов или мегакариоцитов, включает в себя идиопатическую тромбоцитопеническую пурпуру, гипопластическую анемию, синдром приобретенного иммунодефицита (СПИД), синдром рассеянной внутрисосудистой коагуляции и тромботическую тромбоцитопению. Кроме того, белок можно также применять в случае аутотрансфузии тромбоцитов, при которой ТРО вводят больному перед хирургической операцией для увеличения числа собственных тромбоцитов больного и, следовательно, увеличенное количество тромбоцитов используется в качестве переливаемых тромбоцитов в момент операции.
Другим применением белка данного изобретения является лечение болезней, связанных с временным снижением или разрушением тромбоцитов, вызванным другими химическими или фармацевтическими лекарственными средствами или лечебными обработками. ТРО можно применять в целях усиленного выделения новых "интактных" тромбоцитов в таких больных.
Далее, данное изобретение касается антитела, специфического для ТРО. Белок данного изобретения можно использовать в качестве антигенов, и соответствующие антитела включают как моноклональные, так и поликлональные антитела и химерные антитела, а именно, "рекомбинантные" антитела, полученные общепринятыми способами. Для получения таких антител против ТРО можно использовать сам человеческий ТРО в качестве антигена или, альтернативно, частичный пептид человеческого ТРО может быть использован в качестве антигена. При использовании антитела к антигену такого пептида эпитоп может быть определен путем спецификации антигенного района. С другой стороны, при использовании антитела к самому антигенному белку(ТРО) антигенный район может быть выяснен анализом эпитопа антитела. В таких случаях эти антитела, каждый из которых имеет выясненный таким образом зпитоп, могут применяться для фракционирования, детектирования, количественного определения и очистки различных ТРО, которые отличаются по их свойствам, например, по типу добавленного сахара, длине пептидных цепей и т.д.
Пептид ТРО, содержащий выбранную таким образом аминокислотную последовательность, синтезируют и связывают с подходящим белком-носителем, например, альбумином, KLH (гемоцианином фиссурелли) или т.п., путем ковалентного связывания для приготовления иммунореактивного антигена. Альтернативно, пептид типа пептида с множеством антигенов (MAP) получают для применения в качестве антигена по способу Tam (Proc.Natl. Acad. Sci. (USA) 85:5409-5423, 1983). Затем приготовленный таким образом антиген вместе с адъювантом или т.п. используют для иммунизации млекопитающих, птиц и т.д., которые обычно применяются для продуцирования антител, таких как кролики, мыши, крысы, козы, овцы, цыплята, хомяки, лошади, морские свинки и т.д. Из иммунизированного таким образом животного была получена его антисыворотка и клетки, из которых получали поликлональные антитела. При использовании пептида в качестве антигена эпитоп можно определить спецификацией антигенного района. В этом случае районы различных молекул ТРО, имеющих разные мол. массы, подвергают скринингу и идентифицируют при помощи иммунологической инженерии. Например, можно скринировать и идентифицировать район с делецией пептида. Кроме того, ген антитела или часть этого гена клонируют из клеток, экспрессирующих целевое антитело, для получения молекулы антитела, которая была экспрессирована с применением генной инженерии.
По сравнению с поликлональным антителом, которое содержит различные антитела, специфические для различных антигенных детерминант (эпитопов), моноклональное антитело специфично только для одной антигенной детерминанты на антигене. Специфическое для ТРО антитело применимо для улучшения селективности и специфичности диагностики, основанной на реакции антиген-антитело, и аналитического тест-способа, а также в проведении отделения и очистки ТРО. Кроме того, такие антитела можно применять для нейтрализации или удаления ТРО из сыворотки. Моноклональные антитела также применимы для детектирования и количественного определения ТРО, например, в сыворотке цельной крови.
Антитело против ТРО человека данного изобретения можно использовать в качестве лиганда в аффинной хроматографии для очистки и выделения ТРО человека. Для фиксирования антитела таким образом, чтобы его можно было применять в аффинной хроматографии, могут быть использованы любые общепринятые способы фиксации различных ферментов. Например, применим способ с использованием носителя СNВr - активированной Sepharose 4В (производимой Pharmacia Fine Chemicals Co. ) или т.п. Для очистки ТРО человека с применением фиксированного антитела против ТРО человека фиксированное антитело загружают в колонку и содержащую ТРО человека жидкость пропускают через эту колонку. В результате этой операции большое количество ТРО человека адсорбируется на носителе в колонке. В качестве растворителя для элюции применимы, например, буфер глицин-НС1 (рН 2.5), раствор хлорида натрия, пропионовая кислота, диоксан, этиленгликоль, хаотропная соль, гидрохлорид гуанидина, мочевина и т.д. Путем элюции подобным растворителем элюируют высокоочищенный ТРО человека.
Антитело данного изобретения можно применять для определения ТРО человека иммунохимическим количественным анализом, в частности, иммуноферментным анализом, проводимым по способу твердофазного "сэндвича".
Преимуществом моноклональных антител является то, что они могут продуцироваться гибридомными клетками в среде, не содержащей каких-либо других молекул иммуноглобулинов. Моноклональные антитела можно получать из культуральных супернатантов гибридомных клеток или из мышиных асцитов, индуцированных интраперитонеальной инъекцией гибридомных клеток. Гибридомный способ, исходно раскрытый Kohler Milstein (Eur.J. Immunol. 6:511-519, 1976), может быть широко использован для получения линий гибридных клеток, которые обладают высоким уровнем моноклональных антител для ряда специфических антигенов.
Для выделения целевого антитела из полученного таким образом содержащего антитело материала, такого как антисыворотка и т.д., могут комбинироваться одна или несколько стадий, которые обычно применяют для очистки белка (аффинная хроматография, например, протеин А-аффинная хроматография, протеин G-аффинная хроматография, Avid гель-хроматография, гель-хроматография с фиксированным анти-иммуноглобулином, и т.д. а также катионообменная хроматография, анионообменная хроматография, аффинная хроматография с применением лектина, адсорбционная хроматография с красителем, гидрофобная хроматография, гельпроникающая хроматография, хроматография с обращенной фазой, гидроксил-апатитная хроматография, фтор-апатитная хроматография, хроматография с применением хелатов металла, хроматография с изоэлектрической фокусировкой, распределительная хроматография, электрофорез с изоэлектрической фокусировкой и т.д.). Кроме этих способов, может быть также применен способ аффинной очистки с использованием антигена, в котором готовят гель-носитель или мембрану, к которым химически присоединен сам белок ТРО человека или пептид, содержащий район антигена или часть этого района, т.е. молекула, способная узнавать целевое антитело, к носителю или мембране, полученным таким образом, добавляют содержащий антитело материал, так что целевое антитело адсорбируется на носителе или мембране. После этого адсорбированное таким образом антитело элюируют и извлекают при подходящих условиях.
Данное изобретение также позволяет применять эндогенные последовательности ДНК, кодирующие ТРО полилептид, для получения больших количеств полипептидных продуктов. Например, могут быть применены способы гомологичной рекомбинации in vitro и ex vivo для трансформации клеток хозяина с целью обеспечения экспрессии или усиленной экспрессии полипептидов. Предпочтительными клетками хозяина являются человеческие клетки (например, печени, костного мозга и т.п.), в которые вводят промотор или анхансерную последовательность, применяя фланкирующие последовательности, которые гомологичны целевому району в клеточном геноме, в результате чего происходит или усиливается экспрессия ТРО полипептида. См., например, U.S. Letters Patent 5 272 071, опубликованный РСТ WO 90/14092, WO 91/06666, WO 91/09955.
Это изобретение обеспечивает также способы получения ТРО полипептида, описанного выше, предусматривающие стадии экспрессии полипептида, кодируемого ДНК данного изобретения, в подходящем хозяине и выделение этого ТРО полипептида. Если экспрессируемым ТРО полипептидом является Met-2 - Lys-1 полипептид, такие способы могут предусматривать далее стадию отщепления Met-2 - Lys-1 от выделенного ТРО полипептида.
Также обеспечены способы получения ТРО полипептидов, имеющих [Gly-1]-структуру, предусматривающие стадии введения в подходящую клетку хозяина ДНК, кодирующей полипептид глутатион- S-трансферазу, 5' по отношению к последовательностям, кодирующим ТРО полипептид, причем GST и ТРО полипептид кодирующие последовательности разделены ДНК, кодирующей полипептид, узнающий тромбин, выделения продукта экспрессии GST-TPO и обработки экспрессированного полипептида тромбином для удаления аминокислот GST. Полученные ТРО полипептиды имеют [Gly-1]-структуру.
Данное изобретение описано далее в деталях.
(А) Очистка крысиного ТРО, анализ частичных аминокислотных последовательностей очищенного крысиного ТРО и анализ биологических характеристик очищенного крысиного ТРО.
Авторы данного изобретения сначала пытались очистить белок (крысиный ТРО), который обладает активностью усиления пролиферации и дифференцировки крысиных CFU-МK. В этом исследовании по очистке были сделаны многие ошибки в процедурах очистки, таких как выбор различных природных источников питания, подбор гелей для хроматографии и способов разделения. В результате авторам удалось очистить белок, имеющий ТРО активность, из плазмы крови тромбоцитопенических крыс, индуцированных облучением рентгеновскими или γ-лучами, с использованием ТРО активности в качестве маркера в тесте с крысиными CFU-МK, который будет описан ниже в "Стандартном Примере", и определить частичные аминокислотные последовательности очищенного белка ("Примеры 1 и 2").
Биологические характеристики полученного из плазмы крысиного ТРО также определяли в "Примере 3".
Основы процедур от очистки крысиного ТРО до определения частичных аминокислотных последовательностей очищенного белка изложены далее.
(i) Пробу плазмы крови получали из приблизительно 1100 тромбоцитопенических крыс, индуцированных рентгеновскими или γ-лучами, и подвергали хроматографии на Sephadex G-25, анионообменной хроматографии (Q-Sepharose FF) и лектиновой хроматографии (WGA-Agarose) в указанном порядке, получая адсорбированную на WGA-Agarose ТРО-активную фракцию.
(ii) Затем полученную адсорбированную ТРО-активную фракцию подвергали аффинной хроматографии с триазиновым красителем (TSK AF-BLUE 650 МН), гидрофобной хроматографии (Phenyl Sepharose 6 FF/LS) и гель-хроматографии (Sephacryl S-200 HR) в этой последовательности. Поскольку ТРО активность разделилась на 4 пика (F1 (фракция 1) в качестве самой высокомолекулярной фракции, затем F2, F3 и F4) при гель-фильтрации на Sephacryl S-200 HR, каждую из ТРО-активных фракций F2 и F3 концентрировали для получения пробы F2 ТРО с высокой мол. массой и пробы F3 с низкомолекулярным ТРО, которые использовали отдельно в последующих стадиях очистки.
(iii) Пробу F3 низкомолекулярного ТРО подвергали препаративной хроматографии с обращенной фазой (YMC-Pack PRO-TEIN-RP), хроматографии с обращенной фазой (YMC-Pack CN-AP), хроматографии с обращенной фазой (Capcell Pack CI) в указанной последовательности. Когда полученную таким образом ТРО-активную фракцию наносили на ПААГ-ДСН (SDS-PAGE) для электрофореза и ТРО-активное вещество экстрагировали из геля, присутствие ТРО активности обнаруживали в полосе, соответствующей средним мол. массам приблизительно 17000-19000 при невосстанавливающих условиях.
(iv) Впоследствии все ТРО-активные фракции наносили на гель SDS-PAGe при невосстанавливающих условиях и переносили на PVDF мембрану. Путем проведения систематического ограниченного ферментного гидролиза белка нa PVDF мембране до пептидных фрагментов были определены частичные аминокислотные последовательности крысиного белка ТРО. На основе информации об аминокислотных последовательностях двух пептидных фрагментов проводили клонирование гена крысиного ТРО.
(v) Отдельно от этого, пробу F2 высокомолекулярного ТРО, полученную при помощи Sephacryl S-200 HR, подвергали очистке таким же способом, которым очищали пробу F3 низкомолекулярного ТРО. Когда ТРО-активную фракцию, полученную конечной хроматографией с обращенной фазой (Capcell Pack C1), наносили на гель SDS-PAGE и ТРО-активное вещество экстрагировали из геля, присутствие ТРО активности обнаруживали в полосе, соответствующей средним мол. массам приблизительно 17000-22000 при невосстанавливающих условиях.
(В) Специализация продуцирующих крысиный ТРО клеток, получение мРНК и конструирование библиотеки кДНК крыс
Поскольку очищенный таким образом крысиный ТРО был получен из плазмы крови, было необходимо скринировать органы или клетки в качестве источников мРНК для применения в клонировании кДНК. В результате ТРО активности в различных органах и супернатантах культур клеток были скринированы на основе биохимических и биологических свойств крысиного полученного из плазмы ТРО. В результате ТРО активности, почти равные активности крысиного полученного из плазмы ТРО были найдены в культурном супернатанте клеточных линий гепатоцитов крыс McA-RH8994, HTC и Н4-11-Е, а также в супернатанте культуры крысиных первичных гепатоцитов (пример 4).
С другой стороны, сконструировали экспрессирующий вектор pEF18S из 2 экпрессируюцих векторов рМЕ189 и рЕFВOS (пример 5). Конструирование этого вектора сделало возможным легкое клонирование кДНК с применением высокоэффективного экспрессирующего вектора, имеющего множественные сайты клонирования, которые могут быть использованы для интегрирования инсерций.
Кроме указанного выше экспрессирующего вектора, для конструирования библиотек кДНК применяли в основном плазмидные векторы рUC и pBR и векторы на основе фага λ.
МсА-RН8994 клетки культивировали, гомогенизировали с добавлением раствора тиоцианата гуанидина и подвергали затем центрифугированию в градиенте плотности CSC1, получая общую РНК (пример 6).
Получение РНК можно также проводить фенольным способом (с горячим фенолом), способом с применением кислого гуанидиния-фенола-хлороформа и т.п.
После отделения поли (А)+РНК от обшей РНК при помощи олиго-dT -иммобилизованных частиц латекса, синтезировали первую цепь кДНК при помощи обратной транскриптазы с применением олиго- dT в качестве праймера, к которому была добавлена последовательность узнвания рестриктазы Notl, с последующим синтезом второй цепи кДНК при помощи РНКазы Н и ДНК-полимеразы 1 E.coli. EcoRl добавляли к полученной таким образом двухцепочечной кДНК, полученную кДНК лигировали с экспрессирующим вектором клеток млекопитающих pEF18S, сконструированным в примере 5, который был расщеплен Notl и EcoRl, и затем лигированную таким образом кДНК трансформировали в компетентные клетки штамма DН5 E.coli для конструирования библиотеки кДНК (пример 7).
(С) Получение (клонирование) фрагмента кДНК крысиного ТРО при помощи PCR
Последовательность ДНК выводили из частичной аминокислотной последовательности крысиного ТРО, очищенного из плазмы крови крыс, для синтеза вырожденных праймеров, применяемых в полимеразной цепной реакции (РСR). Праймеры для использования в РСR можно получить также на основе другой аминокислотной последовательности, чем положение праймеров, примененных здесь. Праймеры с высокой вырожденностью можно также использовать без применения инозина. Кроме того, праймеры с уменьшенной вырожденностью могут быть сконструированы путем применения кодона, высоко применимого в крысах (Wada et al., Nucleic Acids Res., vol. 18, p. 2367-2411, 1990).
Когда плазмидную ДНК экстрагировали из всей части библиотеки кДНК, полученной, как описано выше, и проводили PCR с использованием экстрагированной ДНК в качестве матрицы, обнаружили полосу приблизительно 330 п.н., которая была затем определена секвенированием нуклеотидов как фрагмент ДНК (А1 фрагмент), кодирующий часть крысиного ТРО (пример 8).
(Д) Скрининг кДНК крысиного ТРО при помощи PCR, последовательность кДНК крысиного ТРО и подтверждение ТРО активности
Библиотеку кДНК, полученную, как описано выше, разделяли на пулы, каждый из которых содержал приблизительно 10000 клонов, и экстрагировали плазмидную ДНК из каждого из 100 пулов. При проведении РСR с применением каждого пула плазмидной ДНК в качестве матрицы и праймеров, вновь синтезированных на основе нуклеотидной последовательности А1 фрагмента, в 3 пулах были обнаружены полосы, которые считали специфическими. Один из этих 3 пулов разделили на субпулы, каждый из которых содержал приблизительно 900 клонов. Плазмидную ДНК очищали из 100 субпулов для проведения PCR описанным способом. В результате специфическую полосу обнаружили в 3 субпулах. Один из этих пулов делили на субпулы, каждый из которых содержал 40 клонов, и наконец каждый клон скринировали при помощи PCR описанным способом. В результате был выделен клон pEF18S-A2α, который, по-видимому, кодирует кДНК крысиного ТРО (примеры 9 и 10).
При анализе нуклеотидной последовательности этого клона обнаружили, что она кодирует частичную аминокислотную последовательность белка, очищенного из плазмы крови крысы, подтвердив, следовательно, сильную вероятность того, что этот клон содержит кДНК крысиного ТРО (пример 10).
При очистке плазмидной ДНК из клона pEF18S-A2α, полученного, как описано выше, и трансфекции в COS 1 клетки обнаружили ТРО активность в супернатанте культуры трансфицированных клеток. Это подтвердило, что клон pEF18S-A2α содержит кДНК, кодирующую крысиный ТРО (пример 11).
(Е) Обнаружение мРНК ТРО в различных тканях крыс
Экспрессию мРНК ТРО в тканях крыс анализировали при помощи РСR. Специфическую экспрессию обнаружили в мозгу, печени, тонких кишках и почках (пример 12).
(F) Конструирование библиотеки кДНК человека
На основании результатов примеров 4 и 12 печень выбрали в качестве исходной ткани для клонирования кДНК человеческого ТРО. Впоследствии библиотеку кДНК конструировали с применением коммерческой мРНК, полученной из печени здорового человека. С применением pEF-18S в качестве вектора, как и в случае библиотеки кДНК крысы, синтезировали кДНК аналогичными способами, получая библиотеку направленно клонированных кДНК с использованием рестриктаз Not1 и EcoR1. Библиотека, полученная введением лигированных с вектором кДНК в E. coli DH5, содержала приблизительно 1200000 клонов (пример 13).
(G) Получение (клонирование) фрагмента кДНК человеческого ТРО при помощи PCR
Несколько праймеров для PCR синтезировали на основе нуклеотидной последовательности клона pEF18S-A2α, кодирующей кДНК крысиного ТРО. При синтезе кДНК с применением коммерческой мРНК, полученной из нормальной печени человека, и проведении РСR с применением этих праймеров и кДНК в качестве матрицы наблюдали полосу приблизительно 620 п.н. При анализе нуклеотидной последовательности обнаружили, что этот клон содержит фрагмент ДНК, который имеет гомологию приблизительно 86% с кДНК крысиного ТРО, подтверждая, таким образом, сильную вероятность того, что это часть гена, кодирующего ТРО человека (пример 14).
(Н) Скрининг кДНК ТРО человека при помощи РСR, последовательность кДНК ТРО человека и подтверждение ТРО активности
Библиотеку кДНК человека, полученную, как описано выше, амплифицировали, разделяли на пулы, каждый из которых содержал приблизительно 10000 клонов, и плазмидную ДНК экстрагировали из каждого из 90 пулов. При проведении PCR с применением каждого пула молекул плазмидной ДНК в качестве матрицы и праймеров, вновь синтезированных на основе нуклеотидной последовательности фрагмента ТРО человека, полученного в примере 14, возможные полосы обнаружили в 3 пулах. Один из этих пулов делили на субпулы, каждый из которых содержал 5000 клонов, и плазмидную ДНК очищали из каждого из 90 субпулов. При проведении РСR с применением каждого пула плазмидной ДНК в качестве матрицы тем же способом возможные полосы обнаруживали в 5 субпулах. При разделении одного из этих пулов на субпулы, каждый из которых содержал 250 клонов, очистке плазмидной ДНК из каждого из 90 субклонов и проведении PCR возможные полосы были обнаружены в 3 субклонах. При разделении одного из этих пулов на субпулы, каждый из которых содержал 30 клонов, очистке плазмидной ДНК из каждого из 90 субпулов и проведении РСR возможные полосы были обнаружены в 3 субпулах. После этого выделили 90 колоний из одного из этих субпулов и плазмидную ДНК, очищенную из каждой из этих колоний, подвергали РСR, получая наконец клон, названный HL34 (пример 15).
При анализе нуклеотидной последовательности плазмидной ДНК в этом клоне обнаружили, что этот клон содержит кДНК, имеющую приблизительно 84% гомологии с кДНК крысиного ТРО (пример 16).
При очистке клонированной плазмидной ДНК и трансфекции ее в COS1 клетки ТРО активность была обнаружена в культуральном супернатанте трансфицированных клеток. Таким образом, было подтверждено, что этот плазмидный клон содержит кДНК, кодирующую ТРО человека (пример 17).
Однако, по-видимому, эта кДНК является искусственным продуктом клонирования, поскольку в этом клоне не был обнаружен терминирующий кодон и на его 3'-конце была обнаружена последовательность, подобная поли(А)-хвосту. Впоследствии был сконструирован экспрессирующий вектор, который кодировал аминокислотную последовательность, за исключением части, соответствующей последовательности, подобной поли(А)-хвосту. При экспрессии сконструированного таким образом вектора в COS 1 клетках ТРО активность была обнаружена в полученном супернатанте культуры (пример 18).
При помощи РСR был получен ДНК фрагмент в 3'-концевом районе ТРО человека для анализа структуры полноразмерной кДНК.
Определение нуклеотидной последовательности этого фрагмента обнаружило, что он частично перекрывается с кДНК, которую несет клон HL34, полученный в примере 15. Также ожидали, что полноразмерная кДНК ТРО человека может содержать открытую рамку считывания и кодировать белок, состоящий из 353 аминокислот, в том числе сигнальную последовательность из 21 аминокислот (пример 19).
Кроме описанного выше способа клонирования, клонирование кДНК ТРО человека может быть достигнуто гибридизацией колоний или гибридизацией бляшек с применением фрагмента кДНК крысиного ТРО в качестве зонда, с использованием библиотеки, сконструированной при помощи векторов на основе рUС или рBR вектора на основе фага λ или т.п. При конструировании вырожденного зонда можно применять инозин для уменьшения степени вырожденности. При наличии тест-способа, способного специфически детектировать ТРО активность или обладающего высокой чувствительностью, можно проводить экспрессионное клонирование с использованием библиотеки, как это было сделано в данном изобретении.
Однако, поскольку содержание кодирующей ТРО РНК человека, по-видимому, чрезвычайно мало в нормальной печени человека, как будет описано далее в примере 15 (содержание, рассчитанное из результатов этого примера, было в отношении 1: 3000000), гибридизационный скрининг, в котором применяют синтезированный олигонуклеотид или фрагмент кДНК ТРО крысы или человека в качестве зонда, будет очень трудно выполнить, поскольку количество клонов или бляшек, которые должны быть обработаны, становится очень большим и чувствительность и специфичность способа гибридизации являются более низкими, чем чувствительность и специфичность способа PCR. На самом деле авторы данного изобретения проводили гибридизацию колоний двух миллионов клонов в библиотеке кДНК нормальной печени человека, полученной, как описано в примере 13, с применением фрагмента кДНК ТРО крысы в качестве зонда, но не смогли получить клон кДНК ТРО человека.
(I) Реконструкция кДНК, полученной из нормальной печени человека
Поскольку клон HL34, полученный в примере 15, по-видимому, содержит неполную кДНК, библиотеку кДНК реконструировали с применением коммерческого препарата полученной из нормальной печени человека поли (А) +РНК для получения полной кДНК ТРО человека. Эта библиотека (hTPO-F1), полученная введением лигированной с вектором кДНК в E.coli DH5, содержала 1,0•106 трансформантов (пример 20).
(J) Скрининг клона кДНК ТРО, определение последовательности и экспрессия кДНК ТРО человека и подтверждение ТРО активности
Праймеры для PCR синтезировали на основе нуклеотидной последовательности (SEQ 1D 3) частичной кДНК, полученной в примере 14, и нуклеотидной последовательности (SEQ 1D 196) полной кДНК ТРО человека, предсказанной в примере 19.
Библиотеку кДНК печени человека (hTPO-F1), сконструированную в примере 20, делили на 3 пула (пулы 1-3). РСR проводили с применением плазмидной ДНК, полученной из каждого пула, в качестве матрицы и синтезированных праймеров. В результате при использовании плазмидной ДНК, полученной из пула 3, был амплифицирован фрагмент ДНК, имеющий ожидаемый размер. Затем пул 3 делили на субпулы, каждый из которых содержал 15000 трансформантов, и проводили скрининг при помощи PCR, как описано выше. В результате амплификация ДНК, имеющей ожидаемый размер, была обнаружена в 6 из 90 пулов. При делении одного из этих положительных пулов на субпулы, каждый из которых содержал 1000 клонов, экстракции плазмидной ДНК и проведении PCR тем же способом не наблюдали амплификации ДНК. Считали, что это вызвано низким извлечением плазмидной ДНК из-за более слабого роста целевого клона по сравнению с ростом других клонов. Поэтому исходный пул 3 размножали на чашках 100L В с таким размером инокулята, что на каждой чашке L В росли 4100 колоний, и из каждой из инокулированных таким образом чашек получали чашку-реплику. При проведении РСR этих ДНК, экстрагированных из колоний, росших на этой чашке, амплификацию ожидаемой полосы наблюдали в одном из 100 субпулов.
Из каждой чашки этого субпула готовили два фильтра-реплики и гибридизацию колоний проводили при помощи меченого зонда (фрагмента EcoR1/BamH1 плазмиды рЕF18S-НL34). В результате только один сигнал был положительным. Колонии собирали из исходной чашки и вновь инокулировали на L В чашку. Пробы ДНК получали из 50 колоний, росших на этой чашке, и подвергали их РСR, получая наконец клон, названный pHTF1 (пример 21). В скрининге клона кДНК применяли в основном такие способы, как гибридизация, PCR и экспрессионное клонирование, и комбинации этих способов увеличила эффективность и чувствительность скрининга или уменьшила затраты труда для скрининга. При определении нуклеотидной последовательности плученного таким образом клона рНТF1 обнаружили, что клон pHTF1 имел открытую рамку считывания и аминокислотная последовательность белка, кодируемого (потенциально) этой открытой рамкой считывания, полностью совпадала с дедуцированной аминокислотной последовательностью (SEQ 1D 6) ТРО человека. Нуклеотидная последовательность отличалась от дедуцированной (SEQ 1D 196) при 3 положениях, но эти отличия не вызывали изменения аминокислот. Подтверждено, что белок ТРО человека содержит 353 аминокислотных остатка, в том числе сигнальную последовательность из 21 аминокислот. При получении плазмидной ДНК из полученного таким образом клона pHTF1 и трансфекции ее в СОS 1 клетки ТРО активность обнаружили в культуральном супернатанте (пример 23).
(К) Скрининг хромосомной ДНК ТРО человека при помощи гибридизации пятен, определение последовательности и экспрессия хромосомной ДНК ТРО человека и подтверждение ТРО активности
Геномную библиотеку, полученную от проф. Т. Ямамото ((Gene Research Center, Tohoku University) высевали на чашки 18 NZYМ в таком размере инокулята, что одна чашка содержала 30000 фаговых частиц, и из каждой из приготовленных таким образом 18 чашек получали по два фильтра-реплики. Фрагмент кДНК ТРО человека (номера положений оснований 178-1025 в SEQ 1D 7) амплифицировали при помощи PCR и очищали. При помощи этого очищенного фрагмента, меченого 32Р, в качестве зонда проводили гибридизацию пятен. В результате получили 13 положительных пятен. Пятна собирали из исходных чашек и инокулировали опять на чашки NZYM в таком размере инокулята, что на каждой чашке образовались 1000 пятен. Из каждой из полученных чашек готовили два фильтра-реплики для проведения гибридизации пятен при условиях, описанных выше. В результате положительные сигналы были обнаружены на всех фильтрах 13 групп. Одно пятно (бляшку) извлекали из каждой из полученных чашек для получения фаговой ДНК. Пробы фазовой ДНК, полученной таким образом из 13 клонов, проверяли на присутствие кодирующего района кДНК при помощи РСR. Пять из 13 клонов, по-видимому, содержат полный кодирующий аминокислоты район, предсказанный из этой кДНК. Впоследствии один из этих клонов (клон λ HGT1) отбирали и анализировали Саузерн-блоттингом (с применением указанного выше зонда). Поскольку в случае расщепления Hind 111 наблюдали одну полосу приблизительно 10 т.п.н., ДНК этого клона расщепляли и Hind 111 подвергали электрофорезу в агарозном геле. Полосу 10 т.п.н. вырезали из геля, очищали и субклонировали в клонирующий фектор pUC13, получая в конце концов клон, названный pHGT1 (пример 24).
При определении нуклеотидной последовательности полученного таким образом клона pHGT1 было обнаружено, что хромосомная ДНК, несомая этим клоном, содержит весь кодирующий район для белка, предсказанного в примере 19, а нуклеотидная последовательность этого кодирующего района полностью совпадает с последовательностью предсказанной нуклеотидной последовательности (SEQ 1D 196).
Кроме того, район, соответствующий кодирующему аминокислоты экзону, содержал 4 интрона и нуклеотидная последовательность отличалась от последовательности полной кДНК клона pHTF1, полученного в примере 21 (SEQ 1D 7).
При определении нуклеотидных последовательностей 4 оставшихся клонов среди 5 хромосомных клонов ДНК, независимо отобранных скринингом, при помощи скрининга обнаружили, что 2 из 4 клонов были идентичны клону pHGT1, а другие 2 клона были сходны с клоном рНGT1, за исключением того, что они имели отличающийся нуклеотид внутри 3'-некодирующего района, как наблюдали с клоном pHTF1 (пример 25).
Фрагмент EcoRI в полученном таким образом плазмидном клоне pHGT1 лигировали с экспрессирущим вектором PEF18S, получая экспрессионную плазмиду pEFHGTE ТРО человека. После получения этой плазмидной ДНК (pEFHGTE) и трансфекции ее в COS 1 клетки в супернатанте культуры была обнаружена TPO активность (пример 26).
(L) Получение делеционных производных TPO человека, их экспрессия в СOS1 клетках и подтверждение TPO активности
Результаты примеров 18 и 22 выявили, что белок TPO человека мог бы проявлять его биологическую активность даже после удаления его карбокси-концевой трети. Таким образом, для дальнейшего анализа биологически активных частей проводили эксперименты с делеционными производными. Ряд экспрессирующих плазмид готовили при помощи РСR с использованием ДНК плазмидного клона рНТ1-231, полученного в примере 18, в качестве матрицы и синтезированных олигонуклеотидов в качестве праймеров.
Были получены экспрессионные плазмиды, которые содержали ДНК, кодирующую делециоиные производные TPO человека, не имеющие карбоксил-концевого района белка TPO, т.е. делеционные производные, кодирующие положения 1-211, 1-191, 1-171 и 1-163 аминокислотной последовательности. При приготовлении плазмидной ДНК из каждого из этих клонов и трансфекции ее в COS 1 клетки TPO активность была обнаружена в каждом культуральном супернатанте (пример 27).
Когда были сконструированы производные, имеющие ряд делеций С-концевых аминокислотных остатков до положения 151, и другие производные, имеющие соответственную делецию N-концевых аминокислотных остатков до 6-го, 7-го и 12-го положений, и активность после их экспрессии в СОS 1 клетках была измерена для анализа несущего ТРО активность района в больших деталях, оказалось, что активность становилась недетектируемой при делеции аминокислотных остатков N-концевой стороны до положения 7 или при делеции аминокислотных остатков С-концевой стороны до положения 151 (примеры 28 и 29).
Производные белка, имеющего ТРО активность, могут быть получены путем модификации (делеции, замены, инсерции или добавления) кДНК, кодирующей этот белок. Для такой модификации можно применять такие способы, как PCR, сайт-направленный мутагенез и химический синтез.
(М) Экспрессия кДНК ТРО человека в СНО клетке и очистка ТРО
Был сконструирован экспрессирующий вектор, pHTP1, который может экспрессировать кДНК, кодирующую дедуцированную аминокислотную последовательность ТРО человека, показанную в SEQ 1D 6, в клетках животных (пример 30).
Для конструирования вектора pDEF202-hTPO-P1 использовали район кДНК в pHTP1 для его экспресии в СНО клетке (пример 31).
В результате трансфекции этого вектора в СНО клетки и последующего отбора трансфицированных клеток были получены трансфицированные клетки, в которых экспрессирующий вектор, несущий кДНК ТРО человека, был интегрирован в хромосомы СНО клеток (пример 32).
В примере 32 проводили широкомасштабное культивирование клеточной линии СНО (СНО 28-30 клетки, устойчивые к 25 нМ МТХ), продуцирующей ТРО человека. Клетки были получены трансфицированием экспрессирующей ТРО человека плазмиды pDEF202-hTPO-P1 в СНО клетки (пример 55).
Из культурального супернатанта 100 л очищали ТРО человека (пример 56).
Альтернативно, ТРО человека очищали из культурального супернатанта в примере 55 другим способом (пример 57).
(N) Экспрессия кДНК ТРО человека в Х63.6.5.3. клетках и подтверждение его активности
С использованием района кДНК ТРО в плазмиде pBLTEN, полученной в примере 30, был сконструирован экспрессирующий вектор BMСGSneo-hTPO-P1 для применения в Х63.6.5.3. клетках (пример 33).
После трансфекции Х63.6.5.3. клеток этим вектором были получены трансформанты, в которых кодирующий кДНК человека экспрессирующий вектор был интегрирован в их хромосомы. При культивировании этих клеток ТРО активность была обнаружена в культуральном супернатанте (пример 34).
(О) Экспрессия больших количеств ТРО человека в COS 1 клетках и очистка, измерение мол. массы и биологические характеристики ТРО человека
Экспрессирующий вектор pHTP1, приготовленный в примере 30, трансфицировали в COS 1 клетки для получения большого количества (в целом приблизительно 40 л) культурального супернатанта, содержащего экспрессируемый продукт (пример 35).
Очистку ТРО проводили из приблизительно 7 л бесклеточного культурального супернатанта COS 1 клеток, полученного в примере 35, содержащего полученный из экспрессирующего вектора pHTP1 ТРО. Высокую активность ТРО можно было получить при помощи стадий катионообменной хроматографии, WGA - колоночной хроматографии и колочной хроматографии с обращенной фазой (пример 36).
Частично очищенный таким образом ТРО из культурального супернатанта COS 1 клеток применяли для измерения мол. массы и анализа биологической активности (примеры 37 и 38).
(Р) Экспрессия ТРО человека в E.coli
Был сконструирован вектор pGEX-2T/h(1-174) для применения в экспрессии слитого белка (называемого "GST-TPO-(1-174)") глутатион-S -трансферазы и ТРО человека (аминокислотные остатки 1-174) в E.coli. В этом случае часть нуклеотидной последовательности кДНК ТРО человека (примерно половина зоны 5'-стороны) была заменена предпочтительными кодонами E.coli (пример 39).
GST -ТРО(1-174) экспрессировали в Е.coli, полученные клетки разрушали и затем солюбилизировали GST-TPO(1-174), содержащийся в осажденной фракции. Затем в результате комбинирования различных стадий, в том числе испытания условий укладки ТРО, испытания условий очистки (аффинная колонка с глутатионом, катионообменная колонка и т.п.) и расщепления района GST тромбином, было возможно выполнить частичную очистку белка, содержащего аминокислотную последовательность ТРО. Было подтверждено, что этот белок обнаруживает ТРО активность в тест-системе крысиных CFU-MK (примеры 40 и 41).
Был сконструирован также вектор pCFM536/h6T(1-163) для применения в экспрессии в E. coli белка ТРО мутированного типа человека (называемого "h6T(1-163")), в котором остаток Ser в положении 1 и остаток Аlа в положении 3 ТРО человека (аминокислотные остатки 1-163) были соответственно заменены остатком Аlа и остатком Val и одновременно остаток Lys и остаток Met были соответственно добавлены в положении -1 и -2. Вся часть нуклеотидной последовательности кДНК ТРО человека (соответствующая аминокислотным остаткам 1-163), несомая этим вектором, была заменена предпочтительными для E.coli кодонами (пример 42).
h6T(1-163) экспрессировали в E.coli, полученные клетки лизировали и затем солюбилизировали h6T(1-163), содержащийся в осажденной фракции. Испытывали условия для складывания белка и частично очищали белок, содержащий аминокислотную последовательность ТРО. Было подтверждено, что этот белок обнаруживает ТРО активность в тест-системе крысиных CFU-МK (примеры 43 и 44).
Кроме того, был сконструирован вектор рСРМ536/hМК(1-163) для применения в экспрессии в E.coli мутированного белка ТРО человека (называемого "hMКТ (1-163)"), в котором остаток Lys и остаток Met были соответственно добавлены к положениям -1 и -2. Этот hMKT(1-163) экспрессировали в E.coli, как описано в примере 43, и экспрессируемый белок подвергали электрофорезу в SDS-PAGE, переносили на PVDF мембрану и затем подвергали анализу N-концевой аминокислотной последовательности, подтвердив, что этот белок содержит описанную аминокислотную последовательность (пример 52).
Кроме того, был сконструирован вектор pCFM536/hMKT(1-332) для экспрессии в E. coli мутированного белка ТРО человека, в котором остаток Lys добавлен при положении -1 и остаток Меt при положении -2 ТРО человека (аминокислотные положения 1-332) (называемого "hMKT (1-322)"). Этот hМКТ(1-332) экспрессировали внутри E.coli так же, как в примере 42, и его экспрессию подтверждали Вестерн-блоттингом с применением антител против пептида ТРО человека, полученных в примере 45, изложенном ниже (пример 66).
(Q) Получение антител против пептида ТРО и приготовление колонки с антителами против пептида ТРО
Кроличьи поликлональные антитела против ТРО пептида получали при помощи синтезированных пептидов, соответствующих трем частичным районам аминокислотной последовательности крысиного ТРО, определенной в примере 10. Было подтверждено, что эти антитела могут узнавать молекулы ТРО крыс и человека. Кроме того, были синтезированы пептиды, соответствующие 6 частичным районам аминокислотной последовательности ТРО человека, показанной в SEQ 1D 6 (или SEQ 1D 7), и затем применены для получения кроличьих поликлональных антител против ТРО пептидов. Было показано, что полученные антитела могут узнавать ТРО человека (пример 45).
ТРО можно очистить аффинной колоночной хроматографией с применением колонки, упакованной определенными молекулами, имеющими связывающую аффинность в отношении ТРО, такими как антитела против ТРО, рецепторы ТРО и т.п. Колонка с антителами против ТРО была сначала приготовлена путем связывания антител против ТРО пептида, полученных в примере 45 (пример 46).
(R) Очистка ТРО человека, экспрессируемого в COS 1 клетках, с применением колонки с антителами против ТРО-пептида и мол. масса и биологические характеристики ТРО
При помощи культурального супернатанта из трансфицированных COS 1 клеток (экспрессирующим вектором рНТР1) в качестве исходного материала был получен частично очищенный ТРО, который был нанесен на колонку с антителами против ТРО. Поскольку ТРО активность была обнаружена в адсорбированной фракции, эту фракцию затем подвергали колоночной хроматографии с обращенной фазой для очистки белка и определения его мол. массы и биологических характеристик (пример 47).
(S) Подтверждение активности частично очищенной пробы ТРО человека, экспрессированного в COS 1 клетках
ТРО-активную фракцию, очищенную в примере 36, а именно, пробу ТРО, очищенную до стадии колонки Capcell Pak Cl 300А из культурального супернатанта, полученного трансфекцией COS 1 клеток экспрессирующим вектором pHTP1, проверяли на ее биологическую активность для определения, способна ли она увеличивать число тромбоцитов в живом теле (пример 48).
Также неочищенную фракцию ТРО, полученную на катионообменной колонке из 33 л культурального супернатанта, полученного путем трансфекции COS 1 клеток экспрессирующим вектором pHTP1, проверяли на ее биологические активности,
чтобы определить, увеличивает ли она число тромбоцитов в теле (пример 49).
(Т) Экспрессия хромосомной ДНК ТРО человека в СНО клетках и подтверждение активности ТРО
Был сконструирован вектор pDEF202-ghTPO для применения в экспрессии хромосомы ТРО человека в СНО клетках (пример 50).
При введении этого вектора в СНО клетки получали клетку-трансформант, в которой экспрессирущий хромосомную ДНК вектор был интегрирован в ее хромосому. При культивировании этого клеточного клона ТРО активность обнаруживали в культуральном супернатанте (пример 51).
(U) Частичная очистка и подтверждение активности человеческого ТРО мутированного типа, зкспрессиванного в E.coli
Человеческий ТРО мутированного типа, полученный из кодирующего нуклеотидную последовательность ТРО человека клона pCFМ536/h6T(1-163), и экспрессированного в E.coli, подвергали повторной укладке с применением гуанидингидрохлорида и глутатиона, чтобы определить, проявляет ли полученный таким образом h6T(1-163) увеличивающее число тромбоцитов действие в живом теле (пример 53).
Человеческий ТРО мутированного типа, полученный из кодирующего нуклеотидную последовательность ТРО человека клона, pCFМ536/h6T(1-163), и зкспрессированного в E.coli, подвергали переукладке с применением N-лауроилсаркозината натрия и сульфата меди и затем катионообменной хроматографии, чтобы определить, увеличивает ли очищенный таким образом h6Т(1-163) число тромбоцитов в живом теле (пример 54).
Кроме того, укладку и очистку человеческого ТРО мутированного типа, h6T(1-163) проводили с применением других процедур (примеры 60 и 61).
(V) Экспрессия кДНК ТРО человека в клетках насекомых и идентификация ТРО активности
Рекомбинантный вирус для экспрессии ТРО человека в клетках насекомых получали, как описано в примере 58, и экспрессировали в клетках насекомых Sf 21, после чего ТРО активность идентифицировали в культуральном супернатанте (пример 59).
(W) Экспрессия ТРО человека (положения аминокислот 1-163) в СНО клетках и его очистка
Был сконструирован вектор pDEF202-hTPО163 для экспрессии белка ТРО человека (называемого "hTPО163"), имеющего аминокислотную последовательность 1-163 в аминокислотной последовательности ТРО человека, представленной в SEQ 1D 7. Экспрессирующий вектор pDEF202-hTPО163 трансфицировали в СHО клетки и полученную таким образом, продуцирующую hTPO 163 СНО клеточную линию культивировали в широкомасштабном производстве. Из супернатанта этой культуры очищали hTPО163 (примеры 62-65).
(X) Получение замещенных производных ТРО человека
Было получено производное (называемое "N3/TPO"), в котором Аrg-25 и Glu-231 ТРО человека были заменены соответственно на Asn и Lys, а также производное (называемое "09/ТРО"), в котором His-33 был заменен на Thr.
Плазмиду, кодирующую каждое производное, трансфицировали в COS 7 клетки, которые затем культивировали. В супернатанте этой культуры была обнаружена ТРО активность.
Были получены производные, в которых одна аминокислота h6T(1-163) была заменена другой аминокислотой, с применением системы экспрессии Е.coli ТРО активность была обнаружена в каждом производном (пример 94).
(Y1) Получение инсерционных или делеционных производных ТРО человека
Аминокислоту вводили в hTPO163 или делетировали из него для приготовления инсерционных или делеционных производных, после чего СOS7 клетки трансфицировали плазмидами, кодирующими полученные производные. В супернатанте культур этих COS 7 клеток была детектирована ТРО активность (пример 68).
Способ измерения ТРО активности (тест-система in vitro), применяемый в данном изобретении, описан далее в виде "Стандартного примера".
(Z) Получение и очистка антител против ТРО человека
Поликлональные антитела получают с применением пептидных фрагментов ТРО человека (пример 69) и затем используют в Вестерн-блоттинге (пример 70) и в конструировании аффинных колонок с антителами против ТРО (пример 71).
(АА) Активность in vivo TPO человека
Очищенный TPO человека вводят мышам с индуцированной тромбоцитопенией и изменения в числе тромбоцитов наблюдают по сравнению с контрольной группой (пример 72) - (пример 79). Описаны фармацевтические композиции, которые потенциально могут быть применимы в лечении тромбоцитопении (пример 80) - (пример 89).
(ВВ) TPO активность как функция связывания рецептора
Обеспечен тест, в котором TPO активность определяют в единицах специфического связывания с рецептором Мр1 (пример 90) - (Пример 93).
Стандартный пример
А. Тест для определения клеток-предшественников мегакариоцитов крыс (тест-система крысиных CFU-МK) (система жидкой культуры)
Мегакариоциты включают и хранят серотонин в цитоплазматических плотных гранулах при помощи активного энергозависимого процесса (Fedorko, Lab, Invest., vol.36, pp.310-320, 1977). Этот феномен можно наблюдать уже в небольших одноядерных ацетилхолинэстераза-положительных клетках, которые, как считают, находятся между CFU-МK и различаемыми мегакариоцитами (Bricker and Zuckerman, Exp. Hematol., vol. 12, p. 672, 1984). Количество включенного серотонина увеличивается в ответ на увеличение размера мегакариоцитов (Schick and Weinstein, J. Lab. Clin. Мed. vol. 98, pp.607-615, 1981).
Кроме того, известно, что такой феномен специфичен только для мегакариоцитов, находящихся среди клеток костного мозга (Schick and Weinstein, J. Lab. Clin. Med. , vol. 98, pp. 607-615, 1981). В данной тест-системе высокообогащенные крысиные CFU-MK клетки (Gр11b/111а+CFU-МК фракция, которая будет описана ниже) культивируют в присутствии тестируемых проб и измеряют включение 14С-серотонина (14С-гидрокситриптамин-креатинсульфата, 14C-5HT) в мегакариоциты, вырастающие из CFU-MK.
Преимуществами этой тест-системы является то, что косвенные влияния примесных клеток (например, образование Меg-CSF активности примесными клетками, стимулируемыми каким-либо веществом, другим, чем целевой фактор, или образование какого-либо фактора примесными клетками, которые подвергаются комбинированному действию интересующего исследователя фактора) могут быть уменьшены, так как процент СFU-MК в клетках фракции Gр11b/111а+CFU-MК очень высок (сравни следующий далее [тест-метод]), тогда как число загрязняющих (примесных) клеток мало. Кроме того, подходящие условия культивирования могут поддерживаться в течение относительно длительного периода, поскольку общее число клеток, посеянное в одну лунку, очень мало. Другим преимуществом является то, что число зрелых мегакариоцитов большого размера, выросших из CFU-MK в присутствии активной пробы во время периода культивирования, можно визуально детектировать под фазово-контрастным микроскопом, что делает возможным качественную оценку присутствия и степени активности. Результаты качественной оценки хорошо согласуются с результатами количественного определения, основанного на включении 14С-серотонина. Таким образом, надежность количественного определения может быть улучшена далее одновременным применением качественной оценки.
Тест-метод:
Высокообогащенные крысиные CFU-МK клетки (фракцию С-р11b/111а+CFU-MК), используемую в этом тесте, готовили по слегка модифцированному методу (Miyazaki et al. (Exp. Hematol., vol. 20, pp. 855-861, 1992).
Вкратце, бедренную кость и большеберцовую кость удаляли из Wistаr крыс (самцов, 8-12-недельного возраста) для приготовления суспензии клеток костного мозга, как описано. Для приготовления суспензии клеток костного мозга применяли суспендирующую среду (раствор, состоящий из 13,6 мМ тринатриевой соли лимонной кислоты, 11,1 мМ глюкозы, 1 мМ аденозина, 1 мМ теофиллина, 10 мМ HEPES (рН 7,25), 0,5% бычьего сывороточного альбумина (далее называемого BSA) (сбалансированный раствор Hanks: далее называемый "HATCH раствор"), который является слегка модифицированной версией среды для отделения мегакариоцитов, сообщенной Levine Fedoroko (Levine and Fedoroko, Blood, vol. 50, pp. 713-725, 1977). Полученную таким образом суспензию клеток костного мозга наслаивают на прерывистый градиент плотности Перколла (плотность: 1,050 г/мл/1,063 г/мл/1,082 г/мл), который готовят разведением раствора Перколла (Pharmаcia) НАТСН раствором, и центрифугируют при 20oС при 400 x g в течение 20 минут. После центрифугирования извлекают клетки между слоями с плотностью 1,063 и 1,082 г/мл. После промывания извлеченные клетки суспендируют в культуральной среде Дульбекко, модифицированной Исков (называемой далее культуральной средой 1МDМ), содержащей 10% фетальную сыворотку теленка (далее называемую FCS), помещают в пластиковые чашки для культуры ткани (100 мм) и культивируют при 37oС в течение 1 часа в инкубаторе с 5% СО2. После инкубации неприлипшие клетки выделяют и культивируют опять в пластиковых чашках при 37oС в течение 1 часа. Неприлипшие клетки суспендируют в HATCH растворе и инкубируют в течение 1 часа на бактериологических чашках Петри (100 мм), на которых предварительно были адсорбированы мышиные моноклональные антитела против крысиных тромбоцитов Gp11b/111a+, P55 антитела (Мiyazaki et al., Thromb. Res., vol. 59, pp. 941-953, 1990).
После инкубации неприлипшие клетки удаляют тщательным промыванием HATCH раствором и оставшиеся клетки, адсорбированные иммобилизованными P55 антителами, отделяют пипетированием и собирают. Обычно получают 3-4•105 клеток из одной крысы. Полученная таким образом фракция клеток содержит высокообогащенные крысиные СFU-MК (далее называемые фракцией Gр11b/111a+CFU-MК). Известно, что эта клеточная фракция содержит приблизительно 5-10% СFU-MК на основании измерения при помощи системы анализа колоний в присутствии насыщающей концентрации крысиного IL-3. Гибридома, способная продуцировать вышеупомянутые P55 антитела, была депонирована под депозитным FERМ BP-4563 National Institute of Bioscience and Human Technology, Agency of Industrial Science and Technology. Ministry of International Trade and Industry, 14 февраля 1994 года.
Затем клетки полученной таким образом фракции Gp11b/111a+CFU-МK суспендировали в 1MDМ культуральной среде, содержащей 10% FCS, и распределяли порциями по 104 клеток на лунку 96-луночного планшета для культуры ткани. В каждую лунку затем добавляли стандартную пробу (это будет детально обсуждаться ниже) или испытуемую пробу, с доведением конечного объема среды до 200 мкл на лунку. Приготовленный таким образом планшет помещают в инкубатор с СО2 и инкубируют в течение 4 дней при 37oС. На 4-й день инкубации к каждой лунке добавляют 0,1 мкКи (3,7 КВq) 14С-серотонина за 3 часа перед завершением культивирования и конечную инкубацию продолжают при 37oС. После 3 часов инкубации планшет центрифугируют при 1000 об/мин, в течение 3 минут и полученный супернатант удаляют отсасыванием. К каждой лунке добавляют 200 мкл РВS, содержащего 0,05% ЭДТА, и планшет центрифугируют для промывания путем удаления полученного супернатанта. Эту стадию промывания повторяют еще один раз. По 200 мкл 2% Тритона Х-100 добавляют к полученным таким образом осадкам в каждую лунку и планшет качают на смесителе для планшетов приблизительно в течение 5-10 минут для полного лизиса клеток. Части по 150 мкл полученных таким образом клеточных лизатов переносят в коммерческий твердый сцинтиллятор в форме колпачка (Ready Cap; Beckman), который затем оставляют на ночь при 50oС в сушильном шкафу для высушивания лизата. На следующий день Ready Cap помещают в стеклянный флакон для измерения радиоактивности 14С при помощи жидкостного сцинтилляционного счетчика.
В этом случае почти одни и те же результаты могут быть получены в тесте включения 14С-серотонина и при измерении ацетилхолинэстеразной активности в упомянутом клеточном лизате по способу Ishibashi и Burstein (Blood, vol. 67, pp. 1512-1514, 1986).
Стандартные пробы:
Во-первых, плазму крови тромбоцитопенических крыс готовили для использования в приготовлении стандартных проб следующим образом.
Нормальных самцов крыс Wistar (7-8-не дельных) превращали в тромбоцитопенических путем внутривенного введения Р55 антител в дозе 0,5 мг на животное, дважды с интервалом приблизительно 24 часа. Примерно через 24 часа после второго введения собирали кровь из брюшной аорты под эфирным наркозом при помощи шприца на 10 мл, в который был введен 1 мл 3,8% (об./об.) тринатриевой соли лимонной кислоты в качестве противосвертывющего средства. Собранную таким образом пробу крови разливали в пластиковые центрифужные пробирки и центрифугировали при 1200 g в течение 10 минут для получения фракции плазмы крови. Извлеченную таким образом фракцию плазмы крови опять центрифугировали при 1200g в течение 10 минут и полученную фракцию плазмы крови собирали, избегая взятия состоящего из клеток и тромбоцитов осадка и т.п., и объединяли (полученную таким образом плазму крови далее называют TRP). Обработанную кальцием TRP (стандартную пробу с), активную фракцию колонки WGA-агарозы (стандартную пробу W) или активную фракцию Phenyl Sepharose 6 FF/LS (стандартную пробу Р), полученные из TRP в соответствии с процедурой примера 1, диализовали экстенсивно против достаточного объема культуральной среды 1MDM и использовали в качестве тест-стандартов.
В процессе очистки крысиного ТРО, описанном в примере 1, на ранней стадии применяли стандартную пробу С, но затем ее заменяли стандартной пробой W на полпути после этого и затем стандартной пробой Р. Удельную активность стандартной пробы С определяли как 1 и относительные активности стандартных проб W и Р рассчитывали на основе этой установки. Относительную активность каждой испытуемой пробы определяли путем сравнения кривой зависимости от дозы ответной реакции стандартной пробы с кривыми испытуемых проб. Относительную активность испытуемой пробы определяли как n, если ее активность в n раз выше, чем активность стандартной пробы С.
В. Тест система с подсчетом колоний
В этой тест-системе клетки костного мозга культивировали в полутвердой культуральной среде в присутствии испытуемой пробы и активность Meg-CSF измеряли подсчетом числа колоний мегакариоцитов, образовавшихся в результате пролиферации и дифференцировки CFU-МK.
Тест-метод
(а) В случае применения неразделенных клеток костного мозга крыс и т.п.
1 мл конечного объема культуральной среды 1MDM, содержащей неразделенные клетки крысиного костного мозга, клетки, полученные в каждой стадии разделения Gp11b/111a+CFU-MK тест-системы А или клетки Gp11b/111a+CFU-MK фракции, и 10% FСS, 2 мM глутамин, 1 мМ пируват натрия, 50 мкМ 2-меркаптоэтанол и 0,3% агар (AGAR NOBLE, изготовленный D1FCO), помещали в пластиковую чашку для культуры ткани (35 мм) и после отверждения при комнатной температуре культивировали при 37oС в инкубаторе с СО2. Обычно число клеток на одну чашку доводили до 2-4•105 в случае неразделенных клеток костного мозга, 2-5•104 в случае клеток на стадии градиента плотности Перколла или истощения прикреплением или 0,5-2•103 в случае клеток фракции Gp11b/111a+CFU-MK. После 6-7 дней культивирования диски агара отделяли от чашек и помещали на стеклянные предметные стекла (76 мм • 52 мм). Для высушивания каждого агарового диска кусок 50-м найлоновой сетки и фильтровальную бумагу помещали на диск в этой последовательности. Высушенные таким образом агаровые стекла фиксировали нагреванием 5 минут при 50oС на горячей пластине и пропитывали в течение 2-4 часов в красящем растворе ацетилхолинэстеразы по способу Jackson (Blood, vol. 42, pp. 413-421, 1973). Если достаточное окрашивание мегакариоцитов наблюдалось, агаровые стекла промывали водой, сушили, подвергали пост-окрашиванию раствором гематоксилина Харриса в течение 30 секунд, промывали водой и затем высушивали на воздухе. Колониями мегакариоцитов считали 3 или более тесно сгруппированных ацетилхолинэстераза-положительных клеток.
(в) В случае применения неразделенных клеток костного мозга мыши
Тест с подсчетом колоний проводили, как описано в (а) с применением 2-4•105 клеток на одну чашку.
(с) В случае применения клеток костного мозга человека или клеток крови спинного мозга человека
Можно применять клетки костного мозга человека или клетки крови спинного мозга человека как таковые или в виде CFU-МK фракций, обогащенных следующим образом.
Во-первых, жидкость костного мозга или кровь спинного мозга настаивают на Lymphoprep (производимый Daiichi Kagaku Co., Ltd.) и центрифугируют, после чего извлекают полученную в интерфазе фракцию лейкоцитов. Из извлеченной таким образом клеточной фракции клетки, с которыми связываются биотинилироваиные моноклональные антитела, специфические для поверхностных антигенов человека (CD2, CD11, CD19), удаляют при помощи соединенных с авидином бусин. Клетки, удаляемые магнитными бусинами, представляют собой в основном В-клетки, Т-клетки, макрофаги и часть гранулоцитов. Оставшиеся клетки окрашивают FITC-мечеными антителами против CD34 и РЕ-мечеными антителами против HLA-DR. Затем CD34- и HLA-DR -положительную фракцию извлекают при помощи клеточного сортера (например, ELITE, изготовляемого COULTER). CFU-MK клетки концентрируются в этой фракции (далее называемой XD34+DR+CFU-MK фракцией). Тест с подсчетом колоний клеток человека проводят так же, как в случае клеток костного мозга крыс, за исключением того, что применяют 3-5•103 клеток CD34+DR+CFU-MK фракции на одну чашку и используют смесь 12,5% плазмы АВ человека и 12,5% FCS вместо 10% FCS. Период культивирования для образования колоний мегакариоцитов равен 12-14 дням. Для обнаружения мегакариоцитов проводят иммунологическое окрашивание мегакариоцитов по способу с применением щелочной фосфатазы-антител против щелочной фосфатазы с мышиными моноклональными антителами, специфическими для поверхностного антигена Gp11b/111a мегакариоцитов (например, Teramura et al. , Exp. Hematol., vol. 16, pp. 843-848, 1988) и считают колонии, состоящие из 3 или более мегакариоцитов.
(С) Тест-система с клеточной линий мегакариобластов (М-07е тест)
М-07е клетки, клеточная линия мегакариобластов человека, пролиферирует в ответ на GМ-CSF, IL-3, SCF, IL-2 и т.п. (Avanzi et al., J. Cell. Physiol., vol. 145, pp.458-464, 1990). Поскольку эти клетки также отвечают на ТРО, они применимы в заменяющей тест-системе для тест-системы с крысиными CFU-МK.
Тест-метод:
М-07е клетки, сохраняемые в присутствии GМ-CSF, извлекают, основательно промывают и затем суспендируют в культуральной среде 1MDM, содержащей 10% FCS. Полученную суспензию М-07е клеток разливают по частям из 104 клеток в лунки 96-луночного планшета для культуры ткани и в каждую лунку добавляют затем стандартную пробу или испытуемую пробу, доводя конечный объем до 200 мкл на лунку. Полученный таким образом планшет помещают в инкубатор с 5% СО2 и инкубируют в течение 3 дней при 37oС. После 3 дней культивирования к каждой лунке добавляют 1 мкКи (37 КВq) 3H-тимидина за 4 часа до завершения культивирования. После завершения культивирования клетки собирают на фильтры из стеклянного волокна при помощи харвестера (сборщика) клеток для измерения 3H-радиоактивности с применением жидкостного сцинтилляционного счетчика (например, Beta Plate, изготовляемого Pharmacia).
Д. Тест-система (Ва/F3 тест), в котором применяют линию про-В-клеток мышей
Мышиная линия про-В-клеток Ва/F3 известна как клеточная линия, которая пролиферирует в ответ на IL-3, IL-4 и т.п. ( Palacios et. al., Cell, vol. 41, pp. 727-734). Поскольку ее подштамм ВF-ТЕ22 способен к пролиферации клеток в ответ не только на IL-3 и IL-4, но и на ТРО, его можно применять в тест-системе, заменяющей тест с крысиным CFU-MK или тест с клеточной линией мегакариобластов человека (M-07е тест), следующим образом. Вкратце, BF-TE22 клетки, растущие в среде 1MDM (1MDM:G1ВСО) в присутствии 1 нг/мл мышиного IL-3, извлекают, промывают трижды 1MDM и суспендируют в среде 1MDM, содержащей 10% FCS. Затем клеточную суспензию разливают в лунки 96-луночного планшета для культуры ткани при плотности 1•104 клеток на лунку, в каждую лунку затем добавляют стандартный раствор ТРО или тестируемую пробу и доводят конечный объем до 200 мкл на лунку. Полученный микропланшет инкубируют в течение 2-3 дней в инкубаторе с 5% CО2. На 2-й и 3-й день к каждой лунке добавляют 1 мкКи (37 KBq) 3H-тимидина и культивирование продолжают еще 4 часа. Наконец, клетки собирают на фильтр из стеклянного волокна при помощи харвестера клеток для измерения 3H-радиоактивности с применением жидкостного счетчика. В этой тест-системе почти все клетки, культивируемые в средах без ТРО активности, умирают и, следовательно, не включают 3Н-тимидин. Однако, клетки, культивируемые в средах, содержащих ТРО активность, обнаруживают активную пролиферацию, зависимую от концентрации ТРО, и включают 3Н-тимидин. Кроме того, результаты этого теста параллельны результатам тестов М-07е и CFU-МK.
Следующие ниже примеры иллюстрируют далее данное изобретение.
Пример 1-1. Очистка ТРО крысы из плазмы крови тромбоцитопенических крыс, индуцированных введением антител против тромбоцитов. Получение плазмы крови тромбоцитопенических крыс, индуцированных введением антител против тромбоцитов
ТRР получали в качестве источника очистки из примерно 1000 крыс согласно процедуре, описанной в "Стандартном примере" А выше, описывающем тест-систему клеток-предшественников мегакариоцитов крыс (CFU-MK).
Очистка ТРО крыс из TRP
Исходно, очистку проводили с применением ТRР примерно из 1000 крыс, предполагая, что содержание ТРО в TRP должно быть максимально 1 на 3 млн, и было получено немного менее чем 1 пмоль частично очищенного ТРО крысы. Хотя это и трудно, была сделана попытка анализа его частичных аминокислотных последовательностей и были получены три частичных аминокислотных последовательности, но с низкой точностью. Эти последовательности совпадали или были близки к аминокислотной последовательности ингибитора сериновой протеазы (SP1), продуцируемого в печени. Из этих данных неясно, имеет ли ТРО структуру, сходную со структурой ингибитора сериновой протеазы, или эти последовательности получены из последовательностей примесных белков, иных чем ТРО. С применением этих неопределенных последовательностей проводили клонирование гена ТРО из библиотеки кДНК крысы, но не смогли получить потенциальных кодирующих ТРО генов. После этого проводили интенсивные исследования на основе предположения, что эта неудача была обусловлена низкой степенью чистоты и недостаточным количеством анализируемой пробы. Для получения очищенной конечной пробы, необходимой для аминокислотного анализа, очистку проводили в соответствии с процессом, описанным в следующем примере 1-2. Неожиданно результаты примера 1-2 показали, что содержание ТРО в ТRР или XRP (это будет описано ниже) был таким чрезвычайно низким, как 1:100000000 - 1 блн. общих белков плазмы, так что полная очистка ТРО была крайне трудной.
Пример 1-2. Очистка ТРО крысы из плазмы крови тромбоцитопенических крыс, индуцированных облучением всего тела рентгеновскими или γ-лучами [Получение плазмы крови тромбоцитопенических крыс, индуцированных облучением всего тела рентгеновскими или γ лучами]
Нормальных самцов Wister крыс (7-8-недельных) превращали в тромбоцитопенических облучением всего тела рентгеновскими или γ-лучами при сублетальной дозе (6-7 Gy). Кровь собирали из крыс на 14-й день после облучения и фракцию плазмы крови (называемую далее XRP) готовили так же, как описано выше для приготовления TRP.
В этом случае XRP (приблизительно 8 л) получали из 1100 крыс и использовали в качестве исходного материала (источника) для очистки.
Очистка ТРО крыс из ХRР
Проба плазмы крови из 1100 крыс была слишком большой для одноразовой обработки, так как ее содержание общего белка достигало 493000 мг. Поэтому пробу плазмы крови делили на 11 серий, каждая соответствовала примерно 100 крысам, в следующих стадиях очистки (1)-(4). В дальнейших стадиях очистки (5)-(7) пробу делили на 6 групп. В и после стадии очистки (8) применяли пробу, грубо очищенную, из общей плазмы крови примерно 1100 крыс. В дальнейшем описан типичный пример каждой стадии очистки в случае серии (XW9) и группы (ХВ6).
Во всех стадиях очистки ТРО активность измеряли с применением тест-системы крысиных CFU-MK, описанной в "Стандартном примере". Если нет иных указаний, все стадии очистки проводили при 4oС, за исключением хроматографии с обращенной фазой и гель-фильтрации на Superdex 75 pg в присутствии поверхностно-активного вещества, которые проводили при комнатной температуре. Определения белка проводили в тесте связывания красителя Кумасси (система реагентов, изготовляемая PIERCE, кат. 23236Х) или методом с применением бицинхониновой кислоты (система реагентов, изготовляемая PIERCE, кат. 23225).
Схема очистки показана в таблице 1.
(1) Обработка хлоридом кальция, центрифугирование и обработка ингибитором протеазы плазмы крови крыс
В случае серии XW9
XRP (742 мл, концентрация белка 54,8 мг/мл, общий белок 40 686 мг), соответствующую примерно 100 крысам, хранящуюся при -80oС, оттаивали и разливали в полипропиленовые центрифужные пробирки (изготовляемые Nalgene). Порошок хлорида кальция добавляли в каждой пробирке до конечной концентрации 100 мМ. После инкубирования в течение ночи при 4oС полученную смесь центрифугировали при 8000 об/мин, в течение 60 минут. К полученному ТРО-активному супернатанту (742 мл, концентрация белка 54,9 мг/мл, общий белок 40 740 мг) добавляли ингибитор протеазы p-APMSF ((п-аминодифенил)метансульфонилфторид, гидрохлорид, изготовляемый WAko Pure Chemical Industries, Ltd., кат. 010-10393) до конечной концентрации 1 мМ. Полученную таким образом пробу использовали в дальнейшей стадии буферного обмена на колонке Sephadex G-25.
Описанным образом проводили обработку хлоридом кальция/ p-APMSF всей ХRР, полученной из 1100 крыс (общий объем 8184 мл, общий белок 493000 мг) разделением на 11 серий, каждая из которых соответствовала примерно 100 крысам, и проведением обработки этих серий по отдельности. Полученные 11 проб обрабатывали отдельно в последующей стадии очистки на колонке Sephadex G-25.
(2) Sephadex G-25 (смена буфера)
В случае серии XW9
Супернатант, полученный после кальциевой обработки стадии (1) (742 мл, концентрация белка 54,9 мг/мл, общий белок 40 740 мг), наносили при скорости течения 40-70 мл/мин на колонку Sephadex G-25 (изготовляемую Pharmacia Biotech, кат. 17-0033-03, диаметр 11,3 см, высота упакованной колонки 47 см), уравновешенную заранее 20 мМ Трис-HC1, рН 8. Элюцию проводили тем же самым буфером. Порцию 1300 мл элюата перед элюцией белка выбрасывали. Когда УФ поглощение обнаруживали в элюатах, фракции собирали, пока электропроводность не достигала 500 мкS/см, извлекая таким образом ТРО-активную белковую фракцию, содержащую теперь 20 мМ Трис-HC1, рН 8 (1377 мл, концентрация белка 27,56 мг/мл, общий белок 37882 мг, выход белка 93%). Относительная активность ТРО в этой фракции была 2,3.
В результате обработки на колонке Sephadex G-25 всех серий проб было получено в целом 21117 мл ТРО-активной фракции (общий белок 480300 мг, средняя относительная активность 1, общая активность 864600).
(3) Q-Sepharose FF (сильная анионообменная хроматография)
В случае серии XW9
ТРО-активную фракцию, полученную в стадии (2) обработкой Sephadex G-25 (1377 мл, концентрация белка 27,5 мг/мл, общий белок 37841 мг, относительная активность 2,3), наносили на Q-Sepharose FF (Pharmacia Biotech, кат. 17-0510-01, диаметр 5 см, высота колонки 27 см) при скорости течения 40 мл/мин, элюировали 20 мМ Трис-HC1 (рН 8) и фракцию F1, проходящую через колонку, собирали (3949 мл, концентрация белка 0,98 мг/мл, общий белок 3870 мл, относительная активность 0).
Затем буфер заменяли 20 мМ Трис-НС1 (рН 8), содержащим 175 мМ NaСl, для элюции ТРО-активной фракции F2 (4375 мл, концентрация белка 5,36 мг/мл).
Наконец фракцию F3 (1221 мл, концентрация белка 3,9 мг/мл, общий белок 4783 мг, относительная активность 3,8) элюировали 20 мМ Трис-НС1 (рН 8), содержащим 1000 мМ NaCl. Общий белок в ТРО-активной фракции F2 был 23440 мг и выход белка F2 на этой стадии был 61,9%. Кроме того, относительная активность ТРО увеличилась до 6,8.
В результате нанесения всех серий ТРО-активных фракций после Sephadex G-25 Q-Sepharose FF получили в целом 35842 мл ТРО-активной фракции F2 (общий белок 314384 мг, средняя относительная активность 9, общая активность 2704000).
(4) Агглютинин зародышей пшеницы (WGA)-Agarose (пектиновая аффинная хроматография)
В случае серии XW9
ТРО-активную фракцию F2, полученную в стадии (3) при помощи Q-Sepharose FF, делили на 3 части и наносили на WGA-Agarose (изготовляемую Honen Corp. кат. 800273, диаметр 5 см, высота 22,5 см) при скорости течения 5 мл/мин и элюировали изотоническим фосфатным буфером Дульбекко (DPBS). фракцию F1, проходящую через колонку, собирали (9336 мл, концентрация белка 2,30 мг/мл, общий белок 21407 мг, относительная активность 6,9).
Затем буфер заменяли на 20 мМ буфер, содержащий фосфат натрия (рH 7,2), содержащий 0,2 М N-ацетил-D-глюкозамин (GlcNAc, изготовленный Nacalai tesque кат. 005-20), 150 мМ NaCl и 0,02% азид натрия, и полученные элюаты объединяли и концентрировали при помощи ультрафильтрационного элемента (Omega Ultrasette, номинальное отсечение мол. массы 8000, изготовленный Filtron), получая в результате ТРО-активную фракцию F2 (2993 мл, концентрация белка 0,376 мг/мл).
Общий белок в ТРО-активной фракции был 1125 мг и выход белка F2 этой стадии был 4,8%. Кроме того, относительная активность ТРО увеличивалась до 101. Полученную таким образом фракцию F2 хранили при -80oС.
В результате нанесения всех серий ТРО-активных фракций Q-Sepharose FF на WGA-Agarose получали в целом 33094 мл WGA-Agarose ТРО-активной фракции F2 (общий белок 15030 мг, средняя относительная активность 132, общая активность 1987000).
(5) TSK - гель AF-BLUE 650 МН (аффинная хроматография с триазиновым красителем)
В случае группы ХВ6
WGA-Agarose, поглощающую ТРО-активную фракцию серии XW9, и WGA-Agarose, поглощающую ТРО-активную фракцию F2 серии XW9, полученные в стадии (4), соответствующие исходным 215 крысам, объединяли в виде группы ХВ6 (5947 мл, концентрация белка 0,388 мг/мл, общий белок 2319 мг, относительная активность 150).
5974 мл из объединенной пробы смешивали с 0,85 моля NaCl на 1000 мл (296,76 г в целом), получая 6132 мл раствора, имеющего конечную концентрацию NaCl 0,822 М, и полученный раствор наносили при скорости течения 7 мл/мин, на TSK - гель AF-BLUE 650 МН-колонку (TOSOH CORP. кат. 08705, диаметр 5 см, высота колонки 23 см), которая была заранее уравновешена 20 мМ натрий-фосфатным буфером (рН 7,2), содержащим 1 М NaCl.
После завершения нанесения белок элюировали 20 мМ натрий-фосфатным буфером (рН 7,2), содержащим 1 М НаСl при скорости течения 10 мл/мин. Полученные элюаты объединяли и концентрировали с применением ультрафильтрационного элемента (Omega Ultrasette, номинальное отсечение мол. массы 8000), получая проходящую фракцию F1 (543 мл, концентрация белка 2,05 мг/мл), общий белок 1112 мг, относительная активность 31).
Затем элюирующий буфер заменяли на 2 М NaSCN для получения элюированной TSK-gel AF-BLUE 650 МН поглощенной ТРО-активной фракции F2 (1427 мл, концентрация белка 0,447 мг/мл).
Общий белок в ТРО-активной фракции F2 был 638 мг и выход белка F2 в этой стадии был 27,5%. Кроме того, относительная активность ТРО увеличивалась до 1500.
В результате нанесения всех групп WGA-Agarose поглощенных ТРО-активных F2 фракций на TSK-gel AF-BLUE 650 МН получали в целом 10655 мл ТРО-активной фракции F2 (общий белок 4236 мг, средняя относительная активность 905, общая активность 3834000).
(6) Phenyl Sepharose 6 FF/LS (гидрофобная хроматография)
В случае группы ХВ6
1424 мл из ТРО-активной фракции F2 из TSK-gel AF-BLUE 650 МН (1424 мл, концентрация белка 0,447 мг/мл, общий белок 638 мг, относительная активность 1500) смешивали с 1,5 моля порошка сульфата аммония на 1000 мл (282,2 г в целом), получая раствор с конечной концентрацией сульфата аммония 1,35 М.
Полученный раствор наносили при скорости течения 7 мл/мин на Phenyl Sepharose 6 FF (Low Sub) - колонку (Pharmacia Biotech, кат. 17-0965-05, диаметр 5 см, высота колонки 10 см), которая была уравновешена заранее 50 мМ натрий-фосфатным буфером (рН 7,2), содержащим 1,5 М сульфата аммония. После завершения нанесения элюцию проводили при помощи 36 мМ натрий-фосфатного буфера, содержащего 0,8 М сульфат аммония, при скорости течения 10 мл/мин. Полученные элюаты (примерно 3160 мл) объединяли и концентрировали при помощи ультрафильтрационного элемента (Omega Ultrasette, номинальное отсечение мол. массы 8000), получая фракцию F1 (485 мл, концентрация белка 0,194 мг/мл, общий белок 94,2 мг, относительная активность 0).
Затем элюирующий буфер заменяли 20 мМ натрий-фосфатным буфером (рН 7,2) для получения элюированной ТРО-активной фракции F2 (приблизительно 2500 мл). Элюированную фракцию концентрировали при помощи ультрафильтрационного элемента (Omega Ultrasette) и брали пробу для анализа. На этой стадии концентрация белка и общий белок ТРО-активной фракции F2 (220 мл) были 1,45 мг/мл и 319 мг, соответственно, и выход белка F2 на этой стадии был 50,0%. Относительная активность ТРО была 1230.
В результате нанесения всех групп ТРО-активных F2 фракций из TSK-gel AF-BLUE 650 МН на Рhеnуl Sepharose 6 FF/LS получили всего 1966 мл фракций F2 (общий белок 2762 мг, средняя относительная активность 847, общая активность 2339000).
(7) Sephacryl S-200 HR (гель-хроматография)
В случае группы ХВ6 (см. фиг.1)
ТРО-активную фракцию F2 из Рhеnуl Sepharose 6 FF/LS (217 мл, концентрация белка 1,45 мг/мл, общий белок 315 мг, относительная активность 1230) смешивали с 144,8 мл 5 М раствора NaCl, получая 362 мл раствора с конечной концентрацией NaCl 2 М, и полученный раствор концентрировали до приблизительно 50 мл при помощи ультрафильтрационного устройства с УМ3 мембраной (76 мм в диаметре, Amicon Corp.).
К этому раствору добавляли равный объем (50 мл) 8 М мочевины, получая примерно 100 мл раствора, содержащего 1 М NaCl и 4 М мочевину в виде конечных концентраций. Его концентрировали примерно до 80 мл, брали пробу 88,78 мл и наносили на колонку Sephacryl S-200 HR (Pharmacia Biotech, кат. 17-0584-01, 7,5 см в диаметре и 100 см высотой).
После этого проводили элюцию PBS при скорости течения 3 мл/мин, и элюаты после пустого объема 1200 мл собирали порциями по 45 мл в 60 полипропиленовых пробирок. Тест проводили через каждые две пробирки и остальные пробирки хранили при -85oС до тех пор, пока не заканчивали обработку всех проб, полученных из 1100 крыс. На основании полученных результатов анализов фракции группы ХВ6 были сгруппированы следующим образом.
(F1) номера пробирок 1-15 (фракции вблизи пустого объема, мол. массы 94000 или более)
(F2) номера пробирок 16-26 (мол. массы 94000-33000)
(F3) номера пробирок 27-44 (мол. масса 33000-3000)
(F4) номера пробирок 45-55 (мол. масса 3000 или менее)
Таким образом все группы F2 фракций (ТРО-активных), полученных при помощи Phenyl Sepharose 6 FF/LS, отдельно наносили на Sephacryl S-200 HR, анализировали и хранили при -85oС. После завершения этой обработки всех групп и непосредственно перед последующей операцией хроматографией с обращенной фазой (YMC-Pack PROTEIN-RP) эти хранящиеся пробы оттаивали и концентрировали при помощи ультрафильтрационного устройства с УМ 3 мембраной (76 мм в диаметре, Amicon Corp.), получая следующие две пробы. Эту концентрированную пробу ТРО-активной фракции F2, полученную после Sephacryl S-200 HR, далее называют "пробой F2 высокомолекулярного ТРО", а F3 - "пробой F3 низкомолекулярного ТРО".
Для удобства проба F2 высокомолекулярного ТРО и проба F3 низкомолекулярного ТРО являются объединенными фракциями различных зон элюции в гель-хроматографии, и термины "высокомолекулярный" и "низкомолекулярный", следовательно, не могут обозначать их истинных мол. масс. (см. табл.I)
Пробу F3 низкомолекулярного ТРО и пробу F3 высокомолекулярного ТРО отдельно подвергали следующим стадиям очистки.
Очистка пробы F3 низкомолекулярного ТРО описана в следующих далее стадиях (8)-(11).
(8) YMC-Pack PROTEIN-RP (хроматография с обращенной фазой)
Пробу F3 низкомолекулярного ТРО (общий белок 50,3 мг, концентрация белка 0,184 мг/мл, относительная активность 20000, общая активность 1007000, общий объем 274 мл), полученную в стадии (7), смешивали с растворителем А (0,025% трифторуксусная кислота (TFA)) и растворителем В (1-пропанол, содержащий 0,025% TРA), получая раствор, имеющий общий объем 508,63 мл и конечные концентрации пропанола, TFA и белка приблизительно 20%, 0,012% и 0,0989 мг/мл, соответственно. Нерастворимый материал, образованный во время приготовления этого раствора, отделяли центрифугированием и полученный супернатант делили на две части по 254,3 мл (25,2 мг белка) и наносили при скорости течения 2 мл/мин на колонку, упакованную YMC-Pack PROTEIN-RP (УМС, кат. А-РRRР-33-03-15, 3 см в диаметре, 7,5 см высота колонки), которая была предварительно уравновешена 30% В. Осадок, образовавшийся при центрифугировании, растворяли в 20 мл 20 мМ ацетата натрия (рН 5,5), содержащего 5 мМ CHAPS (сульфоната 3-[3-холамидопропил)диметиламмонио] -1-пропана, изготовляемого Dojindo Laboratories, кат. 75612-03-3) и также наносили на эту колонку.
После нанесения пробы приблизительно 50 мл растворителя (растворители А: В= 3: 1) пропускали через колонку для получения проходящей через колонку фракции. Затем начинали проявляющую программу (120 минут линейного градиента от 30% В до 45% В) и элюаты собирали в 36 полипропиленовых пробирках по 10 мл в каждой пробирке. Этот процесс повторяли опять для оставшейся пробы с применением тех же самых пробирок для сбора фракций, получая таким образом в целом 36 пробирок, каждая из которых содержит 20 мл элюата. Проходящую через колонку фракцию сразу же концентрировали до 20 мл при помощи ультрафильтрационного устройства с УМ 3 мембраной (76 мл в диаметре, изготовленном Amicon Corp.).
0,1 мл проходящей через колонку фракции и каждой из 20 мл фракций пробирок с номерами 1-36 смешивали с 20 мкл 5% В А, сушили выпариванием, растворяли в 0,25 мл тест-среды 1MDM и затем анализировали для идентификации ТРО-активных фракций. В результате ТРО активность была обнаружена во фракциях пробирок с номерами 17-27 (диапазон концентраций пропанола 36,0-43,0%), которые объединяли и использовали как YMC-Pack PROTEIN-RP ТРО-активную фракцию F2, полученную из пробы F3 низкомолекулярного ТРО. Эту фракцию хранили при -85oС до ее применения в последующей стадии с использованием YMC-Pack CN-AP.
YMC-Pack PROTEIN-RP ТРО-активная фракция F2, полученная из пробы F3 низкомолекулярного ТРО
Общий объем - 220 мл
Концентрация белка - 0,0130 мг/мл
Общий белок - 2,85 мг
Относительная активность - 130000
Общая активность - 371000
(9) YMC-Pack CN-AP (хроматография с обращенной фазой)
214,9 мл YMC-Pack PROTEIN-RP ТРО-активной фракции F2, полученной из пробы F3 низкомолекулярного ТРО (общий белок 2,79 мг, концентрация белка 0,0130 мг/мл, относительная активность 130000, общая активность 36300), полученной из стадии (8), смешивали с 0,6 мл 50% глицерина и концентрировали до 1,8 мл. Этот концентрат затем доводили до объема 5 мл, содержащего 20% или менее пропанола и приблизительно 6% глицерина.
Концентрат делили на 5 частей для применения в следующей колоночной операции (0,555 мг белка и 1 мл объема для каждой операции). Каждую из разделенных таким образом проб наносили при скорости течения 0,6 мл/мин на колонку, упакованную YMC-Pack CN-AP (умс, кат. АР-513, 6 мм в диаметре и 250 мм высотой), которая была предварительно уравновешена 15% В, с применением 0,1% TFA в качестве растворителя А и 0,05% TFA содержащего 1-пропанола в качестве растворителя В. После нанесения концентрацию пропанола увеличивали с 15% В до 25% В и проводили 65 минут линейного градиента от 25% В до 50% В. После завершения конечной (5-й) операции 1 мл раствора, имеющего тот же самый состав, за исключением белка, наносили на колонку и обрабатывали так же, для извлечения ТРО активности, удержанной колонкой. Поскольку использовали те же самые полипропиленовые пробирки для сбора элюатов в 6 операциях, получали в целом 44 пробирки, каждая из которых содержала 7,2 мл элюата.
30 мкл каждой из полученных таким образом фракций (1/240 часть каждой фракции) смешивали с 20 мкл 5% BSA, сушили выпариванием, растворяли в 0,24 мл тест-среды 1MDM и затем анализировали для идентификации ТРО-активных фракций. В результате высокая ТРО активность была обнаружена во фракциях
пробирок с номерами 28-33 (37,0-42,0% - диапазон концентраций пропанола), которые объединяли и использовали как YMC-Pack CN-AP основную ТРО-активную фракцию FА, полученную из пробы F3 низкомолекулярного ТРО.
YMC-Pack CN-AP ТРО-активная фракция FА, полученная из пробы F3 низкомолекулярного ТРО
Общий объем - 43,20 мл
Концентрация белка - 0,00863 мг/мл
Общий белок - 0,373 мг
Относительная активность - 800000
Общая активность - 298400
(10) Capcell Pak C1 300 (конечная хроматография с обращенной фазой)
43,12 мл из 43,20 мл ТРО-активной фракции FА, полученной из пробы F3 низкомолекулярного ТРО (общий белок 0,372 мг, концентрация белка 0,00863 мг/мл, относительная активность 800000, общая активность 297500), полученной в стадии (9), смешивали с 0,2 мл 50% глицерина и концентрировали для получения 0,1 мл глицеринового раствора.
Этот раствор смешивали с 2 мл раствора растворителя А (0,1% ТFА): растворителя В (1-пропанол, содержащий 0,05% ТFА) = 85:15 (15% В), для получения 2,1 мл пробы, содержащей приблизительно 14% пропанола, приблизительно 4,8% глицерина и 0,177 мг/мл белка. Полученную таким образом пробу наносили на колонку, упакованную Capcell Pak C1 300A (изготовляемую Shiseido Co., Ltd., кат. C1 TYPE:SG300A; 4,6 мм в диаметре и 250 мм высоты), которая была предварительно уравновешена 15% В, и проявляли 65 минут линейным градиентом от 27% В до 38% В при скорости тока 0,4 мл/мин.
Элюаты собирали в 72 полипропиленовые пробирки по 0,6 мл в каждую пробирку.
3 мкл каждой из полученных таким образом фракций (1/200 часть каждой фракции) смешивали с 20 мкл 5% BSA, среду заменяли до 225 мкл тест-среды 1MDM и анализировали разведенную в 75 раз фракцию.
1 мкл каждой фракции (1/600 фракции) брали для применения в электрофорезе, сушили выпариванием и обрабатывали при 95oС 5 минут 10 мкл невосстанавливающим буфером для проб для SDS-PAGE. Обработанную таким образом пробу подвергали электрофорезу SDS-PAGE с применением 15-25% SDS-полиакриламидного преформированного геля (изготовляемого Daiichi Pure Chemicals Co., Ltd) и затем окрашивали серебром из набора 2D-Silver Stain-11 "DАIIСНI" (изготовляемого Daiichi Рurе Chemicals Co., Ltd., кат. 167997, называемый далее набором для окрашивания серебром). В качестве маркеров мол. массы использовали /DAIICHI/-111 низкомолекулярные маркеры (изготовляемые Daiichi Pure Chemicals Co., Ltd., кат. 181061, далее называемые "DPC111").
В результате этого анализа значительная ТРО активность была обнаружена во фракциях пробирок с номерами 35-43 (диапазон концентраций пропанола 30,0-32,5%). Из них фракции с номерами пробирок 36-42 (диапазон концентраций пропанола 30,5-32,0%) объединяли и использовали в качестве основной ТРО-активной фракции FА. Эти результаты показаны на фиг.2.
Когда содержание белка, установленное из хроматограммы, сравнивали с этими результатами анализа, было обнаружено, что полученная таким образом фракция имела общий белок 39,6 мкг, концентрацию белка 9,4 мкг/мл, относительную активность 4890000 и общую активность 193600. Исследование распределений SDS-PAGE ТРО-активных фракций с номерами 36-42 обнаружило присутствие полосы, плотность окрашивания которой коррелирует с силой активности. Кроме того, кажущаяся мол. масса этой невосстановленной полосы была 17000-19000 при сравнении с мол. массами стандартных белков с известными мол. массами на том же самом геле, которые были восстановлены, что показывает, что эта полоса является в сильной степени кандидатом ТРО.
(11) Экстракция ТРО-активного белка из геля для электрофореза (15% SDS-PAGE)
Пример анализа ТРО-активной фракции FА
Из 4200 мкл ТРО-активной фракции FА, полученной из пробы F3 низкомолекулярного ТРО (общий белок 39,6 мкг, концентрация белка 9,4 мкг/мл, относительная активность 4890000, общая активность 193600), полученной в стадии (10), 5,5 мкл (1/764 фракции) для применения в экстракции активного белка и 2,5 мкл (1/1680 фракции) для применения в окрашивании серебром переносили в соответствующие пробирки для проб, сушили выпариванием, обрабатывали 10 мкл невосстанавливающего буфера для проб для SDS-PAGE при 37oС в течение 1 часа и затем оставляли стоять до 18 часов при комнатной температуре, что способствовало реакции SDS.
Предварительно окрашенный маркер низкого диапазона (Bio-Rad Laboratories, Inc., 161-0305) и вышеупомянутые DPC111 применяли в качестве маркеров мол. массы. Применяя микропластинки гелей, электрофорез на 15% SDS-PAG этих проб проводили при 4oС согласно общепринятому способу (Laemmli, Nature, vol. 227, pp.680-685, 1970). После завершения электрофореза части геля, предназначенные для окрашивания серебром, сразу вырезали скальпелем из геля, помещали в фиксирующий раствор и затем окрашивали с применением упомянутого выше набора для окрашивания серебром.
Отдельно от этого весь диапазон мол. масс геля, предназначенный для детектирования активности, разрезали скальпелем на 34 среза и каждый срез, имеющий ширину 1,5-2,5 мм, разбивали по слегка модифицированному способу Kobayashi (Kobayashi, M. , Seikagaku (Biochemistry), vol.59, N9, 1987, опубликованный Японским Биохимическим Обществом). Каждый срез геля, разбитый таким образом на мелкие фрагменты, смешивали с 0,3 мл буфера для экстракции (20 мМ Трис- НСl, рН 8, 500 мМ NaCl, 0,05% BSA) и качали в течение 6 часов при 4oС для усиления экстракции.
К этой суспензии добавляли 500 мМ фосфат калия (рН 6,8) до конечной концентрации 20 мМ. После 1 часа качания при 4oС полученную смесь переносили в Ultra Free C3GV 0,22 мкм фильтрационное устройство (Millipore Corp., UFC3 OGVOS) и центрифугировали при 1000 g (4000 об/мин) 15 минут для удаления осажденного SDS и извлечения образующегося супернатанта. Фильтрат переносили в Ultra Free C3-LGC ультрафильтрационное устройство (номинальное отсечение мол. массы 10000, Millipore Corp., UFC3 LGC 00) и центрифугировали при 3000 g (7000 об/мин). Когда объем концентрируемого раствора достигал приблизительно 50 мкл, добавляли 300 мкл 20 мМ натрий-фосфатного буфера (рН 7,2) и опять подвергали ультрафильтрации.
Ультрафильтрацию с применением 300 мкл 20 мМ натрий-фосфатного буфера повторяли дважды для удаления оставшегося SDS. После этого подобную стадию повторяли для обмена на культуральную среду для приготовления пробы (300 мкл), которую затем стерилизуют и подвергают измерению ТРО активности.
Три белковые полосы были обнаружены четко при окрашивании серебром, которые показали средние мол. массы приблизительно 17000-19000, 14000 и 11000, согласно DPC111 маркерам мол. масс.
В предшествующей стадии (10) полосу, имеющую корреляцию между силой активности и плотностью окрашивания и имеющую среднюю мол. массу приблизительно 17000-19000, наблюдали в электрофорезе ТРО-активной фракции Capcell Pak C1 колонки. В эксперименте этой стадии (11) ТРО активность была обнаружена только в полосе со средней мол. массой приблизительно 17000-19000 (см. фиг.3).
На основании изложенных выше результатов было подтверждено, что ТРО-активный белок был очищен в активной фракции Capcell Pak C1 300А колонки до детектируемого уровня на геле для электрофореза. На основании плотности окрашивания серебром этой полосы, полученной на 15% SDS-PAGE, было определено, что количество кандидата на белок ТРО (средняя мол. масса приблизительно 17000-19000) в обшей ТРО-активной фракции составляет приблизительно 1,7 мкг.
Очистка пробы F2 высокомолекулярного ТРО описана в следующих стадиях (12)-(15).
(12) YMC-Pack PROTEIN-RP (хроматография с обращенной фазой)
Пробу F2 высокомолекулярного ТРО (общий белок 257 мг, концентрация белка 0,894 мг/мл, относительная активность 7840, общая активность 2015000, общий объем 287 мл), полученную в стадии (7), смешивали с 95,8 мл (1/3 объема пробы) растворителя В (1-пропанол, содержащий 0,025% ТFА) для получения раствора, имеющего общий объем 383 мл и конечные концентрации пропанола, ТFА и белка приблизительно 25%, 0,006% и 0,671 мг/мл, соответственно. Раствор 0,025% ТFА применяли для растворителя А. Нерастворимый материал, образованный во время приготовления наносимой пробы, отделяли центрифугированием и полученный супернатант делили на шесть частей по 62,3 мл (42,8 мл белка) и наносили при скорости течения 2 мл/мин на колонку, упакованную YMC-Pack PROTEIN-RP (УМС, кат. А-РRRР-33-03-15, 3 см в диаметре и 7,5 см в высоту), которая была предварительно уравновешена 30% В. Осадок, полученный при центрифугировании, растворяли в 10 мл 20 мМ ацетата натрия (рН 5,5), содержащего 5 мМ CHAPS, и тоже наносили на эту колонку.
После нанесения пробы приблизительно 50 мл растворителя (растворители А: В= 3:1) проходили через колонку для получения проходящей через колонку фракции. Затем начинали проявляющую программу (120 мин линейного градиента от 30% В до 45% В), и элюаты собирали в 24 полипропиленовые пробирки по 15 мл в каждую пробирку. Этот процесс повторяли для 6 разделенных проб использованием тех же самых пробирок для сбора фракций, получая, таким образом, в целом 24 пробирки, каждая из которых содержала 90 мл элюата. Проходящую через колонку фракцию и фракцию пробирки 1 объединяли и сразу концентрировали до 90 мл при помощи ультрафильтрационного устройства с УМ3 мембраной (76 мм в диаметре, Amicon Corp.).
0,3 мл проходящей через колонку фракции плюс фракции пробирки 1 и каждой из фракций пробирок 2 - 24 смешивали с 10 мкл 5% BSA, сушили выпариванием, растворяли в 0,30 мл тест-культуральной среды IMDM и затем анализировали для идентификации ТРО-активных фракций. В результате ТРО активность была обнаружена во фракциях пробирок с номерами 10-15 (диапазон концентраций пропанола 34,0-39,5%), которые объединяли и использовали как YMC-Pack PROTEIN-RP ТРО-активную фракцию F2, полученную из пробы F2 высокомолекулярного ТРО. Эту фракцию хранили при -85oС до применения в последующей стадии YMC-Pack CN-AP.
YMC-Pack PROTEIN-RP ТРО-активная фракция F2, полученная из пробы F2 высокомолекулярного ТРО
Общий объем - 540 мл
Концентрация белка - 0,021 мг/мл
Общий белок - 11,4 мг
Относительная активность - 227000
Общая активность - 2588000
(13) Superdex 75 рg (гель-хроматография в присутствии CHAPS)
538,2 мл происходящей из пробы F2 высокомолекулярного ТРО YMC-Pack PROTEIN-RP ТРО-активной фракции F2 (общий белок 11,3 мг; концентрация белка 0,021 мг/мл; относительная активность 227000, общая активность 2565000), полученной в стадии (12), смешивали с 0,6 мл 50% глицерина и концентрировали выпариванием. Затем добавляли 18 мл 20 мMCHAPS с последующим перемешиванием и инкубацией 41 час при 4oС. После этого первую пробу наносили на колонку, упакованную Нiload 26/60 Superdex 75 рg (изготовляемую Pharmacia Biotech, кат. 17-1070-01, 2,6 см в диаметре и 60 см высотой) и элюировали DPBS, содержащим 5 мМ CHAPS при скорости течения 1 мл/мин. Части по 4 мл каждой пробы (концентрация белка 0,466 мг/мл, белок 1,86 мг) наносили на эту колонку.
В этом случае операцию колонки Superdex 75 повторяли 6 раз путем разделения всей YМС-Pack PROTEIN-RP ТРО-активной фракции на 6 частей. Элюаты каждой операции собирали в 45 пробирок, получая в конце 45 фракций, каждая из которых содержала 30 мл элюата после завершения 6 колоночных операций.
0,1 мл каждой из полученных таким образом фракций смешивали с 10 мкл 5% BSA, сушили выпариванием, растворяли в 0,25 мл IMDM и затем анализировали для идентификации ТРО-активных фракций. В результате ТРО активность была обнаружена во фракциях пробирок с номерами 13-31 (диапазон мол. масс от 78000 до 3000), которые объединяли и использовали в качестве полученной из пробы F2 высокомолекулярного ТРО Superdex 74 рg ТРО-активной фракции F2.
Полученная из пробы F2 высокомолекулярного ТРО Superdex 75 рg ТРО-активная фракция F2
Общий объем - 540 мл
Концентрация белка - 0,00216 мг/мл
Общий белок - 1,17 мг
Относительная активность - 1750000
Общая активность - 2041000
(14) YMC-Pack CN-AP (хроматография с обращенной фазой)
513,2 мл из 540 мл полученной из пробы F2 высокомолекулярного ТРО Superdex 75 рg ТРО-активной фракции F2 (мол. масса 78000-3000) (общий белок 1,11 мг, концентрация белка 0,00216 мг/мл, относительная активность 1750000, общая активность 1943000), полученной в стадии (13), смешивали с 1/10 объема растворителя В (1-пропанол, содержащий 0,05% ТFА) и наносили при скорости течения 0,6 мл/мин на колонку, упакованную YMC-Pack CN-AP (изготовляемым УМС, кат. АР-513, 6 мм в диаметре и 250 мм высотой), которая была предварительно уравновешена 15% В, с применением растворителя А (0,1% ТFА) и растворителя В. После нанесения концентрацию пропанола увеличивали от 15% В до 25% В и проводили 65 минут линейный градиент от 25% В до 50% В.
Перед началом хроматографии на описанной колонке 1/20 часть общей вносимой пробы подвергали испытанию для подтверждения правильного извлечения активности. После этого оставшиеся 19/20 пробы делили на 2 пробы для проведения колоночной операции дважды. Другими словами, операцию с колонкой повторяли три раза. Поскольку одни и те же пробирки применяли для сбора элюатов в 3 операциях, всего получали 44 пробирки, каждая из которых содержала 3,6 мл элюата.
5 мл из каждой из полученных таким образом фракций (1/720 часть каждой фракции) смешивали с 0,25 мл IMDM и затем анализировали для идентификации ТРО-активных фракций. В результате ТРО активность (достаточно сильная) была обнаружена во фракциях пробирок с номерами 24-30 (диапазон концентраций пропанола 36,0-42,0), которые объединяли и использовали как полученную из пробы F2 высокомолекулярного ТРО YMC-Pack CN-AP основную ТРО-активную фракцию FА.
Полученная из пробы F2 высокомолекулярного ТРО YMC-Pack CN-AP ТРО-активная фракция FА
Общий объем - 25,20 мл
Концентрация белка - 0,0246 мг/мл
Общий белок - 0,620 мг
Относительная активность - 700000
Общая активность - 434000
(15) Capcell раk C1 300А (конечная хроматография с обращенной фазой)
24,66 мл из 25,20 мл полученной из пробы F2 высокомолекулярного ТРО YMC-Pack CN-AP TPO-активной фракции FA (общий белок 0,606 мг, концентрация белка 0,0246 мг/мл, относительная активность 700000, общая активность 424000), полученной в стадии (14), смешивали с 0,4 мл 50% глицерина и концентрировали выпариванием.
Таким путем были получены 2 мл концентрированной пробы с концентрацией пропанола несколько %, концентрацией глицерина 10% и концентрацией белка 0,303 мг/мл. Ее наносили на колонку, упакованную Capcell Pak C1 300А (изготовляемым Shiseido Co., Ltd., кат. C1 TYPE:SG300A, 4,6 мм в диаметре и 250 мм высотой), которая была предварительно уравновешена 15% В, с применением растворителя А (0,1% ТFА) и растворителя В (1-пропанол, содержащий 0,05% ТFА) и проявляли 65 минут линейным градиентом от 27% В до 38% В при скорости течения 0,4 мл/мин. Элюаты собирали в 72 полипропиленовые пробирки по 0,6 мкл в каждую пробирку.
По 0,75 мкл из каждой из полученных таким образом фракций (1/800 часть каждой фракции) смешивали с 20 мкл 5% BSA, среду заменяли до 225 мкл IMDM и анализировали разбавленные таким образом в 300 раз фракции.
Из 2 мкл каждой фракции (1/300 фракции) готовили пробы для электрофореза, сушили выпариванием и обрабатывали при 95oС в течение 5 минут 10 мкл невосстанавливающего буфера для проб для SDS-PAGE. Обработанную таким образом пробу подвергали SDS-PAGE с применением 15-25% SDS -полиакриламидным преформированным гелем (изготовляемым Daiichi Pure Chemicals Co., Ltd.) и затем окрашивали с применением набора для окрашивания серебром. Описанные выше DPCIII применяли в качестве маркеров мол. массы.
В результате описанного анализа значительная ТРО активность была обнаружена во фракциях пробирок с номерами 33-39 (диапазон концентраций пропанола 29,5-31,5%). Фракции пробирок 34-39 (диапазон концентраций пропанола 30,0-31,5%) объединяли и использовали в качестве основной ТРО-активной фракции А. Использование SDS-PAGE распределения основной ТРО-активной фракции обнаруживало присутствие полосы, плотность окрашивания которой коррелирует с силой активности, в пределах средних мол. масс 17000-19000 при невосстанавливающих условиях, которая была сходна с полосой в случае полученной из пробы F3 низкомолекулярного ТРО ТРО-активной фракции, описанной в стадии (10).
Пример 2. Анализ частичных аминокислотных последовательностей очищенного ТРО крыс
Аминокислотный анализ последовательностей кандидата-белка ТРО крыс в Capcell Pak C1 300А ТРО-активной фракции FА, полученной в стадии очистки (10) примера 1, проводили согласно процедуре Iwamatsu (Iwamatsu et al., "Shin Kiso Seikagaku Jikken-ho (New Basic Biochemical Experiments)", vol.4, pp. 33-84, pub. Maruzen; Iwamatsu, A, Seikagaku (Biochemistry): vol. 63, No 2, pp.139-143, 1991; Iwamatsu, A., Electrophoresis, vol. 13, pp.142-147, 1992): Т. Е. пробу подвергали SDS-PAGE и переносили электрическим способом на поливинилидендифторидную (PVDF) мембрану. После восстановления и S-алкилирования белка на PVDF мембране обработанный таким образом белок расщепляли на пептидные фрагменты систематическим и ступенчатым гидролизом при помощи рестриктирующих ферментов in situ тремя протеазами и полученные пептидные фрагменты на каждой стадии расщепления отделяли и очищали хроматографией с обращенной фазой и анализировали их аминокислотные последовательности при помощи высокочувствительного способа определения аминокислот. Далее этот процесс описан детально.
Пример анализа белка-кандидата ТРО в полученной из пробы F3 низкомолекулярного ТРО Capcell Pak C1 300А фракции ТРО FА
(1) Концентрирование Capcell Pak C1 300A фракции ТРО FА
Из 4200 мкл полученной из пробы F3 низкомолекулярного ТРО ТРО-активной фракции FА с колонки Capcell Pak C1 300А (номера пробирок 36-42), полученной в стадии (10), (общий белок 39,6 мкг, концентрация белка 9,4 мкг/мл, относительная активность 4890000, общая активность 193600), 4151 мкл (98,8% всей фракции) применяли в анализе последовательностей аминокислот. Общий белок, рассчитанный из хроматограммы, был 39,1 мкг, из которых 1,6 мкг был предполагаемый белок ТРО, окрашенный серебром после SDS-PAGE и имеющий среднюю мол. массу приблизительно 17000-19000.
Эту пробу смешивали с глицерином и концентрировали выпариванием, получая 5 мкл глицеринового раствора. К нему добавляли невосстанавливающий буфер для SDS-РАGЕ и 1 М Трис-НС1 (рН 8) для корректировки его рН, получая приблизительно 25 мкл пробы, содержащей 200 мМ Трис-НС1 (рН 8), 50 мМ Трис-НС1 (рН 6,8), 1,1% SDS, 2 мМ ЭДТА, 0,02% бромфенол синий и 30 г глицерин.
Полученную таким образом пробу выдерживали при комнатной температуре в течение 14 часов и затем обрабатывали при 60oС в течение 5 минут для проведения реакции с SDS без избыточного нагревания.
(2) Электрофорез
Готовили микропластинки гелей (4,0% акриламидный концентрирующий гель и 15% акриламидный разделяющий гель) и SDS-PAGE проводили при комнатной температуре в течение 2 часов при постоянном токе 12,5 мА и затем при 17,5 мА. Предварительно окрашенный Low Range Marker (Bio-Rad, 161-0305) и DOCIII применяли в качестве маркеров мол. масс. Сразу после электрофореза полученные белковые молекулы переносили на PVDF мембрану (см. следующую стадию).
Часть этой пробы использовали также в другом электрофорезе с применением 15-25% полиакриламидного преформированного геля (Multi Gel 15/25, изготовляемый Daiichi Pure Chemicals Co., Ltd., 211072) при невосстанавливающих условиях или после восстановления пробы дитиотреитолом (DTT). Когда полученный гель окрасили при помощи описанного выше набора для окрашивания серебром, было подтверждено, что мол. масса полосы белка предполагаемого ТРО была приблизительно 19000 при восстанавливающих условиях и чистота предполагаемого ТРО белка с колонки Capcell Pak C1 300А была всего несколько процентов. Кроме того, поскольку подвижность этой полосы вариировала при невосстанавливающих и восстанавливающих условиях, было сделано предположение, что белок-кандидат содержит по меньшей мере одну дисульфидную связь.
(3) Перенос белка нa PVDF мембрану электроблоттингом и обнаружение полосы
Перенос белка на PVDF мембрану ProBlott, Applied Biosystems; кат. 400994) проводили в течение 1 часа при постоянном токе 160 мА (11-17 В) с применением полусухого устройства для переноса (Model KS-8460, изготовляемого Marysol). Раствор, состоящий из 0,3 М Триса и 20% метанола (рН 10), применяли в качестве анолита, раствор, состоящий из 25 мМ Триса и 20% метанола (рН 10,4), применяли в качестве раствора для мембраны и раствор, состоящий из 25 мМ Триса, 40 мМ аминокапроновой кислоты и 20% метанола (рН 10,4) в качестве католита.
При окрашивании перенесенной таким образом мембраны окрашивающим раствором Роnсеаu S (0,1) г Ponceau S и 1 мл уксусной кислоты в 100 мл воды) обнаружили множество полос и было подтверждено, что предполагаемый ТРО белок имел мол. массу 19000. Эту полосу вырезали для применения в последующих стадиях образования пептидных фрагментов.
(4) Образование пептидных фрагментов, пептидное картирование и анализ аминокислотных последовательностей
Для выполнения систематической (направленной) фрагментации предполагаемого ТРО белка, перенесенного на мембрану и S-алкилированного после восстановления на PVDF мембране, ступенчатый ограниченный ферментативный гидролиз проводили с применением следующих трех протеаз.
Первое расщепление: лизилэндопептидаза (Achromobacter lyticus m497-1, изготовляемый Wako Pure Chemical Industries, Ltd., кат. 129-02541).
Второе расщепление: эндопротеиназа ASp-N (изготовляемая Boeringer-Mannheim Corp., кат. 1054 589)
Третье расщепление: трипсин-ТРСК (изготовляемый Worthington Biochemical кат. 3740)
Пептидные фрагменты, полученные в результате каждого ферментативного расщепления, наносили на колонку Wakosil-11 5С18 С18 с обращенной фазой (Wako Pure Chemical Industries, Ltd., 2,0 мм в диаметре и 150 мм в длину) и элюировали при помощи растворителя А (0,05% ТFА) и растворителя В (изопропанол: ацетонитрил = 7:3, 0,02% ТFА) 30 мин линейным градиентом от 1% В до 50% В при скорости тока 0,25 мл/мин и при температуре колонки 30oС. Таким способом были сделаны пептидные картирования (см. фиг.4). Полученные пептидные фрагменты извлекали и подвергали расщеплению по Эрдману с применением аминокислотного секвенатора с газовой фазой (PPSQ-2, изготовляемого Shimadzu Corp. ). После этого каждую аминокислоту, получаемую по очереди из секвенатора, N-конец которой был соединен с РТН, идентифицировали с применением С18 колоночной хроматографии с обращенной фазой при помощи изократической элюции. Результаты представлены в табл.II.
В последовательностях, представленных в табл.II, последовательности, показанные в скобках в N-концах, являются аминокислотными остатками, которые могут быть дедуцированы из направленного ферментативного расщепления.
(5) Анализ гомологии полученных аминокислотных последовательностей (поиск гомологии)
Для того чтобы узнать, содержатся ли полученные таким образом аминокислотные последовательности в известном сообщенном белке или существует ли белок, имеющий сходные последовательности, их анализировали при помощи анализирующей последовательности программы Mac Vector (Kodak International Biotechologies, Inc. ). Для информации об известных белках или известных генах применяли базу данных Entrez Release 6 (National Center of Biotechnology Information, National Library of Medicine, National Inctitute of Health, USA, published on August 15, 1993).
В ней включены следующие базы данных:
.Entrez Release 6 database
.NCB1-GenBank, August 15, 1993 (Release 78.0)
.EMBL, July 15, 1993 (Release 35,0 plus updates)
.DDBL, July 15, 1993
.SWISS-PROT, April, 1993 (Release 25,0)
.PIR, June 30, 1993 (Release 37,0)
.PDB, April, 1993
.PRF, May, 1993
.dbEST, July 15, 1993 (Release 1,10)
.U.S. and European Patents
В результате последовательность (K) DSFLADVK AP12 полностью совпадала с внутренней последовательностью KDSFLADVK крысиного кортикостероидсвязывающего предшественника глобулина (CBG) [PIR data base accession No A40066; Smith and Hammond; "Крысиный кортикостероидсвязывающий глобулин: первичная структура и уровни мРНК в печени при различных физиологических условиях", Mol. Endocrinol., (1989), 3, 420-426].
Дальнейшее подробное исследование выявило, что последовательность KQYYESE (SEQ 1D No 193), имеющая высокое сходство с последовательностью (K) XYYESZ (X=A, S, G, M или Q и Z=Е или К) АР3, содержится в крысиной СВG-аминокислотной последовательности. В крысином СВG последовательности, соответствующие последовательностям АР12 и АР3, соединены друг с другом, образуя внутреннюю аминокислотную последовательность KDSFLADVKQYYESE (SEQ 1D No 190).
Что касается аминокислотных последовательностей других фрагментов, чем АР12 и АР3, не были обнаружены белок или ген, сходство которых могло бы обсуждаться.
Пример 3. Анализ биологических характеристик ТРО, полученного из плазмы крови тромбоцитопенических крыс
(1) В тест-системе крысиных CFU-MK (жидкой культуральной системе)
В качестве типичного примера фиг.5 показывает кривую зависимости ответа от дозы в случае применения пробы ТРО, частично очищенного из плазмы тромбоцитопенических крыс (ТРО-активная фракция F2 из колонки YMC PackProtein-RP, описанная в стадии очистки (8) примера 1-2). При периодическом наблюдении культивируемых клеток под микроскопом обнаруживали генерирование и созревание мегакариоцитов, а именно увеличение клеточного размера, возможно, вместе с увеличением числа мегакариоцитов. В качестве особенно значительного изменения процесс образования мегакариоцитов был обнаружен на 4-й день, который был последним днем культивирования (они были трудно распознаваемыми на 3-й день). Такой процесс образования называют цитоплазматическим процессом образования (Leven and Yee, Blood, vol.69, pp.1046-1052, 1987) или процессом образования протромбоцитов (Торр et al., Blood, vol. 76, pp. 912-924, 1990), которые считают структурой предшественников тромбоцитов, дифференцированных далее из зрелых мегакариоцитов, и эти протромбоциты считают конечной стадией дифференцировки мегакариоцитов, наблюдаемой до сих пор in vitro. Поскольку такое морфологическое изменение наблюдали с высокой частотой при применении только пробы ТОР, возможно, что этот фактор один может стимулировать пролиферацию и дифференцировку CFU-MK, может продуцировать зрелые мегакариоциты и может в конце концов высвобождать тромбоциты.
(2) В тест-системе подсчета колоний
При испытании пробы ТРО, частично очищенного из плазмы крови тромбоцитопенических крыс, при помощи тест-системы подсчета колоний с применением неразделенных клеток костного мозга крыс, клеток каждой стадии разделения/концентрирования или Gp11b/111a+ CFU-MK фракции ТРО, полученный из плазмы крыс, стимулировал образование колоний мегакариоцитов. По сравнению с колониями мегакариоцитов, индуцируемыми другими цитокинами, такими как IL-3, мышиный GM-CSF или человеческий ЕРО, индуцированные ТРО колонии мегакариоцитов отличались тем, что каждая колония состояла из небольшого числа мегакариоцитов, но каждый мегакариоцит был больше по размеру, что означало большую зрелость. Кроме того, поскольку ТРО не вызывал образования колоний или вызывал небольшое образование колоний других клеточных линий, Meg-CSF активность ТРО может рассматриваться как мегакариоцит-специфическая. На основании этих фактов очевидно, что ТРО имеет отличающиеся биологические свойства по сравнению со свойствами других цитокинов, таких как крысиный IL-3, мышиный GM-CSF и человеческий ЕРО, и проявляет уникальную Meg-CSF активность.
Проба ТРО, частично очищенного из плазмы крови тромбоцитопенических крыс, также проявляла Meg-CSF активность на CD34+, DR+ клеточных фракциях, полученных из клеток костного мозга человека или клеток крови спинного мозга человека, и индуцировала значительные количества колоний мегакариоцитов человека, что свидетельствует о том, что этот фактор не имеет видовой специфичности.
Пример 4. Специализация крысиных ТРО продуцирующих клеток
(1) Скрининг продуцирующих ТРО органов крыс
Скрининг и специализацию продуцирующих ТРО органов крыс проводили для того, чтобы обеспечить источник мРНК для применения в клонировании гена ТРО крыс. Во-первых, из крыс, сделанных тромбоцитопеническим путем введения Р55 антител, периодически брали костный мозг, легкие, печени и селезенки, их клетки (срезы органов в случае легких и печени) культивировали и активности в полученном культуральном супернатанте тестировали при помощи тест-системы с крысиными CFU-MK. Однако, эта первоначальная попытка не дала ясных результатов. Затем была сделана дальнейшая попытка культивирования клеток печени (гепатоцитов), полученных коллагеназной перфузией из печеней тромбоцитопенических крыс, индуцированных введением Р55 антител, с учетом сообщения, указывающего на связь между печенью и образованием ТРО в крысах (Siemensma et al., J. Lab. Clin. Med., vol.86, pp.817-833, 1975).
При нанесении полученного культурального супернатанта на колонку WGA-Agarose с последующим фракционированием адсорбированной фракции на Vydac phenyl колонке с обращенной фазой была обнаружена очень сходная активность при том же самом положении полученного из плазмы крыс ТРО в тест-системе с крысиными CFU-MK. Слабая, но та же самая активность была обнаружена в культуральном супернатанте нормальных гепатоцитов крыс. Эти результаты предполагают, что печень, вероятно, является одним из продуцирующих ТРО органов.
(2) Скрининг продуцирующих крысиный ТРО клеточных линий
На основании приведенных выше результатов проводили скрининг крысиных продуцирующих ТРО клеточных линий. Во-первых, каждую из 20 крысиных полученных из печени клеточных линий культивировали в соответствующей субкультуральной жидкой среде до тех пор, пока клетки почти не достигали стадии слияния. Затем культуральную среду заменяли той же самой соответствующей средой с добавлением 5% FCS и культивирование продолжали в течение 3 дней. Когда каждый из полученных культуральных супернатантов был частично очищен в соответствии с процедурой, описанной в стадии (1), для исследования наличия образования ТРО, ТРО активность была отчетливо обнаружена в трех полученных из паренхимных печеночных клеток крыс клеточных линий, а именно МсА-RН8994 клетках (АТСС CRL 1602, Becker et al., "0ncodevelopmental Gene Expression", ed. by Fishman and Sell, Academic Press, NY, pp.259-270, 1976; приобретенных из Dainippon Pharmaceutical Co. , Ltd.), H4-11-E клетках (АТСС CRL 1548, Pitot et al. , Nat. Cancer Int. Monogr., vol.13, pp. 229-245, 1964; приобретенных из Dainippon Pharmaceutical Co., Ltd.) и НТС клетках (Thompson et al. , Proc. Natl. Acad. Sci, USA, vol. 56, pp. 296-303: 1966, приобретенных из Dainippon Pharmaceutical Co., Ltd.).
(3) Детальный анализ ТРО активностей в McA-RH8994 клетках, Н4-11-Е клетках и НТС клетках
Биохимические и биологические свойства ТРО-активных белков, секретируемых из этих трех крысиных клеточных линий, исследовали детально и сравнивали со свойствами полученного из плазмы крыс ТРО.
McA-RH8994 клетки суспендировали в альфа-МЕМ(-) жидкой культуральной среде, содержащей 10% FCS, и переносили в пластиковую колбу для микрокультивирования (175 см2) при 1•106 клеток на колбу. После 3 дней культивирования при 37oС в инкубаторе с CO2 (5%) эту среду заменяли культуральной средой IMDM, содержащей 5% FCS, и культивирование продолжали еще 3 дня для получения образующегося культурального супернатанта. Н4-11-Е клетки суспендировали в модифицированной по способу Дульбекко среде Игла (жидкой среде) (глюкоза 4,5 г/л, далее эту среду называют DMEM), содержащей 10% FCS, и переносили в пластиковые колбы (175 см2) при 5•105 клеток на колбу. После 3 дней культивирования при 37oС в инкубаторе с 5% СO2 среду заменяли на среду IMDM, содержащую 5% FCS, и культивирование продолжали еще 3 дня для получения образующегося культурального супернатанта. Также НТС клетки суспендировали в DМЕМ жидкой культуральной среде, содержащей 5% FCS, и переносили в пластиковые колбы (175 см2) при 2,5•105 клеток на колбу. После 3 дней культивирования при 37oС в инкубаторе с 5% СО2 среду заменяли культуральной средой IMDM, содержащей 5% FCS, и культивирование продолжали еще 3 дня для извлечения полученного культурального супернатанта.
С использованием 2 л полученного таким образом культурального супернатанта из каждой из этих трех клеточных линий проводили частичную очистку полученного из клеточных линий ТРО в соответствии с процедурой очистки ТРО из XRP, описанной в примере 1-2. Полученные результаты описаны далее.
Во-первых, каждый культуральный супернатант концентрировали приблизительно в 6 раз при помощи ультрафильтрационного устройства и среду заменяли 20 мМ Трис-НС1 буфером (рН 8,0) при помощи колонки Sephadex G-25. Полученный элюат наносили на колонку Q-Sepharose FF, колонку промывали 20 мМ Трис-НС1 (рН 8,0) и затем адсорбированную фракцию элюировали 20 мМ Трис-НС1 (рН 8,0), содержащим 175 мМ NaCl. Элюированную таким образом фракцию наносили на WGA-Agarose- колонку, колонку промывали PBS и адсорбированную фракцию элюировали 20 мМ фосфатом натрия (рН 7,2), содержащим 0,2 М GlcNAc и 0,15М NaCl. Полученный элюат наносили на колонку TSK-gel AF-BLUE 650MH, колонку промывали 20 мМ фосфатом натрия (рН 7,2), содержащим 1 М NaCl, и затем адсорбированную фракцию элюировали 2 М NaSCN. Полученный элюат наносили на колонку Phenyl-Sepharose 6 FF/LS, колонку промывали 50 мМ фосфатом натрия (рН 7,2), содержащим 1,5 М сульфат аммония, с последующим промыванием 0,8 М сульфатом аммония, содержащим 36 мМ фосфат натрия, и затем адсорбированную фракцию элюировали 20 мМ фосфатом натрия (рН 7,2). Полученную таким образом фракцию концентрировали и затем фракционировали при помощи колонки Vydac Protein C4 с обращенной фазой (The Separation Group, кат. 214ТР51015, диаметр 1 см, высота 15 см). Т.е. эту пробу наносили на C4 колонку, предварительно уравновешенную 20% В, и затем проводили элюцию 90 минут линейным градиентом от 20% В до 40% В при скорости течения 1 мл/мин, с применением 0,1% ТFА в качестве проявляющего растворителя А и 1-пропанола, содержащего 0,05% ТFА, в качестве проявляющего растворителя В. В результате каждый из этих ТРО-активных белков, полученных из этих клеточных линий, был элюирован при концентрации 1-пропанола 30-43%.
При измерении ТРО активностей в пробах каждой стадии очистки при помощи тест-системы с крысиными CFU-MK результаты показали, что ТРО активности этих 3 клеточных линий давали сходные распределения с активностями полученного из XRP ТРО (сравни пример 1-2). Относительная активность, выход активности и т. п. на каждой стадии очистки, измеренные в тест-системе с крысиными CFU-MK, показаны в таблице 2. Кроме того, при измерении ТРО активностей в элюатах из конечной колонки с обращенной фазой в тест-системе с крысиными CFU-MK ТРО активностями 3 клеточных линий и активность полученного из XRP ТРО были обнаружены в одних и тех же пиках. При проведении теста с подсчетом колоний ТРО-активных фракций из колонки с обращенной фазой с применением неприкрепленных клеток, полученных в стадии истощения прикреплением для отделения и концентрирования крысиных Ср11b/111а+CFU-МК, распределения элюции, MEG-CSF активности были очень сходными с распределениями ТРО активности, измеренной в тест-системе с крысиными CFU-MK (см. таблицу 3).
Кроме того, подобно полученному из XRP ТРО, каждая из ТРО активностей этих 3 клеточных линий не образовала (или образовала очень мало) колоний других клеточных линий.
Каждую из объединенных активных фракций из колонки с обращенной фазой подвергали SDS-PAGE согласно процедуре, описанной в примере 1-2. При экстракции белка из полученного геля и измерения активности ТРО в тест-системе с крысиными CFU-MK было обнаружено, что средние мол. массы полученного из XRP ТРО, полученного из клеток МсА-RН8994 ТРО и полученного из клеток Н4-11-Е ТРО, были 17000-22000, 33000-39000 и 31000-38000, соответственно, а полученный из НТС клеток ТРО обнаружил средние мол. массы 17000-22000 и 28000-35000.
Таким образом, эти результаты показали, что ТРО белки, продуцируемые McA-RH8994 клетками, Н4-11-Е клетками и НТС клетками, равноценны полученному из XRP ТРО по их биологическим свойствам, хотя их биохимические свойства слегка отличаются, в частности, в отношении мол.масс. Примечательным открытием в данном изобретении является возможность того, что молекула ТРО-активного белка, содержащаяся в крови, может быть продуктом частичного расщепления в ее специфическом или в неупорядоченном сайте в продуцирующих ее клетках или после ее секреции из продуцирующих клеток. Это также предполагает возможное существование генов ТРО (мРНК и кДНК), имеющих разную длину.
Пример 5. Конструирование экспрессирующего вектора (pEF18S) для библиотеки кДНК
Вектор, в который может быть легко интегрирован фрагмент ДНК и который может влиять на высокую эффективность экспрессии клонированной кДНК, был сконструирован для применения в клонировании и экспрессии кДНК ТРО. Т.е. экспрессирующий вектор pEF18S был сконструирован из экспрессирующего вектора pME18S путем замещения SRα промотора промотором фактора элонгации Iα (EFIα), который известен как промотор, вызывающий высокую экспрессию (см. фиг.6). Промотор фактора элонгации Iα был получен частичным расщеплением 1 мкг экспрессирующего вектора pEF-BOS (Mizushima et al., Nucleic Acids Res., vol. 18, p. 5322, 1990) рестрикционными ферментами Hind111 и EcoRI, после чего продукт расщепления подвергали электрофорезу в 2% агарозном геле (FMC BioProducts) для выделения фрагмента ДНК приблизительно 1200 п.н. и очистки этого фрагмента ДНК с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad Laboratories, Inc.; набор для применения в селективной очистке ДНК, которая выполняется путем адсорбции ДНК пористым матриксом из диоксида кремния, разработанным на основе процедуры, сообщенной Willis et al., in Bio Tehniques, vol.9, pp.92-99, 1990), 100 нг полученного таким образом фрагмента ДНК лигировали с 50 нг экспрессирующего вектора pME18S (Liu et al., Proc. Natl. Acad. Sci. USA, vol.90, pp. 8957-8961, 1993), который был предварительно расщеплен HindIII и EcoRI. Клетки коммерческого штамма-хозяина, высококомпетентного штамма DН 5 E.coli (Toyobo Co., Ltd.; компетентный штамм E. coli получен модифицированным способом Hanahan et al., J. Mol.Biol. vol.166, pp. 557-580, 1983), трансформировали сконструированным таким образом вектором. После культивирования трансформированных клеток 12 колоний отбирали произвольно и из каждой колонии очищали плазмидную ДНК. В целом получали 10 клонов, содержащих целевую плазмиду (pEF18S), анализом распределений расщепления рестриктазами этих молекул ДНК. После этого клон, выбранный из них, культивировали для получения большого количества этой плазмидной ДНК.
Очистку плазмидной ДНК проводили в основном в соответствии с процедурой, описанной в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989). Т.е. клон. рEF18S, полученный, как описано выше, культивировали в течение ночи при 37oС в 50 мл среды LВ (1% Bacto-триптон, 0,5% Васtо-дрожжевой экстракт, 0,5% NaCl), содержащей 50 мкг/мл ампициллина, и полученные клетки собирали центрифугированием и суспендировали в 4 мл раствора ТЕG-лизоцима (25 мМ Трис-HCl, рН 8, 10 мМ ЭДТА, 50 мМ глюкоза, 0,5 лизоцим). К нему добавляли 8 мл 0,2N NaOH/1% SDS (раствор) и затем 6 мл 3 М раствор калий /5 М ацетат для основательного суспендирования клеток. После центрифугирования этой суспензии, полученную супернатантную жидкость обрабатывали смесью фенол/хлороформ (1:1), смешанной с таким же объемом изопропанола, и затем центрифугировали. Полученный осадок растворяли в ТЕ растворе (10 мМ Трис-HCl, рН 7,5, 1 мМ ЭДТА) и обрабатывали РНКазой и затем смесью фенол/хлороформ (1:1) с последующей преципитацией этанолом. Полученный осадок опять растворяли в ТЕ растворе, к которому последовательно добавляли NaCI и полиэтиленгликоль 3000 до их конечной концентрации 0,63 М и 7,5%, соответственно. После центрифугирования осадок растворяли в ТЕ растворе и осаждали этанолом. Таким путем получали приблизительно 300 мкг плазмидной ДНК. 100 мкг этой ДНК полностью расщепляли рестриктазами EcoRl и Notl и подвергали электрофорезу на 0,8% агарозном геле (изготовляемым FМС BioProducts) и извлеченные таким образом фрагменты вектора очищали при помощи набора для очистки ДНК Prep-AGene (Bio-Rad Laboratories, Inc.), получая приблизительно 55 мкг плазмидной ДНК, которую использовали в последующем конструировании библиотеки кДНК.
Пример 6. Очистка мРНК из МсА-RН8994 клеток
МсА-RН8994 клетки, обнаружившие относительно высокую активность в тесте с подсчетом колоний примера 4, отбирали в качестве материала для применения в клонировании кДНК ТРО крыс и подвергали следующим экспериментам.
Выделение общей РНК проводили в основном в соответствии с процедурой, описанной в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989). Т.е. McA-RH8994 выращивали до слияния в 15 культуральных чашках 90 мм в диаметре. После удаления жидкой среды клетки в каждой чашке тщательно суспендировали в 0,8 мл 5 М раствора гуанидина (5 М гуанидинтиоцианат, 5 мМ цитрат натрия (рН 7,0), 0,1 М β-меркаптоэтанол, 0,5% саркозилсульфат натрия) и полученные суспензии во всех чашках собирали в одну пробирку и общий объем доводили до 29 мл гуанидиновым раствором. Полученную смесь, которая становилась вязкой из-за разрушения клеток, подвергали приблизительно 20 повторениям засасывания/ выброса шприцем на 20 мл, снабженным иглой 21G, до тех пор, пока смесь не переставала быть вязкой. 18 мл 5,7 М CSC1-0,1 М ЭДТА (рН 7,5) применяли в качестве подушки в полиалломерной центрифужной пробирке для применения в роторе SW28 Beckman, и вышеупомянутую смесь в количестве приблизительно 20 мл осторожно наслаивали на эту подушку таким образом, чтобы слои не были потревожены и пробирка была почти заполнена. Полученную таким образом пробирку центрифугировали в течение 20 часов при 25000 об/мин при 20oС. Полученный осадок промывали дважды небольшим количеством 80% этанола, растворяли в ТЕ растворе, экстрагировали смесью фенол/хлороформ (1:1) и затем осаждали этанолом с 1/10 объема 3 М ацетата натрия и 25 объемами этанола. Таким путем получали приблизительно 2,5 мг общей РНК из приблизительно 108 клеток.
Очистку поли (А)+РНК из обшей РНК проводили с применением ОligotexТМ-dT30 (Super) (изготавливаемых Japan Synthetic Rubber/Nippon Roche; олигоdT молекулы иммобилизованы на частицах латекса ковалентным связыванием, и очистка поли (A)+PHK может быть достигнута сходно с тем, как это достигается с применением колонки олиго dT, которую обычно используют для этой цели). В результате получили 20 мкг поли(А) +РНК из приблизительно 500 мкг общей РНК.
Пример 7. Конструирование библиотеки кДНК крысы
Двухцепочечную кДНК, имеющую сайт узнавания EcoRl на ее 5'-конце и сайт узнавания Notl на ее 3'-конце, синтезировали из 5 мкг поли (А)+РНК, полученной, как описано в примере 6, с применением набора для синтеза кДНК Time SaverTM (изготовляемого Pharmacia; набор для синтеза кДНК, основанного на модифицированном способе Okayama and Berg, Mol. Cell. Biol., vol.2, pp. 161-170, 1982) и DIRECTIONAL CLONING TOOLBOX (изготовляемого Pharmacia; набор из
праймера 5'- AACTGGAAGAATTCGCGGCCGCAGGAA(T)18-3' (SEQ 1D No 15), содержащего последовательность узнавания Notl, для применения в синтезе кДНК, и адапторов 5'- AATTCGGCACGAG-3' (SEQ 1D no 16) 5'-CTCGTGTGCCG-3' (SEQ 1N No 17) для применения в добавлении последовательности узнавания EcoRl). Синтезированную таким образом кДНК лигировали с 1,2 мкг экспрессирующего вектора pEF18S (см. пример 5), который был предварительно расщеплен EcoRl и Notl, и трансформировали в 8,4 мл вышеупомянутой высококомпетентной E.coli DH5 (изготовляемой Toyobo Co., Ltd.). В результате было получено 5,3•105 трансформантов.
Пример 8. Получение (клонирование) кДНК фрагмента ТРО крысы при помощи PCR
530000 клонов кДНК библиотеки Mca-RH8994, сконструированной в примере 7, культивировали в течение ночи в 50 мл LB среды, содержащей 50 мкг/мл ампициллина, и плазмида МсА-RН8994 библиотеки кДНК очищали из полученных клеток, собранных центрифугированием, с применением QIAGEN-tip 100 (изготовляемого DIAGEN; анионообменная колонка с диоксидом кремния для очистки ДНК). Получили приблизительно 200 мкг плазмидной ДНК.
Синтезировали два антисмысловых нуклеотидных праймера AP18-1R и АР8-2R, которые соответствуют аминокислотной последовательности пептидного фрагмента АР8, описанного в примере 2, и два смысловых нуклеотидных праймера ЕFIα-1 и EFIα-2, которые соответствуют первому интрону фактора элонгации Iα человека плазмидного вектора pEF-18S (см. фиг.6), применяемых для получения библиотеки кДНК. Синтез этих праймеров выполняли с применением 394DNA/RNA синтезатора (изготовляемого Applied Biosystems, синтезатор основан на β-цианоэтиламидитном способе), и их очистку выполняли с применением колонки ОРС для очистки синтетической ДНК (изготовляемой Applied Biosystems; колонка с силикагелем с обращенной фазой для применения в очистке синтетической ДНК, имеющей тритильные группы). Каждую из таким образом синтезированных и очищенных молекул ДНК растворяли в ТЕ растворе до конечной концентрации 50 мкМ и хранили при -20oС до их использования. Синтетические олигонуклеотиды, применяемые в следующей процедуре, синтезировали и очищали таким же образом.
Праймеры AP8-1R и АР8-2R являются смешанными праймерами, состоящими из 17 непрерывных нуклеотидов, в которые включен дезоксиинозин, как сообщалось Takahashi et al. (Proc. Natl. Acad. Sci. USA, vol. 82, pp.1231-1935, 1985).
Праймеры EFIα-1 и EFIα-2, состоящие из 21 и 20 нуклеотидов, соответственно, синтезировали на основе нуклеотидных последовательностей положений 1491-1512 и 1513-1532 геномной последовательности, сообщенной Uetsuki et al. (J.Biol.Chem., vol. 264, pp. 5791-5798, 1989). (см. табл.III)
С 3 мкг МсА-RН8994 библиотеки кДНК плазмиды в качестве матрицы, АР8-1R (500 пмолей) и EFIα-1 (100 пмолей) в качестве праймеров и набора реагентов GeneAmpТМ PCR с ДНК-полимеразой AmpliTaqТМ (изготовляемой Таkara Shuzo Co., Ltd., набор из термостабильной Taql полимеры, реакционного буфера и dNTP для применения в PCR) проводили PCR при помощи GeneAmpТМ PCR системы (Perkin-Elmer; термоблок для PCR) (100 мкл объем, нагревание при 95oС 2 мин, всего 35 циклов, каждый цикл состоит из денатурации при 95oС в течение 1 минуты, гибридизации (отжига) при 40oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, и конечное инкубирование при 72oС в течение 7 минут). Для улучшения специфичности амплифицированных таким образом фрагментов ДНК дальнейшую PCR проводили (100 мкл объем, нагревание при 95oС в течение 2 минут, всего 35 циклов, каждый цикл состоит из денатурации при 95oС в течение 1 минуты, гибридизации при 45oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, и конечное инкубирование при 72oС в течение 7 минут) с применением 1 мкл полученного раствора реакции PCR в качестве матрицы и EFlα-2 (100 пмолей) и АР8-IR (500 пмолей) в качестве праймеров.
Полученный таким образом реакционный раствор подвергали электрофорезу в 2% агарозном геле (изготовляемом FМC BioProducts) для выделения фрагмента ДНК приблизительно 330 т. п.н. в качестве основного продукта PCR, который затем очищали с применением указанного выше набора для очистки ДНК Prep-A-Gene (изготовляемого Bio-Rad Laboratories, Inc.) с применением Т4 ДНК-лигазы (изготовляемой Life Technologies), очищенный таким образом фрагмент ДНК субклонировали в вектор pCRТМ11 (вектор для использования в ТА клонировании продуктов PCR, изготовляемый Invitrogen). Поскольку термостабильная полимераза, применяемая в PCR, имеет терминальную трансферазную активность, амплифицированный фрагмент ДНК непосредственно субклонировали в вектор pCRTM11, который имеет 5'-dT липкий конец, используя свойство этого фермента добавлять одну дезоксиадениловую кислоту к 3'-концу амплифицированной при помощи PCR ДНК. Плазмидные молекулы ДНК очищали из 28 клонов, выбранных произвольно из полученных клонов с применением QIAGEN-tip100 (DIAGEN), и их нуклеотидные последовательности определяли при помощи секвенатора ДНК 373А (Флуоресцентный секвенатор, изготовленный Applied Biosystems) с применением набора Таq Dye DeoxyТМ Terminater Cycle Seguening Kit (Applied Biosystems; набор для использования в определении нуклеотидной последовательности с применением флуоресцентных красителей, на основе дидезокси-способа Сенгера и др. (Proc. Natl. Acad. Sci. USA, vol. 74, pp. 5463-5467, 1977). Когда фрагмент ДНК, кодирующий три аминокислотных остатка (Ile/Thr/Ser)-Val-Pro аминокислотной последовательности АР8, показанной выше, смежной с праймером AP8-2R, отбирали из полученных таким образом фрагментов ДНК и секвенировали его полную длину, он содержал кДНК из 261 т.п.н. Положения 173-175 этого фрагмента кДНК кодировали метионин и рамка кодирования совпадала с аминокислотной рамкой АР8. Поскольку последовательность, вклинивающаяся в положение 173-175 фрагмента ДНК, совпадала с последовательностью Kozak (Kozak, M., Cell, vol. 44, pp. 238-292, 1986), можно думать, что этот фрагмент ДНК состоит из района инициации трансляции, кодирующего N-конец белка ТРО крыс. Этот фрагмент ДНК был назван А1. Нуклеотидная последовательность А1 фрагмента, включающая векторную последовательность, и выведенная из нее аминокислотная последовательность показаны в списке последовательностей (SEQ 1D No 1), прилагаемом здесь.
Пример 9. Скрининг клона кДНК ТРО крысы при помощи PCR
Библиотеку кДНК делили на пулы приблизительно по 10000 клонов, культивировали в течение ночи в 1 мл вышеупомянутой LB среды, содержащей 50 мкг/мл ампициллина, и затем экстрагировали плазмидную ДНК при помощи автоматизированного устройства для экстракции плазмидной ДНК P1-100 (VER-3,0, Kurabo Industries, Ltd. , автоматизированное устройство для экстракции плазмидной ДНК, основанное на модифицированном способе щелочного SDS способа, описанного в Molecular Cloning, Sambrook et. al., Cold Spring Harbor Laboratory Press, 1989). Отдельно синтезировали и очищали следующие два олигонуклеотида на основе фрагмента A1 кДНК.
5'-CGAGGGTGTACCTGGGTCCTG-3' (SEQ 1D No 31) (смысловая последовательность положений 1-17 последовательности SEQ 1D No 1; CGAG обозначает адапторную последовательность 5'-CAGAGTTAGTCTTGCGGTGAG-3' (SEQ 1D No 32) (антисмысловая последовательность положений 212-232 последовательности SEQ 1D No 1).
С применением 1/30 объема каждой пробы плазмидной ДНК, полученной, как описано выше, в качестве матрицы, синтезированных таким образом олигонуклеотидов в качестве праймеров и набора реагентов GeneAmpТМ PCR с AmpliTaqТМ ДНК-полимеразой (изготовляемые Takara Shuzo Co., Ltd.) проводили PCR при помощи GeneAmpТМ PCR System 9600 (PERKIN-ELMER) (всего 30 циклов, каждый цикл состоит из денатурации при 94oС в течение 30 секунд, гибридизации при 66oС в течение 30 секунд и синтеза при 72oС в течение 1 минуты). В результате обнаружили специфическую полосу 236 т.п.н. в 3 из 100 пулов. При разделении одного из этих пулов на субпулы приблизительно по 900 клонов для экстракции плазмидной ДНК и проведения PCR описанным образом обнаружили специфическую полосу в 3 из 100 субпулов. При дальнейшем разделении этих субпулов на пулы из 40 клонов и проведении того же процесса скрининга специфическую полосу обнаружили в 3 из 100 испытанных пулов. Один из пулов-кандидатов культивировали на LВ планшете (LB среда, содержащая 15% агар) с добавлением 50 мкг/мл ампициллина и каждую из образовавшихся колоний подвергали экстракции плазмидной ДНК и PCR описанным образом. В результате положительную полосу детектировали в 2 из 100 испытанных клонов.
Пример 10. Секвенирование кДНК ТРО крысы
Очистку плазмидной ДНК проводили в основном в соответствии с процедурой, описанной в Molecular Cloning (Sambrook et. al., Cold Spring Harbor Laboratory Press, 1989). Каждый из двух полученных клонов примера 9 культивировали в течение ночи в 50 мл LB среды, содержащей 50 мкг/мл ампициллина, и приблизительно 300 мкг плазмидной ДНК получали после очистки, описанной в примере 6.
Полученную таким образом плазмидную ДНК секвенировали по способу, описанному в примере 8 с использованием набора Taq Dye DeoxyTM Terminater Cycle Sequening Kit (Applied Biosystems) для определения полной нуклеотидной последовательности, содержащей А1 фрагмент. В результате нуклеотидные последовательности этих двух клонов полностью совпадали друг с другом, свидетельствуя о том, что они являются одним и тем же клоном. Выбирали один из этих клонов и плазмиду этого клона назвали pEF18S-A2α. Ее нуклеотидная последовательность и выведенная из нее аминокислотная последовательность показаны в списке последовательностей (SEQ 1D No 2).
Последовательность, представленная в списке последовательностей (SEQ 1D No 2), имеет четкие характеристики. Последовательность в направлении 5'-3' от 172 т. п.н. 5'-нетранслирующего района кодирует аминокислотную последовательность, богатую гидрофобными аминокислотами, которая предположительно является сигнальной последовательностью секреции белка, состоящей из 21 аминокислот, начинающихся с метионина. Этот белок содержит 126 аминокислотных остатков. 3'-нетраслируемая последовательность содержит 1025 нуклеотидов и поли А-хвост после терминирующего кодона (ТАА). Этот белок содержит последовательность, которая соответствует аминокислотной последовательности АР8, анализируемой в примере 2 (аминокислотные остатки 1-12 в SEQ 1D No 2), но не содержит консенсусной последовательности для N-гликозилирования. Нуклеотидная последовательность 1624-1629, расположенная вблизи конца 3'-нетранслируемой последовательности, отличается от консенсусной последовательности и является, по-видимому, потенциальной последовательностью полиаденилирования.
Вектор рЕF18S-А2α, несомый штаммом E.coli DH5, депонирован авторами данного изобретения 14 февраля под депозитным FERM BP-4565 В National Institute of Bioscience and Human Technology, Agency of Industrial Science and Techology, Ministry of International Trade and Industry, Japan.
Пример 11. Экспрессия кДНК ТРО крысы в COS 1 клетках и подтверждение ТРО активности
Трансфекцию плазмиды pEF18S-A2α В COS 1 клетки проводили следующим образом путем небольшой модификации ДЭАЭ-декстранового способа, предусматривающего обработку хлорохином (Sompayrac et al., Proc. Natl. Acad. Sci. USA, vol. 78, pp. 7575-7578, 1981; Luthman et al., Nucl. Acids Res., vol. 11, pp. 1295-1308, 1983). COS 1 клетки (АТСС CRL 1650) суспендировали в описанной выше среде DМEМ, содержащей 10% FCS, помещали в пластиковую чашку для культуры ткани (100 мм) и культивировали при 37oС в инкубаторе с 5% СО2 до тех пор, пока клетки не достигали стадии приблизительно 40% слияния. Отдельно плазмиду pEF18S-A2α (10 мкг), растворенную в 30 мкл HBS (21 мМ HEPES, 145 мМ NaCl, рН 7,1), добавляли к 4 мл культуральной среды DMEM, добавляли 500 мкг/мл ДЭАЭ-декстрана (Pharmacia), 80 мкМ хлорохина (Sigma Chemical Co.), 8% (об. /об.) HBS и 9% (об./об.) Nu - сыворотки (Collaborative Research, Inc.). Приготовленную таким образом смесь добавляли к культивируемым COS 1 клеткам, которые промывали дважды культуральной средой ДМЕМ, и культивирование продолжали еще в течение 5 часов в инкубаторе с 5% СО2. После этого культуральный супернатант в этой чашке удаляли отсасыванием и оставшиеся в чашке клетки промывали два раза культуральной средой ДМЕМ, добавляли 15 мл среды ДМЕМ, содержащей 10% FCS, и культивировали при 37oС в течение 3-5 дней в инкубаторе с 5% СО2 для извлечения полученного культурального супернатанта.
Полученный таким образом культуральный супернатант экстенсивно диализовали против культуральной среды IMDM и оценивали в тест-системе с крысиными CFU-MK. ТРО активность была обнаружена в зависимом от дозы виде в культуральном супернатанте COS 1 клеток, в которых экспрессировалась плазмида pEF18S-A2α (фиг.7). Подобно случаю с полученным из ХRP ТРО, много мегакариоцитов образовали удлиненные цитоплазматические образования на 4-й день культивирования. В противоположность этому, ТРО активность не была обнаружена в культуральном супернатаите COS 1 клеток, в которые вводили только плазмиду pEF18S (фиг. 7). В M-07е тест-системе активность пролиферации М-07е клеток также была найдена в зависимости от дозы виде в культуральном супернатанте COS 1 клеток, в которых экспрессировалась плазмида pEF18S-A2α, но не в культуральном супернатанте COS 1 клеток, в которых экспрессировалась плазмида pEF18S. Эти результаты показывают, что А2α содержит кДНК, кодирующую белок, имеющий ТРО активность.
Затем, пробу частично очищенного ТРО готовили для испытания способности этой ТРО активности усиливать образование тромбоцитов in vivo. Во-первых, COS 1 клетки, трансфицированные плазмидой pEF18S-A2α, культивировали в течение 3 дней в бесссывороточной культуральной среде, в которой добавляли 0,2 мг BSA, получая приблизительно 5,8 л бессывороточного культурального супернатанта. К нему добавляли ингибитор протеазы pAPМSF до конечной концентрации 1 мМ и смесь фильтровали при помощи фильтра 0,22 мкм. К 5793 мл полученного фильтрата (концентрация белка 0,229 мг/мл, общий белок 1326 мг, относительная активность 1000, общая активность 1326000) добавляли 0,85 моля NaCl на 1000 мл (288 г всего), получая 5849 мл раствора, содержащего 0,822 М NaCl. Полученный таким образом раствор наносили при скорости течения 7 мл/мин на колонку TSK-gel AF-BLUE 650 МН (Tosoh Corp., кат. 08705, диаметр 5 см и высота 6 см), которая была предварительно уравновешена 20 мМ фосфатом натрия (рН 7,2), содержащим 1 М NaCl. После завершения нанесения пробы приблизительно 7900 мл элюата проходили через колонку при элюции 20 мМ фосфатом натрия (рН 7,2), содержащим 1 М NaCl. Этот элюат собирали вместе и концентрировали при помощи ультрафильтрационного устройства (Omega Ultrasette, номинальное отсечение мол. массы 8000), изготовляемого Filtron), получая проходящую через колонку фракцию F1 (460 мл, концентрация белка 1,11 мг/мл, общий белок 973 мг, относительная активность 16,3). Затем элюирующий раствор заменяли на 2 М NaSCN и элюированную таким образом поглощенную TSK-gel AF-BLUE 650 МН ТРО-активную фракцию F2 (2840 мл) концентрировали до 6,81 мл при помощи ультрафильтрационного устройства с УМ 3 мембраной (76 мм в диаметре, Amicon Corp.). Общий белок в этой ТРО-активной фракции F2 составлял 12,5 мг и выход белка F2 на этой стадии был 0,62%. Затем F2 наносили на колонку HiLoad 26/60 Superdex 200 pg (Pharmacia Biotech, кат. 17-1071-01, диаметр 2,6 см и высота 60 см) и проявляли 20 мМ ацетатом натрия (рН 5,5), содержащим 50 мМ NaCl, при скорости течения 1 мл/мин. После начала проявления ТРО активность обнаружили в элюатах в диапазоне от 194 до 260 мл после нанесения. Эти элюаты объединяли, получая Superdex 200 pg ТРО активную фракцию F2 (66 мл, концентрация белка 0,112 мг/мл, общий белок 7,41 мг, относительная активность 142860, общая активность 1058600). Затем, готовили буфер А (20 мМ ацетат натрия, рН 5,5) и буфер В (20 мМ фосфата натрия, рН 7,2, содержащий 500 мМ NaCl) и ТРО-активную фракцию наносили при скорости течения 1 мл/мин на сильную катионообменную колонку RESOURCE S (Pharmacia Biotech, кат. 17-1178-01, 0,64 см в диаметре и высотой 3 см), которая была предварительно уравновешена 100% А. После этого проводили элюцию при скорости течения 0,3 мл/мин, с 40 мин линейного градиента от 100% А до 100% В. При анализе активности ТРО активность обнаружили в широком диапазоне элюатов (от 5% В до 32% В). Эти ТРО-активные элюаты объединяли и концентрировали при помощи ультрафильтрационного устройства с УМ 3 мембраной (диаметр 25 мм, Amicon Corp. ), получая RESOURCE S ТРО-активную фракцию F2 (1,65 мл, концентрация белка 4,74 мг/мл, общий белок 7,82, относительная активность 71400, общая активность 558600).
Пробу частично очищенного ТРО вводили подкожно в течение последовательных 5 дней (474 мкг (100 мкл)/мышь/день) ICR самцам мышей (9-недельного возраста), количества тромбоцитов у которых измеряли в день перед введением. В качестве контроля таким же образом вводили BSA (200 мкг (100 мкл)-/мышь/день) или буфер, применяемый в качестве растворителя частично очищенного ТРО (100 мкл/мышь/день). На следующий день после последнего введения собирали кровь из сердца каждой мыши для измерения числа тромбоцитов при помощи гемоцитометра (F800, Тоа Iyo Denshi). В мышах, которым вводили частично очищенный ТРО, наблюдали число тромбоцитов, приблизительно в 2,14 раза большее по сравнению с числами тромбоцитов перед введением. При сравнении с контрольными группами числа тромбоцитов в мышах, которым вводили частично очищенный ТРО, были приблизительно в 1,74 раза выше, чем числа тромбоцитов в получавших BSА мышах и приблизительно в 1,90 раз выше, чем в получавших буфер мышах. Эти результаты показали, что ТРО усиливает образование тромбоцитов in vivo. Кроме того, поскольку количество иммуносупрессивного кислого белка (1AP), известного как белок острой фазы мышей, не повышалось в мышах, которым вводили частично очищенный ТРО, это дает сильное основание предполагать, что ТРО отличается от IL-6 и IL-11 в отношении индукции белка острой фазы.
Пример 12. Обнаружение мРНК ТРО в тканях крыс
Для локализации экспрессируюцей мРНК ТРО ткани в теле крысы РНК экстрагировали из различных тканей крысы. Всего 6 крыс подвергали облучению рентгеновскими лучами, как описано в примере 1, и различные ткани (мозг, вилочковую железу, легкие, сердце, селезенку, тонкий кишечник, почки, яички и клетки костного мозга) вырезали из этих крыс на 11-й - 14-й день после облучения и сразу же замораживали в жидком азоте. Экстракцию общей РНК выполняли с применением реагента для выделения РНК ISOGEN (изготовляемого Wako Pure Chemical Industries, Ltd.). Количество ISOGEN определяли согласно весу замороженной пробы ткани и пробу с добавленным реагентом обрабатывали гомогенизатором (PhyscotronR NS-60, N1T1-ON Medical & Physical Instruments MFG.Co., Ltd) до полного разрушения ткани (приблизительно 45-60 секунд при 10000 об/мин). Гомогенат ткани подвергали затем экстракции общей РНК с применением процедуры, основанной на способе со смесью кислый гуанидиний фeнол-xлopoфopм (Chomczynski et al. (Anal. Biochem., vol.162, pp. 156-159, 1987). В результате получали 1,1-5,6 мг общей РНК из соответствующих тканей.
С применением OligotexТМ-dT30 (Super) (изготовляемого Japan Synthetic Rubber/Nippon Roche) 20 мкг поли(А)+ РНК очистили приблизительно из 500 мкг общей РНК.
С применением произвольного праймера первую цепь кДНК синтезировали на 1 мкг поли(А)+ РНК, полученной из каждой ткани. Т.е. 1 мкг поли (А)+ РНК растворяли в 10 мкл стерильной воды, инкубировали при 70oС в течение 15 минут и затем быстро охлаждали. К этому раствору добавляли 75 пмолей произвольного праймера (Takara Shuzo Co., Ltd.), 10 Е ингибитора РНКазы (Boehringer-Mannheim Corp.), 50 мМ Трис-НСl (рН 8,3), 75 мМ КСl, 3мМ МgСl2 и 200 E Super ScriptТМ11 (обратная транскриптаза, изготовляемая Life Technologies). Полученный в результате раствор (общий объем 20 мкл) инкубировали при 37oС в течение 1 часа и полученный реакционный раствор инкубировали при 70oС в течение 10 минут для инактивации фермента и хранили при -20oС до применения.
Новые праймеры для РСR синтезировали на основе последовательности кДНК ТРО крысы, полученной в примере 10. Последовательности синтезированных праймеров были следующими.
rТРО-1: 5'-ССТ GTC CTG CTG CCT GCT GTG-3' (SEQ 1D No 33) (положения 347-367 в SEQ 1D No 2).
rTPO-N: 5'-TGA AGT TCG TCT ССА АСА ATC-3' (SEQ 1D No 34) (антисмысловой праймер, соответствующий положениям 1005-1025 в SEQ 1D No 2).
С применением 1/10 объема каждого из раствора синтезированных кДНК в качестве матрицы, 1 мкМ каждого из синтезированных rТРО-1 и rTPO-N в качестве праймеров и GeneAmpTM PCR Reagent Kit с AmpliTaqTM ДНК-полимеразой (изготовляемой Takara Shuzo Co., Ltd.) проводили PCR в объеме 100 мкл с применением GeneAmpTM PCR System 9600 (PERKIN-ELМER) и нагреванием при 95oС в течение 2 минут, повторением в целом 30 циклов, каждый цикл состоял из денатурации при 95oС в течение 1 минуты, гибридизации при 57oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты с последующим конечным инкубированием в течение 7 минут. При электрофорезе полученных реакционных растворов на 2% агарозном геле (FМC BioProducts) и испытании амплифицированных полос полосы, которые казались специфическими для мРНК ТРО, были найдены на гелях, происходящих из мозга, печени, тонкого кишечника и почек. Эти результаты показывают, что экспрессия мРНК ТРО в крысах происходит в тканях этих органов, хотя ее количественное выражение не может быть оценено. При одновременном обсуждении возможности существования подобного способа экспрессии в теле человека и результатов примера 4 можно предполагать, что печень является подходящим исходным материалом для получения кДНК ТРО человека.
Пример 13. Конструирование библиотеки кДНК, полученных из нормальной печени человека
На основании результатов примера 12 печень была выбрана как исходный материал для применения в клонировании кДНК ТРО человека. Таким же образом, как описано в примере 7, двухцепочечную кДНК, имеющую сайт узнавания EcoRl на ее 5'-конце и сайт узнавания Notl на ее 3'-конце, синтезировали из 5 мкг коммерческой полученной из нормальной печени человека поли(А)+ РНК (изготовляемой Clontech; продукт экстрагировали на основе способа со смесью кислый гуанидиний-фенол-хлороформ Chomczynski et al.), с применением Time SaverТМ кДНК synthesis Kit (набора) (изготовляемого Pharmacia) и DIRECTIONAL CLONING TOOLBOX (изготовляемого Pharmacia). Полученную таким образом кДНК лигировали с 1,2 мкг экспрессирующего вектора pEF18S, который был расщеплен заранее EcoRl и Notl, и трансформировали в 8,4 мл Компетентного штамма E. coli DH5 (Toyobo Co., Ltd.). В результате получили 1,2•106 трансформантов.
Пример 14. Получение (клонирование) фрагмента кДНК ТРО человека при помощи PCR
При помощи произвольных праймеров первую цепь кДНК синтезировали из 1 мкг коммерческой полученной из нормальной печени человека поли(А)+ РНК (изготовляемой Clontech). Т.е. 1 мкг поли(А)+ РНК растворяли в 10 мкл стерильной воды, инкубировали при 70oС в течение 15 минут и затем быстро охлаждали. К этому раствору добавляли 75 пмолей произвольных праймеров (Takara Shuzo Co., Ltd.), 10 Е ингибитора РНКазы (Boehringer-Mannheim Corp.), 50 мМ Трис-HСl (рН 8,3), 75 мМ КСl, 3 мМ MgCl2 и 200 Е обратной транскриптазы Super ScriptТМ (изготовляемой Life Technologies). Полученный таким образом раствор (общий объем 20 мкл) инкубировали при 37oС в течение 1 часа и образующийся реакционный раствор инкубировали при 70oС в течение 10 минут для инактивации фермента и хранили при -20oС до использования.
Праймеры для РСR синтезировали на основе кДНК последовательности кДНК ТРО крысы (SEQ 1D 2). Последовательности синтезированных праймеров были следующими.
rTPO-AIN: 5'-ATG GAG CTG ACT GAT TTG CTC -3'- (SEQ 1D 35) (положения 173-193 в SEQ 1D 2).
rTPO-N: 5'-TGA AGT TCG TCT ССА АСА АТС -3' (SEQ 1D 36) антисмысловой праймер, соответствующий положениям 1005-1025 в SEQ 1D 2).
С применением 1/10 объема раствора синтезированной кДНК в качестве матрицы, 1 мкМ каждого из синтезированных таким образом rTPO-A1N и rTPO-N в качестве праймеров и набора реагентов GeneAmpТМ PCR с ДНК-полимеразой AmpliTaqТМ (изготовленной Takara Shuzo Co., Ltd.) проводили PCR с объемом 100 мкл, с применением GeneAmpТМPCR System 9600 (PERKIN-ELMER) и нагреванием при 95oС в течение 2 минут, повторяя всего 35 циклов, состоящих из денатурации при 95oС в течение 1 минуты, гибридизации при 40oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут.
Полученный таким образом реакционный раствор подвергали электрофорезу в 2% агарозном геле (изготовляемом FMC BioProducts) для выделения фрагмента ДНК приблизительно 620 т. п.н. в качестве основного продукта PCR, который затем очищали с использованием указанного выше набора для очистки ДНК Prep-A-Gene (Bio-Rad Laboratories, Inc.). Нуклеотидную последовательность очищенного таким образом фрагмента ДНК сразу же определяли ДНК секвенатором 373А (Applied Biosystems) с применением вышеупомянутого набора Taq Dye DeoxyТМ Terminater Cycle Sequening Kit (изготовляемого Applied Biosystems). Определенная таким образом нуклеотидная последовательность, за исключением праймерной, части, и выведенная из нее аминокислотная последовательность показаны в списке последовательностей (SEQ 1D 3).
Этот фрагмент ДНК, за исключением праймерных последовательностей, имеет длину 580 т.п.н. При сравнении эта кДНК человека обнаружила гомологию 86% с нуклеотидной последовательностью кДНК крысы, что свидетельствует о том, что этот фрагмент ДНК кодирует часть ТРО человека.
Пример 16. Скрининг клона кДНК ТРО человека при помощи PCR
Соответствующие ТРО человека праймеры для РСR синтезировали на основе SEQ 1D 3. Последовательности синтезированных таким образом праймеров были следующими.
hTPO-I: 5'-TTG TGA CCT CCG AGT CCT CAG - 3' (SEQ 1D 37) (положения 60-80 в SEQ 1D 3).
hTPO-j: 5'-TGA CGC AGA GGG TGG ACC СТС-3' (SEQ 1D 38) (антисмысловой праймер, соответствующий положениям 479-499 в SEQ 1D 3).
Библиотеку кДНК человека, сконструированную в примере 13, амплифицировали и делили на пулы (каждый пул содержал приблизительно 100000 клонов), культивировали в течение ночи в 1 мл LB среды, содержащей 50 мкг/мл ампициллина, и затем из них экстрагировали плазмидную ДНК при помощи автоматизированного устройства для выделения плазмид P1-100 (VER-3,0, изготовляемого Kurabo Industries, Ltd.). Экстрагированную ДНК растворяли в ТЕ растворе.
С применением 5% экстрагированной пробы ДНК в качестве матрицы, 1 мкМ каждого из синтезированных олигонуклеотидов (hTPO-I и hTPO-j) в качестве праймеров и набора реагентов для PCR GeneAmpTM с AmpliTaqTM ДНК-полимеразой (Tukara Shuzo Co., Ltd) проводили PCR в 20 мкл объема при помощи системы РСR 9600 GeneAmpTM (изготовляемой PERKIN-ELMER) (всего 35 циклов, каждый цикл состоит из денатурации при 95oC в течение 1 минуты, гибридизации при 59oC в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут). В результате специфическая полоса была обнаружена в 3 из примененных 90 пулов. Один из этих 3 пулов делили на субпулы, каждый из которых содержал приблизительно 5000 клонов, и плазмидную ДНК очищали из 90 субклонов для проведения PCR описанным способом. В результате специфическую полосу обнаружили в 5 субпулах. Один из этих 5 пулов делили на субпулы, каждый из которых содержал 250 клонов, и плазмидиую ДНК экстрагировали из 90 субпулов, причем специфическую полосу обнаруживали в 3 субпулах. Один из этих 3 пулов делили на субпулы, каждый из которых содержал 30 клонов, и плазмидную ДНК очищали из 90 субпулов для проведения РСR описанным способом. В результате специфическую полосу обнаруживали в 3 субпулах. Один из пулов-кандидатов культивировали на описанном LB планшете, содержащем 50 мкг/мл ампициллина, и каждую из полученных таким образом 90 колоний подвергали экстракции плазмидной ДНК и PCR, как описано. В результате в конце был получен клон HL34.
Пример 16. Секвенирование кДНК ТРО человека
Очистку плазмидной ДНК проводили в основном в соответствии с процедурой, описанной в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989). Клон HL 34 культивировали в течение ночи в 50 мл LВ среды, содержащей 50 мкг/мл ампициллина, и полученные клетки собирали центрифугированием и суспендировали в 4 мл вышеупомянутого ТЕG-лизоцим-раствора. К ним добавляли 8 мл 0,2 N NaOH/ 1% SDS и затем 6 мл раствора 3 M калий/5 М ацетата для тщательного суспендирования клеток. После центрифугирования этой суспензии полученный супернатант обрабатывали смесью фенол/хлороформ (1:1), смешанное с таким же объемом изопропанола, и затем центрифугировали. Полученный осадок растворяли в ТЕ растворе и обрабатывали РНКазой и затем смесью фенол/хлороформ (1: 1) с последующим осаждением этанолом. Полученный осадок опять растворяли в ТЕ растворе, к которому добавляли NaCl и полиэтиленгликоль 3000 до конечных концентраций 0,63 М и 7,5%, соответственно. После центрифугирования осадок растворяли в ТЕ растворе и осаждали этанолом. Таким путем получали приблизительно 300 мкг рЕF18S-НL34 плазмидной ДНК.
Очищенную таким образом плазмидную ДНК применяли в секвенаторе ДНК 373А (Applied Biosystems) для определения ее полной нуклеотидной последовательности с применением Таq Dye DeoxyТМ Terminater Cycle Sequening Kit (Applied Biosystems). Определенная таким образом нуклеотидная последовательность и выведенная из нее аминокислотная последовательность показаны в списке последовательностей (SEQ 1D 4). В этом случае в качестве праймеров при определении нуклеотидной последовательности использовали олигонуклеотиды, синтезированные на основе нуклеотидной последовательности SEQ 1D 3, и синтетические олигонуклеотиды, сконструированные на основе внутренней последовательности, полученной анализом секвенкрования.
В результате было доказано, что плазмидный клон pEF18-S-HL-34 содержит фрагмент кДНК из 861 п.н. и содержит высокогомологичные последовательности с аминокислотными последовательностями АР8 (номера аминокислот 1-12 в SEQ 1D 4) и ТР2/ТР3 (аминокислоты 157-162 в SEQ 1D 4), анализированными в примере 2. По-видимому, этот фрагмент ДНК кодирует открытую рамку считывания, начинающуюся при положении 25, но не имеет терминирующего кодона и содержит поли А-хвосту подобную последовательность из 76 оснований на его 3'-конце. Его аминокислотная последовательность, состоящая из 253 аминокислотных остатков, обнаружила 84% гомологии с соответствующей частью кДНК ТРО крысы (частью аминокислотной последовательности, состоящей из 147 остатков, показанной в SEQ 1D 2). Впоследствии было обнаружено, что этот клон является фрагментом ДНК, кодирующим часть кДНК человека, которая соответствует кДНК ТРО крысы. Вследствие отсутствия терминирующего кодона и присутствия поли А-хвосту подобной последовательности на его 3'-конце этот клон, по-видимому, не является полной кДНК, а представляет собой искусственный продукт, получаемый во время процедуры конструирования библиотеки кДНК.
Пример 17. Экспрессия кДНК ТРО человека в COS 1 клетках и подтверждение ТРО активности
Трансфекцию полученного описанным способом плазмидного клона pEF18S-HL-34 в COS 1 клетки проводили согласно способу примера 11. Т.е. трансфекцию проводили с 10 мкг плазмидной ДНК по ДЭАЭ-декстрановому способу, предусматривающему обработку хлорохином. Эти COS 1 клетки культивировали в течение 3-5 дней и затем получали супернатант.
Полученный культуральный супернатант экстенсивно диализовали против культуральной среды IMDM и оценивали его активность в тест-системе с крысиными CFU-MK. ТРО активность обнаруживали в зависимом от дозы виде в культуральном супернатанте COS 1 клеток, в которых экспрессировалась плазмида pEF18S-HL-34 (фиг.8). После 4 дней культивирования многие мегакариоциты образовали удлиненные цитоплазматические выступы. В противоположность этому, ТРО активность не была обнаружена в культуральном супернатанте COS 1 клеток, в которых экспрессировалась только плазмида pEF18S (фиг.8). В тест-системе с М-07e клетками активность усиления пролиферации М-07е клеток была обнаружена только в супернатанте COS 1 клеток, в которых экспрессировалась плазмида pEF18S-HL-34 в результате ее трансфекции. Эти результаты показали, что рEF18S-HL34 содержит ген, который кодирует белок, имеющий ТРО активность. Кроме того, было показано, что ТРО человека действует на клетки-предшественники мегакариоцитов крысы (т.е. отсутствует видовая специфичность).
Пример 18. Экспрессия кДНК делеционного типа ТРО человека и подтверждение ТРО активности
Клон HL34, полученный в примере 15, содержал кДНК, имеющую поли А-хвосту подобную непрерывную последовательность на ее 3'-конце, которого нет в кДНК ТРО крысы, и поэтому является, по-видимому, искусственным продуктом этого эксперимента. Вследствие этого получали кДНК молекулу, из которой была делетирована последовательность, подобная поли А-хвосту, чтобы проверить, имеет ли белок, экспрессируемый этой делеционной кДНК, ТРО активность. Делеционную кДНК получали при помощи РСR. Последовательности праймеров, применяемых для РСR, были такими, в которых сайт узнавания рестриктазой добавляли к 5'-концу (ЕcoRI k hТРO5 и Notl и два стоп-кодона ТАА и TGA k hТРО3).
hTPO5: 5'-TTT GAA TTC GGC CAG ССА GAC ACC CCG GCC-3' (SEQ 1D 170) (положения 1-21 в SEQ 1D 4).
hТРО3: 5'-ТТТ GCG GCC GCT CAT TAT TCG TGT ATC CTG TTC AGG TAT CC-3' (SEQ 1D 171) (антисмысловой праймер, соответствующий положениям 757-780 в SEQ 1D 4).
С применением 1 мкг плазмидной ДНК плазмидного клона pEF18S-HL34, полученного в примере 16, в качестве матрицы, 10 мкМ каждого из синтезированных hTРO5 и hТРО3 в качестве праймеров и набора реагентов для PCR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (Takara Shuzo Co., Ltd.,) PCR проводили с объемом 100 мкл с использованием РСR системы 9600 GeneAmpТМ (PERKIN-ELMER) и повторяли всего 15 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, гибридизации при 65oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием в течение 7 минут. Полосу приблизительно 800 п.н., полученную таким образом, расщепляли рестриктазами EcoRl и Notl, очищали и затем субклонировали в экспрессирующий вектор pEF18S, который был обработан предварительно теми же рестриктазами. Из полученных трансформантов 5 клонов, содержащих фрагмент ДНК из приблизительно 800 п.н., отбирали для получения большого количества плазмидной ДНК в соответствии с процедурой, описанной в примере 5. Полную длину амплифицированного района приблизительно из 800 л.н. каждой плазмиды подвергали анализу нуклеотидной последовательности, получая ее полную идентичность с нуклеотидной последовательностью, анализированной в примере 16 (положения 1-780 в SEQ 1D 4).
Полученный таким образом клон был назван pHT1-231. Вектор pHT1-231, несомый штаммом E. coli DH5, был депонирован авторами данного изобретения 14 февраля 1994 под депозитным FERM BP-4564 В National Institute of Bioscience and Human Technology, Agency of Industrial Science and Technology, Ministry of International Trade and Industry, Japan. Трансфекцию полученной таким образом плазмиды в COS1 клетки проводили согласно способу примера 11. Т.е. трансфекцию проводили с 10 мкг плазмидной ДНК по ДЭАЭ-декстрановому способу, предусматривающему обработку хлорохином. COS 1 клетки культивировали в течение 3-5 дней при 37oС и затем собирали супернатант.
Полученный таким образом культуральный супернатант экстенсивно диализовали против культуральной среды IMDM и оценивали в тест-системе с крысиными CFU-MK. ТРО активность была обнаружена в культуральном супернатанте COS 1 клеток, в которых экспрессировалась плазмида pHT1-231 (фиг.9). На 4-е дни культивирования многие мегакариоциты образовали удлиненные цитоплазматические выступы. В противоположность этому, ТРО активность не была обнаружена в культуральном супернатанте COS 1 клеток, в которых экспрессировалась только pEF18S (фиг. 9). В M-07e тест-системе активность усиления пролиферации М-07е клеток была обнаружена только в культуральном супернатанте COS 1 клеток, в которых экспрессировалась плазмида pHT1-231. Эти результаты показали, что pHT1-231 содержит кДНК, которая кодирует белок, имеющий ТРО активность.
Пример 19. Получение 3'-концевого района кДНК ТРО человека при помощи РCR
Клон HL34, полученный в примере 15, содержал кДНК, имеющую подобную поли А-хвосту последовательность, на основании чего было сделано предположение, что ее 3'-концевой район был неполным. Поэтому была сделана попытка получить полноразмерный 3'-концевой район при помощи РСR. Следующие 4 типа праймеров 5 стороны для PCR были синтезированы на основе последовательностей, определенных в примере 16:
hTPO-H: 5'-AGC AGA АСС ТСТ СТА GTC СТС - 3' (SEQ 1D 39) (положения 574-594 в SEQ 1D 4).
hTPO-K: 5'-АСА CTG AAC GAG CTC CCA AAC-3' (SEQ 1D No 4) (положения 595-615 в SEQ 1D 4).
hTPO-N: 5'-AAC TAC TGG CTC TGG GCT TCT-3' (SEQ 1D No 41) (положения 660-680 в SEQ 1D 4).
hTPO-O: 5'-AGG GAT TCA GAG CCA AGA TTC-3' (SEQ 1D No 42) (положения 692-712 в SEQ 1D 4).
Следующие праймеры 3'-стороны, содержащие смешанные нуклеотиды при 4 основаниях 3'-конца, синтезировали для амплификации кДНК от начала поли А-части. Был синтезирован также якорный праймер, без смешанного нуклеотида.
hTPO3mix: 5'-TAG CGG CCG C(T)17G GGG-3' (SEQ 1D No 43).
A AAA-3' (SEQ 1D No 44).
С ТТТ-3' (SEQ 1D No 45).
CCC-3' (SEQ 1D No 46).
hTPO3anchor: 5'-TAG CGG CCG С(Т)11-3' (SEQ 1D No 47).
Первую цепь кДНК синтезировали с 1 мкг коммерческой полученной из нормальной печени человека поли (А)+ РНК (Clontech), как в примере 14, но с применением 0,5 мкг олиго dT праймера (он включен в набор для синтеза кДHК Time SaverTM, изготовляемый Pharmacia). С применением 1/10 объема раствора синтезированной кДНК в качестве матрицы, 20 мкМ hТРО-Н праймера и 10 мкМ hTPO3mix праймера и набора реагентов для PCR CeneAmpTM c AmpliTaqTM ДНК-полимеразой (изготовляемой Takara Shuzo Co., Ltd.) проводили PCR с объемом 50 мкл при помощи PCR системы 9600 GeneAmpTM (PERKIN-ELMER) с нагреванием при 96oС в течение 2 минут, повторением в целом 10 циклов, каждый из которых состоит из тепловой денатурации при 96oС в течение 1 минуты, гибридизации при 48oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут.
Вторую РСR проводили с применением 1/10 объема раствора, полученного в первой РСR, в качестве матрицы, 20 мкМ hTPO-K праймера и 10 мкМ hTPO3mix праймера с объемом 50 мкл, с нагреванием при 96oС в течение 2 минут и повторением в целом 10 циклов, каждый из которых состоял из тепловой денатурации при 96oС в течение 1 минуты, гибридизации при 63oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут.
Третью PCR проводили с применением 1/10 объема раствора, полученного при второй PCR, в качестве матрицы, 20 мкМ hTPO-N праймера и 10 мкМ hTPО3mix праймера с объемом 50 мкл, с нагреванием при 96oС в течение 2 минут и повторением в целом 10 циклов, каждый из которых состоял из тепловой денатурации при 96oС в течение 1 минуты, гибридизации при 63oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут.
Четвертую РСR проводили с применением 1/10 объема раствора, полученного в третьей РСR, в качестве матрицы, 20 мкМ hTPO-O праймера и 10 мкМ hTPO3mix праймера с объемом 50 мкл и нагреванием при 96oС в течение 2 минут, с повторением в целом 10 циклов, каждый из которых состоял из тепловой денатурации при 96oС в течение 1 минуты, гибридизации при 63oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут.
Пятую PCR проводили с применением 1/10 объема раствора, полученного в четвертой PCR, в качестве матрицы, 20 мкМ hTPO-O праймера и 20 мкМ hТРО3anchor праймера с объемом 50 мкл с нагреванием при 96oС в течение 2 минут, повторением в целом 10 циклов, каждый из которых состоял из тепловой денатурации при 96oС в течение 1 минуты, гибридизации при 58oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут.
Полученный таким образом реакционный раствор подвергали электрофорезу в 2% агарозном геле (FMC BioProducts) для выделения фрагмента ДНК приблизительно 600 п.н. в качестве основного продукта PCR, который затем очищали с применением вышеупомянутого набора для очистки ДНК Prep-A-Gene (изготовляемого Bio-Rad Laboratories, Inc.). Нуклеотидную последовательность очищенного таким образом фрагмента ДНК сразу же определяли при помощи секвенатора ДНК 373 А (Applied Biosystems) с применением Taq Dye DeoxyТМ Terminator Cycle Sequening Kit (Applied Biosystems). Определенная нуклеотидная последовательность и выведенная из нее аминокислотная последовательность показаны в списке последовательностей (SEQ 1D 5).
Этот фрагмент ДНК имеет нуклеотидную последовательность, кодирующую 130 аминокислот, начинаясь с праймера hTPO-О, с последующей последовательностью из более 180 нуклеотидов при 3'-конце (нуклеотиды после положения 577 не могли быть определены). Аминокислотная последовательность, начинающаяся с глицина, совпадает с аминокислотной последовательностью положений 203-232 в SEQ 1D 4. Нуклеотидная последовательность положений 1-94 также совпадает с нуклеотидной последовательностью положений 692-785 в SEQ 1D 4.
Пример 20. Реконструкция библиотеки кДНК, полученной из нормальной печени человека
Поскольку клон HL34, полученный в примере 15, содержал подобную поли А-хвосту последовательность непосредственно на его 3'-конце его открытой рамки считывания без терминирующих кодонов и поэтому был, по-видимому, неестественным продуктом синтеза кДНК, библиотеку кДНК реконструировали с использованием 5 мкг коммерческой полученной из нормальной печени человека поли (А)+ РНК (препарата, изготовляемого Clontech). Синтез кДНК проводили с применением SuperScriptТМ Lambda System для синтеза кДНК и λ Cloning Kit SuperScriptТМ 11 РНКазы Н- (обе изготовлены LIFE TECHNOLOGIES). Поли (А)+ РНК денатурировали нагреванием и затем добавляли к 20 мкл реакционного раствора, содержащего включающего в себя последовательность узнавания Notl олиго dT в качестве праймера, включенного в набор (50 мМ Трис-HСl, рН 8,3, 75 мМ КСl, 3 мМ MgСl2, 1 мМ ДТТ, 1 мM dNTP-микс, 200 Е SuperScriptТМ 11 РНКазы Н-), с последующей инкубацией при 37oС в течение 60 минут. После синтеза второй цепи кДНК (2 часа инкубации при 16oС в 150 мкл реакционного раствора, содержащего 25 мМ Трис-HСl, рН 8,3, 100 мМ КСl, 5 мМ MgСl2, 250 мкМ dNTP-микс, 5 мМ ДТТ, 40 Е ДНК-полимеразы 1E.coli, 2 Е РНКазы H E.coli и 10 Е ДНК-лигазы E.coli) добавляли 10 Е Т4 ДНК-полимеразы и полученную смесь инкубировали при 16oС в течение 5 минут. Реакционный раствор нагревали при 65oС в течение 10 минут и экстрагировали таким же объемом смеси фенол/хлороформ. Молекулы кДНК, имеющие длину менее 400 п.н., удаляли при помощи SizeSepТМ400 вращающейся колонки (колонка для удаления низкомолекулярной ДНК, присоединенная к набору для синтеза кДНК TimeSaverТМ, Pharmacia). После добавления EcoRl Адаптора (присоединенного к Directional Cloning Toolbox, Pharmacia) обработанную таким образом пробу расщепляли Notl и опять наносили на ту же колонку для удаления низкомолекулярной ДНК. Синтезированную таким образом двухцепочечную кДНК (1,3 мкг), имеющую последовательность узнавания EcoRl на ее 5'-конце и последовательность узнавания Notl на ее 3'-конце, лигировали с экспрессирующим вектором pEF18S, расщепленным предварительно EcoRl и Notl, и затем трансформировали в 9,2 мл Компетентного штамма E.coli DH5 (Toyobo Co., Ltd.).
Полученная таким образом библиотека кДHК печени человека (hTPO-F1) содержала 1,0•106 трансформантов.
Пример 21. Скрининг клона кДНК ТРО из библиотеки hTPO-F1 кДНК печени человека
Праймеры, соответствующие кДНК ТРО человека, для РСR синтезировали на основе SEQ 1D 3 и 6, представленных в списке последовательностей. Последовательности синтезированных праймеров были следующими.
hTPO-1: 5'-TTG TGA CCT CCG AGT CCT CAG-3' (SEQ 1D No 48) (положения 60-80 в SEQ 1D 3).
hTPO-KU: 5'-AGG ATG GGT TGG GGA AGG AGA-3' (SEQ 1D No 49) (антисмысловой праймер, соответствующий последовательности положений 901-921 в SEQ 1D 6).
Библиотеку hTPO-F1 кДНК печени человека (1,0•106 трансформантов), сконструированную в примере 20, делили на 3 пула (пулы 1 - 3) и эти пулы замораживали. PCR проводили с применением 2 мкг плазмидной ДНК, полученной из каждого пула, в качестве матрицы и 1 мкМ каждого из синтезированных олигонуклеотидов (hTPO-1 и hTPO-KU) в качестве праймеров. С применением набора реагентов для РСR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (Takara Shuzo Со. Ltd. ) PCR проводили в 100 мкл объема при помощи системы PCR 9600 GeneAmpТМ (PERKIN-ELMER) (всего 35 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, гибридизации при 59oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут). В результате фрагмент ДНК, имеющий ожидаемый размер, амплифицировали в ток случае, когда применяли плазмидную ДНК, полученную из пула 3. Пул 3 затем делили на субпулы, каждый из которых содержал 15000 трансформантов, и эти субпулы культивировали в течение ночи в 1 мл LВ среды, содержащей 50 мкг/мл ампициллина, с последующей экстракцией плазмидной ДНК при помощи автоматизированного устройства для выделения плазмид Р1-100. Экстрагированную таким образом ДНК растворяли в ТЕ растворе и 5% полученных растворов использовали в качестве матриц для проведения РСR с применением тех же самых праймеров и при условиях, описанных выше. В результате амплификации ДНК с ожидаемым размером была обнаружена в 6 из 90 пулов. Один из этих положительных пулов разделяли на субпулы, каждый из которых содержал 1000 клонов, экстрагировали плазмидную ДНК и проводили РСR, как описано выше. При этом же наблюдали амплификации ДНК. Плотность полос фрагментов ДНК на гелях для электрофореза после ряда PCR становилась все меньше, по мере дальнейших разделений на пулы, что, по-видимому, связано с низким извлечением плазмидной ДНК из-за слабого роста целевого клона. Поэтому другой скрининг проводили посредством гибридизации колоний с применением первоначального пула 3.
Пул 3 распределяли на 100 LB агаровых чашках 15 см в диаметре с таким размером инокулята, что на каждой чашке выросли 4100 колоний. После получения чашки-реплики из каждой из инокулированных таким образом чашек одну из чашек-дупликатов культивировали при 37oС 6 часов для извлечения выросших на этой чашке колоний и экстракции проб плазмидной ДНК. При проведении PCR этих проб ДНК, как описано выше, амплификацию полосы, эквивалентной ожидаемому размеру, наблюдали в одном из 100 субпулов. Из чашки этого субпула готовили два фильтра-реплики с применением BIODYNEТМ TRANSFER MEMBRANE (PALL). Денатурацию этих фильтров проводили пропитыванием их в 10% SDS в течение 10 минут, в 0,5N NaOH/1,5 M NaCl в течение 10 минут и в 0,5 M Трис-HCl (рН 8,0)/1,5 M NaCl в течение 10 минут в указанной последовательности, с последующими 30 мин высушивания на воздухе и последующим нагреванием в течение 1 часа при 80oС в вакуумном сушильном шкафу. Полученные фильтры промывали 6 х SSC (полученного разведением 20 х SSC исходного раствора, состоящего из 175,3 г NaCl и 88,2 г цитрата Na, растворенных в 1 л воды, рН 7,0) с добавлением 1% SDS. Предгибридизацию промытых таким образом фильтров проводили инкубированием их при 42oС в течение 30 минут с качанием в 30 мл реакционного раствора, состоящего из 50% формамида, 5 х SSC, 5 х раствора Денхардта (полученного из 50 х раствора Денхардта, содержащего 5 г Фиколла, 5 г поливинилпирролидона и 5 г фракции V бычьего сывороточного альбумина в 500 мл воды), 1% SDS и 20 мкг/мл ДНК спермы лосося.
После предгибридизации реакционный раствор обменивали с 30 мл гибридизационного раствора того же состава, смешивали с зондом, меченым [α-32Р] - СТР (Amersham) и затем инкубировали с качанием при 42oС в течение 20 часов. Меченый зонд, примененный в этом эксперименте, представлял собой EcoRl/BamH1 фрагмент плазмиды pEF18S-HL34, который был получен очисткой части SEQ 1D 4 от ее 5'-конца до положения 458 и мечением этой очищенной части способом произвольных праймеров с Megaprime DNA Labelling System (набор, изготовляемый Amersham, основанный на способе, описанном в Anal. Biochem., 132, 6-13, 1983). Обработанные таким образом фильтры промывали в 2 х SSC/0,1% SDS растворе при 42oС в течение 30 минут и затем в 0,2 х SSC/0,1% SDS - растворе при 42oС в течение 30 минут. Затем эти фильтры подвергали 16 часам радиоавтографии при -70oС с использованием интенсифицирующего экрана Х-ОМАТТМ пленки AR5 (Eastman Kodak). В результате наблюдали один сигнал, рассматриваемый как положительный. Колонии, приблизительно соответствующие этому сигналу, собирали с исходной чашки и высевали опять на LВ агаровую чашку 10 см. 50 колоний, выросших на этой чашке, культивировали по отдельности и их пробы ДНК подвергали РСR с использованием праймеров hТРО-1 и hТРО-KU при описанных выше условиях. В результате амплификация полосы, эквивалентной ожидаемому размеру, была обнаружена только в одном клоне, который был назван pHTF1.
Пример 22. Определение нуклеотидной последовательности клона рНТF1 кДНК ТРО человека
Очистку плазмидной ДНК проводили в основном согласно процедуре, описанной в Molecular Cloning (Sambrook et al. Cold Spring Harbor Laboratory Press, 1989). Клон pHTF1 культивировали в течение ночи в 50 мл LB среды, содержащей 50 мкг/мл ампициллина. Полученные клетки собирали центрифугированием и суспендировали в 4 мл растворе ТЕG-лизозима. К ним добавляли 8 мл раствора 0,2 N NaOH/1% SDS и затем 6 мл раствора 3 М калия/ 5 М ацетата для тщательного суспендирования клеток. После центрифугирования этой суспензии полученный супернатант обрабатывали смесью фенол/хлороформ (1:1), смешанным с таким же объемом изопропанола, и затем центрифугировали. Полученный осадок растворяли в ТЕ растворе и обрабатывали РНКазой и затем со смесью фенол/хлороформ (1:1) с последующим осаждением этанолом. Полученный осадок опять растворяли в ТЕ растворе, к которому затем добавляли NaCl и полиэтиленгликоль 3000 до их конечных концентраций 0,63 М и 7,5%, соответственно. После центрифугирования осадок растворяли в ТЕ растворе и осаждали этанолом. Таким путем получали приблизительно 300 мкг плазмидной ДНК pHTF1.
Очищенную таким образом плазмидную ДНК применяли в вышеупомянутом секвенаторе ДНК 373А (Applied Biosystems) для определения ее полной нуклеотидной последовательности с применением Taq Dye DeoxyТМ Terminator Cycle Sequening Kit (Applied Biosystems). Определенная таким образом нуклеотидная последовательность и выведенная из нее аминокислотная последовательность показаны в списке последовательностей (SEQ 1D 7). Олигонуклеотиды, синтезированные на основе нуклеотидной последовательности SEQ 1D 6 и на внутренней последовательности, полученной анализом их секвенирования, применяли в определении нуклеотидной последовательности в качестве праймеров.
В результате было подтверждено, что клон рНТF1 содержит фрагмент кДНК из 1721 п. н. и имеет высокую гомологию с аминокислотными последовательностями АР8 (номера аминокислот 1-12 в SEQ 1D 7) и ТР2/ТР3 (номера аминокислот 157-162 в SEQ 1D 7), анализированных в примере 2. Этот фрагмент ДНК, по-видимому, имеет 5'- некодирующую область из 101 основания, открытую рамку считывания, состоящую из 353 аминокислотных остатков, начинающуюся при остатке метионина, кодируемого при положениях нуклеотидов 102-104, и кончающуюся при остатке глицина, кодируемого при положениях нуклеотидов 1158-1160, следующего далее терминирующего кодона (ТАА) и 3'- некодирующей области из 531 оснований и последовательности поли А-хвоста из 30 оснований. Аминокислотная последовательность белка, который может кодироваться этой открытой рамкой считывания, полностью совпадала с предсказанной аминокислотной последовательностью ТРО человека, показанной в SEQ 1D 6. Последовательность кДНК pHTF1 была большего размера, чем вероятная последовательность кДНК, показанная в SEQ 1D 6, и содержит 77 дополнительных оснований при 5'-конце и 347 дополнительных оснований при 3'-конце перед последовательностью поли А-хвоста. Также эта нуклеотидная последовательность отличалась от SEQ 1D 6 при трех положениях. Т. Е. А (положение 84), А (положение 740) и G (положение 1198) в SEQ 1D 7 были С, Т и А в SEQ 1D 6, соответственно. Только мутация в положении 740 входила в кодирующий белок район, но эта мутация не вызывала изменения аминокислоты, поскольку основания А и Т были третьим основанием кодонов треонина. Хотя причина этих изменений в основаниях в настоящее время неясна, анализ плазмидного клона pHTF1 подтвердил, что белок ТРО человека содержал 353 аминокислотных остатка, в том числе сигнальную последовательность, состоявшую из 21 аминокислот. Мол. масса зрелого белка без сигнальной последовательности была определена как 35466.
Вектор рНТF1 в штамме E.coli DH5 был депонирован авторами данного изобретения 24 марта 1994 под FERM BP-4617 при National Institute of Bioscience and Human Technology, Agency of Industrial Science and Techology, Ministry of International Trade and Industry, Japan.
Пример 23. Экспрессия клона pHTF1 кДНК ТРО человека в COS 1 клетках и подтверждение ТРО активности
Трансфекцию СOS1 клеток полученным таким образом плазмидным клоном pHTF1 проводили согласно способу примера 11. Т.е. трансфекцию проводили с 10 мкг плазмидной ДНК ДЭАЭ-декстрановым способом, предусматривающим обработку хлорохином. Трансфицированные COS 1 клетки культивировали в течение 3 дней при 37oС и собирали культуральный супернатант.
Культуральный супернатант экстенсивно диализовали против культуральной среды IMDM и оценивали в тест-системе с крысиными CFU-MK. ТРО активность была обнаружена в зависимом от дозы виде в супернатанте COS 1 клеток, в которых экспрессировалась плазмида pHTFl (фиг.10а). В противоположность этому, ТРО активность не была обнаружена в культуральном супернатанте COS 1 клеток, в которых экспрессировалась только плазмида pEF18S (фиг.10а). Подобные результаты были получены в М-07е тест-системе. Супернатант COS 1 клеток, в которых экспрессировалась плазмида pHTF1, значительно усиливал пролиферацию М-07е клеток зависимым от дозы образом (фиг. 10b). Эти результаты показали, что pHTF1 содержит ген, кодирующий белок, имеющий ТРО активность.
Пример 24. Клонирование хромосомной ДНК ТРО человека
Клонирование хромосомной ДНК ТРО человека проводили с применением кДНК ТРО человека в качестве зонда. Геномная библиотека, используемая в клонировании, была подарком от профессора Т. Ямамото из Gene Research Center, Tohoku University (библиотека, сообщенная Sakal et al. в J. Biol. Chem, 269, 2173-2182, 1994, сконструированная частичным расщеплением хромосомной ДНК человека рестриктазой Sau3A1 и лигированием продукта частичного расщепления с сайтом BamH1 фагового вектора Lambda EMBL3, изготовляемого Stratagene). Скрининг из этой библиотеки с применением кДНК ТРО человека в качестве зонда проводили в основном согласно способу, описанному в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989). С применением E. coli LE392 в качестве хозяина эту библиотеку инокулировали на чашку (15 см), содержащую NZYM (10г NZ амина, 5 г NaCl, 5 г Bacto - дрожжевого экстракта, 2 г МgSO4•7Н2О и 15 г агара в 1 л воды (рН 7,0) в таком размере инокулята, что одна чашка содержала 30000 частиц фага. Два фильтра-реплики получали из каждой из полученных 18 чашек с применением BIODYMETMA TRANSFER MEMBRANE (изготовляемой PALL.). Денатурацию фильтров выполняли пропитыванием их в 0,5 N NaOH/1,5 M NaCl в течение 10 минут и затем в 0,5 M Трис-HCl (рH 8,0) /1,5 M NaCl в течение 10 минут, с последующим высушиванием на воздухе в течение 30 минут и горячей сушкой при 80oС в течение 1,5 часов в вакуумном тормостате. Предгибридизацию проводили инкубированием обработанных таким образом фильтров при 42oС в течение 1 часа в 500 мл реакционного раствора, состоящего из 50% формамида, 5х SSC, 5 х раствора Денхардта, 1% SDS и 20 мкг/мл ДНК спермы лосося. Для применения в качестве зонда фрагмент кДНК ТРО человека (номера положений оснований 178-1025 в SEQ 1D 7) амплифицировали PCR и очищали и очищенный фрагмент метили 32Р с применением набора для мечения произвольных праймеров ДНК (набор для мечения ДНК, изготавливаемый Takara Shuzo на основе способа произвольных праймеров, описанного в Anal.Biochem. 132, 6-13, 1983). Последовательности праймеров, примененные в этой PCR, были следующими.
hTPO-1: 5'-TTG TGA CCT CCG AGT CCT CAG-3' (SEQ 1D 50) (положения 178-198 в SEQ 1D 7).
hTPO-N: 5'-AGG GAA GAG CGT ATA CTG TCC-3' (SEQ 1D 51) (антисмысловой праймер, соответствующий последовательности положений 1005-1025 в SEQ 1D 7).
С применением меченого изотопом зонда гибридизацию проводили при 42oС в течение 20 часов в 500 мл реакционного раствора, имеющего тот же состав, что и состав предгибридизационного раствора. Полученные фильтры промывали 3 раза в растворе 2 х SSC / 0,1% SDS при комнатной температуре в течение 5 минут и затем один раз в растворе 0,1 х SSС /0,1% SDS при 68oС в течение 1 часа. Затем фильтры подвергали радиоавтографии при -79oС с применением интенсифицирующего экрана и Х-ОМАТТМ AR5 пленки (Eastman Kodak). В результате получали 13 положительных сигналов. Бляшки, приблизительно соответствующие каждому из положительных сигналов, собирали из исходных чашек и инокулировали опять на NZYM чашки (15 см) в таком размере инокулята, что на каждой чашке образовались 1000 бляшек. Два фильтра-реплики готовили из каждой из полученных чашек для проведения гибридизации при условиях, описанных выше. В результате положительные сигналы были обнаружены на всех фильтрах из 13 групп. Одну бляшку извлекали из каждой из полученных чашек для получения фаговой ДНК по способу лизата чашек, описанному в Molecular Cloning. Пробы фаговой ДНК, полученные таким образом из 13 клонов, проверяли на присутствие кДНК кодирующего района при помощи РСR с применением следующих праймеров.
hTPO-L: 5'-GGC CAG CCA GAC ACC CCG GCC-3' (SEQ 1D 52) (положения 1-21 в SEQ 1D 6).
hTPO-F: 5'-ATG GGA GTC ACG AAG CAG TTT-3' (SEQ 1D 53) (антисмысловой праймер, соответствующий последовательности положений 137-147 в SEQ 1D 6).
hTPO-V: 5'-AAC CTT ACC CTT CCT GAG АСА-3' (SEQ 1D 55) (антисмысловой праймер, соответствующий последовательности положений 1070-1090 в SEQ 1D 6).
При проведении PCR при помощи этих комбинаций праймеров, 5 из 13 клонов, по-видимому, содержали полный кодирующий аминокислоты район, подсказанный из этой кДНК. Хромосомные молекулы ДНК, содержащие эти 5 клонов, имели сходную длину приблизительно 20 п.н. и обнаружили почти то же самое распределение фрагментов при предварительном рестрикционном анализе при помощи растриктаз. Поэтому один из этих клонов (клон λ HGTl) был выбран и проанализирован блоттингом по Саузерну. Т.е. 1 мкг ДНК клона λ HGTl расщепляли полностью рестриктазой EcoRl или Hind111 и наносили на 0,8% агарозный гель для электрофореза и полосы на геле переносили на BIODYNEТМ A TRANSFЕR MEMBRANE (PALL). Полученный фильтр сушили на воздухе в течение 30 минут и отжигали 2 часа при 80oС в вакуумной лечи. Предгибридизацию проводили инкубированием обработанного таким образом фильтра при 42oС в течение 1 часа в 50 мл реакционного раствора, состоящего из 50% формамида, 5 х SSC, 5 х раствора Денхардта, 1% SDS и 20 мкг/мл ДНК спермы лосося. Для использования в качестве зонда фрагмент кДНК ТРО человека (номера положений оснований 178-1025 в SEQ 1D 7) амплифицировали PCR и очищали и очищенный фрагмент метили 32Р с применением Random Prime DNA Labelling Kit (набора, изготовляемого Takara Shuzo). При помощи меченого изотопом зонда гибридизацию проводили при 42oС в течение 20 часов в 50 мл реакционного раствора, имеющего тот же состав, что и раствор для предгибридизации. Полученный фильтр промывали 3 раза в 2 х SSC/ 0,1% SDS - раствора при комнатной температуре в течение 5 минут и затем 1 раз в растворе 0,1 х SSC/0,1% SDS при 68oС в течение 1 часа. Затем фильтр подвергали в течение 16 часов радиоавтографии при -70oС с применением интенсифицирующего экрана и Х-ОМАТТМ AR5 пленки (Eastman Kodak). в результате наблюдали одну полосу приблизительно 10 т.п.н. в случае расщепления Hindlll. Поэтому 10 мкг ДНК клона λ HGTl расщепляли Hindlll и подвергали электрофорезу в 0,8% агарозном геле и полосу 10 т.п.н. вырезали из геля, очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad) и субклонировали в клонирующий вектор pUC13 (Pharmacia), который предварительно был расщеплен Hind111. В этом случае высококомпетентный штамм E.coli DH5 (ТОУОВО) применяли в качестве штамма-хозяина.
Из полученных таким образом клонов был выбран клон, содержащий Hind111 фрагмент из 10 т.п.н., и был обозначен pHGT1.
Рестрикционная карта фагового клона λ HGT1 показана на фиг.11.
Пример 25. Определение нуклеотидной последовательности хромосомного клона pHGT1 TPO человека
Культивирование клона pHGTl и очистку плазмидной ДНК проводили в основном согласно способу, описанному в Мolecular Cloning (Sambrook et al., Cold Spring Наrbоr Laboratory Press, 1989). Клон рНGТ1 культивировали в течение ночи в 50 мл LB среды, содержащей 50 мкг/мл ампициллина. Полученные клетки собирали центрифугированием и суспендировали в 4 мл описанного выше раствора ТЕG-лизозима. К ним добавляли 8 мл раствора 0,2 N NaOH /0,1% SDS и затем 6 мл раствора 3 М калия/5М ацетата для тщательного суспендирования клеток. После центрифугирования этой суспензии полученный супернатант обрабатывали смесью фенол/хлороформ (1:1), смешанной с равным объемом изопропанола, и затем центрифугировали. Полученный осадок растворяли в ТЕ растворе и обрабатывали РНКазой и затем смесью фенол/хлороформ (1:1) с последующим осаждением этанолом. Полученный осадок опять растворяли в ТЕ растворе, к которому затем добавляли NaCl и полиэтиленгликоль 3000 до их конечных концентраций 0,63 М и 7,5%, соответственно. После центрифугирования осадок растворяли в ТЕ растворе и осаждали этанолом. Таким путем получали приблизительно 300 мкг плазмидной ДНК рНGТ1.
С применением Taq Dye DeoxyТМ Terminater Cycle Sequening Kit (набора, изготовляемого Applied Biosystems) очищенную таким образом плазмидную ДНК вводили в секвенатор ДНК 373А (Applied Biosystems) для определения нуклеотидной последовательности кодирующего белок района, предсказанного из нуклеотидной последовательности кДНК. Определенная таким образом нуклеотидная последовательность и выведенная из нее аминокислотная последовательность показаны в списке последовательностей (SEQ 1D 8). В этом случае олигонуклеотиды, примененные в примере 22 для нуклеотидного анализа кДНК, и синтетические олигонуклеотиды, построенные на основе внутренней последовательности, полученной анализом из последовательности, использовали в качестве праймеров в определении нуклеотидной последовательности.
В результате было обнаружено, что хромосомная ДНК, несомая плазмидным клоном рНGТ1, содержит полный кодирующий район аминокислотной последовательности, выведенной из SEQ 1D 6, и нуклеотидная последовательность этого кодирующего района совпадала полностью с SEQ 1D 6. Кроме того, район, соответствующий кодирующему аминокислоты экзону, содержал 4 интрона, имеющие длины 231 п.н., 286 п.н., 1932 п.н. и 236 п.н. в этом порядке, от 5'-стороны (фиг. 11). Также эта нуклеотидная последовательность отличалась от нуклеотидной последовательности кДНК (SEQ 1D 7) при 3 положениях, тех же, которые были описаны в примере 22 (различные положения между SEQ 1D 6 и 7). Т.е. А (положение 84), А (положение 740) и G (положение 1198) в SEQ 1D 7 было соответственно С, Т и А в SEQ 1D 8. Таким образом, было обнаружено, что нуклеотидная последовательность клона рНТG1 кДНК ТРО человека, полученная в примере 21, отличается от нуклеотидной последовательности клона рНFТ1 хромосомной ДНК человека при 3 положениях. Поэтому для анализа, отражают ли эти мутации в последовательности клона pHFT1 кДНК последовательность хромосомной ДНК, определяли нуклеотидные последовательности других 4 клонов среди 5 клонов хромосомной ДНК, независимо выбранные этим скринингом. Анализ последовательностей проводили прямым способом определения нуклеотидной последовательности с применением пробы ДНК фага, полученной согласно методу лизиса в чашках, описанному в Molecular Cloning. Секвенирование каждого клона проводили при помощи секвенатора ДНК 373А (Applied Biosystems) с применением праймеров, использованных в примере 22, которые были синтезированы на основе таких частей последовательности, что можно было анализировать упомянутые выше 3 положения мутаций в нуклеотидной последовательности. Применяли Taq Dye DeoxyTM Terminater Cycle Sequening Kit (Applied Biosystems). Было обнаружено, что нуклеотидные последовательности всех 4 клонов были идентичны SEQ 1D 6. Положения 84 и 740, соответствующие SEQ 1D 7, были заменены С и Т, соответственно, в этих 4 клонах. Однако положение 1198 было G в 2 клонах и А в двух других клонах. Другими словами, было обнаружено, что имеются два типа нуклеотидных последовательностей, присущих исходной хромосомной ДНК. В настоящее время неясно, получены ли такие различия в нулеотидной последовательности из гомологичной хромосомы или из множественных генов. Кроме того, было сделано предположение, что различия в нуклеотидной последовательности могут быть обусловлены расовым различием, т.к. поли (А)+ РНК, приобретенная из Clontech, имеет Кавказское происхождение, тогда как хромосомная ДНК имеет японское происхождение.
Вектор рн Т1, несомый штаммом E.coli DH5, был депонирован авторами данного изобретения 24 марта 1994 года FERM ВР-4616 при National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology, Ministry of International Trade and Industry, Japan.
Пример 26. Экспрессия хромосомной ДНК ТРО человека в COS 1 клетках и подтверждение ТРО активности
Плазмидный клон рНGТ1, полученный субклонированием, содержал всего 4 последовательности узнавания EcoRl, 3 в инсерционной части и 1 в векторе. Анализ нуклеотидной последовательности выявил, что весь кодирующий белок ТРО человека район был включен в ДНК-фрагмент из приблизительно 4,3 т.п.н., который был вставлен между последовательностью узнавания EcoRl, ближайшей к 5'-стороне в этой инсерции и последовательностью узнавания EcoRl в векторе. Поэтому этот фрагмент лигировали с обработанным EcoRl экспрессирующим вектором pEF18S и получили 4 экспрессирующих ТРО человека плазмиды 1 - 4 (см. фиг.11). Получив эти плазмидные ДНК, проводили эксперимент по их экспрессии. Очистку плазмидной ДНК проводили в основном согласно способу, описанному в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989), получая приблизительно 250 мкг плазмидной ДНК.
Трансфекцию COS 1 клеток полученными таким образом клонами pЕFНGТЕ 1-4 проводили согласно способу примера 11. Т.е. трансфекцию проводили с 10 мкг плазмидной ДНК ДЭАЭ-декстрановым способом, предусматривающим обработку хлорохином. Трансфицированные COS 1 клетки инкубировали 3 дня при 37oС и затем собирали культуральные супернатанты.
Культуральные супернатанты экстенсивно диализовали против культуральной среды IMDM и оценивали в тест-системе с крысиными CFU-MK. ТРО активность обнаружили в зависимом от дозы виде в супернатанте COS 1 клеток, в которых экспрессировался каждый из четырех клонов (pEFHGTE 1 - 4). В противоположность этому, ТРО активность не была обнаружена в супернатанте COS 1 клеток, в которых экспрессировалась только плазмида pEF18S. Характерные результаты с pEFHGTE 1 показаны на фиг.12а. Подобные данные были получены в М-07е тест-системе. Супернатант COS 1 клеток, в которых экспрессировался каждый из клонов (pEFHGTE 1 - 4), значительно увеличивал рост клеток М-07е зависимым от дозы образом. Репрезентативные данные с pEFHGTE 1 показаны на фиг.12.
Эти результаты показали, что плазмидные клоны рEFНGТE 1 - 4 несут функциональную хромосомную ДНК ТРО человека.
Пример 27. Получение ДНК делеционного типа ТРО человека и ее экспрессия и подтверждение ТРО активности
Результаты в примере 18 показали, что ТРО человека мог бы проявлять его биологическую активность, даже после удаления его карбоксильной трети. Поэтому для дальнейшей оценки биологически активных частей проводили эксперименты с его делеционными производными. В этом примере делеционные производные ТРО, с удаленными 29, 40, 60 или 68 аминокислотами от карбоксил-терминального конца белка ТРО (аминокислоты 1-231), кодируемые pHT1-231, исследовали на их способность оказывать влияние на биологическую активность ТРО in vitro. Сaмoe короткое производное (Аминокислоты 1-163) все еще содержали аминокислотные последовательности, соответствующие ТР2/3 ТРО плазмы крысы, описанного в примере 2. Делеционные плазмиды получали PCR с использованием ДНК плазмидного клона рНТ1-231, полученного в примере 18, в качестве матрицы и синтезированных олигонуклеотидов в качестве праймеров. Последовательности праймеров, примененных для PCR, были следующими.
hTPO-5: 5'-TTT GAA TTC GGC CAG ССА GAC ACC CCG GCC-3' (SEQ 1D 56) (полученный добавлением последовательности узнавания EcoRl к последовательности положений 1-21 в SEQ 1D 4, это идентичная последовательность, описанная на фиг.9).
hTPO-S: 5'-TTT GCG GCC GCT CAT TAG CTG GGG АСА GCT GTG GTG GGT-3' (SEQ 1D 57) (антисмысловой праймер, соответствующий последовательности положений 555-576 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-163 аминокислотных остатков,
hТРО-4: 5'-ТТТ GCG GCC GCT CAT TAC AGT GTG AGG ACT AGA GAG GTT CTG-3' (SEQ 1D 58) (антисмысловой праймер, соответствующий последовательности положений 576-600 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-171 аминокислотных остатков),
hTPO-30: 5'-TTT GCG GCC GCT CAT TAT CTG GCT GAG GCA GTG AAG TTT GTC-3' (SEQ 1D 59) (антисмысловой праймер, соответствующий последовательности положений 636-660 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-191 аминокислотных остатков), и
. hTPO-2: 5'-TTT GCG GCC GCT CAT TAC AGA CCA GGA ATC TTG GCT CTG AAT-3' (SEQ 1D 60) (антисмысловой праймер, соответствующий последовательности положений 696-720 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-211 аминокислотных остатков).
С применением 1 мкг плазмидной ДНК клона pHT1-231, полученного в примере 18, в качестве матрицы, 10 мкМ каждого из синтезированных hТРО-5 (для 5'-стороны) и hТРО-2, -3, -4 и S (для 3'-стороны) в качестве праймеров и набора реагентов для PCR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (Takara Shuzo) проводили РСR в объеме 100 мкл при помощи РСR системы 9600 GeneAmpТМ (PERKIN-ELMER) с повторением в целом 20 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, гибридизации при 65oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут. Целевую полосу, полученную каждой РСR, расщепляли рестриктазами EcoRl и Notl и полученный продукт расщепления подвергали электрофорезу в 1% агарозном геле (FМС BioProducts) для выделения основного фрагмента ДНК, имеющего ожидаемый размер, амплифицированного каждой РСR. Выделенный таким образом фрагмент ДНК очищали при помощи набора для очистки ДНК Prep-A-Gene (Bio-Rad) и затем субклонировали в экспрессирующий вектор pEF18S, предварительно расщепленный теми же самыми рестриктазами. В этом случае Высококомпетентный штамм E. coli DH5, изготовляемый ТОУОВО, применяли в качестве штамма-хозяина. Из полученных трансформантов, каждый из которых происходил из своей PCR, были отобраны 4-5 клонов, содержащих инсерцию ожидаемого размера, для получения проб плазмидной ДНК, в основном по способу, описанному в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989). В ряде таких операций были получены пробы плазмидных ДНК из делеционного производного, кодирующего положения 1-163 аминокислотных остатков (pHT1-163 1-5), делеционного производного, кодирующего положения 1-171 аминокислотных остатков (pHT1-171 1-4), делеционного производного, кодирующего положения 1-191 аминокислотных остатков (pHT1-191 1-4) и делеционного производного, кодирующего положения 1-211 аминокислотных остатков (pHT1-211 1-4). Полную длину амплифицированных районов каждой плазмидной ДНК подвергали анализу нуклеотидной последовательности для нахождения ее полной идентичности с нуклеотидной последовательностью, показанной в SEQ 1D 4.
Трансфекцию в COS 1 клетки полученных таким образом клонов проводили согласно способу примера 11. Т.е. трансфекцию проводили с 10 мкг каждой из проб плазмидных ДНК по ДЭАЭ-декстрановому способу, предусматривающему обработку хлорохином. Трансфицированные COS 1 клетки культивировали в течение 3 дней и затем собирали культуральные супернатанты.
Эти культуральные супернатанты экстенсивно диализовали против культуральной среды IМDM и оценивали в тест/системе с крысиными CFU-MK. ТРО активность была обнаружена (зависимая от дозы) в супернатанте COS 1 клеток, в которых экспрессировалась каждая из pHT1-211, pHT1-191, pHT1-171 или pHT1-163. Репрезентативные данные с pHT1-211 1, рНТ1-191 1 и pHT1-171 2 показаны на фиг. 13а и с pHT1-163 2 на фиг.13. Подобные результаты были получены в М-07е-тест-системе. Супернатант COS 1 клеток, в которых экспрессировалась каждая из pHT1-211, pHT1-191, pHT1-171 или pHT1-163, значительно усиливал пролиферацию М-07е клеток. Репрезентативные данные с pET1-211 1, pET1-191 1, pHT1-171 2 или pHT1-163 2 показаны на фиг.14.
Эти результаты показали, что ТРО человека все еще сохраняет его биологическую активность in vitro даже после делеции его карбоксил-терминальной половины за Ser (положение 163), и дали сильное основание для предположения, что биологически активные части ТРО человека локализованы внутри амино-концевой половины, заканчивающейся при Ser (положение 163).
Пример 28. Получение ТРО человека с делецией на С-конце и его экспрессия и активность в COS 1 клетках
Для анализа района, необходимого для того, чтобы белок ТРО человека проявлял его активность, С-концевые аминокислоты делетировали из делеционных производных, полученных в примере 27, и испытывали ТРО активность, экспрессированную полученными производными, в COS 1 клетках. С использованием плазмидного клона pEF18S-HL34, полученного в примере 16, получали экспрессирующие плазмиды путем делеции нуклеотидных последовательностей, кодирующих С-концевые аминокислотные остатки, начиная с серина в положении 163. Конструирование этих делеционных плазмид выполняли с применением PCR. При этом использовали следующие праймеры:
. hTPO-5: 5'-TTT GAA TTC GGC CAG ССА GAC ACC CCG GCC-3' (SEQ 1D 61) (полученный добавлением последовательности узнавания EcoRl к последовательности положений 1-21 в SEQ 1D 4, та же самая последовательность показана в примере 18),
. hTPO-150: 5'-TTT GCG GCC GCT CAT TAG AGG GTG GAC ССT CCT АСА AGC AT-3' (SEQ 1D 62) (антисмысловой праймер, соответствующий последовательности положений 514-537 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl для использования в получении делеционного мутанта, кодирующего положения 1-150 аминокислотных остатков),
. hTPO-151: 5'-TTT GCG GCC GCT CAT TAG CAG AGG GTG GAC CCT CCT АСА A-3' (SEQ 1D 63) (антисмысловой праймер, соответствующий последовательности положений 518-540 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-151 аминокислотных остатков),
.hТРО-153: 5'-TTT GCG GCC GCT CAT TAC CTG ACG CAG AGG GTG GAC СС-3' (SEQ 1D 64) (антисмысловой праймер, соответствующий последовательности положений 526-546 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-153 аминокислотных остатков),
.hTPO-154: 5'-TTT GCG GCC GCT CAT TAC CGC CTG ACG CAG AGG GTG GA-3' (SEQ 1D 65) (антисмысловой праймер, соответствующий последовательности положений 529-549 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-154 аминокислотных остатков),
.hТРО-155: 5'-TTT GCG GCC GCT CAT TAG GCC CGC CTG ACG CAG AGG GT-3' (SEQ 1D 66) (антисмысловой праймер, соответствующий последовательности положений 532-552 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-155 аминокислотных остатков),
.hTPO-156: 5'-TTT GCG GCC GCT CAT TAT GGG GCC CGC CTG ACG CAG AG-3' (SEQ 1D 67) (антисмысловой праймер, соответствующий последовательности положений 535-555 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl, для применения в получении делеционного производного, кодирующего положения 1-156 аминокислотных остатков), и
.hTPO-157: 5'-TTT GCG GCC GCT CAT TAG GGT GGG GCC CGC CTG ACG CA-3' (SEQ 1D 68) (антисмысловой праймер, соответствующий последовательности положений 538-558 в SEQ 1D 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl для применения в получении делеционного производного, кодирующего положения 1-157 аминокислотных остатков).
PCR проводили с применением 1 мкг плазмидной ДНК клона pEF18S-HL34, полученного в примере 16, в качестве матрицы и 10 мкМ каждого из синтезированных таким образом олигонуклеотидов (hTPO-5 для 5'-стороны и hTPO-150' -151, -153, -154, -155, -156, -157 для 3'-стороны) в качестве праймеров. С применением набора реагентов для PCR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (изготовляемой Takara Shuzo) проводили PCR в 100 мкл объема, с использованием системы PCR 9600 GeneAmpТМ (PERKIN-ELMER) и повторением в целом 20 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, гибридизации при 66oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут. Целевую полосу, полученную в каждой PCR, расщепляли рестриктазами EcoRl и Notl и полученный продукт расщепления подвергали электрофорезу в 1% агарозном геле (FMC BioProducts) для выделения основного фрагмента ДНК, имеющего ожидаемый размер, для каждой PCR. Выделенный таким образом фрагмент ДНК очищали с применением набора для очистки ДНК Рrep-A-Gene (Bio-Rad) и затем субклонировали в экспрессирующий вектор pEF18S, обработанный предварительно теми же рестриктазами. В этом случае Высококомпетентный штамм E-coli DH5 (ТОУОВО) применяли в качестве штамма-хозяина. Из полученных трансформантов, происходящих из каждой РСR, отбирали 3-5 клонов, содержащих инсерцию ожидаемого размера, для получения проб плазмидной ДНК по существу в соответствии со способом, описанным в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989). Путем ряда таких операций получили пробы плазмидной ДНК из делеционного производного, кодирующего положения 1-150 аминокислотных остатков (pHT1-150 21, 22 и 25), делеционное производное, кодирующее положения 1-151 аминокислотных остатков (pHT1-151 16, 17 и 18), делеционное производное, кодирующее положения 1-153 аминокислотных остатков (рНТ1-153 1-5), делеционное производное, кодирующее положения 1-155 аминокислотных остатков (pHT1-155 1-5), делеционное производное, кодирующее положения 1-156 аминокислотных остатков (pHT1-156 1-5), и делеционное производное, кодирующее положения 1-157 аминокислотных остатков (pHT1-157 1-5).
Из этих очищенных проб плазмидных ДНК последовательность pНT1-150 21, 22 и 25 и pHT1-151 16, 17 и 18 проверяли при помощи секвенатора ДНК 373А (Applied Biosystems) с использованием Taq Dye DeoxyТМ Terminater Cycle Sequening Kit (Applied Biosystems), подтверждая, что ожидаемые последовательности кДНК ТРО не имели замен во всех нуклеотидных последовательностях.
Трансфекцию COS 1 клеток каждым из полученных таким образом клонов проводили в соответствии со способом примера 11. Т.е. трансфекцию проводили с 10 мкг каждой из проб плазмидных ДНК ДЭАЭ-декстрановым способом, предусматривающим обработку хлорохином, и культуральный супернатант извлекали после 3 дней культивирования. Полученные культуральные супернатанты диализовали против культуральной среды IМDМ, описанной ранее, и оценивали при помощи тест-системы с М-07е. ТРО активности обнаруживали в культуральном супернатанте COS 1 клеток, трансфицированных соответствующими клонами, кодирующими С-концевые делеционные производные, состоящие из положений аминокислот 1-151, положений аминокислот 1-153, положений аминокислот 1-154, положений аминокислот 1-155, положений аминокислот 1-156 или положений аминокислот 1-157. Все эти производные содержат остаток цистеина при положении 151. Однако ТРО активности не были обнаружены в культуральном супернатанте COS 1 клеток, трансфицированных клоном, кодирующим делеционное производное С-концевой стороны, состоящее из положений аминокислот 1-150, в котором остаток цистеина при положении 151 был также делетирован.
Пример 29. Получение ТРО человека с делецией на N-конце и его экспрессия и активность в COS 1 клетках
Для анализа района, необходимого для того, чтобы белок ТРО проявлял его биологическую активность, делеционные производные с делецией аминокислот на N-конце, полученные в примере 28, делетировали далее и активность ТРО в полученных делеционных производных определяли. С применением плазмидного клона рEF18S-HL34, полученного в примере 16, и плазмидного клона pHT1-163, полученного в примере 27, экспрессирующие плазмиды получали делецией нуклеотидных последовательностей, кодирующих N-концевые аминокислотные остатки, идущие после сигнальной последовательности. Конструирование этих делеционных плазмид выполняли с применением PCR. Последовательности праймеров, изготовленных для применения в PCR, являются следующими:
hТРО-5: 5'-ТТТ GAA TTC GGC CAG ССА GAC ACC CCG GCC-3' (SEQ ID 69) (полученный добавлением последовательности узнавания EcoRl к последовательности положений 1-21 в SEQ ID 4, та же самая последовательность показана в примере 18),
hTPO-3: 5'-TTT GCG GCC GCT CAT TAT TCG TGT ATC CTG TTC AGG TFT CC-3' (SEQ ID 70) (антисмысловой праймер, соответствующий последовательности положений 757-780 в SEQ ID 4, полученный добавлением двух терминирующих кодонов ТАА и ТGА и последовательности узнавания Notl; та же самая последовательность синтезирована для применения в получении делеционного производного, кодирующего положения 1-231 аминокислотных остатков),
. hTPO-S: 5'-ТТТ GCG GCC GCT CAT TAG CTG GGG AGA GCT GTG GTG GGT-3' (SEQ ID 71) (антисмысловой праймер, соответствующий последовательности положений 555-576 в SEQ ID 4 полученный для применения в получении делеционного производного, кодирующего положения 1-163 аминокислотных остатков),
.hTPO-13: 5'-AGT AAA CTG CTT CGT GAC TCC CAT GTC СТТ САС AGC AGA CTG AGC CAG TG-3' (SEQ ID 72) (положения 124-173 в SEQ ID 4, полученный для применения в получении производного, в котором делетированы аминокислотные остатки положений 1-12),
. hTPO-13R: 5'-CAT GGG AGT CAC GAA GCA GTT TAC TGG АСА GCG TTA GCC TTG CAG TTA G-3' (SEQ ID 73) (антисмысловой праймер, соответствующий последовательности положений 64-87 и 124-148 в SEQ ID 4, полученный для применения в получении производного, в котором делетированы аминокислотные остатки положений 1-12),
hTPO-7: 5'-TGT GAC CTC CGA GTC CTC AGT AAA CTG CTT CGT GAC TCC CAT GTC CTT C-3' (SEQ ID 74) (положения 106-154 в SEQ IG 4, полученный для применения в получении производного, в котором делетированы аминокислотные остатки положений 1-6),
.hTPO-7R: 5'-TTT ACT GAG GAC TCG GAG GTC АСА GGA CAG CGT TAG CCT TGC AGT TAG -3' (SEQ ID 75) (антисмысловой праймер, соответствующий последовательности положений 64-87 и 106-129 в SEQ ID 4, приготовленный для применения в получении производного, в котором делетированы аминокислотные остатки положений 1-6),
. hTPO-8: 5'-GAC CTC CGA GTC CTC AGT AAA CTG CTT CGT GAC TCC CAT GTC CTT CAC A-3' (SEQ ID 76) (положения 109-157 в SEQ ID 4, полученный для применения в получении производного, в котором делетированы аминокислотные остатки положений 1-7),
.hTPO-8R: 5'-CAG TTT ACT GAG GAC TCG GAG GTC GGA CAG CGT TAG CCT TGC AGT TAG-3' (SEQ ID 77) (антисмысловой праймер, соответствующий последовательности положений 64-87 и 109-132 в SEQ ID 4, полученный для применения в получении производного, в котором делетированы аминокислотные остатки положений 1-7).
(1) Получение производного (рНТ13-231), в котором делетированы аминокислотные остатки положений 1-12
РСR проводили с применением 1,4 мкг плазмидной ДНК клона pEF18S-HL34, полученного в примере 18, в качестве матрицы и 5 мкМ каждого из синтезированных таким образом олигонуклеотидов (hTPO-13 и hТРО-3 в одной комбинации и hTPO-5 и hTPO-13 в другой комбинации) в качестве праймеров. С применением набора реагентов для PCR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (Takara Shuzo) проводили PCR в 100 мкл объема с использованием системы PCR 9600 GeneAmpТМ (PERKIN-ELMER) и, после 5 минут денатурации при 95oС, повторяли в целом 30 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, гибридизации при 65oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующей конечной инкубацией при 72oС в течение 7 минут. Каждый из продуктов PCR подвергали электрофорезу в 1,2% агарозном геле (изготовляемом FMC BioProducts) для выделения основного продукта ДНК, имеющего ожидаемый размер, который затем очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad) и растворяли в 15 мкл ТЕ буфера. После этого проводили вторую PCR с применением 1 мкл каждого из приготовленных таким образом растворов в качестве матрицы.
Вторую PCR проводили с применением 5 мкМ каждого из синтезированных праймеров (hТРО-5 и hTPO-3). С применением набора реагентов для PCR, GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (Takara Shuzo) PCR проводили в 100 мкл объема с применением PCR системы 9600 Gene-AmpТМ (PERKIN-ELMER), и после 5 минут денатурации при 95oС, повторяли всего 30 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, гибридизации при 60oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 1 минуты. Каждый из продуктов РСR подвергали электрофорезу в 1,2% агарозном геле (FMC BioProducts) для выделения основного фрагмента ДНК, имеющего ожидаемый размер, который затем очищали при помощи набора для очистки ДНК Prep-A-Gene (Bio-Rad) и растворяли в 15 мкл ТЕ буфера. После расщепления рестриктазами EcoRl и Notl полученный раствор подвергали экстракции таким же объемом смеси фенол/хлороформ с последующим осаждением этанолом. После центрифугирования полученный осадок растворяли в 15 мкл ТЕ буфера и затем субклонировали в экспрессирующий вектор pEF18S, обработанный предварительно теми же рестриктазами. Высококомпетентную E.coli DH5 (ТОУОВО) применяли в качестве штамма-хозяина. Из полученных трансформантов отбирали 45 клонов, содержащих инсерцию ожидаемого размера для получения проб плазмидных ДНК в основном в соответствии со способом, описанным в Molecular Cloning (Sambrook et al. Cold Spring Harbor Laboratory Press, 1989). Таким путем можно было получить плазмидную ДНК делеционного производного (рНТ13-231), кодирующую белковую молекулу, в которой аминокислотные остатки положений 1-12 были делетированы из исходной аминокислотной последовательности 1-231. При проведении РСR этих 45 клонов с применением праймеров hTPO-5 и hTPO-3 инсерции, имеющие приблизительно ожидаемые размеры, были подтверждены в 8 клонах. Из них последовательность 3 клонов была проверена в ДНК-секвенаторе 373А (Applied Biosystems) с использованием Taq Dye DeoxyТМ Terminater Cycle Sequening Kit (Applied Biosystems) с получением в результате клона рНТ13-231, имеющего последовательность кДНК, которую хотели сконструировать, и делецию, которая ожидалась, без замен и иных изменений всей нуклеотидной последовательности.
(2) Получение производного (рНТ7-163), в котором делетированы аминокислотные остатки положений 1-6
В этом случае аминокислоту положения 163 использовали в качестве С-конца для получения делеционных производных, хотя в предшествующей стадии использовали аминокислоту положения 231 в качестве С-конца белка ТРО для получения делеционного производного. Было обнаружено, что ТРО активность могла бы экспрессироваться, даже при дальнейшем делетировании С-концевых аминокислот. По этой причине положение аминокислоты 163 использовали как С-конец в следующей стадии (3). PCR проводили с применением 1,4 мкг плазмидной ДНК клона pHT1-163, полученного в примере 27, в качестве матрицы и 5 мкМ каждого из синтезированных олигонуклеотидов (hTPO-7 и hTPO-S) в одной комбинации и hTPO-5 и hTPO-7R в другой комбинации) в качестве праймеров. С применением набора реагентов для PCR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразы (изготовляемой Takara Shuzo) PCR проводили в 100 мкл объема с использованием РСR системы 9600 GeneAmpТМ (PERKIN-ELMER) и, после 5 минут денатурации при 95oС, повторяли в целом 30 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, ренатурации при 65oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с конечным инкубированием при 72oС в течение 7 минут. Каждый из продуктов PCR подвергали электрофорезу в 1,2% агарозном геле (изготовляемом FMC BioProducts) для выделения основного фрагмента ДНК, имеющего ожидаемый размер, который затем очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad) и растворяли в 15 мкл ТЕ буфера. После этого проводили вторую PCR с применением 1 мкл каждого из полученных таким образом растворов в качестве матрицы.
Вторую РСR проводили с применением 5 мкМ каждого из синтезированных праймеров (hTPO-5 и hTPO-S). С применением набора реагентов для PCR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (Takara Shuzo) реакции PCR проводили в 100 мкл объема с использованием PCR системы 9600 GeneAmpТМ (PERKIN-ELMER) и, после 5 минут денатурации при 95oС, повторяли в целом 30 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, ренатурации при 60oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут. Каждый из продуктов PCR подвергали электрофорезу в 1,2% агарозном геле (изготовляемом FMC BioRpoducts) для выделения основного фрагмента ДНК, имеющего ожидаемый размер, который затем очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad) и растворяли в 15 мкл ТЕ буфера. После расщепления рестриктазами EcoRl и Notl полученный раствор подвергали экстракции таким же объемом смеси фенол/хлороформ с последующим осаждением этанолом. После центрифугирования полученный осадок растворяли в 15 мкл ТЕ буфера и затем субклонировали в экспрессирующий вектор pEF18S, который был предварительно обработан теми же рестриктазами. В этом случае Высококомпетентный штамм E.coli DH5 (ТОУОВО) использовали в качестве штамма-хозяина. Из полученных трансформантов отбирали 30 клонов, содержащих инсерцию ожидаемого размера, для получения проб плазмидной ДНК в основном согласно способу, описанному в Molecular Cloning (Sambrook et al., Cold Sprining Harbor Laboratory Press, 1989). Таким путем можно было получить плазмидную ДНК делеционного производного (рНТ7-163), кодирующую молекулу белка, в котором аминокислотные остатки положений 1-6 были делетированы из исходной последовательности аминокислотных остатков 1-163 в SEQ ID 4. При проведении с этими клонами PCR с применением праймеров hTPO-5 и hTPO-S инсерции, имеющие приблизительно ожидаемые размеры, были подтверждены во всех клонах. Из них последовательности 3 клонов проверяли при помощи ДНК-секвенатора 373А (Applied Biosystems) с применением Taq Dey DeoxyТМ Terminater Cycle Sequening Kit (Applied Biosystems) с получением 2 клонов, рНТ7-163 4 и 29, каждый из которых имеет кДНК последовательность ТРО, которая была сконструирована, и делецию, которая ожидалась, без замен и каких-либо других изменений во всей нуклеотидной последовательности.
(3) Получение производного (рНТ8-163), в котором делетированы аминокислотные остатки положений 1-7
Производное (рНТ8-163) с делецией аминокислотных остатков положений 1-7 готовили в основном так же, как в приведенном выше случае получения производного (рНТ7-163) с делецией аминокислотных остатков положений 1-6. PCR проводили с применением 1,4 мкг плазмидной ДНК клона pHT1-163, полученного в примере 27, в качестве матрицы и 5 мкM каждого из синтезированных олигонуклеотидов (hTPO-8 и hTPO-S в одной комбинации и hТРО-5 и hTPO-8R в другой комбинации) в качестве праймеров. С применением набора реагентов для PCR GeneAmpТМ с AmpliTaqТМ ДНК-полимеразой (Takara Shuzo) реакцию PCR проводили в 100 мкл объема с применением РСR системы 9600 GeneAmpТМ (PERKIN-ELMER) и, после 5 минут денатурации при 95oС, повторяли в целом 30 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, ренатурации при 65oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием при 72oС в течение 7 минут. Каждый из продуктов PCR подвергали электрофорезу в 1,2% агарозном геле (FMC BioProducts) для выделения основного фрагмента ДНК, имеющего ожидаемый размер, который затем очищали с применением набора для очистки ДНК Prep-A-Gеne (Bio-Rad) и растворяли в 15 мкл ТЕ буфера. После этого проводили вторую PCR с применением 1 мкл каждого из полученных растворов в качестве матрицы.
Вторую РСR проводили с применением 5 мкМ каждого из синтезированных праймеров (hTPO-5 и hTPO-S). С применением набора реагентов для PCR GeneAmpТМ с AnipliTaqТМ ДНК-полимеразой (изготовляемой Takara Shuzo) проводили PCR в 100 мкл объема с использованием PCR системы 9600 GeneAmpТМ (PERKING-ELMER) и, после 5 минут денатурации при 95oС, повторяли в целом 30 циклов, каждый из которых состоял из денатурации при 95oС в течение 1 минуты, ренатурации при 60oС в течение 1 минуты и синтеза при 72oС в течение 1 минуты, с последующим конечным инкубированием в течение 7 минут. Каждый из продуктов PCR подвергали электрофорезу в 1,2% агарозном геле (FMC Bio-Products) для выделения основного фрагмента ДНК, имеющего ожидаемый размер, который затем очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad) и растворяли в 15 мкл ТЕ буфера. После расщепления рестриктазами EcoRl и Notl полученный раствор подвергали экстракции таким же объемом смеси фенол/хлороформ и последующему осаждению этанолом. После центрифугирования полученный осадок растворяли в 15 мкл ТЕ буфера и затем субклонировали в экспрессирующий вектор pEF18S, который был предварительно расщеплен теми же рестриктазами. В этом случае Высококомпетентный штамм E.coli DH5 (TOYOBO)
использовали в качестве штамма-хозяина. Из полученных трансформантов отбирали 30 клонов, содержащих инсерцию ожидаемого размера для получения проб плазмидных ДНК в основном согласно способу, описанному в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press, 1989). Таким путем можно было получить плазмидную ДНК делеционного производного (рНТ8-163), кодирующую молекулу белка, в котором аминокислотные остатки положений 1-7 были делетированы из исходных аминокислотных остатков положений 1-163 в SEQ ID No 4. При проведении с этими клонами PCR с применением праймеров hTPO-5 и hTPO-S инсерции, имеющие приблизительно ожидаемые размеры, были подтверждены во всех клонах. Из них последовательность 3 клонов была проверена при помощи ДНК-секвенатора 373А (Applied Biosystems) с применением Taq Dye DeoxyТМ Terminater Cycle Sequening Kit (Applied Biosystems) с получением 2 клонов, рНТ8-163 33 и 48, каждый из которых имеет последовательность кДНК ТРО, которая была сконструирована, и делецию, которая ожидалась, без замен или подобных изменений во всей нуклеотидной последовательности.
(4) Экспрессия делеционного производного в COS 1 клетках и подтверждение ТРО активности
Трансфекцию COS 1 клеток каждым из полученных таким образом делеционных клонов проводили согласно способу примера 11. Т.е. трансфекцию проводили с применением 10 мкг каждой из плазмидных проб ДНК ДЭАЭ-декстрановым способом, предусматривающим обработку хлорохином, и культуральные супернатанты извлекали после 3 дней культивирования. Полученный таким образом культуральный супернатант основательно диализовали против культуральной среды IMDM, описанной выше, и оценивали при помощи тест-системы в М-07е.
В результате слабую, но значимую, ТРО активность обнаружили в культуральном супернатанте COS 1 клеток, трансфицированных клоном, кодирующим делеционное производное, состоящее из аминокислот положений 7-163. Однако ТРО активность не была обнаружена в культуральном супернатанте COS 1 клеток, трансфицированных клоном, кодирующим делеционное производное, состоящее из аминокислот положений 8-163 или аминокислот положений 13-231.
Пример 30. Получение плазмиды кДНК (pHTP1) ТРО полной длины человека
Экспрессирующий вектор, имеющий все кодирующие аминокислоты районы кДНК ТРО человека, предполагаемые в SEQ ID 6, был сконструирован для экспрессии в клетках млекопитающих.
РСR проводили следующим образом для получения фрагмента ДНК, охватывающего все кодирующие районы кДНК ТРО человека.
Нуклеотидные последовательности примененных праймеров были следующими:
. hTPO-1: 5' TTG TGA CCT CCG CGT CCT CAG-3' (SEQ ID No 78) (положения 105-125 в SEQ ID No 6);
.SA: 5'-CAG GTA TCC GGG GAT TTG GTC GTC-3' (SEQ ID No 79) (антисмысловой праймер, соответствующий последовательности положений 745-765 в SEQ ID No 6);
. hTPO-P: 5'-TGC GTT TCC TGA TGC TTG TAG-3' (SEQ ID No 80) (положения 503-523 в SEQ ID 6);
. hTPO-KO: 5'-GAG AGA GCG GCC GCT TAC CCT TCC TGA GAC AGA TT-3' (SEQ ID No 81) (последовательность, полученная добавлением последовательности узнавания Notl и последовательности GAGAGA (SEQ ID No 82) к антисмысловой последовательности, соответствующей последовательности положений 1066-1086 в SEQ ID No 6).
Первую PCR проводили с применением 300 нг клона pEF18S-HL34, полученного в примере 16, в качестве матрицы. С применением 0,5 мкМ каждого из праймеров hTPO-1 к SA I E Vent RТМ ДНК-полимеразы (изготовляемой New England Biolabs) проводили РСR (повторение в целом 30 циклов, каждый из которых состоял из инкубации при 96oС в течение 1 минуты, инкубации при 62oС в течение 1 минуты и инкубации при 72oС в течение 1 минуты, с последующей конечной инкубацией при 72oС в течение 7 минут). Состав реакционного раствора в конечных концентрациях был следующим: 10 мМ КС1, 10 мМ (NH4)2SO4, 20 мМ Трис-HC1 (рН 8,8), 2 мМ MgSО4, 0,1% Тритон Х-100 и 200 мкМ смесь dNTP.
1 мкг коммерчески доступного препарата поли (А)+ РНК, полученной из нормальной печени человека (производимой Clontech), нагревали при 70oС в течение 10 минут, быстро охлаждали на льду и затем смешивали с 10 мМ ДТТ, 500 мкМ смеси dNTP, 25 нг произвольного праймера (производимого Boehringer-Mannheim) и 200 единиц SuperscriptТМ 11 РНКазы К- (изготовляемой LIFE TECHNOLOGIES) с последующим инкубированием в течение 1 часа при 37oС для проведения синтеза кДНК. После этого проводили вторую PCR (повторение в целом 30 циклов, каждый цикл состоял из инкубаций при 96oС 1 мин, при 58oС 1 мин и при 72oС 1 мин) с применением 1/20 объема реакционного раствора синтезированной кДНК в качестве матрицы и 2,5 мкМ каждого из праймеров hTPO-P и hTPO-KO и 2,5 единицы AmpliTaqТМ ДНК-полимеразы (производимой Takara Shuzo).
Каждый из полученных растворов первой и второй РСR подвергали электрофорезу в 1% агарозном геле для выделения соответствующих основных фрагментов ДНК ожидаемого размера, которые затем очищали с применением набора для очистки ДНК Prep-A-Gene (изготовляемого Bio-Rad). После этого проводили третью PCR с применением 1/20 объема каждого из полученных таким образом растворов в качестве матрицы. Эту реакцию (нагревание при 96oС 2 мин, с последующим повторением всего 3 циклов, каждый из которых состоял из инкубаций при 96oС 2 мин и при 72oС 2 мин, и последующим инкубированием при 72oС в течение 7 минут) с применением 1 единицы Vent RТМ ДНК-полимеразы (изготовляемой New England Biolabs). Полученный реакционный раствор смешивали с 1 мкМ каждого из праймеров hTPO-1 и TPO-KO, нагревали при 96oС 2 минуты, подвергали 25 циклам PCR, каждый из которых состоял из инкубации при 96oС 1 мин, при 62oС 1 мин и при 72oС 1 мин, с последующим инкубированием при 72oС в течение 7 минут. Полученный реакционный раствор экстрагировали тем же объемом насыщенной водой смеси фенол/хлороформ с последующим осаждением этанолом (2,5 объема этанола в присутствии 0,3 М ацетата натрия и 0,5 мкл гликогена, производимого Boehringer-Mannheim, для извлечения ДНК. Полученную таким образом ДНК расщепляли рестриктазами BamH1 и Notl и подвергали электрофорезу в 1% агарозном геле и основную полосу, имеющую ожидаемый размер, выделенную таким образом, очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad), лигировали с вектором pBLUESCRIPT 11 SK+ (Stratagene), который был предварительно расщеплен рестриктазами BamH1 и Notl, и затем трансформировали в Высококомпетентный штамм E. coli DH5 (TOYOBO). Из полученных колоний отбирали 4 клона для получения проб плазмидных ДНК. Последовательность очищенных проб плазмидных ДНК проверяли при помощи ДНК-секвенатора 373А, производимого Applied Biosystems с применением Taq Dye DeoxyТМ Terminater Cycle Sequening Kit (Applied Biosystems), получая в результате клон, pBLTP, имеющий последовательность кДНК ТРО, сконструированную без замены в нуклеотидной последовательности внутри района между BamH1 и Notl.
Этот клон pBLTP расщепляли рестриктазами EcoR1 и BamH1 и подвергали электрофорезу в 1% агарозном геле и полосу с высоким молекулярным весом, выделенную таким образом, очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad). Таким же образом PEF18S-HL34 обрабатывали рестриктазами для очистки фрагмента ДНК приблизительно 450 п.н. Каждую из полученных таким образом проб ДНК подвергали лигированию и трансформировали в Компетентный штамм E.coli DH5 (ТОУОВО). Пробы плазмидной ДНК получали из образующихся колоний для получения клона, pBLTEN, содержащего инсерцию кДНК ТРО человека. Полученный таким образом клон pBLTEN расщепляли рестриктазами EcoR1 и Notl и подвергали электрофорезу в 1% агарозном геле, и фрагмент ДНК приблизительно 1200 п.н., выделенный таким образом, очищали с применением набора для очистки ДНК Prep-A-Gene (Bio-Rad), лигировали с экспрессирующим вектором pEF18S, который был предварительно расщеплен теми же рестриктазами, и затем трансформировали в Высококомпетентный штамм Е.соli DH5 (ТОУОВО). Пробы плазмидной ДНК получали из полученных колоний, получали клон, РНТ1, содержащий кодирующий район кДНК всего ТРО человека. Плазмидную ДНК получали из этого клона в большом количестве и использовали в дальнейших экспериментах. Получение плазмидной ДНК проводили в основном в соответствии со способом, описанным в Molecular Cloning (Sambroоk et al., Cold Spring Harbor Laboratory Press, 1989).
Пример 31. Конструирование экспрессирующей плазмиды млекопитающих, pDEF202-hTPO-P1, для экспрессии ТРО человека в клетках яичника китайского хомяка (СНО)
1 мкг плазмиды рМG1, содержащей миниген мышиной дигидрофолятредуктазы (DHFR), расщепляли рестриктазами EcoR1 и BamH1 и подвергали электрофорезу в агарозном геле для выделения фрагмента (приблизительно 2,5 т.п.н.), содержащего миниген мышиной DHFR. Выделенный фрагмент ДНК растворяли в 25 мл реакционного раствора, состоящего из 50 мМ Трис-НС1 (рН 7,5), 7 мМ MgCl2, 1 мМ 2-меркаптоэтанол и 0,2 мМ смесь dNTP, смешивали с 2 единицами фрагмента Кленова и затем инкубировали при комнатной температуре в течение 30 минут для образования тупых концов на обоих концах этого фрагмента ДНК. После обработки смесью фенол/хлороформ и осаждения этанолом полученный фрагмент растворяли в 10 мл ТЕ раствора (10 мМ Трис-НС1 (рН 8,0)/1 мМ ЭДТА). Экспрессирующий вектор млекопитающих pEF18S расщепляли рестриктазой Sma1, дефосфорилировали щелочной фосфатазой (изготовляемой Takara Shuzo), и затем лигировали с фрагментом ДНК, содержащим миниген мышиной DHFR, с применением Т4 ДНК-лигазы (Takara Shuzo), получая экспрессирующий вектор рDЕF202.
Затем сконструированный экспрессирующий вектор pDEF202 расщепляли рестриктазами EcoR1 и Spel и затем подвергали электрофорезу в агарозном геле для выделения большого фрагмента ДНК. кДНК ТРO человека, которая была получена расщеплением плазмиды рНР1, содержащей кДНК ТРО человека (клон р1), рестриктазами EcoR1 и Spel, лигировали с этим линеаризованным вектором pDEF202 при помощи Т4 ДНК-лигазы (Takara Shuzo), получая плазмиду рЕF202-hТРО-Р1 для экспрессии кДНК ТРО человека. Эта сконструированная плазмида содержит начало репликации SV40, 1-α- промотор фактора элонгации человека и ранний сигнал полиаденилирования SV40 для транскрипции кДНК ТРО человека, минигена мышиной DHFR и начало репликации pUC18 и ген β-лактамазы (Ampr).
Пример 32. Экспрессия ТРО человека в СНО клетках
СНО клетки (DHFR - штамм, Urlaub and Chasin, Proc. Natl. Acad. Sci. USA, vol.77, p.4216, 1980) поддерживали в α-минимальной основной среде (α-МЕМ(-), с добавлением гипоксантина и тимидина), содержащей фетальную сыворотку теленка (FCS), с применением чашки для культуры ткани 6 см в диаметре (Falcon). Трансформацию СНО клеток плазмидной pDEF202-hTPO-P1 проводили кальций-фосфатным способом (CellPhect, Pharmacia), как описано ниже. 10 мкг плазмиды pDEF202-hTPO-P1, полученной в примере 31, смешивали с 120 мл буфера А и 120 мл Н2О и полученную смесь инкубировали при комнатной температуре в течение 10 минут. Этот раствор затем смешивали с 120 мл буфера В и давали ему стоять при комнатной температуре еще 30 минут. Этот раствор смеси ДНК затем добавляли в культуру и инкубировали в течение б часов в инкубаторе с СО2. После 6 часов культуру промывали дважды бессывороточной α-МЕМ(-), обрабатывали α-МЕМ(-), содержащей 10% диметилсульфоксид, в течение 2 минут и затем инкубировали в течение 2 дней в неселективной среде (α-MEM (-) с добавлением гипоксантина и тимидина), содержащей 10% диализованную FСS. После 2 дней культивирования клетки отбирали в избирательной среде (α-МЕМ(-)), содержащей 10% диализованную FCS. Для отбора трансформированных клеток с DHFR - положительным фенотипом эти клетки обрабатывали раствором типсин/ЭДТА и ресуспендировали в избирательной среде. Клетки в чашке для культуры ткани (6 см) разделяли на 5 чашек (10 см) или на 12-24-луночных культуральных планшетов и затем культивировали в этой избирательной среде. Культуральную среду заменяли при интервалах 2 дня. Активность ТРО человека в культуральной среде СНО клеток с DHFR -положительным фенотипом измеряли CFU-MK - тестом, М-07е-тестом или Ва/Р3 тестом. Клетки, секретирующие ТРО человека в культуральную среду, обрабатывали 25 нМ метотрексатом для выделения клеточного клона, продуцирующего высокое количество ТРО человека.
В этом случае трансфекцию СНО клеток можно выполнять также проведением контрансфекции СКО клеток плазмидами рНТР1 и рМG1.
СНО штамм (СНО- DUKXB11), трансфицированный плазмидой pDEF202-hTPO-P1, был депонирован автором данного изобретения 31 января 1995 г., под FERM BP-4988 National Institute of Bioscience and Human Technology, Agency of Industrial Science and Technology, Ministry of International Trade and Industry, Japan.
Пример 33. Конструирование рекомбинантного вектора, BMCGSneo-hTPO-P1, для применения в Х 63.6.5.3. клетках
Один микрограмм экспрессирующего вектора млекопитающих pBMCGSneo расщепляли рестриктазами Xhol и Notl и подвергали электрофорезу в агарозном геле для выделения части векторной ДНК. Этот фрагмент ДНК и кДНК ТРО человека (Р1 клон), который был получен расщеплением плазмиды pBLTEN, содержащей кДНК ТРО человека, рестриктазами Xhol и Notl лигировали при помощи Т4 ДНК-лигазы, получая целевой экспрессирующий вектор, pBMCGSneo-hTPO-P1. Эта экспрессирующая плазмида содержит ранний промотор цитомегаловируса, части полученного из гена β-глобина кролика интрона и полиаденилированный район для транскрипции кДНК ТРО человека, ген β-глобина человека, 69% гена вируса 1 бычьей папилломы, промотор тимидинкиназы и полиаденилированный район тимидинкиназы, ген фосфотрансферазы 1 (устойчивый к неомицину ген) и начало (ориджин) репликации рВR322 и ген β-лактамазы (Аmрr) и кДНК ТРО человека была лигирована в сайте, расположенном в направлении 5'-3' от промотора цитомегаловируса.
Пример 34. Экспрессия в X 63.6.5.3. клетках
X 63.6.5.3 клетки культивировали в минимальной основной среде Дульбекко (DME), содержащей 10% фетальную бычью сыворотку. Трансфекцию X.63.6.5.3. клеток плазмидной BMCGSneo-hTPO проводили методом электропорации, как описано ниже.
20 мкг плазмиды BMCGSneo-hTPO-P1, полученной в примере 33, добавляли к 750 мкл DME среды, содержащей 107 клеток, и смесь помещали в кювету для электропорации, имеющую щель 4 мм, и давали стоять в течение 10 минут при 4oС. Затем кювету помещали в устройство для электропорации (ВТХ 600, изготовляемое ВТХ) для проведения переноса гена при условиях 380 В, 25 мФ и 24 ангстремах. После переноса гена кювете опять давали стоять в течение 15 минут при 4oС. Затем клетки промывали один раз DEM, содержащей 10% фетальную бычью сыворотку. Трансфицированные клетки суспендировали в 50 мл DEM, содержащей 10 FCS, разливали в лунки 96-луночного культурального планшета и затем культивировали в течение 2 дней в СО2-инкубаторе. После 2 дней культивирования культуральную среду заменяли DEM, содержащей 10% FCS и 1 мг/мл G418 (производимого GIBCO) и смену среды проводили после этого каждые 3 дня. Активность ТРО человека в культуральной среде трансформантов измеряли тестами с CFU-MK, М-07е или Ва/F3. Трансформанты, секретирующие ТРО человека в культуральную среду, клонировали дважды лимитирующим разбавлением для установления продуцирующих ТРО человека клеточных линий.
Пример 35. Широкомасштабная экспрессия ТРО человека в COS 1 клетках
Трансфекцию COS 1 клеток проводили согласно ДЭАЭ-декстрановому способу, предусматривающему обработку хлорохином, как описано в примере 11. COS 1 клетки (АТСС CRL 1650) культивировали в IMDM, содержащей 10% (об./об.) FCS с применением покрытой коллагеном 175 см2 культуральной колбы при 37oС в 5% СО2-инкубаторе до тех пор, пока клетки не сливались на 100%. Для покрытия коллагеном культуральиой колбы 25 мл раствора коллагена (Cellmatrix type 1-C, изготовляемого Iwaki), концентрацию которого доводили до 0,3 мг/мл при помощи 1 мМ НС1, добавляли к каждой 175 см2 культуральной колбе, раствор коллагена удаляли после 1 часа стояния при комнатной температуре и затем обработанную таким образом колбу промывали 1 раз 20-50 мл PBS. Трансфекцию выполняли смешиванием 20 мл раствора IMDM, содержащего 250 мкг/мл ДЭАЭ-декстрана (Pharmacia), 60 мкМ хлорохина (Sigma) и 10% (об./об.) Nu-сыворотки (Collaborative) с рНТР1 (40 мкг), который был растворен в 500 мкл HBS, добавлением полученной смеси к COS 1 клеткам, описанным выше, содержащимся в одной 175 см2 культуральной колбе, которые были промыты один раз IMDM непосредственно перед трансфекцией. Затем эти клетки культивировали в течение 3 часов при 37oС в 5% СО2-инкубаторе. После этого культуральный супернатант удаляли отсасыванием и клетки промывали один раз IMDM, смешивали в 50 мл бессывороточной среды и затем культивировали в течение 5 дней при 36oС в 5% СО2-инкубаторе для извлечения культурального супернатанта. В одной операции использовали 100-260 культуральных колб и извлекали 5-13 л культурального супернатанта. Бессывороточную среду готовили добавлением к IMDM 5 мкг/мл инсулина (производимого Sigma), 5 мкг/мл трансферрина (Sigma), 10 мкМ моноэтаноламина (Wako Pure Chemicai Industries), 25 нМ селенита (Sigma) и 200 мкг/мл ВSА (не содержащий жирных кислот высокоочищенный бычий альбумин, производимый Nichirei).
Пример 36. Очистка полноразмерного ТРО человека (полученного из экспрессирующей плазмиды рНТР1, P1 клона), экспрессируемого в СОS 1 клетках, и его биологическая активность
Далее описаны примеры активности и очистки полноразмерного ТРО человека (полученного из экспрессирующей плазмиды pHTP1), экспрессируемого в COS1 клетках. Частично очищенный продукт получали сначала, для испытания его увеличивающей тромбоциты активности и затем проводили очистку, получая продукт высокой чистоты. Для измерения ТРО активности в последующих стадиях применяли в основном М-07е тест-систему. Подобным образом ТРО активность определяли в тест-системе с крысиными CFU-МК. При проведении этих тестов к каждой пробе добавляли сывороточный альбумин человека до конечной концентрации 0,02-0,05%.
Во-первых, COS 1 клетки, трансфицированные плазмидой pHTP1 согласно способу Ohashi and Sudo (Hideya Ohashi and Tadashi Sudo, Biosci. Biotech. Biochem. , 58 (4), 758-759, 1994) культивировали в течение 5 дней при 37oС в 5% СО2 с применением бессывороточной культуральной среды 1MDM, содержащей 0,2 г BSA, 5 мг бычьего инсулина, 5 мг человеческого трансферрина, 0,02 мМ моноэтаноламина и 25 нМ селенита натрия в 1000 мл, получая в результате приблизительно 7 л бессывороточного культурального супернатанта. К нему добавляли ингибиторы протеолитических ферментов p-APMSF Pefabloc SC гидрохлорид (4-(2-аминоэтил)-бензолсульфонилфторида, изготовляемый Merk кат. 24839) до конечной концентрации приблизительно 1 мМ, с последующим фильтрованием с применением фильтра 0,2 мкм для получения супернатанта. Супернатант концентрировали приблизительно в 10 раз при помощи ультрафильтрационного устройства (Omega Ultrasette, с отсечением номинальной мол. массы 8000, изготовляемого Filtron; или PLGC Pellicon Cassette, отсечением мол. массы 10000, Millipore). 723 мл этого концентрата (концентрация белка 3,38 мг/мл, общий белок 2445 мг, относительная активность 43000, общая активность 105100000) смешивали с 1,6 молями на 1000 мл сульфата аммония (288 г в целом), получая 804 мл раствора, содержащего 1,5 М аммоний сульфат (в конечной концентрации). Затем полученный таким образом раствор наносили при скорости течения 10 мл/мин, на колонку Macro-Prep Methyl HIC (диаметр 5 см и высотой 9 см) Bio-Rad, кат. 156-0080), которая была предварительно уравновешена 20 мМ цитратом натрия (буфер) (рН 5,2), содержащим 1,5 М сульфат аммония. После загрузки пробы проводили элюцию 20 мМ натрий-цитратным буфером (рН 5,2), содержащим 1,25 М сульфат аммония, получая проходящую через колонку фракцию F1 (2384 мл, концентрация белка 0,864 мг/мл, общий белок 2061 мг, относительная активность 6000).
Затем элюирующий раствор заменяли на 20 мМ цитрат натрия, рН 5,8, получая фракцию F2 (1092 мл, концентрация белка 0,776 мг/мл, общий белок 847 мг, относительная активность 150000). Полученную таким образом фракцию F2 (1081 мл) Macro-Prep Methyl HIC - колонки наносили при скорости течения 10 мл/мин, на колонку SP Sepharose Fast Flow (диаметр 3 см и высота 10 см), изготовляемой Pharmacia Biotech кат. 17-0729-01), которая была предварительно уравновешена 20 мМ буфера из цитрата натрия (рН 5,8). После загрузки пробы проводили элюцию 20 мМ натрий-цитратным буфером (рН 5,6), содержащим 110 мМ NaCl, получая фракцию F1 (2262 мл, концентрация белка 0,270 мг/мл, общий белок 610 мг, относительная активность 30000). Затем элюирующий раствор заменяли на 20 мМ цитрат натрия (рН 5,4), содержащий 400 мM NaCl, собирая фракцию F2 (856 мл, концентрация белка 0,189 мг/мл, общий белок 162 мг, относительная активность 300000). Затем элюирующий раствор заменяли на 20 мМ натрий-цитратный буфер (рН 5,2), содержащий 1000 мМ NaCl, собирая элюированную фракцию F3 (370 мл, концентрация белка 0,034 мг/мл, общий белок 12,6 мг, относительная активность 150000).
Основную ТРО-активную фракцию F2 (845 мл) стадии колонки SP Sepharose Fast Flow смешивали приблизительно с 10% в конечной концентрации 1-пропанолом и наносили при скорости течения 3 мл/мин на колонку LA-WGA (диаметр 2 см, высота 14 см, Honen кат. WG-007), предварительно уравновешенную 20 мМ натрий-цитратным буфером (рН 5,4), содержащим 400 мM NaCl. После загрузки колонки проводили элюцию смесью буфера 20 мМ цитрата натрия (рН 5,4), содержащего 400 мМ NaCl с 1-пропанолом (9:1), получая фракцию F1 (64,4 мл, концентрация белка 0,0178 мг/мл, общий белок 1,15 мг, относительная активность 17220). Затем элюирующий раствор заменяли на 20 мМ натрий-цитратный буфер (рН 6,1), содержащий 0,4 М GlcNAc и 10% 1-пропанол, собирая элюированную фракцию F2 (45 мл, концентрация белка 0,0104 мг/мл, общий белок 0,470 мг, относительная активность 675000).
Основную ТРО-активную фракцию F2 (340 мл) операции колонки LA-WGA смешивали приблизительно с 0,005% в конечной концентрации ТFА и подвергали колоночной хроматографии на YMC-Pack CN-AP (6 мм в диаметре и 250 мм высотой), изготовляемой УМС, кат. АР-513. С применением 0,1% ТFА в качестве проявляющего растворителя А и 1-пропанола, содержащего 0,05% ТFА, в качестве проявляющего растворителя В пробу наносили при скорости течения 0,6 мл/мин на колонку YMC-Pack CN-AP, уравновешенную предварительно 15% В. После загрузки пробы концентрацию пропанола увеличивали с 15% В до 25% В и проводили элюцию линейным градиентом от 25% В до 50% В в течение 65 минут, собирая элюаты в полипропиленовые пробирки по 1,5 мл (2,5 мин).
0,5 мкл (1/3000 фракции) элюата в каждой пробирке смешивали с HSA, и концентрировали ультрафильтрацией, получая 0,25 мл IMDM тест-культурального раствора, содержащего 0,05% НSА, для использования в идентификации ТРО-активных фракций при помощи этого теста. В результате высокая ТРО активность (относительная активность приблизительно 630000-4800000) была обнаружена в пробирках с номерами 24-30 (в диапазоне концентраций пропанола 35,0 и 42,0%), которые объединяли в виде ТРО-активной фракции FА (13,5 мл).
Фракцию FА, полученную на стадии колонки YMC-Pack CN-AP, подвергали колоночной хроматографии на Cepcell Pak C1 300А (диаметр 4,6 мм, высота 150 мм, изготовляемого Shiseido, кат. C1 TYPE: SG 300А) с применением 0,1% ТFА в качестве проявляющего растворителя А и 1-пропанола, содержащего 0,05% ТFА, в качестве проявляющего растворителя В. Часть (8,9 мл) фракции FА, полученной в колоночной операции с YMC-Pack CN-AP, смешивали с 0,3 мл глицерина, концентрировали выпариванием с центрифугированием и затем смешивали приблизительно с 2,5 мл 10% В. Полученный раствор наносили затем при скорости течения 0,4 мл/мин на колонку Capcell Pak C1 300А, уравновешенную предварительно 20% В. После загрузки колонки проводили элюцию сначала 20% В в течение 15 минут и затем линейным градиентом от 20% В до 40% В в течение 50 минут, собирая элюаты в полипропиленовые пробирки по 1 мл (2,5 мин) в каждую пробирку.
Часть 1 мкл (1/1000 фракции) элюата в каждой пробирке смешивали с HSА и концентрировали ультрафильрацией, получая 0,25 мл IMDM тест-культурального раствора, содержащего 0,05% HSA для применения в идентификации ТРО-активных фракций этим тестом. В результате высокая активность ТРО (относительная активность приблизительно 3000000-22500000) была обнаружена в пробирках с номерами 20-23 (в пределах концентрации пропанола 28,5 и 32,5%), которые объединяли в виде высокоактивной фракции.
Пример 37. Измерение мол. массы ТРО человека, экспрессируемого в COS 1 клетках
Было сделано предположение, что мол. масса полноразмерного ТРО человека (полученного из экспрессирующей плазмиды pHTP1, клон F1), экспрессируемого в COS 1 клетках, должна быть больше, чем мол. масса, предполагаемая на основании размера пептидной цепи, вследствие добавления сахарной цепи. Поэтому в качестве первой стадии получали частично очищенную фракцию ТРО следующим образом из культурального супернатанта, содержащего полноразмерный ТРО человека (полученный из экспрессирующей плазмиды pHTP1, F1 клон), экспрессируемого в COS 1 клетках. Т. е. с применением проявляющего растворителя А (0,1% трифторуксусная кислота (ТFА) и проявляющего растворителя В (1-пропанол, содержащий 0,05% ТFА) порцию 0,3 мл культурального супернатанта наносили на колонку YMC-Pack PROTEIN-RP (диаметр 0,46 см, высота слоя 15 см, УМС, кат. А-РRRР-33-46-25), уравновешенную предварительно 25% В. 25% В пропускали через колонку в течение 5 минут при скорости течения 0,4 мл/мин и затем выполняли фракционирование при помощи линейного градиента плотности от 25% до 50% В в течение 50 минут. ТРО активность была обнаружена в элюатах в диапазоне концентраций пропанола 34,5% и 43,5%. Эти элюаты выпаривали досуха выпариванием с центрифугированием, получая частично очищенную пробу.
Затем ТРО активность определяли М-07е тест-системой после экстракции белка из геля SDS-PAGE электрофореза, проводимого при невосстанавливающих условиях, как описано в примере 1, или мол. массу измеряли на основе восстановленных DPC111 маркеров посредством вестерн-анализа, который будет описан ниже в примере 45. В результате ТРО активность была найдена в широком диапазоне средних мол. масс приблизительно от 69000 до 94000, свидетельствуя о вариировании в мол. массе. Также было измерено, что средняя мол. масса ТРО на основании восстановленных ДРС111 маркеров после гидролиза сахарной цепи типа N-гликозидной связи при помощи N-гликаназы (Genzyme, кат. 1472-00) была 36000-40000, т. е. больше, чем мол. масса приблизительно 35000, предполагаемая из размера пептидной цепи, что является сильным указанием на присутствие также сахарной цепи типа О-гликозидной связи.
Таким же образом было найдено, что средняя мол. масса полноразмерного ТРО человека (полученного из экспрессирующей плазмиды pHTP1, P1 клон), экслрессируемого в COS 1 клетках, равна 63000-83000.
Пример 38. Биологические характеристики ТРО человека
Влияния ТРО человека на гемопоэтические клетки человека и крыс исследовали в основном с применением культурального супернатанта от COS 1 клеток, трансфицированных pHTF1.
В тесте с подсчетом колоний с применением клеточной фракции CD34+DR+, полученной из крови спинного мозга человека, ТРО человека стимулировал образование значительного числа колоний мегакариоцитов. Например, 11,5 колоний мегакариоцитов образовались 6000 CD34+DR+ клеток в присутствии 10% (об./об.) культурального супернатанта из COS 1 клеток, трансфицированных pHT1-231. Для испытания действия ТРО человека на клетки-предшественники мегакариоцитов человека получали CD34+ клетки из периферической крови человека следующим образом. Фракцию лейкоцитов, полученную из периферической крови человека при помощи градиента плотности Ficoll-Paque, истощали от клеток, прикрепившихся к пластиковым чашкам. Неприкрепленные клетки затем очищали от SBA (агглютинин сои)-положительных клеток пэннингом с применением AIS Microselecter SBA (Asahi Medical). Полученные клетки инкубировали на AIS Microselecter CD 34 и адсорбированные клетки, CD34+ клетки, собирали после культивирования CD34+ клеток в течение 10 дней в присутствии культурального супернатанта из трансфицированных COS 1 клеток в жидкой среде, наблюдали селективную пролиферацию клеток Gp11b/111a+ большого размера и обнаружили увеличение в плоидности в этих Gp11b/111a+ клетках. Эти результаты дают сильное основание для предположения, что ТРО человека оказывает избирательное действие на клетки-предшественники мегакариоцитов человека и стимулирует их пролиферацию и дифференцировку.
В тесте подсчета колоний с применением Gp11b/111a+ клеток, высокообогащенных CFU-MK из клеток костного мозга крыс, и клеток, не прикрепившихся к пластику, полученных на предшествующей стадии Gp11b/111a+ клеток, ТРО человека индуцировал образование значительного числа колоний мегакариоцитов. Например, 34,5 колоний мегакариоцитов образовались из 1000 Gp11b/111a+ клеток и 28,5 колоний мегакариоцитов были образованы 20000 не прикрепившимися к пластику клетками в присутствии 20% (об./об.) культуральным супернатантом из трансфицированных СОS 1 клеток. Кроме того, хотя фракция не прикрепленных к пластику клеток содержит различные гематопоэтические клетки-предшественники, иные, чем CFU-MК, в присутствии ТРО человека образовались только колонии мегакариоцитов, что предполагает, что ТРО человека проявляет его избирательное действие на клетки-предшественники мегакариоцитного происхождения. При культивировании Gp11b/111a+ клеток, полученных из костного мозга крыс, в течение 3-5 дней в присутствии 20% (об./об.) культурального супернатанта от трансфицированных COS 1 клеток в жидкой культуре, на 5-м дне культивирования наблюдали увеличение плоидности клеток.
Пример 39. Конструирование вектора для применения в экспрессии слитого белка (далее называемого - "GST-TPO (1-174)" глутатион-S-трансферазы (GST) и ТРО человека (аминокислотные остатки 1-174) в E.coli
Для облегчения экспрессии ТРО человека в E.coli получали искусственный ген, кодирующий ТРО человека и содержащий кодон предпочтения E.coli. Нуклеотидная последовательность этой ДНК содержит амино-концевой кодон метионина (ATG) в положении -1 для применения в инициации трансляции в E.coli.
Синтетические олигонуклеотиды 1-12, представленные в табл.IV, были приготовлены и каждый из синтетических олигонуклеотидов 2-11 фосфорилировали Т4 киназой (изготовляемой Pharmacia) в растворе, состоящем из 1 мМ АТР, 10 мМ Трис-ацетата, 10 мМ Мg-ацетата и 50 мМ К-ацетата. Для превращения этих синтетических олигонуклеотидов в 6 двухцепочечных фрагментов ДНК их объединяли в комбинации 1 и 2, 3 и 4, 5 и 6, 7 и 8, 9 и 10 и 11 и 12 и каждую комбинацию подвергали отжигу в растворе, состоящем из 10 мМ Трис-НС1 (рН 7,5), 10 мM MgCl2 и 50 мМ NaCl. Затем 3 двухцепочечных фрагмента ДНК 1 и 2, 3 и 4 и 5 и 6 и 3 других двухцепочечных фрагмента ДНК 7 и 8, 9 и 10 и 11 и 12, соответственно, обрабатывали Т4 лигазой (изготовляемой Life Technology) и эти 2 реакционных раствора опять обрабатывали Т4 лигазой таким же образом. Фрагмент ДНК, полученный реакцией лигирования, расщепляли BamH1 (Boehringer-Mannheim) и подвергали электрофорезу в 2% агарозном геле, извлекая фрагмент приблизительно 390-400 п. н. по размеру, который затем очищали при помощи набора для очистки ДНК Prep-A-Gene и субклонировали в pUC18, расщепленную предварительно Xbal и BamH1 (E.coli DH5 использовали в качестве хозяина). Из полученных таким образом клонов клон, имеющий искусственный ген, кодирующий ТРО человека и содержащий следующие предпочтительные кодоны E.coli, был выбран анализом нуклеотидной последовательности и назван pUC18(ХВ) (1-123). Последовательность ДНК кодирующей цепи показана в списке последовательностей (SEQ ID No 9) (см. табл.V).
Одноцепочечную кДНК синтезировали из 1 мкг полученной из нормальной печени человека поли (А)+ РНК с применением олиго dT-праймера, и PCR проводили с применением 0,1 объема реакционного раствора для синтеза кДНК в качестве матрицы. PCR (инкубирование при 96oС в течение 2 минут, повторение и в целом 30 циклов, каждый из которых состоял из инкубаций при 96oС в течение 1 минуты, при 58oС в течение 1 минуты и при 72oС в течение 1 минуты, и конечное инкубироваиие при 72oС в течение 7 минут) проводили с объемом 100 мкл с применением hTPO-C и hTPO-Z (EcoR1) в качестве праймеров. Фрагмент кДНК ТРО человека, полученный таким образом, расщепляли BamHI и EcoR1 и подвергали электрофорезу в 2% агарозном геле, извлекая фрагмент размером приблизительно 600 п. н., который затем очищали при помощи набора для очистки ДНК Prep-A-Gene и субклонировали в pUC18, расщепленную предварительно EcoR1 и BamH1 (E. coli DH5α использовали в качестве хозяина). Клон, кодирующий правильную нуклеотидную последовательность ТРО человека, отбирали анализом нуклеотидной последовательности и он был назван рUC18(BE)(124-332). Олигонуклеотидные последовательности праймеров, примененных в этой РСR, были следующими:
. hTPO-C: 5'-GGA GGA GAC CAA GGC АСА GGA-3' (SEQ ID No 95) (положения 329-349 клона hHTF1); и
. hTPO-Z(EcoR1): 5'-CCG GAA TTC TTA CCC TTC CTG AGA CAG ATT-3' (SEQ ID 96) (полученный добавлением последовательности узнавания к антисмысловому праймеру, соответствующему положениям 1143-1163 клона pHTF1).
Клон pUC18(BE) (124-332) расщепляли ВаmНl и EcoR1 и подвергали электрофорезу в 2% агарозном геле для извлечения фрагмента С-концевой стороны кДНК ТРО человека размером приблизительно 600 п.н., который затем очищали при помощи набора для очистки ДНК Prep-A-Gene и субклонировали в pUC18(XB) (1-123), расщепленную предварительно рестриктазами EcoR1 и BamHl (E.coli DH5α использовали в качестве хозяина). Клон, полученный таким образом, был назван pUC18(XE)(1-332).
Поскольку присутствие ТРО активности человека в пептидном фрагменте ТРО человека из положений 1-163 аминокислотных остатков было подтверждено примером, в котором были получены различные С-концевые делеционные конструкции кДНК ТРО человека и экспрессировали для измерения in vitro ТРО активности, был сконструирован экспрессирующий вектор, содержащий этот пептидный фрагмент. В этом случае проводили экспрессию последовательности ДНК, кодирующей GST-TPO (1-174).
Так как нуклеотидная последовательность, соответствующая положениям 681-686 (аминокислотные остатки 173 и 174) клона pHTF1, узнается рестриктазой Sас1, два концевых кодона вводили при помощи 2 синтетических олигонуклеотидов с использованием сайта узнавания этой рестриктазы. Т.е. два синтетических олигонуклеотида SSE1 и SSE2 отжигали в растворе, состоящем из 10 мМ Трис-HС1 (рН 7,5), 10 мМ MgCl2 и 50 мМ NaCl, и с применением набора для лигирования ДНК (Takara Shuzo) полученный таким образом двухцепочечный фрагмент вводили в pUC18(ХЕ)(1-332), из которой С-концевой фрагмент размером приблизительно 480 п.н. кДНК ТРО человека был удален расщеплением его Sacl и EcoR1, получая клон рUС18(ХS)(1-174) (E.coli DH5α использовали в качестве хозяина). Последовательности синтетических олигонуклеотидов, примененных при этом, были следующими:
SSE1: CTAATGAG (SEQ ID No 97)
SSE2: AATTCTCATTAGAGCT (SEQ ID No 98);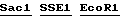
. 5'-CTAATGAG-3' (SEQ ID No 99)
3'-TCGAGATTACTCTTAA-5' (SEQ ID No 100)
. 
- Клон pUC18 (XS) (1-174), полученный выше, расщепляли Xbal и EcoR1 и подвергали электрофорезу в 2% агарозном геле для извлечения фрагмента размером приблизительно 600 п.н., кодирующего аминокислотные остатки 1-174 ТРО человека. Затем этот фрагмент очищали при помощи набора для очистки ДНК Prep-A-Gene и субклонировали в pBluescript 11SK+ (изготовляемый Stratagene), предварительно расщепленный Xbal и EcoR1 (E.coli DH5α использовали в качестве хозяина). Клон, полученный таким образом, был назван pBL(XS)(1-174). Таким же образом клон pBL(XS) (1-174) расщепляли Xbal и Hind111, извлекая фрагмент размером приблизительно 550 п.н., кодирующий аминокислотные остатки 1-174 ТРО человека, и затем этот фрагмент очищали при помощи набора для очистки ДНК Prep-A-Gene и субклонировали в рСFМ536 (EР-А-136490), расщепленный предварительно Xbal и Hind111 (E.coli DH5α использовали в качестве хозяина). Клон, полученный таким образом, был назван рСFM536/hТ (1-174).
Следующий эксперимент проводили для экспрессии GST-TPO(1-174) с применением pGEX-2T (Pharmacia), который является экспрессирующим вектором слитого с глутатион- S-трансферазой (GST) белка. РСR (инкубация при 96oС в течение 2 минут, повторение в целом 22 циклов, каждый из которых состоял из инкубаций при 96oС в течение 1 минуты, при 41oС в течение 1 минуты и при 72oС в течение 1 минуты, и конечная инкубация при 72oС в течение 7 минут) проводили с применением рСFМ536/hТ(1-174) в качестве матрицы и двух PCR праймеров GEZ1 и GEX3. Кодирующий ТРО человека фрагмент, полученный таким образом, расщепляли Nаеl и EcoR1 и подвергали электрофорезу в 2% агарозном геле, извлекая фрагмент размером приблизительно 550 п.н., этот фрагмент затем очищали при помощи набора для очистки ДНК Prep-A-Gene и клонировали в рGЕХ-2Т, расщепленный заранее EcoR1 и Smal (E.coli DH5 использовали в качестве хозяина) и затем клон, названный pGEX-2T/hT(1-174) был отобран анализом нуклеотидной последовательности для получения трансформанта для использования в экспрессии GST-ТРО (1-174). Олигонуклеотидные последовательности примененных праймеров в PCR были следующими:
GEX1: 5'-АТС GCC GGC TCC GCC AGC TTG TGA C-3'-(SEQ ID No 101) (полученный добавлением последовательности узнавания Nael к последовательности положений 21-39 в SEQ ID 10) и
. GЕХ3: 5'-GCC GAA TTC TCA TTA GAG CTC GTT CAG TGT-3' (SEC ID 102) (антисмысловой праймер, соответствующий последовательности положений 523-549 в SEQ ID 10).
Эта экспрессирующая плазмида содержит последовательность ДНК, кодирующую GST белок, за которым следует пептид узнавания тромбина и ТРО человека (аминокислотные остатки 1-174). Последовательность ДНК, кодирующая пептид узнавания тромбина и ТРО человека (аминокислотные остатки 1-174), показана в списке последовательностей (SEQ ID 10).
Пример 40. Экспрессия GST-TPO (1-174) в E.coli
Трансформант, полученный в примере 39, культивировали в течение ночи при 37oС на шейкере с применением 60 мл LB среды, содержащей 50 мкг/мл ампициллина, и 25 мл полученного культурального бульона добавляли к 1000 мл LВ среды, содержащей 50 мкг/мл ампициллина, и культивировали при 37oС на качалке, пока CD при А600 не достигала 0,7-0,8. После этого 1PTG добавляли к культуральному бульону до конечной концентрации 0,1 мM и качание культуры продолжали еще 3 часа для индуцирования экспрессии GST-TPO (1-174).
Пример 41. Очистка GST-TPO (1-174), экспрессированного в E.coli, и подтверждение его биологической активности
5,9 г замороженных клеток рекомбинантного штамма, продуцирующего GSТ-ТРО (1-174), полученного из клона pGEX-2T/hT (1-174), кодирующего нуклеотидную последовательность ТРО человека, суспендировали в 10 мл воды и разрушали дезинтегратором высокого давления. После центрифугирования полученной суспензии GST-ТРО (1-174) извлекали в осажденной фракции, большую часть примесных белков, клеточных компонентов и т.п. удаляли. Затем извлеченную таким образом осажденную фракцию, содержащую GST-TPO (1-174), суспендировали в 5 мл воды, к которой затем добавляли при перемешивании 6 мл 1М Трис-буфера (рН 8,5), 120 мл 10 М мочевины и 16 мл воды. После 5 минут перемешивания для растворения содержимого полученный раствор равномерно делили на 4 части для проведения следующих стадий (1)-(4).
(1) Одну из проб разбавляли в 10 раз 20 мМ Трис-буфером (рН 8,5). Затем добавляли глутатион восстановленного типа и глутатион окисленного типа до соответствующих конечных концентраций 5 мМ и 0,5 мМ с последующим инкубированием в течение ночи при 4oС. Полученную смесь центрифугировали для извлечения GST-TPO (1-174) в виде супернатантной жидкости, которую затем разбавляли в 2 раза 20 мМ натрий-цитратным буфером (рН 5,5) и затем доводили рН до 5,5 уксусной кислотой. GST- ТРО (1-174) в растворе наносили на SP Sepharose Fast Flow (Pharmacia Biotech, кат. 17-0729-01) катионообменную колонку, уравновешенную предварительно 20 мМ буфером из цитрата натрия (рН 5,5). После промывания колонки 20 мМ цитратом натрия рН 5,5, элюцию GST- ТРО (1-174) проводили 20 мМ буфером, содержащим цитрат натрия, рН 5,5, содержащим 500 мМ хлорид натрия. Часть 129 мл этого элюата смешивали с 2,6 мл 1 М Трис-буфера, рН 8,5 и полученную смесь с рН 8,1 наносили на колонку Glutathione Sepharose 4B (Pharmacia Biotech, кат. 17-0756-01) для aдcopбции GST-TPO (1-174). После промывания колонки PBS проводили элюцию GST-TPO (1-174) при помощи Трис-буфера с рН 8,5, содержащего 10 мМ глутатиона восстановленного типа. Полученный элюат смешивали с 37 NlH единицами тромбина, давали стоять в течение 4 часов при комнатной температуре, разбавляли в 10 раз PBS и затем наносили на колонку Glutathione Sepharose 4B для адсорбции отщепленного GST и извлечения ТРО (1-174) в неадсорбированной фракции. Извлеченную неадсорбированную фракцию разбавляли в 3 раза 20 мМ содержащим цитрат натрия буфером с рН 5,5 и наносили на катионообменную колонку SP Sepharose Fast Flow, уравновешенную предварительно тем же самым буфером, и целевое соединение элюировали линейным градиентом плотности 0-500 мМ хлорида натрия, растворенного в том же буфере.
(2) Все операции стадии (1) проводили в присутствии 0,1% полисорбата 80.
(3) Одну из проб разбавляли в 10 раз 20 мМ Трио-буфером с рН 8,5. К ней добавляли глутатион восстановленного типа и глутатион окисленного типа до соответствующих конечных концентраций 5 мМ и 0,5 мМ с последующим стоянием в течение ночи при 4oС. Полученную таким образом смесь центрифугировали для извлечения GST-TPO (1-174) в виде супернатантной жидкости, которую затем разбавляли в 2 раза 20 мМ содержащим цитрат натрия буфером с рН 5,5 и затем доводили рН до 5,5 уксусной кислотой. GST-TPO (1-174) в этом растворе наносили на катион-обменную смолу SP Sepharose Fast Flow, предварительно урановешенную 20 мМ содержащим цитрат натрия буфером с рН 5,5. После промывания смолы 20 мМ содержащим цитрат натрия буфером с рН 5,5 проводили элюцию GST-TPO (1-174) 20 мМ содержащим цитрат натрия буфером с рН 5,5, содержащим 500 мМ хлорид натрия. Элюат доводили до рН 8 1 М Трис-буфером с рН 8,5, смешивали с 320 NlH единицами тромбина для удаления полипептидных последовательностей GST путем расщепления при пептидном линкере узнавания тромбина и давали стоять в течение 4 часов при комнатной температуре, разбавляли в 5 раз 20 мМ содержащим цитрат натрия буфером с рН 5,5 и затем наносили на катионообменную смолу SP Sepharose Fast Flow, уравновешенную заранее тем же самым буфером, и целевое соединение элюировали затем линейным градиентом плотности 0-500 мМ хлорида натрия, растворенного в том же буфере.
(4) Все операции стадии (3) проводили в присутствии 0,1% полисорбата 80.
Каждую из фракций, элюированных при плотностях хлорида натрия приблизительно 200-400 мМ при колоночной хроматографии на катионообменной колонке SP Sepharose Fast Flow, основательно диализовали против IMDM культуральной среды и затем оценивали в тест-системе с крысиными CFU-MK. При анализе фракции электрофорезом SDS-PAGE в присутствии восстанавливающего агента обнаружили полосу, соответствующую мол. массе 19 кД (чистота 1-20%) в качестве одной из основных белковых полос этой фракции. При анализе N-концевой последовательности этой полосы в SDS-PAGE и переносе на PVDF мембрану согласно способу, описанному в примере 1, было подтверждено, что эта полоса содержит последовательность ТРО, экспрессируемую в виде полученного из pGEX-2T/hT(1-174) слитого белка. Дедуцированная (выведенная) аминокислотная последовательность этого полученного ТРО была [Gly-1] TPO вследствие последовательности пептидного линкера узнавания тромбина и известной активности тромбина на этом линкере.
Пример 42. Конструирование вектора для применения в экспрессии в Е.соli ТРО человека (аминокислотные остатки 1-163), имеющего замещения в положении 1 (Ser-->Ala) и положении 3 (Ala-->Val)
Клон pUC18(XE) (1-332), полученный в Примере 39, расщепляли Xbal и EcoR1 и подвергали электрофорезу в 2% агарозном геле, выделяя фрагмент размером приблизительно 1000 п. н., кодирующий аминокислотные остатки 1-332 ТРО человека, и этот фрагмент затем очищали с применением набора для очистки ДНК Prep-A-Gene и субклонировали в pBlues-cript 11SK+ (изготовляемый Stratagene), который был расщеплен заранее рестриктазами Xbal и EcoR1 (E.coli DH5α использовали в качестве хозяина). Клон, полученный таким путем, был назван pBL(XE)(1-332). Затем район этого клона pBL (XE)(1-332) от его последовательности узнавания BamH1 до положения 163 аминокислоты (положения 366-489) заменяли на доминантные кодоны E.coli. Были получены синтетические олигонуклеотиды 13-20, показанные ниже, и каждую пару синтетических олигонуклеотидов 13 и 14, 15 и 16 и 17 и 18 фосфорилировали в той же самой пробирке, содержащей раствор, состоящий из 0,1 мМ АТР, 10 мм Трис-ацетата, 10 мМ Мg-ацетата и 50 мМ К-ацетата. После дальнейшего добавления 1/10 объема раствора, состоящего из 100 мМ Трис/НСl (рН 7,5), 100 мМ MgCl2 и 500 мМ NaCl, полученный раствор нагревали в кипящей бане в течение 3 минут и затем давали остыть до комнатной температуры, получая двухцепочечный фрагмент ДНК. Затем полученные таким образом три пары двухцепочечных фрагментов, состоящих из 13 и 14, 15 и 16 и 17 и 18, лигировали с применением набора для лигирования ДНК (Takara Shuzo) и проводили PCR с использованием лигированного продукта в качестве матрицы и синтетических олигонуклеотидов 19 и 20 в качестве праймеров. Полученный продукт PCR расщепляли BamH1 и Hind111 и подвергали электрофорезу в 2% агарозном геле, извлекая фрагмент размером приблизительно 130 п. н. с применением набора для очистки ДНК Рrер-A-Gene. Этот фрагмент субклонировали в pBL(XE)(1-332), расщепленный предварительно ВаmН1 и Hind111 (E. coli ДН5 использовали в качестве хозяина). Клон, имеющий нуклеотидные последовательности, представленные в табл. VI, был выбран из полученных клонов анализом нуклеотидной последовательности и назван pBL (XH)(1-163).
C целью увеличения количества экспрессированного ТРО человека (положения 1-163) и предотвращения гидролиза N-конца протеазой был сконструирован экспрессирующий вектор для применения в экспрессии мутированного типа ТРО человека (положения 1-163) (далее называемого "h 6Т(1-163)"), имеющего замены при положении 1 (Ser на Ala) и положении 3 (Ala на Val) ТРО человека (положения 1-163) ([Аlа1, Val3] ТРО (1-163)) и, в то же самое время, кодирующего Lys при -1 положении и Met при -2 положении ([Меt-2, Lys-1, Ala1, Val3] TPO (1-163)). Получали четыре синтетических олигонуклеотида, показанных ниже, и синтетические олигонуклеотиды 2-9 и 3-3 фосфорилировали Т4 киназой (изготовляемой Pharmacia) в растворе, состоящем из 1 мМ АТР, 10 мМ Трис-ацетата, 10 мМ Мg-ацетата и 50 мМ К-ацетата. Для того чтобы превратить эти синтетические олигонуклеотиды в двухцепочечные фрагменты ДНК, каждую пару одноцепочечных фрагментов ДНК 1-9 и 2-9 и 3-3 и 4-3 отжигали в растворе, состоящем из 10 мМ Трис-НС1 (рН 7,5), 10 мМ МgСl2 и 50 мМ NаСl. Затем полученные таким образом пары двухцепочечных фрагментов ДНК, состоящие из 1-9 и 2-9 и 3-3 и 4-3, обрабатывали при помощи набора для лигирования ДНК (Takarа Shuzo). Фрагмент ДНК, полученный в реакции лигирования, субклонировали в pBL(XH)(1-163), расщепленный предварительно Xba1 и Nru1, и клон, правильно замещенный следующим синтетическим олигонуклеотидом, представленным в табл. VII, был отобран анализом нуклеотидной последовательности (E.coli ДН5α использовали в качестве хозяина) и назван рBL(ХН)h6Т(1-163).
Клон рВL (XH)(1-163) расщепляли Xba1 и Hind111, извлекая фрагмент размером приблизительно 500 п.н., кодирующий аминокислотные остатки 1-163 ТРО человека мутированного типа, и этот фрагмент затем очищали при помощи набора для очистки ДНК Prep-A-Gene и клонировали в рСFМ536 (ЕР-A-136490), расщепленный предварительно Xba1 и Hind111 (Е. coli JM109, который был предварительно трансформирован pMW1 (АТСС 39933), использовали в качестве хозяина). Клон, полученный таким путем, был назван рСFМ536/h6Т (1-163), и штамм E. coli, имеющий этот экспрессирующий вектор, использовали в качестве трансформанта для применения в экспрессии ТРО человека мутированного типа, а именно h6T(1-163). Эта экспрессирующая плазмида содержит последовательность ДНК, показанную в списке последовательностей (SEQ ID 11).
Пример 43. Экспрессия h6Т (1-163) в Е.соli
Экспрессия экспрессирующей плазмиды рСFМ536 контролируется промотором λPL, который сам находится под контролем репрессорного гена с1857. Трансформант, полученный в Примере 42, культивировали в течение ночи при 30oС на шейкере с применением 60 мл LB среды, содержащей 50 мкг/мл ампициллина и 12,5 мкг/мл тетрациклина, и часть 25 мкл полученного культурального бульона добавляли к 1000 мл LВ среды, содержащей 50 мкг/мл ампициллина, и культивировали при 37oС на шейкере до тех пор, пока OD при A600 не достигала 1,0-1,2. После этого приблизительно 330 мл LВ среды, нагретой до 65oС, добавляли к культуральному бульону, доводя конечную температуру среды до 42oС, и качание культуры продолжали в течение 3 часов при 42oС для индуцирования экспрессии h6Т(1-163).
Пример 44. Очистка h6Т(1-163), экспрессированного в Е.соli, и подтверждение его биологической активности
Порцию 3,6 г замороженных клеток h6T(1-163) продуцирующего рекомбинантного штамма суспендировали в 10 мл воды и разрушали при помощи дезинтегратора высокого давления. Центрифугированием этой суспензии получали осажденную фракцию, а большую часть примесных белков, клеточных компонентов и т. п. удаляли. Извлеченную осажденную фракцию, содержащую h6Т(1-163), суспендировали в 7 мл воды и к суспензии при перемешивании добавляли 3 мл Трис-буфера (рН 8,5) с последующим добавлением мочевины (конечная концентрация 8 М), гуанидинхлорида (конечная концентрация 6 М) и N-лауроилсаркозината (конечная концентрация 2%). После 5-20 минут перемешивания при комнатной температуре для растворения компонентов полученный раствор разбавляли в 10 раз 20 мМ Трис-буфером с рН 8,5 и подвергали повторной белковой укладке цепи посредством инкубирования в течение ночи при 4oС. В этом случае применяли глутатион и сульфат меди в качестве добавок, кроме окисления воздухом. При помощи центрифугирования 6Т(1-163) извлекали из супернатантной жидкости. При анализе каждой из извлеченных фракций при помощи SDS -PAGE в присутствии восстанавливающего агента обнаружили полосу, соответствующую мол. массе 18 кД (чистота 30-40%) в качестве основной белковой полосы каждой фракции. При проведении анализа N-концевой последовательности при помощи SDS-PAGE и переноса на PVDF мембрану по способу, описанному в Примере 1, было подтверждено, что эта полоса содержит экспрессируемую последовательность ТРО в виде полученного из рСМF536/h6Т(1-163) ТРО мутированного типа. При тщательном диализе каждой фракции против 1MDM культуральной среды и оценки при помощи тест-системы с крысиными СFU-МК ТРО активность была обнаружена во всех фракциях и она зависела от дозы.
Пример 45. Получение антитела против ТРО пептида и обнаружение ТРО при помощи электрофореза в ПААГ-ДСН (SDS-PAGE) и вестерн-анализа
Успех в получении антитела против ТРО пептида сделает возможным обнаружение белка ТРО иммунологическим способом. В связи с этим три района, которые, по-видимому, являются относительно применимыми в качестве антигенов (см. ниже), были выбраны из доказанных аминокислотных последовательностей ТРО и пептид типа квадрупольно-цепочечного пептида с множественными антигенами (MAP) был синтезирован по способу Tam (Proc.Natl Acad.Sci.USA, 85, 5409-5413, 1988) с использованием каждого из выбранных районов и затем каждого из 2 кроликов иммунизировали 8 раз по 100 мкг каждого из синтезированных таким образом пептидов для получения соответствующих проб антисывороток.
Аминокислотные последовательности, содержащиеся в синтезированных пептидных антигенах
а) Крысиный ТРО (9-28) (Пептидный район R T1)
( при положении 24 представляет собой исходно S в аминокислотных остатках, определенных на основании гена)
при положении 24 представляет собой исходно S в аминокислотных остатках, определенных на основании гена)
b) Крысиный ТРО (46-66) (Пептидный район RT2)
FSLGЕWKTQTEQSKAQDILGA (SEQ 1D 117)
с) Крысиный ТРО (163-180) (Пептидный район RT4)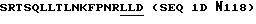
( в положениях 178-180 исходно представляет собой TSG в аминокислотных остатках, определенных на основании этого гена)
в положениях 178-180 исходно представляет собой TSG в аминокислотных остатках, определенных на основании этого гена)
Затем эти пробы антисывороток, анти-RT1 пептид и анти-RT2 пептид подвергали сначала колоночной хроматографии на Protein A (PROSEP-A, Bioprocessinq Ltd. , кат. 8427) для получения 1gG фракций (2344 мг и 1920 мг, соответственно). 54 мг и 32 мг из этих соответствующих фракций биотинилировали связыванием их с активным биотином (NHS -LC - Biotin 11; изготовляемым P1ER CE, кат. 21336). Рекомбинантный ТРО-содержащую пробу подвергали SDS - PAGE и затем электроблоттингу на PVDF или нитроцеллюлозную мембрану, как описано в Примере 1. Вестерн-анализ проводили обычным способом с применением этих биотинилироваиных антител в качестве первых антител.
Т. е. мембрану после блоттинга промывали раствором, состоящим из 20 мМ Триc-HC1 и 0,5 NаС1 (рН 7,5) (ТВS), в течение 5 минут и дважды 0,1% Твин 20-содержащим ТВS (ТТВS) в течение 5 минут каждый раз и затем обрабатывали блокирующим агентом (Block Асе; изготовленным Dainippon Pharmaceutical, кат. UК-В25) в течение 60 минут. Затем мембрану обрабатывали в течение 60 минут раствором ТТВS, содержащим 10 мкг/мл биотинилированных антител против пептида ТРО, 0,05% ВSА и 10% BlockAce и затем промывали дважды ТТВS каждый раз по 5 минут. После этого мембрану обрабатывали в течение 30 минут раствором, полученным путем разбавления меченого щелочной фосфатазой авидина (Leinco Technoloqies, кат. А108), в 5000 раз раствором ТТВS, содержащим 10% BlockAce, и промывали дважды по 5 минут TTBS и ТВS в течение 5 минут. После этого выполняли развитие окраски при помощи субстрата щелочной фосфатазы (изготовленного Bio-Rad, кат. 170-6432). Описанный вестерн-анализ проводили при комнатной температуре.
В результате удалось узнавать не только различные рекомбинантные крысиные белки ТРО, экспрессируемые в COS 1 клетках, но также рекомбинантные человеческие белки ТРО, экспрессируемые в COS 1 клетках и клетках E.coli (например, полученный из плазмид рНТР1 или pHTF1 ТРО человека, экспрессируемый в СOS 1 клетках, и его продукт, полученный расщеплением N-гликозидной связи, GSТ-ТРО (1-174), экспрессируемый в Е.сoli, и его расщепленный тромбином продукт и h6Т(1-163), который является ТРО мутированного типа, экспрессируемый в E. coli, которые были описаны в Примерах). Кроме того, эти результаты дали возможность анализировать эти различные типы рекомбинантного ТРО человека.
Кроме вышеописанного, была подтверждена возможность получения антител против пептида ТРО человека. Антитела, полученные этими способами, можно применять не только для вестерн-анализа, но также для очистки ТРО при помощи колонок с антителами и любых общепринятых иммунологических способов с применением антител.
6 районов в аминокислотной последовательности ТРО человека, показанной в SEQ 1D 7, которые, как предполагалось, имеют относительно подходящую антигенность, были выбраны (см. Таблицу 4). Были синтезированы пептиды типа тетрамерного пептида с множественными антигенами (MAP), соответствующие этим районам. Каждым пептидом иммунизировали 2 кроликов 8 раз в количестве 100 мг на каждую иммунизацию.
Отдельно синтезировали также мономерный пептид, в котором остаток цистеина был связан с С-концом каждого пептидного района, показанного в Таблице 4. Этот пептид затем использовали в качестве тест-антигена для определения титра антител с применением иммуноферментного анализа. В результате для приготовленных сывороток было показано увеличение титра антител, так что эти сыворотки применяли в качестве антисывороток.
Антитела были получены иммунизацией каждым антигенным пептидом двух кроликов для выработки антисывороток, направленных против такого пептида. Полученные два антитела против пептида НТ1 для различения называют антитело против пептида НТ1-1 и антитело против пептида НТ1-2, как полученные в двух отдельных кроликах.
Пример очистки антител против пептида НТ1 1 будет показан ниже.
Сначала 30 мг мономерного пептида НТ1, соединенного с остатком цистеина, связывали с 12 мл Sulfo-Link связывающего геля (Pierce, кат. 44895). Вкратце, раствор пептида, содержащий антиген, связывали в течение 15 минут с гелем, уравновешенным связывающим буфером (50 мМ Трис, 5 мМ ЭДТА-Na рН 8,5) в количестве 6 объемов к объему геля. Затем после инкубирования в течение 30 минут гель промывали свызывающим буфером в количестве 3 объемов на объем геля. Связывающий буфер, содержащий 0.05 ML -цистеин -НС1, добавляли к гелю в количестве 1 мл/мл геля для блокирования непрореагировавших радикалов в течение 15 минут. После инкубирования в течение 30 минут гель промывали связывающим буфером (8 объемов на 1 объем геля). Все реакции связывания проводили при комнатной температуре. Таким образом, пептид был ковалентно связан с гелем с эффективностью связывания 28,3%, и была приготовлена колонка с антигеном, содержащая 0,8 мг пептида на 1 мл геля.
76,7 мл (содержание белка 3620 мг) антисыворотки, содержащей антитело против пептида НТ1-1, которую получали из одного из кроликов в общем количестве 78,4 мл, наносили на колонку с антигеном, предварительно уравновешенную 50 мМ фосфатным буфером (рН 8), содержащим 150 мМ NаС1 и 0,05% азид натрия, и затем промывали тем же буфером, получая 105,9 мл проходящей через колонку фракции (содержание белка 3680 мг). Затем элюировали адсорбированную фракцию 0,1 М цитратным буфером (рН 3,0) и сразу же нейтрализовали 21,1 мл 0,1 М карбонатного буфера (рН 9,9) с последующим концентрированием посредством ультрафильтрации (с применением мембраны Amicon УМ 30), получая очищенное антитело против пептида НТ1-1 в растворе 50 мМ фосфатного буфера (рН 8), содержащего 150 мМ NаСl и 0,05% NaN3.
Подобным образом получали следующие антитела: антитело против пептида НТ1-2 (60,0 мг); антитело против пептида НТ2-1 (18,8 мг); антитело против пептида НТ2-2 (8,2 мг); и т.п. Так же можно получить антитела против пептидов НТ3-НТ6.
3 мг аффинно-очищенных антител против пептида НТ1-1, против пептида НТ1-2, против пептида НТ2-1 или против пептида НТ2-2 биотинилировали связыванием с активированным биотином (Pierce, NHS-LC-Biotin 11, кат. 21336).
Было обнаружено, что все указанные выше очищенные антитела узнают и детектируют ТРО человека при анализе Вестерн-блоттингом (на нитроцеллюлозном фильтре или на PVDF) после SDS-PAGE рекомбинантного стандартного ТРО человека, частично очищенного из культурального супернатанта СНО клеток, в которые был введен ген, кодирующий аминокислотную последовательность, показанную в Таблице 4, и в которых этот ген экспрессировался.
Таблица 4
Аминокислотные последовательности ТРО человека, включенные в синтетический пептидный антиген типа тетрамерного MAP
Пептидный район НТ1 ТРО человека (номера аминокислот 8-28): DLRVLSKLLRDSHVLHSRLSQ (SEQ 1D 119)
Пептидный район НТ2 ТРО человека (номера аминокислот 47-62): SLGEWKTQMEETKAQD (SEQ 1D 120)
Пептидный район НТ3 ТРО, человека (номера аминокислот 108-126): LGTQLPPQGRTTAHKDPNA (SEQ 1D 121)
Пептидный район НТ4 ТРО человека (номера аминокислот 172-190): NELPNRTSGLLETNFTASA (SEQ 1D 122)
Пептидный район НТ5 ТРО человека (номера аминокислот 262-284): SLPPNLQPGYSPSPTHPPTGQYT (SEQ 1D 123)
Пептидный район НТ6 ТРО человека (номера аминокислот 306-332): PSAPTPTPTSPLLNTSYTHSQNLSQEG (SEQ 1D 124)
Пример 46 Приготовление колонки с антителами против пептидов ТРО
Поскольку антитело против пептида ТРО крысы, полученное в Примере 45, было способно узнавать молекулы крысиного и человеческого ТРО, фракции иммуноглобулина (1gG) антитела против пептида RT1 и антитела против пептида RT2 были соединены с химически активированным гелевым носителем для приготовления колонок с антителами против пептидов ТРО следующим образом. Каждое из двух антител, используемых в качестве таких материалов, получали из сывороток двух кроликов отдельно. Т.е., антитела против пептида RT1, полученные из двух кроликов, были соответственно названы антителом против пептида RТ1-1 и антителом против пептида RТ1-2, а антитела против пептида RТ2, полученные из двух других кроликов, были соответственно названы антителом против пептида RT2-1 и антителом против пептида RT2-2. Поскольку эти антитела использовали отдельно для приготовления колонок с антителами, получили всего 5 колонок, а именно, две колонки с антителами против пептида RT1 (называемые далее колонкой с анти-RT1-1 антителами и колонкой с анти-RT1-2 антителами), две колонки с антителами против пептида RT2 (называемые далее колонкой с анти-RТ2-1 антителами и колонкой с анти-RT2-2 антителами) и одну колонку (колонку со смесью антител анти-RT1--2+2-1), которую готовили смешиванием анти-RТ1-2 антител и анти-RТ2-1 антител и связыванием этой смеси с гелем.
Каждое антитело растворяли в растворе 50 мМ фосфата натрия и 0,15 М NаС1 (рН 8,0) до конечной концентрации 5 мг/мл или 2,5 мг/мл каждого из равномерно смешанных анти-RT1-2 антитела и анти-RТ2-1 антитела в случае колонки со смесью анти-RT1-1+2 антител) и 2,31 мл полученного раствора смешивали с 1,54 мл в объеме набухшего формил-активированного геля (Formyl-Cellulofine, Chisso) и подвергали 2-часовой реакции связывания при 4oС. Затем добавляли 1,1 мл раствора 10 мг/мл восстанавливающего агента (триметиламинборана (ТМАВ); изготовляемого Seikagaku Kogyo, кат. 680246), с последующими 6 часами дополнительной реакции связывания и с последующим центрифугированием для извлечения геля. 10 мл очищенной воды добавляли к этой порции геля, опять центрифугировали и непрореагировавшие молекулы антител удаляли повторением этой стадии 4 раза. Затем полученный гель смешивали с 4,6 мл блокирующего раствора (0,2 М фосфат натрия и 1 М этаноламин, рН 7,0) и 1,1 мл раствора восстанавливающего агента и обрабатывали при 4oС по меньшей мере в течение 2 часов для блокирования активных групп непрореагировавшего геля. После этого гель промывали очищенной водой и DPBS с использованием центрифуги, упаковывали в трубку для небольшой колонки, промывали 3 М раствором тиоцианата калия и 0,1 М раствором глицина-НС1 (рН 2,5), уравновешивали DPBS и затем хранили.
В 5 гелях с антителами против пептидов ТРО колонки с анти-RT1-1 антителами, колонки с анти-RT1-2 антителами, колонки с анти-RT2-1 антителами, колонки с анти-RТ2-2 антителами и колонки со смесью анти RТ1-2+2-1 антител, эффективность связывания соответствующих фракций lgG достигала 97,4%, 95,4%, 98,4%, 98,3% и 99,4%. Кроме того, количества фракций lgG, связанные на объем геля, были 5,6 мг/мл геля, 5,8 мг/мл геля, 5,7 мг/мл геля, 5,7 мг/мл геля и 2,9 мг/мл геля, соответственно. Поэтому, т.к. было подтверждено, что эффективность связывания и количество связывания не изменялось значительно в зависимости от происхождения антител и различия в антигенах, эксперименты, показанные в следующем Примере 47, проводили с использованием этих колонок с антителами.
Пример 47. Очистка ТРО из культурального супернатанта, полученного трансфекцией СOS 1 клеток экспрессирующим вектором рНТР1, при помощи колонки с анти-ТРО антителами с последующей колоночной хроматографией с обращенной фазой и подтверждение его биологической активности
Для оценки следующих очищенных проб применяли М-07е тест-систему в качестве теста in vitro. Частично очищенные пробы ТРО из культурального супернатанта, полученного в Примере 35 с применением СОS 1 клеток, трансфицированных экспрессирующим вектором рНТР1 (фракции, иные, чем основная ТРО фракция, а именно проходящая фракция F1 и фракция F3, элюированная после F2 промыванием 20 мМ цитрата натрия, рН 5,8, колонки Масго-Рrер Methyl H1C и фракции F1 и F3 колонки SP Sepharose Fast Flow) объединяли для получения фракции субпула ТРО (5453,79 мл; концентрация белка 0,490 мг/мл; общий белок 2676 мг; относительная активность 12100 и общая активность 32380000) и концентрировали с применением ультрафильтрационного устройства (Omega Ultraset, номинальное отсечение мол. массы 8000, изготовляемого Filtron), растворитель концентрированной фракции заменяли на DPBS и затем полученную пробу малого объема обрабатывали на ультрафильтрационном устройстве с УМ-10 мембраной (Amicon), получая в конце концов пробу в 120,2 мл раствора DPBS, содержащего 0,05% азида натрия. Затем все 5 гелей с антителами против пептидов ТРО, полученные в Примере 46, смешивали и готовили единую колонку с антителами (диаметр 1,6 см и высота 4,8 см, называемую колонкой смеси анти-RТ1+2 антител). Фракцию ТРО субпула наносили на эту колонку при скорости течения 0,033 мл/мин. После завершения загрузки колонки проводили элюцию DPBS для сбора элюатов до тех пор, пока УФ поглощение не становилось достаточно низким. Собранные таким образом элюаты концентрировали посредством ультрафильтрации с УМ-10 мембраной (Amicon), получая проходящую через колонку фракцию F1 (86,62 мл; концентрация белка 14,3 мг/мл; общий белок 1184 мг; относительная активность 67500; общая активность 79900000). Затем адсорбированную колонкой фракцию F2 (92,97 мл; концентрация белка 0,12 мг/мл; общий белок 11,1 мг; относительная активность 257000 и общая активность 2860000) элюировали кислым элюирующим раствором (0,1 М глицин-НС1, рН 2,5). Хотя проходящая через колонку фракция F1 все еще содержала ТРО активность, адсорбированный колонкой с антителами ТРО очищали далее, т.к. он обнаруживал увеличение относительной активности примерно в 20 раз, т.е. с применением 0,1% TFA в качестве проявляющего растворителя А и 1-пропанола, содержащего 0,05% ТFА в качестве проявляющего растворителя В проводили колоночную хроматографию на Capcell Pak Сl 300А (Shiseido, кат. С1 ТУРЕ: SG 300А; диаметр 4,6 мм и высота 150 мм, соединенной с пред-колонкой с диаметром 4,6 мм и высотой 35 мм) путем смешивания фракции F2, полученной из колонки с антителами, с 1/10 объема проявляющего растворителя В и нанесения смеси при скорости течения 0,4 мл/мин на колонку Capcell Pak С1 300А, уравновешенную заранее 20% В. После завершения загрузки колонки проводили элюцию в течение 5 минут 20% В и затем в течение 50 минут линейным градиентом плотности от 20% В до 40% В и элюаты собирали в полипропиленовые пробирки порциями по 1 мл (2,5 мин).
Порции 2 мкл (1/500 фракции) элюата в каждой пробирке смешивали с HSA и концентрировали ультрафильтрацией, получая 0,25 мл 1МDМ тест-культурального раствора, содержащего 0,02% НSА, для применения в идентификации ТРО-активных фракций этим способом. В результате высокая ТРО активность была обнаружена в пробах пробирок с номерами 20-23 (в диапазоне концентрации пропанола 28,5 и 32,5%). Кроме того, эти пробы подвергали вестерн-анализу согласно способу, описанному в Примере 45, и было подтверждено присутствие ТРО, который имел среднюю мол. массу 60000-70000 при измерении при восстанавливающих условиях на основании ДРС111 маркеров мол. масс, а также присутствие других молекул, имеющих соответствующие мол. массы 32000-43000 и 20000-30000.
Пример 48 Подтверждение биологической активности ТРО из культурального супернатанта, полученного трансфекцией COS 1 клеток экспрессирующим вектором рНТР1, и очищенного до стадии колонки Саpcеl Pak С1 300А
ТРО-активные фракции, очищенные в Примере 36, а именно пробирки с номерами 20-23 (в диапазоне концентрации пропанола 28,5 и 32,5%) колонки Capcell Pak C1 300А, объединяли, смешивали с 0,21 мл глицерина и затем концентрировали выпариванием при центрифугировании. Концентрат смешивали с 0,21 мл 6 М раствора гуанидингидрохлорида, разбавляли до объема 1 мл DРВS, буфер заменяли DРВS, содержащим 0,01 % HSА при помощи колонки Sephadex G-25 (NAP-10, Pharmacia Biotech, кат. 17-0854-01) и затем смешивали с 1,1 мл раствора DРВS, содержащего 0,01% НSА, получая в результате ТРО-активную фракцию FA (2,6 мл). Для испытания биологической активности этой пробы проводили тест in vivo. Эту активную фракцию вводили подкожно 1СR самцам мышей (8-недельного возраста), причем каждая группа включала 4 животных, ежедневно в течение 5 дней при дозе 100 мкл (имеющей общую активность 110000, измеренную тест-системой с М-07е). В качестве контроля так же вводили 100 мкл DРВS, содержащего 0,01% НSА, подкожно. Непосредственно перед введением и на следующий день после последнего введения собирали кровь из глазного дна для измерения чисел тромбоцитов при помощи микроцитометра (F800, Toa 1yo Denshi). В обработанной ТРО группе числа тромбоцитов увеличивались в среднем примерно в 1,42 раза после завершения введения по сравнению с величиной перед введением со значимой разницей между величинами (р<0,05, Student t-тест). На основании этих результатов было получено подтверждение, что вышеупомянутый ТРО человека, продуцируемый клетками млекопитающих, способен увеличивать количества тромбоцитов in vivo.
Пример 49. Подтверждение биологической активности неочищенной фракции ТРО, полученной из 33 л культурального супернатанта, полученного трансфекцией COS 1 клеток экспрессирующим вектором рНТР1, и частично очищенной катионообменной колонкой
1) М-07е тест-систему применяли в качестве in vitro теста для оценки следующих очищенных проб. Так же, как описано в Примере 36, в целом 33 л бессывороточного культурального супернатанта получали трансфекцией СOS 1 клеток экспрессирующим вектором рНТР1 и фильтровали через фильтр 0,22 мкм для получения супернатанта. Его концентрировали приблизительно в 10 раз при помощи ультрафильтрационного устройства (PLGC pellicon Cassette, номинальное отсечение мол. массы 10000, изготовляемого Millipore) и смешивали с 1 мМ в конечной концентрации ингибитором протеазы p-APMSF, получая пробу 2018 мл по объему (концентрация белка 3,22 мг/мл; общий белок 6502 мг; относительная активность 66000 и общая активность 429100000). Затем этот объем концентрировали повторно при помощи ультрафильтрационного устройства (Omega Ultraset, отсечение мол. массы 30000; Filtron), получая в результате фракцию с мол. массой 30000 или более (объем 1190 мл; концентрация белка 2,54 мг/мл; общий белок 3020 мг; относительная активность 82500 и общая активность 249000000) и фракцию с мол. массой 30000 и менее (объем 2947 мл; концентрация белка 0,471 мг/мл; общий белок 1402 мг; относительная активность 4500 и общая активность 6310000). Присутствие ТРО активности во фракции с мол. массой 30000 или менее указывает на возможность того, что этот культуральный супернатант содержит ТРО, мол. масса которого уменьшилась во время его экспрессии в клетках животных или во время его секреции из клеток животных. С другой стороны, фракцию 30000 или более после замены буфера на 20 мМ Na-цитратный буфер (6,1) наносили при скорости течения 5 мл/мин на колонку SP Sepharose Fast Flow (диаметр 2,6 см и высота 29 см), изготовляемую Pharmacia Biotech, кат. 17-0729-01), уравновешенную заранее 20 мМ натрий-цитратным буфером (рН 6,1). После завершения загрузки пробы колонку промывали и элюировали 20 мМ нитратным буфером (рН 6,1). Затем, с применением 20 мМ натрий-цитратного буфера (рН 6,1) в качестве проявляющего растворителя А и раствора, состоящего из проявляющего растворителя А и 1 М NаС1, в качестве проявляющего растворителя В проводили элюцию при скорости течения 3 мл/мин линейным градиентом от 0% В до 50% В в течение 215 минут и затем от 50% В до 100% В в течение 20 минут. Элюаты собирали по 30 мл (10 мин) в полипропиленовые пробирки. При проверке порции каждого из этих элюатов в М-07е-тест-системе для определения распределения активности элюция ТРО активности была обнаружена в широком диапазоне. Поэтому фракции, элюированные концентрацией NаС1 50 мМ или ниже, в том числе проходящую через колонку фракцию, объединяли в виде фракции F1, а основные ТРО фракции, элюированные 50-1000 мМ NаС1, объединяли в виде фракции F2. Объем фракции F1 был 1951 мл (концентрация белка 2,05 мг/мл; общий белок 3994 мг; относительная активность 13500 и общая активность 53900000). Объем фракции F2 был 649,8 мл (концентрация белка 1,11 мг/мл; общий белок 721 мг; относительная активность 268000 и общая активность 193000000).
2) Подтверждение биологической активности фракции F2 SP Sepharose Fast Flow
Порцию 2,5 мл фракции F2 SP Sepharose Fast Flow, отделенной в стадии (1), после обмена буфера с 3,5 мл раствора DPBS, содержащего 0,01% НS А, при помощи колонки Sephadex G-25 (NAP-25, Phаrmacia Biotech, кат. 17-0852-01) испытывали на биологическую активность ТРО в тесте in vivo. Т.е. эту активную фракцию вводили подкожно 1CR самцам мышей (8-недельного возраста), причем каждая группа содержала 4 животных, ежедневно в течение 5 последовательных дней при дозе 100 мкл (имеющих общую активность 123000, как измерено в М-07е-тест-системе). В качестве контроля 100 мкл DPBS, содержащего 0,01% НSА, вводили подкожно таким же образом. Непосредственно перед введением и на следующий день после последнего введения кровь собирали из глазного дна для измерения числа тромбоцитов при помощи микроцитомера (F800, Тоа 1 yо Denshi). В группе с введением ТРО числа тромбоцитов увеличились приблизительно в 1,29 раз в среднем после завершения введения по сравнению с величиной перед введением со значимой разницей (р<0,05, Student t-тест) между ними. Эти результаты показали, что вышеупомянутый ТРО человека, продуцируемый клетками млекопитающих, способен увеличивать количество тромбоцитов.
Пример 50. Конструирование рекомбинантного вектора, рDEF202- gh ТРО, для применения в экспрессии хромосомной ДНК ТРО человека в СНО клетках
Вектор pDEF 202 расщепляли рестриктазами Kpn1 и Sре 1 и затем подвергали электрофорезу в агарозном геле для выделения фрагмента, содержащего мышиный DHFR миниген и сигнал полиаденилирования SV40, и экспрессирующий вектор рDЕF 202-gh ТРО получали лигированием при помощи Т4 ДНК-лигазы (Takara Shuzo) полученного таким образом фрагмента с вектором ДНК, который был получен удалением содержащего сигнал полиаденилирования SV40 района из плазмиды рЕFНGТЕ путем ее обработки рестриктазами Крn1 и Sре1. Эта плазмида содержит начало (ориджин) репликации SV40, 1-α промотор фактора элонгации человека, район раннего полиаденилирования SV40 для транскрипции гена ТРО человека, мышиный DHFR миниген, начало репликации pUC 18 и ген β-лактамазы (Аmрr). Хромосомная ДНК ТРО человека соединена с сайтом в направлении 5'-3' от 1-α промотора фактора элонгации человека.
Пример 51. Экспрессия хромосомной ДНК ТРО человека в СНО клетках
СНО клетки (штамм dhfr-; Urlaub and Chasin, Proc. Natl. Acad.Sci.USA, vol. 77, p. 4216, 1980) выращивали культивированием их в α-минимальной основной среде (α-МЕМ(-), добавленной тимидином и гипоксантином), содержащей 10% фетальную бычью сыворотку, с применением чашки с диаметром 6 см (изготовляемой Falcon) и полученные клетки трансфицировали кальций-фосфатным способом (CellPhect, Pharmacia).
Т. е. 10 мкг плазмиды pDEF202-gh ТРО, полученной в Примере 50, смешивали с 120 мкл буфера А и 120 мкл Н2О и полученную смесь инкубировали при комнатной температуре в течение 10 минут. Этот раствор затем смешивали с 120 мкл буфера В и инкубировали при комнатной температуре еще в течение 30 минут. Полученный таким образом раствор ДНК помещали в чашку и культивировали в течение 6 часов в СO2-инкубаторе. После удаления среды и промывания полученной чашки дважды α-МЕМ(-) добавляли α-МЕМ(-), содержащую 10% диметилсульфоксид, к чашке и обрабатывали ее 20 минут при комнатной температуре. Затем добавляли неизбирательную среду (α-МЕМ(-) с добавлением гипоксантина и тимидина), содержащую 10% диализованную FCS, и культивировали в течение 2 дней и затем проводили отбор с применением избирательной среды (α-МЕМ(-), не содержащей гипоксантина и тимидина), содержащей 10% диализованную FCS. Отбор проводили обработкой клеток трипсином, распределением обработанных клеток в одной чашке (с диаметром 6 см) на 5 чашек с диаметром 10 см или 20 чашек типа 24-луночных планшетов и продолжением культивирования этих клеток со сменой избирательной среды каждые 2 дня. Присутствие ТРО активности человека подтверждалось в культуральном супернатанте в этих чашках или лунках, в которых наблюдалась пролиферация клеток при измерении активности ТРО человека при помощи Ва/F3 теста.
В этом случае трансфекцию СНО клеток можно также проводить котрансфекцией СНО клеток плазмидами рЕFНGТЕ и pMG1.
Пример 52. Конструирование вектора для применения в экспрессии в Е.соli белка ТРО человека (аминокислотные остатки положений 1-163) (называемого далее "hМКТ(1-163)", в котором остаток Lus и остаток Met были соответственно добавлены к положениям -1 и -2, и подтверждение его экспрессии
Для экспрессии белка, аминокислотные остатки которого в положениях 1 и 3 такие же, что и в белке ТРО человека (аминокислотные остатки 1-163), был сконструирован вектор рСFМ536/hМКТ(1-163), также называемый [Met-2, Lys-1] ТРО (1-163), для применения в экспрессии hМКТ (1-163) в E.coli тем же способом, который описан в Примере 42, с использованием следующих синтетических олигонуклеотидов 1-13, 2-13, 3-3 и 4-3, представленных в табл.VIII.
Эта экспрессирующая плазмида содержит последовательность ДНК, показанную в списке последовательностей (SEQ 1D 12). После этого экспрессию hМКТ ( 1-163) индуцируют так же, как описано в Примере 43.
При обработке полученного таким образом экспрессированного белка SDS-РАGЕ, переносе на PVDF мембрану и анализе N-концевой аминокислотной последовательности было подтверждено, что этот белок содержит аминокислотную последовательность hМКТ(1-163), которая должна была экспрессироваться.
Пример 53. Укладывание цепи ТРО человека, h6Т(1-163), полученного из ТРО человека, экспрессируемого в Е.coli, с применением гуанидингидрохлорида и глутатиона и очистка и подтверждение его биологической активности
Порции 1,2 г замороженных клеток продуцирующего h6T(1-163) рекомбинантного штамма, полученного в Примере 43, суспендировали в 3 мл воды и разрушали при помощи дезинтегратора высокого давления. Центрифугированием этой суспензии осажденную фракцию извлекали и большую часть примесных белков, клеточных компонентов и т.п. удаляли. Полученную таким образом осажденную фракцию, содержащую h6T(1-163), суспендировали в 4 мл конечного объема воды, 1 мл 1 М Триc-буфера (рН 8,5) добавляли к этой суспензии при перемешивании с последующим добавлением 20 мл 8 М гуанидинхлорида с последующим перемешиванием в течение 5 минут при комнатной температуре для растворения содержимого. Полученный раствор разбавляли в 10 раз 20 мМ Трис-буфера (рН 8,5), 5 мМ глутатион восстановленного типа и 0,5 мМ глутатион окисленного типа растворяли в этом разведенном растворе с перемешиванием и затем приготовленный таким образом раствор инкубировали в течение ночи при 4oС. Посредством центрифугирования h6Т(1-163) извлекали в супернатантной жидкости. Порцию 160 мл полученной супернатантной жидкости концентрировали при помощи УМ10 ультрафильтрационной мембраны (Amicon) и буфер обменивали на DPBS, получая 3,4 мл в конечном объеме фракции (концентрация белка 2,18 мг/мл). При тщательном диализе этой фракции против 1 MDM культуральной среды и оценке при помощи тест-системы с крысиными CFU-MK была обнаружена высокая ТРО активность (относительная активность 58000).
Полученную ТРО-активную фракцию подвергали тестированию in vivo. Эту фракцию вводили подкожно 1СR самцам мышей (7-недельного возраста), причем каждая группа состояла из 4 животных ежедневно в течение 5 последовательных дней при дозе 170 мкл (имеющую общую активность 22000, как измерено в тест-системе с крысиными CFU-МК) на 1 животное. В качестве контроля 170 мкл DPBS подкожно вводили каждому из 6 животных контрольной группы таким же способом. Непосредственно перед введением и на следующий день после последнего введения кровь собирали из глазного дна для измерения числа тромбоцитов при помощи микроцитометра (F800, изготовляемого Toa Iyo Denshi). В группе с введением ТРО количества тромбоцитов увеличились в среднем приблизительно в 2,15 раза на следующий день после завершения введения по сравнению с величиной перед введением, и такой эффект оставался неизменным даже после 3 дней после завершения введения, показывая в 2,13 раз более высокую величину. Количества тромбоцитов на следующий день и на 3-й день после завершения введения были в 1,73 и в 1,80 раз выше в группе, обработанной ТРО, чем в контрольной группе, со значимой разницей между ними (р<0,001, Student t-тест). Эти результаты показали, что ТРО человека, продуцируемый E.coli и полученный описанным выше способом, способен увеличивать количества тромбоцитов in vivo.
Пример 54. Укладывание цепи ТРО человека, h6Т(1-163), экспрессируемого в E. coli, с применением N-лауроилсаркозината натрия и сульфата меди и очистка и подтверждение его биологической активности.
Порции по 0,6 г замороженных клеток продуцирующего h6Т(1-163) рекомбинантного штамма, полученного в Примере 43, суспендировали в 3 мл воды, разрушали дезинтегратором высокого давления и затем центрифугировали для извлечения осажденной фракции. После суспендирования осажденной фракции в 3,1 мл воды, 0,19 мл 1 М Трис-буфера (рН 9,2), 11,25 мкл 1 М ДТТ, 38 мкл 0,5 М ЭДТА и 0,38 мл 10% раствора дезоксихолата добавляли к суспензии с перемешиванием и полученную смесь перемешивали при комнатной температуре в течение 40 минут. Затем смесь центрифугировали для извлечения осажденной фракции и удаления большей части примесных белков, клеточных компонентов и т.п. Полученный осадок суспендировали в 4 мл воды и центрифугировали для извлечения осажденной фракции, содержащей h6Т(1-163). Полученную таким образом осажденную фракцию суспендировали в 3,8 мл воды и к этой суспензии при перемешивании добавляли 0,2 мл 1 М Трис-буфера (рН 8) и 1 мл 10% N-лауроилсаркозината с последующим перемешиванием в течение 20 минут при комнатной температуре для растворения содержимого. Полученный раствор смешивали с 5 мкл 1% сульфата меди и перемешивали в течение ночи (около 20 часов) при комнатной температуре. Полученный раствор центрифугировали для извлечения супернатантной фракции, содержащей h6Т(1-163) и порцию 5 мл этого супернатанта смешивали с 5 мл воды и 10 мл 20 мМ Трис-буфера (рН 7,7) и затем с 2,6 г ионообменной смолы 20-50 меш Dowex 1-х4 хлоридной формы с последующим перемешиванием в течение 90 минут. Эту ионообменную смолу удаляли при помощи стеклянного фильтра для извлечения неадсорбированной смолой фракции, которую затем центрифугировали для извлечения супернатантной жидкости. Ее наносили на катионообменную колонку SP Sepharose Fast Flow, уравновешенную предварительно 20 мМ Трис-буфером (рН 7,7), и элюировали тем же буфером при помощи линейного градиента 0-500 мМ NaC1. После тщательного диализа каждой из элюированных фракций из этой колонки против 1MDM культуральной среды и оценки их в тест-системе с крысиными CFU-МК высокая ТРО активность (относительная активность 19000000) была обнаружена во фракции, элюированной с концентрацией NаС1 приблизительно 100 мМ. Эту фракцию концентрировали в 1,6 раз (2,5 мл; концентрация белка 25 мкг/мл) при помощи ультрафильтрационного устройства (Ultra Free CL, номинальное отсечение мол. массы 5000, изготовляемого Millipore, кат. UFC4LCC25). При анализе концентрированной фракции при помощи SDS-РАGЕ в присутствии восстанавливающего агента полосу, соответствующую ТРО, обнаруживали в виде основной полосы (чистота 70-80%).
ТРО-активную фракцию, полученную описанным выше способом, испытывали в тесте in vivo, т.е. эту активную фракцию вводили подкожно 1CR самцам мышей (8-недельного возраста), причем каждая группа состояла из 4 животных, ежедневно в течение 5 последовательных дней при дозе 100 мкл (имеющих общую активность 47500, как измерено в тест-системе с крысиными CFU-MK) на 1 животное. В качестве контроля 100 мкл 20 мМ Tpиc-буфера (рН 7,7), содержащего 100 мМ NaС1, вводили подкожно таким же способом. Непосредственно перед введением и на следующий день после завершения введения пробы крови брали из глазного дна для измерения количеств тромбоцитов при помощи микроцитометра (F800, Toa Iyo Denshi). В обработанной ТРО группе количества тромбоцитов увеличились в среднем примерно в 1,73 раз после завершения введения в сравнении с величиной перед введением и были в 1,59 раз выше, чем в контрольной группе со значимой разницей (р<0,01, Student t-тест) между ними. Эти результаты показали, что ТРО человека, продуцируемый E.coli и полученный описанным выше способом, обладает способностью увеличивать количества тромбоцитов in vivo.
Пример 55. Широкомасштабная культура СНО клеток
Широкомасштабное культивирование продуцирующей ТРО человека линии клеток СНО (СНО 28-30 клетки, устойчивые к 25 нМ МТХ), которая была получена трансфекцией экспрессирующей ТРО человека плазмиды рDЕF202-hTРО-Р1 в СНО клетки в Примере 32, проводили следующим образом. СНО клеточную линию культивировали и размножали в DMEM/F-12 культуральной среде (G1ВСО), содержащей 25 нм МТХ и 10% FCS. После отделения клеток раствором трипсина 1•107 клеток инокулировали в роллерный флакон (ex Falcon, Falcon 3000), содержащий 200 мл той же самой среды, и затем культивировали при 37oС при скорости вращения 1 об/мин в течение 3 дней. После 3 дней культивирования культуральный супернатант удаляли отсасыванием и прилипшие СНО клетки затем ополаскивали 100 мл РВS. 200 мл среды DМЕМ/F-12 (G1ВСО), не содержащей ни 25 нМ МТХ, ни 10% FCS, добавляли к флакону и клетки культивировали при 37oС при скорости вращения 1 об/мин в течение 7 дней. После 7 дней культивирования культуральный супернатант извлекали как исходный материал для последующей процедуры очистки. Описанные выше процедуры проводили в 500 роллерных флаконах, получая 100 л культурального супернатанта.
Пример 56. Очистка ТРО человека из продуцирующей ТРО человека СНО клеточной линии
1) Приблизительно 100 л бессывороточного культурального супернатанта, полученного в Примере 55, фильтровали через фильтр 0,22 мкм, получая фильтрат, который затем концентрировали на ультрафильтрационном устройстве (PLTK Pellicon cassette, номинальное отсечение мол. массы 30000, ex Millipore). После замены растворителя этого концентрата чистой водой рАРМ SF (Wako Pure Chemicals) и Pefabloc SC (Merck), являющиеся ингибиторами протеиназы, добавляли к полученному водному раствору при конечных концентрациях 1 мМ и 0,35 мМ соответственно, получая 3628 мл фракции мол. массы 30000 или более (концентрация белка 1,60 мг/мл; общий белок 5805 мг; относительная активность 1230000 и общая активность 7149000000). Фракцию затем подвергали Вестерн-блоттингу, как описано в Примере 45. В результате присутствие белка ТРО было обнаружено при мол. массе между 66000 и 100000. При более детальном исследовании было обнаружено, что более низкомолекулярные виды ТРО, чем этот ТРО, содержались в проходящей через колонку фракции.
Ультрафильтрат, содержащий ТРО с мол. массой не более 30000, концентрировали отдельно при помощи ультрафильтрационного устройства (PLGC Pellicon cassette, номинальное отсечение мол. массы 10000, Millipore), получая 1901 мл фракции низкомолекулярного ТРО (концентрация белка 0,36 мг/мл; общий белок 684 мг; относительная активность 245500; общая активность 167900000). Это показывает, что во время культивирования были образованы эти низкомолекулярные виды ТРО.
Затем к 3614 мл фракции, содержащей ТРО с мол. массой 30000 или выше, добавляли 764 г сульфата аммония и 144,5 мл 0,5 М содержащего цитрат натрия буфера (рН 5,5), получая 4089 мл раствора, содержащего 1,41 М (конечная концентрация) сульфата аммония и 17,7 мМ (конечная концентрация) содержащего цитрат натрия буфера. После удаления нерастворимых веществ из этого раствора центрифугированием был получен прозрачный раствор.
Этот прозрачный раствор затем наносили на колонку Масrо-Prep Меthyl Н1С (Вiо-Rаd, кат. 156-0080; диаметр 5 см, высота слоя 24,5 см), предварительно уравновешенную 20 мМ цитратным буфером (рН 5,5), содержащим 1,2 М сульфат аммония, при 25 мл/мин скорости течения. После завершения загрузки проводили элюцию 20 мМ содержащим цитрат натрия буфером (рН 5,5), содержащим 1,2 М сульфат аммония. Элюированную фракцию затем концентрировали на ультрафильтрационном устройстве (ex Filtron, Omega Ultrasette, номинальное отсечение мол. массы 8000), получая проходящую через колонку фракцию F1 (4455 мл; концентрация белка 0,400 мг/мл; общий белок 1780 мг; относительная активность 1831000).
Затем 20 мМ цитрат Nа (рН 6,0) в качестве элюирующего буфера пропускали через колонку, получая элюат, который затем концентрировали на том же самом ультрафильтрационном устройстве Omega Ultrasette, номинальное отсечение мол. массы 8000), собирая фракцию F2 (1457 мл; концентрация белка 0,969 мг/мл; общий белок 1411 мг; относительная активность 1715000). Во фракции F2 присутствие белка, имеющего мол. массу от 66000 до 100000, было подтверждено на SDS-PAGE. Вестерн-анализ показал, что этот белок представлял собой ТРО.
Затем F2, элюированную из колонки Масrо-Рrер Methyl Н1С (1443 мл), наносили на колонку SР Sepharose Fast Flow (Parmacia Biotech, кат. 17-0729-01; диаметр 5 см, высота слоя 12 см), уравновешенную предварительно 20 мМ Nа-цитратным буфером (рН 6,0) при скорости течения 15 мл/мин. После завершения нанесения проводили элюцию 20 мМ Nа-цитратным буфером (рН 6,0), содержащим 50 мМ NаС1. Элюат собирали и он был назван фракцией F1 (3007 мл; концентрация белка 0,226 мг/мл; общий белок 679 мг; относительная активность 88830). После этого элюирующий буфер заменяли 20 мМ Nа-цитратным буфером (рН 5,4), содержащим 750 мМ NаС1, для сбора элюированной фракции F2 (931 мл; концентрация белка 0,753 мг/мл; общий белок 710 мг; относительная активность 5558000), которую затем концентрировали до 202 мл при помощи ультрафильтрационного устройства (Filtron, Omega Ultrasette, номинальное отсечение мол. массы 8000; и Omicon УМ 3 мембрана).
Затем концентрированную содержащую ТРО активность фракцию F2 (197 мл) наносили на гель-фильтрационную колонку Sеphacryl S -200H (Parmacia Biotech, кат. 17-0584-05; диаметр 7,5 см, высота слоя 199 см) при скорости течения 3 мл/мин. Элюированные объемы 1200-1785 мл, 1785-2010 мл, 2010-2280 мл и 2280-3000 мл собирали отдельно, получая фракции F1 (585 мл; концентрация белка 1,00 мг/мл; общий белок 589 мг; относительная активность 4118000), F2 (225 мл; концентрация белка 0,263 мг/мл; общий белок 59,2 мг; относительная активность 2509000), F3 (270 мл; концентрация белка 0,119 мг/мл; общий белок 32,1 мг; относительная активность 2535000) и F4 (720 мл; концентрация белка 0,0467 мг/мл; общий белок 33,6 мг; относительная активность 1155000), соответственно. Таким образом, фракционированная ТРО активность имела широкий диапазон мол. масс, как определено гель-фильтрацией. После SDS-РАGЕ фракцией с последующим Вестерн-блот-анализом было найдено, что F1 имела ТРО молекулы с мол. массой 66000-100000 (преимущественно); F2 содержала ТРО молекулы с мол. массой 32000-60000; и F3 содержала ТРО молекулы с мол. массой 32000-42000. Все типы молекул ТРО обладали ТРО активностью.
На основании результата секвенирования N-концевых аминокислот молекулы ТРО с мол. массой 66000-100000 мы подтвердили, что ТРО молекула имела аминокислотную последовательность белка, кодируемого геном ТРО человека.
Кроме того, ТРО молекула, имеющая мол. массу 66000-100000, была подвергнута экспериментам с ферментативным расщеплением с применением гликозидазных ферментов, состоящих из N-гликаназы (ex Genzyme, кат. 1472-00), нейраминидазы (Nakarai-Tesqu, кат. 242-29 Р), эндо-а-N-ацетилгалактоаминидазы (Seikagaku Kogyo, кат. 100453) и О-гликозидазы (Boehriger Mannheim Biochemica, кат. 1347101), отдельно или в комбинации, а затем SDS- PAGE анализу. В результате было обнаружено, что эта полипептидная часть ТРО имеет мол. массу приблизительно 36000, как и ожидалось на основании его теоретической мол. массы, и представляет собой гликопротеин как с N-связанными, так и О-связанными сахарными цепями.
Пример 57. Очистка ТРО человека из продуцирующей ТРО человека СНО клеточной линии
1) К 1 л бессывороточного культурального супернатанта СНО клеток, полученного, как описано в Примере 55, добавляли 211,4 г сульфата аммония, после чего смесь фильтровали через фильтр 0,2 мкм (ех Yelman Science, кат. 12992). Полученный фильтрат наносили на колонку Масrо-Рrер Methyl Н1С (Bio-Rad, кат. 156-0081, диаметр 50 мм, высота слоя 90 мм), уравновешенную предварительно 20 мМ Nа-ацетатным буфером (рН 5,6), содержащим 1,2 сульфат аммония, при скорости течения 15 мл/мин. После завершения нанесения проводили элюцию 450 мл 20 мМ Nа-ацетатным буфером (рН 5,6), содержащим 1,2 М сульфат аммония. Затем элюированную фракцию наносили на колонку Vydac C4 с обращенной фазой (The Separation Grouh, кат. 214ВТР54, диаметр 4,6 мм, высота 250 мм) при скорости потока 0,75 мл/мин. После промывания колонки 10 мМ Трис-буфером (рН 6,4), содержащим 5% этанол (называемым проявляющим растворителем А) в течение 15 минут проводили элюцию в течение 66 минут линейным градиентом от проявляющего растворителя А до 10 мМ Трис-буфера (рН 6,4), содержащего 94% этанол (называемого проявляющим растворителем В). Полученная хроматограмма показана на фиг.15. В результате SDS-РАGЕ анализа молекулы, имеющие мол. массу 65000-100000, т.е. предполагаемые молекулы ТРО, соответствующие фракции со временем удерживания 68-72 минуты после добавления пробы, появились на геле SDS-PAGE в виде единственной полосы (см. фиг.16). Последующий Вестерн-анализ показал, что полученный белок представляет собой ТРО.
Анализ N-концевых аминокислот этой пробы белка обнаружил, что белок имел аминокислотную последовательность белка, кодируемого геном ТРО человека.
Пример 58. Получение рекомбинантного вируса для экспрессии ТРО человека в клетках насекомых
Плазмиду pHTP1, полученную в Примере 30, расщепляли рестриктазами EcoR1 и Notl и затем подвергали электрофорезу в 1% агарозном геле, получая полосу 1200 п.н., которую затем очищали при помощи набора для очистки ДНК Prep-A-Genе. Затем очищенную ДНК лигировали с транспортным вектором рVL 1393 (1nvitrogen), предобработанным теми же рестриктазами, с последующей трансформацией в Высококомпетентные клетки штамма Е.соli (Toyo Boseki). Из полученных колоний был отобран клон pVL 1393/h ТРО, содержащий кодирующий район полной длины кДНК ТРО человека. Затем из этого клона получали плазмидную ДНК способом, описанным в Molecular Cloning (Sambrook et al., Cold Spring Habor Laboratory Press, 1989), с последующей трансфекцией в клетки насекомых Sf 21 1nvitrogen) с использованием набора для трансфекции BaculoGoldТМ (Farmingen). Трансфицированные клетки культивировали в Sf-900 среде (Lifetechnology) при 27oС в течение 4 дней, собирая супернатант, содержащий вирус. Супернатант разбавляли до 1:109-1:105 и приблизительно 7•105 Sf 21 клеток инфицировали 1 мл разбавленного супернатанта при 27oС в течение 1 часа в чашке с диаметром 35 мм. После удаления супернатанта 1% горячую агарозу в Sf-900 среде помещали в эту культуру и давали отвердеть, после чего кульвитировали в течение 6 дней при 27oС во влажной атмосфере. Единственную образовавшуюся бляшку выскребали и из нее вирус выделялся в 200 мкл Sf-900 среды, Sf 21 клетки инфицировали полученным вирусным клоном в 24-луночном планшете для размножения вируса. Часть содержащей вирус жидкости, полученной из единственной бляшки, обрабатывали смесью фенол/хлороформ с последующим осаждением этанолом для выделения вирусной ДНК, которую затем использовали в качестве матрицы для проведения РСR с применением праймеров, специфических для кДНК ТРО человека. Рекомбинантный вирус, содержащий кДНК ТРО человека, отбирали на основе амплификации специфического фрагмента ДНК. Затем Sf 21 клетки инфицировали супернатантом, содержащим этот рекомбинантный вирус, несущий кДНК ТРО человека, для размножения вируса.
Пример 59. Экспрессия ТРО человека в Sf 21 клетках насекомых и идентификация ТРО активности
Sf 21 клетки культивировали в 175 см2 культуральной колбе до достижения 80% слияния с последующим инфицированием рекомбинантным вирусом, несущим кДНК ТРО человека, полученным в Примере 58, при 27oС в течение 1 часа, после чего полученные клетки культивировали в Sf-900 среде при 27oС в течение 4 дней для получения супернатанта культуры. Полученный культуральный супернатант подвергали смене среды на 1MDM культуральную среду на колонке NАРTM-5 (Pharmacia). Значительную ТРО активность в полученных супернатантах обнаружили в зависимом от дозы виде как в тесте с крысиными СFU-МК, так и в М-07е-тесте. Рекомбинантный ТРО человека, экспрессируемый в Sf 21 клетках, идентифицировали также посредством Вестерн-анализа, описанного в Примере 45.
Пример 60. Укладка цепи вариантного ТРО человека, h6Т(1-163), полученного из клона pCFМ536/h6Т(1-163), несущего последовательность оснований ТРО человека, путем его экспрессии в Е.coli, при помощи N-лауроилсаркозината натрия и сульфата меди и очистка h6Т(1-163)
30 г замороженного продуцирующего h6Т(1-163) рекомбинантного микроорганизма, полученного в Примере 43, суспендировали в 300 мл воды, разрушали в дезинтеграторе высокого давления (10000 psi, Rannie High Pressure Laboratory) и затем центрифугировали, извлекая осажденную фракцию. Эту фракцию осадка суспендировали в 90 мл воды и при перемешивании добавляли воду до общего количества 150 мл. Затем к этой смеси добавляли 9 мл 1 М Трис-буфера (рН 9,2), 540 мкл 1 М ДТТ, 1,8 мл 0,5 М ЭДТА и 18 мл 10% деоксихолата натрия при перемешивании с последующим перемешиванием в течение 30 минут при комнатной температуре. Осажденную фракцию извлекали центрифугированием и удаляли большую часть примесных белков и компонентов микроорганизмов. К этому осадку добавляли 180 мл 5 мМ ДДТ, получая суспензию, которую затем центрифугировали, собирая фракцию осадка, содержащую h6Т(1-163). К полученному осадку добавляли 300 мл воды, получая суспензию, к которой затем добавляли воду до количества жидкости 570 мл при перемешивании. 30 мл 1 М Трис-буфера (рН 8), 150 мл 10% N-лауроилсаркозина добавляли к этой смеси при комнатной температуре с 20 мин перемешивания для растворения h6Т(1-163). К раствору добавляли 750 мкл 1% сульфата меди и смесь перемешивали в течение ночи (примерно 20 часов) при комнатной температуре. После извлечения фракции супернатанта, содержащей h6Т(1-163), центрифугированием к 750 мл этого супернатанта добавляли 750 мл воды и 1500 мл 20 мМ Триc-буфера (рН 7,7) с последующим добавлением 3 мл полисорбата 80 (Nikko Chemicals) при перемешивании. К полученному раствору добавляли 600 г ионообменной смолы Dowex 1-X4 (20-50 меш, хлоридная форма) и перемешивали 90 минут при комнатной температуре. После извлечения неадсорбированной фракции при помощи стеклянного фильтра ионообменную смолу промывали 750 мл 20 мМ Трис-буфера (рН 7,7). Объединенный раствор неадсорбированной и промывной фракций доводили до рН 9,2 2N гидроксидом натрия, после чего его наносили на анионообменную колонку Q Sepharose Fast Flow (1D 5 см х 10 см), уравновешенную 20 мМ Трис-буфером (рН 9,2), содержащим 0,1% полисорбат 80, для извлечения неадсорбированной фракции. рН полученной неадсорбированной фракции доводили до 7,2 соляной кислотой, наносили на катионообменную колонку Sp Sepharose Fast Flow (1D 5 см х 10 см), уравновешенную 20 мМ Трис-буфером (рН 7,2), содержащим 0,1% полисорбат 80, и затем элюировали линейным градиентом от 0 до 500 М NаС1 в том же самом буфере. Элюированные фракции подвергали SDS-РАGЕ анализу для сбора фракции ТРО, к которой затем добавляли трифторуксусную кислоту до концентрации 0,1%. После нанесения полученного раствора на колонку Capcell Pak 5 мкм 300А Cl (1D 2,1 см х 5 см х 2, Shiseido) проводили НРLС с обращенной фазой по способу элюции линейным градиентом с увеличивающейся концентрацией 1-пропанола в 0,1% трифторуксусной кислоте. Элюат анализировали на SDS-РАGЕ в отсутствие восстанавливающего агента. В результате фракционировали три фракции на основе положения элюции в HPLC с обращенной фазой и каждую фракцию разбавляли в 3 раза 0,1% трифторуксусной кислотой. Каждую разбавленную фракцию наносили на колонку HPLС с обращенной фазой описанным образом. Полученные три фракции ТРО были обозначены как Fr. S-а, Fr. S-b и Fr. S-c, в зависимости от порядка элюции на НРLС с обращенной фазой (см. фиг.17). Их анализировали на SDS-PAGE в отсутствие восстанавливающего агента. В результате одна полоса была обнаружена при мол. массе приблизительно 18 кДа (Fr. S-а), приблизительно 19 кДа (Fr. s-b) или 18 кДа (Fr. S-с) (см. фиг.18). После их аминокислотного анализа каждый из аминокислотных составов почти совпадал с соответствующей величиной, определенной из информации последовательности. Дополнительно результаты N-концевого аминокислотного анализа показали, что были получены ожидаемые последовательности. Количество белка каждой фракции, определенное аминокислотным анализом, было 0,64 г (Fr.S-а), 1,81 мг (Fr. S-b) и 3,49 мг (Fr. S-с). После полного диализа каждой из фракций против 1МDМ среды проводили М-07е-тест. В результате относительная активность ТРО в каждой фракции была приблизительно 1620000 (Fr. S-a), 23500000 (Fr. S-b) и 746000000 (Fr. S-с).
Пример 61. Укладка цепи вариантного ТРО человека, h6Т(1-163), полученного из клона рСFМ536/ /h6Т, несущего последовательность оснований ТРО человека, через его экспрессию в Е.соli, при помощи гуанидинхлорида и цистеина-цистина и очистка h6Т(1-163)
50 г продуцирующего h6Т(1-163) замороженного рекомбинантного микроорганизма, полученного в Примере 43, добавляли к 500 мл воды и суспендировали, разрушали при помощи устройства высокого давления (10000 рsi, Rannie High Pressure Laboratory) и затем центрифугировали для извлечения фракции осадка. Фракцию осадка суспендировали в воде и количество жидкости доводили затем до 250 мл при перемешивании добавлением воды. К суспензии добавляли 15 мл 1 М Трис-буфера (рН 9,2), 900 мкл 1 М ДТТ, 3 мл 0,5 М ЭДТА и 30 мл 10% деоксихолата натрия и перемешивали при комнатной температуре в течение 30 минут. После центрифугирования извлекали фракцию осадка, оставляя в супернатанте большую часть примесных белков, компонентов микроорганизма и т.п. К осадку добавляли 300 мл 5 мМ ДТТ и суспендировали, после чего осажденную фракцию, содержащую h6Т(1-163), собирали центрифугированием. Полученную осажденную фракцию, содержащую h6Т(1-163), суспендировали в воде в общем объеме 104 мл. К суспензии добавляли 20 мл 1 М Трис-буфера (рН 8,5) и затем 376 мл 8 М гуанидинхлорида при перемешивании и затем перемешивали при комнатной температуре в течение 10 минут для растворения h6Т(1-163). К раствору добавляли 2500 мл 20 мМ Трис-буфера (рН 8,5), содержащего 0,1% полисорбат 80 и затем 2000 мл 20 мМ Трис-буфера (рН 8,5), содержащего 1 М гуанидинхлорид, при перемешивании с последующим добавлением 5 мМ цистеина и 0,5 мМ цистина. Полученный таким образом раствор инкубировали в течение ночи при 4oС и затем центрифугировали, получая h6Т(1-163) в супернатанте, который затем концентрировали при помощи ультрафильтрационной мембраны Prep Scale UF cartridge PLDC (Millipore). Буфер этого концентрата заменяли 20 мМ Трис-буфером (рН 9,2), содержащим 0,1% полисорбат 80, до достижения конечного объема 1000 мл. Полученный раствор наносили на анионообменную колонку Q Sepharose Fast Flow (1D 5 см х 10 см), уравновешенную 20 мМ Трис-буфером (рН 9,2), содержащим 0,1% полисорбат 80, и затем элюировали линейным градиентом от 0 до 500 М NаС1 в том же самом буфере, получая фракцию (240 мл), элюированную приблизительно при 20-150 мМ NаС1. Полученную фракцию разбавляли в 4 раза 20 мМ Трис-буфером (рН 7,2), доводя конечный объем до 960 мл, и затем доводили рН до 7,2 уксусной кислотой, наносили на катионообменную колонку SP Sepharose Fast Flow (1Д 5 см х 10 см), уравновешенную 20 мМ Трис-буфера (рН 7,2), содержащим 0,1% полисорбат 80, и затем элюировали линейным градиентом от 0 до 500 М NаС1 в том же самом буфере. Элюат анализировали на SDS-РАGЕ, собирая ТРО фракцию. К фракции ТРО добавляли трифторуксусную кислоту до конечной концентрации 0,1%. Полученный раствор наносили на колонку Capce11 Pak 5 мкм 300А C1 (1D 2,1 см х 5 см х 2, Shiseido), затем проводили НРLС с обращенной фазой при помощи способа элюции линейным градиентом с увеличивающейся концентрацией 1-пропанола в 0,1% трифторуксусной кислоте. Элюат анализировали SDS-PAGE в отсутствие восстанавливающего агента и фракционировали на две фракции в зависимости от положения при элюции в HPLС с обращенной фазой. Две фракции ТРО были обозначены как Fr. G-а и Fr. G-d на основе порядка их элюции в НРLС с обращенной фазой (см. фиг.17). Каждую фракцию затем анализировали на SDS-РАGЕ в отсутствие восстанавливающего агента. В результате обнаружили одну полосу при мол. массе приблизительно 18 кДа (Fr. G-а) или приблизительно 32 кДа (Fr. G-d) (cм. фиг.18). Далее SDS-РАGЕ анализ этих фракций в присутствии восстанавливающего агента показал, что в обеих фракциях была обнаружена полоса при мол. массе приблизительно 20 кДа. Результат аминокислотного анализа этих фракций показал, что каждый из аминокислотных составов почти совпадал с соответствующей теоретической величиной, определенной из информации последовательности. Дополнительно, результаты N-концевого аминокислотного анализа показали, что эти фракции содержали ожидаемые последовательности. Количество белка каждой фракции, определенное из результатов аминокислотного анализа, было 2,56 мг (Fr. G-а) или 1,16 мг (Fr. G-d ). После полного диализа каждой фракции против 1MDM среды проводили М-07е-тест. В результате эта фракция имела относительную активность ТРО приблизительно 3960000 (Fr. G-а) или приблизительно 7760000 (Fr. G-d).
Пример 62. Конструирование рекомбинантного вектора рDEF202-hТРO163 для экспрессии ТРО человека частичной длины (аминокислоты 1-163) (называемого далее hТРO163) внутри СНО клеток
Вектор pDEF202, сконструированный в Примере 31, обрабатывали рестриктазами ЕсоR1 и Sре1 с последующим извлечением большого векторного фрагмента при помощи электрофореза в агарозном геле. Этот фрагмент лигировали при помощи Т4 ДНК-лигазы (Takara-Shuso) с кДНК hТРO163, которую получали обработкой плазмиды рЕF18S-hТРO163, содержащей кДНК АТРO163, кодирующую аминокислоты -21-163 (SEQ 1D 13), рестриктазами EcoR1 и Spe1, получая экспрессирующий вектор pDEF202-hTPO163. Эта плазмида содержала начало репликации (ориджин) SV 40, 1-α-промотор фактора элонгации человека, сайт раннего полиаденилирования SV 40, мышиный DHFR миниген, начало репликации pUC18 и ген β-лактамазы (Аmpr), в которой кДНК hТРO163 соединен в сайте в направлении 5'-3' от 1-α-промотора фактора элонгации человека.
Пример 63. Экспрессия hТРO163 в СНО клетках
СНО клеточную линию (dhfr- штамм, Urlaub and Chasin, Proc. Natl. Acad. Sci. USA, 77, p. 4216, 1980) выращивали в α-минимальной основной среде (α-MEM(-) с тимидином и гипоксантином), содержащей 10% фетальную телячью сыворотку, в чашке 6 см (Faleon) и затем трансформировали плазмидой pDEF202-hTPO163 по способу трансфектума (Seikaga Ku Kogyo К.К.).
Вкратце, 20 мкг плазмиды рDЕF202-hТРO163, полученной в Примере 62, смешивали с 240 мкл 0,3 M NaC1, затем со смесью 20 мкл трансфектума и 220 мкл воды. Этот раствор ДНК добавляли по каплям к чашке и культивировали в течение 6 часов в СО2-инкубаторе. Среду удаляли из чашки, которую затем промывали дважды α-МЕМ(-) и добавляли содержащую 10% DMS О α-МЕМ(-) перед инкубированием 2 минуты при комнатной температуре. После этого к чашке добавляли 10% диализованную FCS-содержащую неизбирательную среду (α-MEM (-) с гипоксантином и тимидином) культивировали еще 2 дня, затем проводили отбор в содержащей 10% диализованную FCS избирательной среде (α-МЕМ(-) без гипоксантина и тимидина). Отбор проводили трипсинизацией клеток, распределением их на 5 чашек 10 см или 20 24-луночных чашек из 1 чашки (6 см) и продолжали культивирование со сменой среды с интервалами в 2 дня. Супернатанты из чашек или из лунок тестировали на ТРО активность при помощи Ва/F3-теста, наблюдая активность ТРО. Клетки, в которых наблюдалась ТРО активность в культуральных супернатантах, переносили на свежие чашки или в свежие лунки после разделения до концентрации клеток 1:15 избирательной средой, содержащей 25 нМ метотрексата. Затем клетки рекультивировали для роста и клонирования клеток, устойчивых к метотрексату.
Альтернативно, трансформацию СНО клеток можно проводить котрансфекцией рЕF18S-hТРO163 и рМG1 в СНО клетки.
СНО штамм (СНО-DUКХВ11), трансфицированный плазмидой pDEF202-hTPO163, был депонирован автором данной заявки 31 января 1995 г. под accession FERM ВР-4989 при National Institute of Bioscience and Human Technology, Agency of Industrial Science and Technology, Ministry of International Trade and Industry, Japan.
Пример 64. Крупномасштабная культура СНО клеток
Крупномасштабное культивирование продуцирующей hТРO163 СНО клеточной линии (СНО 109 клеток, полученных описанным выше отбором в содержащей 10% диализованную FCS избирательной среде [α-МЕМ(-) без гипоксантина и тимидина] ), которая была получена трансфекцией экспрессирующей hТРO163 плазмиды pDEF202-hTPO163 в СНО клетки в Примере 63, проводили следующим образом. СНО 109 клетки выращивали в DMEM/F-12 среде (G1ВСО) с 10% FCS. После сбора клеток (или трипсинизации) при помощи раствора трипсина 10 миллионов клеток инокулировали в роллерный флакон Фалькона (FALCON 3000), содержащий 200 мл той же самой среды, и культивировали при скорости вращения 1 об/мин, при 37oС в течение 3 дней. Культуральную среду удаляли отсасыванием и поверхность клеточной культуры ополаскивали 100 мл РВS. Затем к клеточной культуре добавляли 200 мл DMEM/F-12 (G1BCO) без 10% FCS и культивировали 7 дней при 37oС при 1 об/мин. Культуральный супернатант собирали и использовали в качестве исходного материала для последующей стадии очистки. Подобные процедуры проводили в 300 роллерных флаконах, получая 60 л бессывороточного культурального супернатанта.
Пример 65. Очистка КТРО163 из продуцирующей hТРO163 СНО клеточной линии
1) 60 л бессывороточного культурального супернатанта, полученного в Примере 64, фильтровали, собирая фильтрат, который затем концентрировали при помощи ультрафильтрационного устройства (Filtron, отсечение мол. массы 10000), получая концентрированную фракцию (600 мл, 11.2 мг/мл, общий белок 6430 мг). При помощи Вестерн-анализа этой фракции с применением антител против пептида НТ1 было показано присутствие белка hТРО163, экспрессированного при диапазоне мол. массы от 20000 до 26000.
Полученный концентрированный культуральный супернатант (537 мл) обрабатывали на колонке Sephadex G-25 Fine (Pharmacia Biotech, кат. 17-0032-02; диаметр 10 см и высота 30 см), предуравновешенной 10 мМ Nа-фосфатным буфером (рН 6,8), получая белковую фракцию F1 (938 мл, концентрация белка 4,9 мг/мл, общий белок 4594 мг) в виде раствора в 10 мМ Nа-фосфатном буфере (рН 6,8).
Эту белковую фракцию (929 мл) из колонки Sephadex G-25 Fine наносили при скорости потока 15 мл/мин на колонку SР Sepharose Fast Flow (Pharmacia-Biotech, кат. 17-0729-01; диаметр 5 см и высота 12 см), уравновешенную предварительно 10 мМ Nа-фосфатным буфером (рН 6,8), с последующей элюцией 10 мМ Nа-фосфатным буфером (рН 6,8), затем тем же буфером, содержащим 10% этанол. Элюаты объединяли в виде фракции F1 (1608 мл, концентрация белка 2,13 мг/мл, общий белок 3426 мг). Затем проводили вторую элюцию 10 мМ Nа-фосфатным буфером (рН 6,8), содержащим 750 мМ NаС1 и 25% этанол, собирая основной элюат hТРO163 фракции F2 (651 мл, концентрация белка 1,67 мг/мл, общий белок 1087 мг).
К содержащей ТРО активность фракции F2 (200 мл) из колонки SР Sepharose Fast Flow добавляли этанол и очищенную воду, получая 300 мл 45% (конеч. конц.) этанольного раствора. Нерастворимые вещества, образующиеся при добавлении этанола, удаляли центрифугированием. Супернатант вводили в колонку SOUR CE 15RPC (Pharmacia-Biotech, кат. 17-0727-02; диаметр 2 см, высота 20 см), уравновешенную предварительно 50% растворителя А (10 мМ Nа-ацетатный буфер, рН 6,7) плюс 50% растворителя В (10 мМ Nа-ацетатный буфер, рН 6,7, содержащий 90% этанол) при скорости течения 2 мл/мин. Затем колонку промывали 55% растворителя А плюс 45% растворителя В до полного удаления неадсорбированных веществ. После этого проводили элюцию при скорости течения 1,5 мл/мин в соответствии со следующим профилем элюции; 50% В в течение 5 минут; линейный градиент от 50% В до 100% В в течение 140 минут и затем 100% В в течение 35 минут. Фракции собирали при интервалах 5 минут (соответствующих 7,5 мл объема). Все фракции подвергали SDS-РАGЕ и затем Вестерн-анализу для испытания диапазона элюирования hТРO163. В результате было обнаружено, что высокоочищенный hТРO163 со средней мол. массой приблизительно от 20000 до 26000 элюируется в диапазоне 66-87% этанола. Среди этих белков hТРО 163 белок hТРO163 с мол. массой более высокой был элюирован ранее из колонки с обращенной фазой, предполагая, что гликозилированная молекула hТРO163 имеет повышенную гидрофильность.
К 88,8 мл элюированной фракции hТРO163 (т.е. фракции hТРO163 (90 мл), элюированной при диапазоне концентраций 68-86,5% этанола) из колонки SOUR CE 15RPC добавляли СНАРS, и эту смесь концентрировали и промывали, получая 2,5 мл концентрата, содержащего приблизительно 5% этанола и 4 мМ СНАPS. Этот концентрат затем наносили на колонку Superdex 75 рg (Pharmacia-Biotech, кат. 17-1070-01; диаметр 2,6 см и высота 60 см), уравновешенную заранее 10 мМ Nа-фосфатным буфером (рН 6,8), содержащим 10% этанол, при скорости течения 1,5 мл/мин. Начиная от времени 60 минут после нанесения, элюированные фракции собирали по 6 мл каждая (т.е. каждые 4 мин). В результате hТРO163 элюировался во фракциях с номерами пробирок 16-31, как определено при помощи SDS-PAGE. Это положение элюирования соответствует диапазону молекулярной массы приблизительно от 44000 до приблизительно 6000, как определено гель-хроматографией с применением стандартных маркеров мол. массы (смесь Bio-Rad, Gel Filtration Standard, кат. 151-1901; и Calbiochem insulin, кат. 407696). Дополнительно, эти молекулы hТРO163, по-видимому, элюируются в порядке уменьшения степени гликозилирования. Все типы hТРO163 пробирок 16-31 (элюирующий объем 180-276) собирали в виде фракции FA. Кроме того, фракции пробирок с номерами 16-18 (элюирующий объем 180-198 мл), 19-24 (элюирующий объем 198-234 мл) и 25-31 (элюирующий объем 234-276 мл) собирали отдельно и обозначали как фракции FH, FМ и FL, соответственно. Также фракцию FA получали объединением частей фракций FН, FМ и FL. Это показано на фиг.19.
2) Затем фракции hТРO163 FН, FM, FL и FА из колонки Superdex 75 рg, изложенные в (1), будут иллюстрированы более детально, N-концевые аминокислотные последовательности типов ТРO163, полученные выше, были определены тем же самым способом, который описан в Примере 1, но большую часть N-концевых остатков Ser не удалось идентифицировать. Это позволяет предположить, что O-связанный сахар добавлен к N-концевому Ser. Далее, было подтверждено, что последовательность после N-концевого Ser является аминокислотной последовательностью, ожидаемой на основании последовательности гена hТРО. Концентрации белков hТРO163 в FH, FM, FL и FA были 10,2 нг/мл, 6,2 нг/мл, 0,84 нг/мл и 3,2 нг/мл, как определено аминокислотным анализом (Асcq. TAQ method, Wasters), соответственно, при условии, что эти концентрации были концентрациями пептидных частей молекул, не содержащих сахарной цепи. Эти фракции (по 100 нг каждая) подвергали SDS -РАGЕ при невосстанавливающих условиях с применением мультигеля 15/25 (Dai-ichi Kagaku Jakuhin, 15-25% преформированный полиакриламидный гель) или при восстанавливающих условиях с применением ДТТ с последующим окрашиванием серебром (Daiichi Pure Chemicals). В результате было обнаружено, что каждая из этих фракций содержит высокоочищенный hТРO163. Средние мол. массы hТРO163 в FН, FM, FL и FA, как рассчитано при помощи DPC111 маркеров мол. массы (Daiichi Pure Chemicals) в качестве стандарта при восстанавливающих условиях, были 24000-21500, 23000-21000, 23000-20500 и 23500-20500, соответственно (см. фиг.20).
Кроме того, средние мол. массы hТРО в FH, FM, FL и FA, рассчитанные с применением биотин-меченого стандарта для SDS-PAGE (Bio-Rad, Biotynylated SDS-PAGE Standards, Broad Range: кат. 161-0319) при восстанавливающих условиях в Вестерн-анализе, были 26000-22000, 25500-22000, 26000-21000 и 26000-21000, соответственно. Гетерогенность среди мол. масс FН, FМ, FL и FA может быть обусловлена гетерогенностью O-соединенных сахарных цепей. Поэтому каждую фракцию FН, FM, FL и FA расщепляли нейраминидазой (Neuraminidase, Nacala tesque, кат. 242-29 Р), восстанавливали ДТТ и затем анализировали на SDS-РАGЕ. В результате во всех фракциях средняя мол. масса была около 19000, свидетельствуя о том, что гетерогенность мол. массы hТРО в основном обусловлена гетерогенностью количества сиаловой кислоты в сахарных цепях, связанных с белком hТРO163, и что hТРO163, полученный таким образом, экспрессируется в виде гликопротеина в СНО клетках.
3) Фракции FН, FМ, FL и FA, полученные в (2), анализировали на их активность in vitro при помощи М-07е-тест-системы. В результате относительные удельные активности были 511000000, 775000000, 1150000000 и 715000000/мг белка hTРO163 на массу пептидной части, не содержащей массы сахара).
Пример 66. Конструирование E.coli вектора для экспрессии ТРО человека (аминокислоты 1-332), в который добавлен Lys при положении -1 и Met при положении -2, соответственно (далее называемого hМКТ (1-332), и экспрессия hMKT(l-332)
Для экспрессии полноразмерной аминокислотной последовательности ТРО человека в Е.соli кодоны от аминокислоты 164 до аминокислоты 332 были заменены кодонами, предпочтительными для Е.соli, как описано ниже.
Синтетические олигонуклеотиды: 21 и 22; 23 и 24; 25 и 26; 27 и 28; 29 и 30; 31 и 32; 33 и 34; 35 и 36; 37 и 38 или 39 и 40, фосфорилировали в растворе 0,1 мМ АТР, 10 мМ Трис-ацетата, 10 мМ Мg-ацетата, 50 мМ К-ацетата при помощи Т4 киназы (Pharmacia) в одной и той же пробирке. Затем 1/10 объемов раствора 100 мМ Трис-НС1 (рН 7,5), 100 мМ MgCl2, 500 мМ NаС1 добавляли к реакционной смеси, кипятили 3 минуты в водяной бане и давали остыть, получая двухцепочечную ДНК. 4 набора двухцепочечных ДНК, т.е. олигонуклеотиды 21 и 22/23 и 24 (комбинация-А); олигонуклеотиды 25 и 26/27 и 28 (комбинация-В); олигонуклеотиды 31 и 32/33 и 34 (комбинация-С) и олигонуклеотиды 35 и 36/37 и 38/39 и 40 (комбинация-D) отдельно лигировали с применением набора для лигирования ДНК (Таkara-Shuso) и затем комбинацию-А лигировали вместе с комбинацией-В и также комбинацию-2 с комбинацией-D, получая соответственно лигирование-1 и лигирование-2. С применением продуктов лигирования 1 и 2 в качестве матриц проводили PCR, в которой в качестве праймеров использовали олигонуклеотиды 41 и 42 для продукта лигирования 1 и олигонуклеотиды -43 и -44 для продукта лигирования 2. Продукты РСR 1 и 2 расщепляли Sас1 и EcoRV и EcoRV и Hind 111, соответственно, с последующим электрофорезом на 2% агарозном геле и очисткой при помощи набора для очистки ДНК Рrер-A-Gene, получая фрагменты приблизительно 240 п.н. и 250 п.н., соответственно. Полученные таким образом два фрагмента субклонировали в pBluescript 11 Sk + (Stratagene), расщепленную предварительно Sас1 и Hind 111 (E.coli ДН5 использовали в качестве хозяина). Из полученных колоний был выбран клон, имеющий последовательность оснований, представленную в табл. 6, посредством секвенирования, и был назван pBL(SH)(174-332) (см. SЕQ 1D 154).
Далее, pBL(XH)h 6T (1-163), полученную в Примере 42, подвергали РСR в качестве матрицы в присутствии следующих синтетических олигонуклеотидов 45 и 46 в качестве праймеров и продукт РСR расцепляли BamH1 и Sac1 с последующим электрофорезом на 6% полиакриламидном геле, получая фрагмент приблизительно 160 п.н. из геля.
45: 5'-ААGGАТССGААСGААСGСТАТСТТССТG-3' (SEQ 1D 155)
46: 5'-GGGАGСТСGТТСАGGGТСАGААССАGА
GAGGTACGAGACGGAACAGCAGTGGTTGG-3' (SEQ 1D 156)
Также pBL(SH) (174-332) расщепляли Sас1 и Hind 111 и продукт расщепления очищали с применением набора для очистки ДНК Рrер-А-Gеnе, получая фрагмент приблизительно 480 п.н. Полученные два фрагмента субклонировали в pBluеscript 11SK+(Stratagene), расщепленную предварительно BamH1 и Hind 111 (E.coli, DH5 использовали в качестве хозяина).
Из полученных клонов секвенированием был выбран клон, имеющий последовательность оснований, представленную в табл. 7, и назван pBL(BH) (123-332) (см. SEQ 1D 157).
pBL (XH)h6T(l-163), полученную в Примере 42, расщепляли BamH1 и Нind 111 и полученную, как описано выше, рВL(ВН)(123-332) также расцепляли, получая фрагмент приблизительно 640 п.н., после чего эти два фрагмента лигировали вместе, получая клон pBL(XH)h6T(1-334). Далее, pCFM536/h МКТ (1-163), полученную в Примере 52, расщепляли Xba1 и Sfi1, получая фрагмент приблизительно 270 п. н. , который затем лигировали с pBL (ХН)-h6Т(1-334), расщепленной теми же рестриктазами, получая клон рВL (ХН)hМКТ(1-334). После расщепления рBL (ХН)hМКТ (1-334) Xba1 и Нind 111 был извлечен и очищен фрагмент приблизительно 1040 п.н., который кодирует ТРО человека мутированного типа (аминокислоты 1-332). Его клонировали в рСFМ536 (ЕР-А-136490), расцепленную предварительно Хbа1 и Нind 111 (E.coli jM109, предварительно трансформированную pMW1 (ATCC 39933), использовали в качестве хозяина). Полученный клон был назван рСFM536/hМКТ(1-332) и штамм E.coli, несущий этот экспрессируюций вектор, применяли в качестве трансформанта для экспрессии белка hМКТ(1-332) мутированного типа. Эта экспрессирующая плазмида рСFМ536/h МКТ(1-332) содержит последовательность ДНК, показанную в SEQ 1D 14.
Этот трансформант культивировали в 60 мл LB среды, содержащей 50 мкг/мл ампициллина и 12,5 мкг/мл тетрациклина, в течение ночи при 30oС и эту культуру (25 мл) затем добавляли к 1000 мл LB среды, содержащей 50 мкг/мл ампициллина, и качали культуру при 36oС, пока ОD600 не достигала 1,0-1,2. Затем, после добавления к культуре приблизительно 330 мл LB среды при 65oС, так что конечная температура культуры стала 42oС, качание культуры продолжали еще 3 часа при 42oС для индуцирования экспрессии вариантного ТРО человека, белка hМКТ(1-332), называемого также [Met-2, Lys-1] ТРО(1-332).
Полученную культуру сразу подвергали SDS-РАGЕ анализу. В нем применяли Multigel 15/25 Dai-ichi Chemical Co.). После электрофореза и окрашивания Кумасси синим был обнаружен белок, специфический для индукции экспрессии, при мол. массе приблизительно 35 кДа на геле в отношении трансформанта. Этот специфический белок подтверждал экспрессию белка hMKT(1-332). Кроме того, после SDS-РАGЕ электрофореза и электроблоттинга (с применением интроцеллюлозной мембраны) экспрессированный белок реагировал с антителами против пептида НТ1, полученными в Примере 45. После окрашивания обнаружили полосу при мол. массе приблизительно 35 кД, свидетельствующую о том, что был экспрессирован hMКТ(1-332).
Пример 67. Получение замещенных производных ТРО человека, их экспрессия в СOS 7 клетках и идентификация их активности
В соответствии со следующими процедурами исследовали, имеют ли ТРО активность некоторые производные, в которых аминокислоты ТРО человека были частично заменены другими аминокислотами.
Вектор рSМТ201 для экспрессии в клетках животных, для применения в этом Примере, конструировали следующим образом.
Сначала экспрессирующую плазмиду pXM-mEPORn, содержащую кДНК мышиного рецептора эритропоэтина (полученную от доктора D'Andrea в Dana Farbor Cancer Institute; Cell: 57, 277-285 (1989)), обрабатывали рестриктазами Kpn1 и EcoR1 и подвергали электрофорезу на агарозном геле, получая рХМ векторный фрагмент, в котором была удалена кДНК мышиного рецептора эритропоэтина. Затем при помощи ДНК-синтезатора (АВ1) синтезировали два олигонуклеотида для введения различных сайтов рестрикции в экспрессирующий вектор. Синтезированные олигонуклеотиды имеют следующие последовательности оснований, представленные в табл.IX.
Эти два олигонуклеотида смешивали и отжигали, получая двухцепочечный олигонуклеотид, который лигировали с полученным, как описано выше, вектором рХМ в присутствии Т4 ДНК-лигазы (Takara-Shuso), получая экспрессирующий вектор рDMТ201. Для удаления мышиной DHFR к ДНК, содержащейся в pDMT201, из pDMT201 векторы рDМТ201 и pEF18S отдельно расщепляли Not 1 и Hpa1 с последующим электрофорезом в агарозном геле, получая большой фрагмент из ДНК вектора рDМТ201 и меньший фрагмент из ДНК вектора pEF18S. Затем эти фрагменты лигировали вместе при помощи Т4 ДНК-лигазы (Takara-Shuso), получая экспрессирующий вектор рDMT201. Этот вектор, как показано на фиг.21, имеет ориджин (начало) репликации SV40, энхансерную последовательность, последовательность основного позднего промотора аденовируса, состоящую из трех частей лидерную последовательность аденовируса, последовательность сигнала сплайсинга, последовательность сайта раннего полиаденилирования SV40, последовательность гена РНК VA аденовируса, начало репликации рUС18 и ген β-лактамазы (Аmpr), вместе со следующими сайтами рестрикции для лигирования желаемого гена: Bgl 11, Pst1, Kpn 1, Xho1, EcoR1, Sma1, Spe1 и Not1. pHTP, иллюстрированный в Примере 30, несущий кДНК ТРО человека полной длины, расщепляли EcoR1 и Sре1, получая фрагмент кДНК полной длины ТРО человека, который затем субклонировали в вектор рSМТ201, расщепленный заранее также, получая плазмиду pSMT201-hTPO.
С применением полученной таким образом плазмиды pSMT201-hTPO в качестве матрицы была сначала сконструирована плазмида βGL-ТРО/рВLuе, которую применяли в качестве матрицы для получения целевых производных, следующим образом.
В βGL-ТРО/рВLue пытались провести добавление лидерной последовательности кроличьего β-глобина к 5'-нетранслируемому сайту hТРО по способу Annweiler (Annweiler et al. , Nucleic Acids Research, 19: 3750, 1991). Для этой цели химически синтезировали две цепи ДНК, представленные в табл.X.
SEQ 1D 160 и 161 лигировали с фрагментом, который был получен расщеплением вектора Bluescript 11SK+ (Toyo-boseki) EcoR1 и Smа1, получая вектор βGL/pBLue, в который была встроена лидерная последовательность.
PCR проводили при помощи следующих праймерных последовательностей:
Sma-ATG: 5'-GGСССGGGАТGGАGСТGАСТGААТТGСТС-3' (SEQ 1D 162) (т.е. 102-122 последовательность SEQ 1D 7, с которой соединена Smа1 последовательность); и (конец): 5'-ТСААGСТТАСТАGТСССТТССТGАGАСАGАТТСТG-3'(SEQ 1D 163) (т.е. антисмысловая последовательность 1140-1160 SEQ 1D 7, с которой были соединены последовательности Sре1 и Hind 111).
РСR проводили с применением 1 нг ДНК плазмиды рSМТ201-hТРО в качестве матрицы с 10 мкМ этих синтетических праймеров. С применением набора для PCR амплификации Takara (Takara-Shuzo) и Программируемого Thermal Controller (MJ Research) PCR проводили в объеме 100 мкл при следующих условиях: 3 цикла 94oС 1 мин, 55oС 2 мин и 72oС 2 мин (цикл); затем 20 циклов: 94oС 45 с, 55oС 1 мин и 72oС 1 мин (цикл). Продукт PCR экстрагировали хлороформом и затем осаждали этанолом дважды с последующим суспендированием в 100 мкл ТЕ буфера.
Затем продукт PCR расщепляли рестриктазами Smа1 и Hind 111, экстрагировали смесью фенол/хлороформ и осаждали этанолом. Полученный осадок растворяли в 10 мкл ТЕ буфера, после чего его субклонировали в вектор βGL /рВlue, расщепленный предварительно теми же рестриктазами (Компетентный штамм E.coli JМ109 (Toyo-boseki) применяли в качестве хозяина). Из полученных трансформированных клеток отбирали 20 клонов, из которых получали плазмидные ДНК, в основном, как описано в Sambrook et al., Molecular Cloning, Cold Spring Harbor Laboratory Press (1989). Очищенную плазмидную ДНК идентифицировали секвенированием с применением набора для секвенирования Таq Dуе DeoxyТМ Terminater Cycle (Applied Biosystems, Inc.) и секвенатора ДНК 373А (Applied Biosystems, Inc.). В результате было подтверждено, что плазмида, кодирующая βGL-TPO/pBLue, имела ожидаемую последовательность кДНК ТРО без каких-либо замен по всей ее длине. Затем после расщепления βGL-TPO/pBLue Bg111 и Spe1 продукт расщепления экстрагировали смесью фенол/хлороформ и осаждали этанолом. Полученный осадок растворяли в 10 мкл ТЕ буфера и субклонировали в вектор pSMT201, который был расщеплен теми же ферментами (Competent-High E. coli JM109 (Toyo-boseki) применяли в качестве хозяина). В соответствии со способом, описанным в Molecular Cloning (Sambrook et al., Supra), из клеток трансформанта получали плазмидную ДНК. Полученный экспрессирующий вектор pSMT/βGL-ТРО имел структуру, в которой лидерная последовательность β-глобина и кДНК ТРО человека были соединены при сайте рестрикции Bg111/Spe1 в направлении 5'-3' от сигнальной последовательности сплайсинга вектора рSМТ201, как показано на фиг. 21. Этот вектор pSМT/βGL-ТРО был применен для трансфекции в С0S 7 клетки.
Для получения замещенных производных ТРО человека применяли PCR, в которой был использован способ Ito (Ito et al., Gene, 102: 67-70, 1991). В частности, получали два производных ТРО человека, в которых Arg-25 и His-33 ТРО человека были заменены АSn и Thr, соответственно. Эти производные могут быть названы, соответственно, [ASn25] ТРО и [Тhr33] ТРО. В РСR применяли следующие праймеры:
Т7: 5'-ТААТАСGАСТСАСТАТАGGGСG-3' (SEQ 1D 164) (соответствующий району Т7 промотора BLuescript 11 S К+); ΔBgl11: 5'-ААТТССААGАТСАСАСАСТTGС-3' (SEQ 1D 165) (для замены последовательности узнавания Bgl 11); (конец): 5'-ТСААGСТТАСТАGТСССТТССТGАGАСАGАТТСТG-3' (SEQ 1D 166) (антисмысловая последовательность 1140-1160 SЕQ 1D 7, с которой соединены последовательности Sре1 и Нind 111); 3: 5'-ТGGGСАСТGGСТСАGGТТGСТGТGААGGАСАТGGG-3' (SEQ 1D 167) (Arg 25 --> Asn SEQ 1D 7); и 09: 5'-ТGТAGGСАААGGGGTААССТСТGGGСА-3' (SEQ 1D 168) (Нis - 33 --> Thr SEQ 1D 7).
Первую стадию PCR проводили с применением 1 нг плазмидной ДНК βGL-TPO/pBLue в качестве матрицы вместе с 10 мкг каждого из синтетических праймеров. Комбинации праймеров были следующими: [1] Δ Bgl11 и "еnd" праймеры; [2] T7 и N3; и [3] T7 и 09. С применением набора для PCR, амплификации (Таkаrа Shuso) и Программируемого температурного контроллера (Мy Research) PCR проводили в объеме 100 мкл при следующих условиях: 3 цикла 94oС 1 мин, 55oС 2 мин и 72oС 2 мин (1 цикл); затем 17 циклов 94oС 45 с, 55oС 1 мин и 72oС 2 мин (1 цикл). Каждый из продуктов PCR экстрагировали хлороформом и осаждали дважды этанолом и осадок растворяли в 100 мкл ТЕ буфера. Полученные продукты PCR (1 мкл каждого) подвергали затем второй PCR. Комбинации матриц были следующими: продукты PCR[1] /[2] и продукты PCR [1]/[3]. В качестве праймеров в обоих случаях применяли комбинацию T7 и "end". После инкубирования при 94oС в течение 10 минут и добавления Таkarа Taq, PCR проводили при следующих условиях: 3 цикла 94oС 1 мин, 55oС 2 мин и 72oС 2 мин (цикл); затем 9 циклов 94oС 45 с, 55oС 1 мин и 72oС 1 мин (цикл). К каждому из полученных продуктов PCR добавляли 1 мкл протеиназы К (5 мг/мл), 2 мкл 0,5 М ЭДТА и 2 мкл 20% SDS, инкубировали при 37oС в течение 30 минут для инактивации Taq, экстрагировали смесью фенол/хлороформ и осаждали этанолом. После этого осадок растворяли в 20 мкл стерильной воды и расщепляли Вgl11 и Sре1 с последующей экстракцией смесью фенол/хлороформ и осаждением этанолом. Полученный таким образом осадок растворяли в 10 мкл ТЕ буфера и затем субклонировали в экспрессирующий вектор рSМТ201, расщепленный предварительно теми же рестриктазами, и обрабатывали щелочной фосфатазой кишечника теленка (Boehringer-Mannheim) (Competent-High E. coli JМ109 применяли в качестве хозяина). Из полученных трансформированных клеток в каждом случае отбирали два клона, из которых плазмидиые ДНК получали в основном, как описано в Molecular Cloning (Sambrook et al., Supra). Очищенные плазмидные ДНК секвенировали при помощи секвенатора ДНК 373А и набора для секвенирования Таq Dye DeoxyTM Terminate Cycle (оба Applied Biosystems, Inc.).
Плазмида, полученная при помощи PCR с применением этих праймеров [1] и [3] , содержала кДНК, кодирующую замещенное производное ТРО (09/ТРО). Замена аминокислоты Нis 33 (САС) на Thr (АСС) и удлинение первоначального С-конца (Gly 332) добавлением аминокислотной последовательности TSIGYPYDVPDYAGVHHHHHH (SЕQ 1D 169) были подтверждены. Это производное называют [Thr33, Thr333, Ser334, Ile335, Gly336, Tyr337, Pro338, Tyr339, Asp340, Val341, Pro342, Asp343, Tyr344, Ala345, Gly346, Val347, His348, His349, His350, His351,His352, His353] ТРО.
С другой стороны, плазмида, полученная при помощи PCR с применением праймеров [1] и [2], содержала кДНК, кодирующую замещенное производное ТРО (N3/ТРО). Были подтверждены замены аминокислот Аrg 25(AGА) на Asn(AAC) и Glu 231 (GAA) на Lys (AAA) и удлинение первоначального С-конца (Gly 332) добавлением аминокислотной последовательности ТS1GУРУDVРD YАGVНННННН. Это производное названо [Asn25, Lys231, Thr333, Sеr334, Ile335, Gly336, Tyr337, Pro338, Tyr339, Asp340, Val341, Pro342, Asp343, Tyr344, Ala345, Gly346, Val347, His348, His349, His350, His351, His352, His353] ТРО.
Трансфекцию в СOS 7 клетки каждого полученного клона проводили ДЭАЭ-декстрановым способом, предусматривающим обработку хлорохином, как описано в Примере 35. Вкратце 40 мкг плазмидных ДНК применяли в трансфекции и после 5 дней извлекали культуральный супернатант, который затем концентрировали до объема 1/20 при помощи Centricon-30 (Аmicon) для оценки их ТРО активности при помощи М-07е-теста. В результате в культуральных супернатантах COS 7 клеток, в которые были соответственно трансфицированы плазмиды, содержащие кДНК, кодирующие производные ТРО человека N3/ТРО и 09/ТРО, была обнаружена ТРО активность, зависящая от дозы (см. фиг.22).
Пример 68. Получение инсерционных или делеционных производных ТРО человека, экспрессия их в СОS 7 клетках и идентификация ТРО активности
Этот Пример показывает, имеют ли ТРО активность инсерционные и делеционные производные hТРO163.
Были получены следующие производные: His 33-делеционное производное ([ΔНis33] ТРО (1-163), dH33); Gly116-делеционное производное ([ΔGly116] TPO(1-163), dG 116); Аrg 117-делеционное производное ([ΔАrg117] TPO(1-163), dR 116); Thr-инсерционное производное ([His33, Thr33, Рro34] TPO(1-163), T33'); Аlа-инсерционное производное ([His33, Ala33', Рro34] ТРО (1-163), А33') и Gly-инсерционное производное ([His33, Gly33', Pro34, Ser38] TPO(1-163), G33'), в которых Тhr, Аlа и Gly были независимо встроены между Нis 33 и Рro 34; и Аsn-инсерционное производное ([Gly116, Аsn116', Arg117] TPO(1-163), N116'). Ala-инсерционное производное ([Gly116, Аlа116', Аrg117] TPO(1-163), А116') и Gly-инсерционное производное ([Gly116, Gly116', Arg117] ТРО (1-163, G116'), в которых Asn, Ala и Gly были независимо встроены между Gly 116 и Arg 117, все последовательности построены на основе SEQ 1D 13.
Среди этих производных T33' и N116 предназначены для введения цепей типа муцина и сахарной цепи типа Asn-связывающей цепи, соответственно.
Указанные выше производные получали при помощи РСR по способу, описанному Jto et al. (Jene, 102: 67-70, 1991).
В PCR использовали следующие праймеры:
dH33: 5'-ТGТАGGСАААGGААССТСТGGGСА-3' (SEQ 1D 172) (для получения His 33-делеционного производного на основе SEQ 1D 7);
Т33': 5'-ТGТАGGСААAGGАGТGТGААССТСТGG-3' (SЕQ 1D 173) (для получения Thr-инсерционного производного с Тhr, встроенным между Нis 33 и Рro 34 в SEQ 1D 7);
А33': 5'-ТGТАGGСАААGGАGСGТGААССТСТGG-3' (SЕQ 1D 174) (для получения Аlа-инсерционного производного с Alа, встроенным между Нis 33 и Рrо 34 в SЕQ 1D 7);
G33': 5'-ТGТАGGСАААGGТССGТGААССТСТGG-3' (SEQ 1D 175) (для получения Gly-инсерционного производного с Gly, встроенным между Нis 33 и Рrо 34 в SEQ 1D 7);
dG116: 5'-АGСТGТGGТССТСТGТGGАGGААG-3' (SEQ 1D 176) (для получения Gly116-делеционного производного на основе последовательности, показанной в SЕQ 1D 7);
dR117: 5'-GТGАGСТGТGGTGСССTGТGGАGG-3' (SEQ 1D 177) (для получения Аrg 117-делеционного производного на основе последовательности, показанной в SEQ 1D 7);
N116: 5'-АGСТGTGGТССТGТТGСССТGТGGАGG-3' (SЕQ 1D 178) (для получения Аsn-инсерционного производного с Аsn, встроенным между Gly116 и Аrg117 в SEQ 1D 7);
А116': 5'-АGСТGТGGТССТАGСGСССТGТGGАGG-3' (SEQ 1D 179) (для получения Аlа-инсерционного производного с Аlа, встроенным между Gly116 и Аrg 117 в SEQ 1D 7);
G116': 5'-АGСТGТGGТССТТССGСССTGТGGАGG-3' (SЕQ 1D 180) (для получения Gly-инсерционного производного с Gly, встроенным между Gly116 и Аrg117 в SEQ 1D 7);
dR1: 5'-СGGGСТGСАGGАТАТССААGАТСТСА-3 (SEQ 1D 181) (для замены последовательности узнавания рестриктазы EcoR1);
Т7: 5'-ТААТАСGАСТСАСТАТАGGGСG-3' (SEQ 1D 182) (соответствующая району Т7 промотора Bluescript 11S К+); и end Not: 5'-GGGCGGССGСТСАGСТGGGGАСАGСТGТGGTGGG-3' (SEQ 1D 183) (антисмысловая нуклеотидная последовательность 633-653 SEQ 1D 7, к которой добавлены терминирующий кодон и последовательность узнавания Not1).
Первую стадию PCR проводили с применением в качестве матрицы 1 нг ДНК плазмиды βGL-TPO/pBlue, полученной в Примере 67, и 10 мкМ синтетических праймеров. Комбинация праймеров была: [1]dR1 и end Not1 или [2] Т7 и мутированные праймеры (dH33, А33', G33', Т33', dG116, dR117, A116', G116' и N116'). В PCR применяли набор для PCR амплификации (Takara Shugo) и Программируемый температурный контроллер (MJ Rescarch) и РСR проводили в конечном объеме 100 мкл. В первой PCR реакции [1] один цикл был 94oС 1 мин, 45oС 2 мин и 72oС 2 мин и 3 цикла проводили с последующими 17 циклами, в которых каждый цикл был 94oС 45 с, 45oС 1 мин и 72oС 1 мин. С другой стороны, первую реакцию [2] проводили при условиях: 94oС 1 мин, 55oС 2 мин и 72oС 2 мин. Для 3 циклов, с последующими 17 циклами, каждый из которых состоял из 94oС 45 с, 55oС 1 мин и 72oС 1 мин. Каждый из продуктов PCR экстрагировали хлороформом перед осаждением дважды этанолом и затем полученный осадок растворяли в 100 мкл ТЕ буфера.
Затем проводили вторую стадию РСR с применением 1 мкл каждого из продуктов РСR [1] и [2]. Применяли комбинацию Т7 и end Not1 в качестве праймеров. После инкубирования при 94oС в течение 10 минут к каждой реакционной смеси PC добавляли Takara Таq. Вторую PCR проводили при условиях: 94oС 1 мин, 55oС 2 мин и 72oС 2 мин в течение 3 циклов, с последующими 94oС 45 с, 55oС 1 мин и 72oС 1 мин в течение 9 циклов. К каждому из полученных таким образом продуктов PCR добавляли 1 мкл 5 мг/мл протеиназы К, 2 мкл 0,5 М ЭДТА и 2 мкл 20% SDS и смесь держали при температуре 37oС в течение 30 мин для инактивации Таq. Продукт PCR экстрагировали смесью фенол/хлороформ с последующим осаждением этанолом и полученный осадок растворяли в 20 мкл стерильной воды. После расщепления EcoR1 и Not1 раствор экстрагировали смесью фенол/хлороформ и затем осаждали этанолом. Полученный осадок растворяли в 10 мкл ТЕ буфера и субклонировали в экспрессирующей вектор pEF18S, расщепленный предварительно теми же рестриктазами, и обрабатывали щелочной фосфатазой (из кишечника теленка, Boehringer-Mannheim). Клетками хозяина были Competent-High E.coli M109 (Toyo-Boseki). Из полученного трансформанта получали плазмидную ДНК по способу, описанному в Molecular Cloning (Sambrook et al., Cold Spring Harbor Laboratory Press (1989)). После этого нуклеотидная последовательность очищенной плазмидной ДНК была подтверждена при помощи Таq Dye DeoxyTM Terminater Cycle набора для секвенироваиия (Applied Biosystems) и секвенатора ДНК 373А той же фирмы. В результате было подтверждено, что все эти плазмиды, кодирующие dН33, А33', Т33', dG116, dR117, А116', G116' и N116', имели ожидаемую последовательность кДНК ТРО без каких-либо замен нуклеотидной последовательности при сайтах иных, чем предназначенные для замен сайты. Более того, в G33 наблюдали замену основания [Рrо38(ССТ) --> Sеr (TCT)], иную, чем замена в намеченном сайте.
Трансфекцию каждого полученного клона в СОS 7 клетки проводили согласно способу в Примере 11. Вкратце, трансфекцию проводили с применением 10 мкг каждой плазмидной ДНК ДЭАЭ-декстрановым способом, предусматривающим обработку хлорохином, и после 4-5 дней извлекали культуральный супернатант, который оценивали при помощи М-07е-тест-системы. В результате в каждом супернатанте культур COS 7 клеток, трансфицированных плазмидами, кодирующими аминокислотные инсерционные или делеционные производные, была обнаружена ТРО активность, зависящая от дозы (см. фиг.23). В противоположность этому, ТРО активность не была обнаружена в супернатанте культуры СОS 7 клеток, экспрессирующая плазмида которых pEF18S, трансфицированная в них и экспрессированная, не содержала кДНК, кодирующей производное ТРО.
Пример 69. Получение и очистка антител против пептида ТРО человека
1) Шесть районов (показанных в табл. 8), которые считаются относительно пригодными в качестве антигенов, были выбраны из аминокислотной последовательности ТРО человека SEQ 1D 191, из которых был синтезирован 4-цепочечный пептид типа пептида с множественными антигенами (MAP) по способу Tam (Proc. Natl. Acad. Sci. USA, 85, 5409-5413, 1988). Двух кроликов иммунизовали 8 раз по 100 мкг этого пептида.
Отдельно получали одноцепочечные пептиды путем связывания цистеинового остатка с С-концом каждого из пептидных районов, показанных SЕQ 1D 119-124 (называемых пептидными районами НТ 1-6, соответственно, каждый из частичных пептидов соответствует 8-28, 47-52, 108-126, 172-190, 262-284 и 306-332 положениям, соответственно, последовательности SEQ 1D 191). Применяя их в качестве тест-антигенов, измеряли количества антител к ним в сыворотках, полученных из иммунизированных таким образом кроликов, при помощи иммуноферментного анализа, причем было подтверждено увеличение в числе антител во всех испытанных сыворотках. Таким образом, эти сыворотки называют далее антисыворотками.
Кроме того, вышеупомянутый одноцепочечный синтетический пептид, имеющий остаток цистеинa, связанный с его С-концом, применяли также в качестве антигена для аффинной очистки антисывороток в следующей стадии (2).
2) Каждого из двух кроликов иммунизировали одним из вышеупомянутых антигенных пептидов, и антитела, полученные из каждого кролика, получали отдельно. Конкретно, для антител против НТ1 пептида были получены антитела против НТ1-1 пептида и антитела против НТ1-2 пептида из двух иммунизированных кроликов. Далее в качестве одного примера проиллюстрирована очистка антитела против пептида НТ1-1.
Сначала 30 мг одноцепочечного пептида НТ1, имеющего цистеиновый остаток, связанный с ним, соединяли с 12 мл Sulfo Link Coupling GE1 (продуцируемым Pierce Co. , кат. 44895). Более точно, раствор пептида, содержащий антиген, соединяли с гелем, уравновешенным буфером связывания (50 мМ Трис, 5 мМ ЭДТА-Nа, рН 8,5) в количестве, в 6 раз большем, чем объем геля, в течение периода 15 минут. Затем, после стояния в течение 30 минут гель промывали буфером связывания в количестве 3 объемов геля. Затем буфер связывания, содержащий 0,05 М L-цистеин-НС1, добавляли к гелю при соотношении 1 мл/мл геля, при помощи которого необработанные группы блокировались в течение 15 минут. После стояния в течение 30 минут гель промывали буфером связывания 8 раз количеством, равным объему геля. Операцию связывания проводили при комнатной температуре. Таким путем содержащий антигенный район пептид присоединялся к гелю посредством ковалентного связывания при эффективности связывания 28,3% с образованием геля с антигенным пептидом, имеющим 0,8 мг пептида, связанного на 1 мл геля, в антигенной колонке. После взятия всей крови из каждого иммунизированного кролика получали 78,4 мл антисыворотки, содержащей антитела против НП-1 пептида. 76,7 мл этой антисыворотки (содержание белка 3620 мг) добавляли к антигенной колонке, уравновешенной предварительно 50 мМ фосфатным буфером (рН 8), содержащим 150 мМ NаС1 и 0,05% азида натрия, затем колонку промывали тем же самым буфером. Таким способом получили 105,9 мл проходящей через колонку фракции (содержание белка 3680 мг). Затем адсорбированную фракцию элюировали 0,1 М нитратным буфером (рН 3,0), которую затем сразу нейтрализовали добавлением к ней 21,1 мл 0,1 М карбонатного буфера (рН 9,9), концентрировали ультрафильтрацией (при помощи УМЗО мембраны, производимой Amicon Cо.) и очищали в 50 мМ фосфатном буфере (рН 8), содержащем 150 мМ NаС1 и 0,05% азид натрия. Так получили 11,2 мл очищенных антител против НТ1-1 пептида (содержание белка 77,7 мг) в буфере. Таким же способом получали антитела против НТ1-2 пептида (60,0 мг), антитела против НТ2-1 пептида (18,8 мг), антитела против НТ2-2 пептида (8,2 мг) и т.д.
Антитела против НТ3-НТ6 пептидов получают так, как описано выше.
Пример 70. Обнаружение ТРО при помощи Вестерн-анализа
1) Для оценки антисывороток, полученных в Примере 69, их тестировали, как описано ниже.
Стандартную пробу рекомбинантного ТРО человека, частично очищенного из супернатанта культуры СНО клеток, в которые был введен ген, кодирующий аминокислоты -21-332 SEQ 1D 6, и в которых он был экспрессирован, подвергали электрофорезу в SDS-полиакриламиде (SDS-РАGЕ) в соответствии с обычным способом и затем проводили электроблоттинг на PVDF или нитроцеллюлозную мембрану. После блоттинга мембрану промывали 20 мМ Трис-НС1 и 0,5 М NаС1 (рН 7,5) (TBS) в течение 5 минут, затем промывали два раза 0,1% Твин 20-содержащим ТВS (ТТВS) в течение 5 минут и обрабатывали блокирующим агентом (Block Асе; производимым Dai Nippon Pharmaceutical Со., кат. UК-В25) в течение 60 минут. Антисыворотки, содержащие (каждая) одно из антител; антитело против НТ1-1 пептида, антитело против НТ2-1 пептида, антитело против НТ3-2 пептида, антитело против НТ4-1 пептида, антитело против НТ5-2 пептида и антитело против НТ6-1 пептида, разбавляли раствором ТТВS, содержащим 0,05% BSA и 10% Вlock Асе до разведений 1/1000. Разведенные таким образом антитела применяли для Вестерн-анализа в качестве первичных антител. В частности, пробу рекомбинантного ТРО человека, обработанную, как описано выше, обрабатывали ТТВS раствором, содержащим антитело против ТРО-пептида, 0,05% BSA и 10% Block Аса, в течение 60 минут и затем промывали ТТВS в течение 5 минут. Затем ее обрабатывали ТТВS раствором, содержащим антикроличьи (аb') козьи биотинилированные антитела (Сaltag Со., кат. 43015), в качестве вторичного антитела, вместе с 0,05% ВSА и 10% Block Асе, в течение 60 минут и затем промывали два раза TTBS в течение 5 минут. Затем ее обрабатывали разведением 1/5000 меченого щелочной фосфатазой авидина (производимого Lеinco Technologies Cо., кат. А108), разведенного содержащим 10% Block Асе раствором ТТВS в течение 30 минут и после этого промывали два раза TTBS в течение 5 минут и затем TBS в течение 5 минут. Затем мембрану проявляли с применением субстрата для щелочной фосфатазы (производимого Bio-Rad Со., кат. 170-6432). Вестерн-анализ проводили при комнатной температуре. Он подтвердил, что ТРО человека узнается и детектируется этими антисыворотками.
2) 3 мг каждого из антител против НТ1-1 пептида, против НТ1-2 пептида, против НТ2-1 пептида и против НТ2-2 пептида, которые были очищены аффинной хроматографией в Примере 69, взвешивали и биотинилировали связыванием их на активированном биотине (NHS-LC-Biotin 11, производимый Pierce Co., кат. 21336). Ту же самую стандартную пробу рекомбинантного ТРО человека, которую использовали в (1), подвергали SDS-PAGE, затем электрически блоттировали на PVDF или нитроцеллюлозную мембрану и подвергали обычному Вестерн-анализу с применением каждого из этих биотинилированных антител в качестве первичных антител. Сразу после блоттинга мембрану промывали ТВS в течение 5 минут и затем промывали два раза TTBS в течение 5 минут и после этого обрабатывали блокирующим агентом (Blосk Асе) в течение 60 минут. Затем ее обрабатывали ТТВS раствором, содержащим 1 мкг/мл биотинилированного антитела против ТРО-пептида, 0,05% ВSА и 10% Block Асе в течение 60 минут и затем промывали два раза ТТВS в течение 5 минут. Затем мембрану обрабатывали разведением 1/5000 меченого щелочной фосфатазой авидина (Leinco Technologies Со., кат. А108), разведенного содержащим 10% Block Асе TTBS раствором, в течение 30 минут и после этого промывали два раза TTBS в течение 5 минут и затем ТВS в течение 5 минут. Затем мембрану проявляли с применением субстрата для щелочной фосфатазы (Вiо-Rad Сo., кат. 70-6432). Описанный Вестерн-анализ проводили при комнатной температуре. Он подтвердил, что ТРО человека узнается и детектируется этими очищенными антителами.
Пример 71. Получение колонок с антителами против ТРО-пептида и обнаружение ТРО человека
Антитела против ТРО-пептида человека, полученные в Примере 69, проверяли на узнавание ТРО человека. Эти антитела по отдельности связывали на хроматографическом носителе для получения колонок с антителами против ТРО-пептида человека. В качестве одного примера далее иллюстрируется приготовление колонки с антителом против НТ1-2 пептида.
1) Готовили раствор, содержащий 50 мМ фосфата натрия и 0,15 М NаС1 (рН 8,0) и содержащий 5 мг/мл антитела, 0,8 мл этого раствора объединяли и связывали с 1,54 мл набухшего формил-активированного геля (Formyl-Gellulofine, производимый Chisso Co.) при 4oС в течение 2 часов. Затем к нему добавляли 1,1 мл раствора, содержащего 10 мг/л восстанавливающего агента (триметиламинборана (ТМАВ), производимого Seikagaku (Kogyo К. К., кат. 680246), и связывание продолжали еще в течение 4 часов. Затем, только часть этого геля брали из реакционной смеси при помощи центрифугирования. 10 мл чистой воды добавляли к нему и только часть геля извлекали из этой смеси центрифугированием. Этот процесс повторяли 4 раза для удаления непрореагировавших антител. Затем полученный таким образом гель обрабатывали 2,1 мл блокирующего буфера (0,2 М Na-фосфат, 1 М этаноламин, рН 7,0) и 1,1 мл раствора восстанавливающего агента, добавленного к нему, при 4oС в течение 2 часов, в результате чего активные группы в непрореагировавшем геле были блокированы. Наконец, полученный гель промывали водой, 20 мМ Трис-НС1 и 0,15 М NаС1 (рН 8,0) с применением центрифуги. Им заполняли небольшую колоночную трубку, которую промывали 3 М раствором тиоцианата натрия и 0,1 М раствором глицин-НС1 (рН 2,5) и затем опять уравновешивали 20 мМ Трис-НС1 и 0,15 М NаС1 (рН 8,0). Уравновешенную таким образом колонку хранили.
Гель с антителами против ТРО-пептида в колонке с антителами против НТ1-2, в частности, имел эффективность связывания до 94,2% для каждой фракции 1gG. Количество фракции 1gG, связанной на геле на единицу объема этого геля, было 1,9 мг/мл геля. Тем же способом, как описано выше, готовили гель, на котором было связано 21,8 мг на мл геля 1gG, не имеющего ТРО в качестве антигена. Этот гель был использован в качестве предколонки (далее называемой предколонкой с антителами) для удаления неспецифически связывающихся молекул из колонок с антителами против ТРО, как упомянуто в (2).
2) Далее представлен другой пример приготовления колонки с антителами против НТ1-2.
Стандартную пробу рекомбинантного ТРО человека, который был частично очищен от супернатанта культуры СНО клеток, в которые был введен ген, кодирующий аминокислотную последовательность любой из SEQ 1D 119-124, который экспрессировался в этих клетках, добавляли к предколонке с антителами (содержащей 1 мл геля), приготовленной в (1), и 10 раз пропускали объем 20 мМ Трис-НС1 (рН 8,0) и 0,15 М NаС1, равный объему геля, через колонку, собирая проходящую через колонку фракцию. Затем элюировали фракцию, адсорбированную на предколонке, 10 раз в объеме геля кислым элюентом (0,1 М глицин-НС1, рН 2,5). Затем фракцию, проходящую через предколонку, добавляли к колонке с антителами против НТ1-2 (содержащей 2 мл геля), приготовленную в (1), и колонку промывали 10 раз в объеме геля 20 мМ Трис-НС1 (рН 8,0) и 0,15 М NаС1, собирая фракцию, проходящую через колонку с антителами против НТ1-2. Затем элюировали фракцию, адсорбированную на колонке, 8 раз в объеме геля кислым элементом (0,1 М глицин-НС1, рН 2,6). Эти фракции анализировали при помощи SDS-PAGE, который подтвердил, что ТРО не адсорбируется на предколонке с антителами, но специфически связывается с колонкой с антителами против НТ1-2, и что почти весь ТРО в добавленной к этой колонке пробе связался с последней колонкой. На основании этого эксперимента было подтверждено, что не менее 200-300 мкг/мл геля ТРО связалось с гелем в последней колонке.
Пример 72. Действие ТРО на увеличение количества тромбоцитов
20 нормальных самцов мышей штамма 1СR, восьминедельного возраста (кровь уже брали из их глазничных вен и количества тромбоцитов измеряли при помощи микроцитометра F-800, изготовляемого Тоа Iyо Denshi) разделяли безвыборочным способом на 4 группы. Одну из 4 групп (контроль-iv-группу) инъецировали 100 мкл РВS внутривенно (iv) один раз в день в течение 5 последовательных дней. Одну из остальных групп (ТРО-iv группу) инъецировали 100 мкл раствора (приготовленного замещением концентрированного раствора ТРО-активной фракции F2 SP Sepharose Fast Flow, полученной в Примере 56, буфером РВS при помощи колонки NАР-25 [Pharmacia Biotech; кат. 17-0852-02] с последующим разбавлением РВS; содержащего относительную активность 211900 в М-07е-тесте) внутривенно один раз в день в течение 5 последовательных дней. Другую группу (контроль-Sс группу) инъецировали 100 мкл РВS подкожно (Sс) 100 мкл PBS 1 раз в день в течение 5 последовательных дней, тогда как последнюю группу (ТРО-Sс группу) инъецировали 100 мкл 1 раз в день в течение 5 последовательных дней, так же как в группе TPO-iv, но подкожно (Sс). После 6, 8, 10, 13 и 15 дней от начала инъекций кровь из глазничной вены брали у каждой из мышей и количества тромбоцитов считали при помощи микроцитометра (F-800; Тоа Iyо Denshi). Изменения в числах тромбоцитов даны на фиг.24. Таким образом, в группе контроль -iv число тромбоцитов увеличилось на 44% по сравнению с числом перед введением даже на 6-й день (день 0 является днем начала введений), когда количества тромбоцитов были наивысшими, а в группе контроль - Sс увеличение только 47% было отмечено даже на 8-й день, когда количества тромбоцитов были наивысшими. С другой стороны, в группе TPO-iv наблюдали увеличение 177% на 6-й день по сравнению с количествами перед введением и в группе ТРО-Sc увеличение 347% было отмечено уже на 6-й день и на 8-й день наблюдали наивысшее увеличение 493%.
Эти результаты подтвердили, что ТРО человека увеличивает число тромбоцитов независимо от различий в типе, а также в пути введения.
Пример 73. Действие ТРО на увеличение количества тромбоцитов
16 нормальных самцов мышей штамма С3Н/НеJ 11-недельного возраста (кровь брали уже из их глазничных вен и количество тромбоцитов измеряли при помощи микроцитометра типа F-800, изготовляемого Тоа Iyо Denshi) разделяли безвыборочным способом на 4 группы. Одну из четырех групп (контрольную группу) инъецировали 100 мкл РВS подкожно 1 раз в день в течение 5 последовательных дней. Другую группу (группу ТРО-1) инъецировали подкожно 100 мкл раствора (приготовленного замещением ТРО-активной фракции Sephacryl S-200Н, полученной в Примере 56, буфером PBS при помощи колонки NAР-25, с последующим разбавлением РВS; содержащего относительную активность 83000 в М-07е-тесте) 1 раз в день в течение 5 последовательных дней. Еще одну группу (группу ТРО-2) инъецировали 100 мкл раствора (полученного разбавлением ТРО-активной фракции, примененной в группе ТРО-1, РВS в 2 раза; содержащего относительную активность 41500 в М-07е-тесте) подкожно 1 раз в день в течение 5 последовательных дней. Последнюю группу (группу ТРО-3) инъецировали 100 мкл раствора (полученного разбавлением ТРО-активной фракции, применяемой в группе ТРО-2, еще в 2 раза; содержащего относительную активность 20750 в М-07е-тесте) подкожно 1 раз в день в течение 5 последовательных дней.
Кровь брали из глазничных вен мышей на 6-й, 8-й, 10-й и 12-й дни после инъекции и количества тромбоцитов считали при помощи микроцитометра (типа F-800; Toa Iyo Denshi).
Изменения в числах тромбоцитов даны на фиг.25. Таким образом, в контрольной группе почти не было изменения в количествах тромбоцитов, тогда, как в группах, которым инъецировали ТРО, увеличения были в количествах тромбоцитов во всех группах, причем наивысшие количества тромбоцитов были на 8-й день после введения. Кроме того, в этих группах были отмечены различия в зависимости ответной реакции от дозы. Так, в группе ТРО-1 примерно 200% увеличение отмечали уже на 6-й день, увеличение возрастало примерно до 270% на 8-й день, а затем, однако, наблюдали уменьшение, даже на 12-й день все еще наблюдали 65% увеличение, между тем наблюдали значимое отличие от контрольной группы (р<0,01; тест множественного сравнения Dunnet). В ТРО-2 группе обнаружили такое же увеличение, и хотя увеличивающее действие было ниже, чем в группе ТРО-1, увеличения 140% и 160% отмечали на 6-й и 8-й дни, соответственно. На 12-й день количества тромбоцитов снижались приблизительно до того же уровня, который был в контрольной группе. Увеличивающее действие в группе ТРО-3 было слабее, чем в группе ТРО-2, и на 8-й день, когда количества тромбоцитов были наивысшими, отмечали 110% увеличение и также было значимое различие (р<0,01) по сравнению с контрольной группой.
Описанные выше результаты показали, что ТРО человека увеличивает количества тромбоцитов независимо от штамма мышей и что его действие обладает характеристикой зависимости ответной реакции от дозы.
Пример 74. Ингибирующее действие ТРО на тромбоцитопению, вызываемую введением противоракового агента
30 самцов мышей 1CR восьминедельного возраста инъецировали 200 мг/кг 5 FU внутривенно и разделяли безвыборочным способом на 2 группы (каждая группа состояла из 15 мышей). Со следующего дня (1-й день) одну из групп (контрольная группа) инъецировали 100 мкл PBS подкожно 1 раз в день в течение 5 последовательных дней, тогда как другую группу (группу ТРО) инъецировали 100 мкл ТРО-активной фракции, применяемой в Примере 72 (содержащими относительную активность 211900 в М-07е-тесте) подкожно 1 раз в день в течение 5 последующих дней. На 4-й, 6-й и 8-й дни по 5 мышей произвольно выбирали из каждой из двух групп, затем брали кровь из глазничных вен и количества тромбоцитов считали при помощи микроцитометра (типа Е-2 500; Toa Iyo Denshi). Изменения в количествах тромбоцитов даны на фиг.26. Так, в контрольной группе количества тромбоцитов после введения 5-FU уменьшались с течением времени и на 6-й день эта величина была самой низкой (приблизительно 28% от величины перед введением 5-FU), хотя на 8-й день отмечали приблизительно 1,3-кратное увеличение как реакцию этого уменьшения. Даже в случае группы ТРО количества тромбоцитов после введения 5-FU уменьшались с течением времени и на 6-й день эта величина была самой низкой (приблизительно 52% от величины перед введением 5-FU). Однако различие между количествами тромбоцитов на 4-й и 6-й дни были очень низкими и на 6-й день сохранялась значимо высокая (р<0,05: тест Dunnet) величина по сравнению с контрольной группой. На 8-й день наблюдали увеличение приблизительно 200% от величины перед введением 5-FU.
Полученный результат подтвердил, что ТРО человека ингибирует снижение числа тромбоцитов, вызываемое противораковым агентом.
Пример 75. Терапевтическое действие ТРО для тромбоцитопении
Гидрохлорид нимустина (АСNU) (50 мг/кг) инъецировали внутривенно каждому из 36 самцов мышей 1CR семинедельного возраста и затем этих мышей разделяли безвыборочным способом на две группы (каждая группа состояла из 18 мышей). Со следующего дня (1-й день) одну из групп (контрольную группу) инъецировали 200 мкл PBS подкожно дважды в день в течение последующих дней, тогда как другую группу (группу ТРО) инъецировали 200 мкл ТРО-активной фракции, применяемой в Примере 73 (без разведения PBS), к которой добавляли 0,04% Твин 80 (относительная активность 380000 в М-07е-тесте), подкожно дважды в день в течение последующих дней.
После инъекции мышей в каждой из групп произвольно разделяли на три группы: из одной группы кровь брали на 5-й и 12-й дни; из другой группы на 8-й и 14-й дни и из третьей группы на 10-й день. Кровь брали из глазничных вен и количества тромбоцитов считали при помощи микроцитометра (типа F-800; производимого Toa Iyo Denshi). Изменения в количествах тромбоцитов даны на фиг. 27. Так, в контрольной группе количества тромбоцитов после введения ACNU уменьшались с течением времени и на 8-10-й дни эта величина была самой низкой (приблизительно 29% от величины перед введением АСNU) и восстановление было только приблизительно 49% и 74% на 12-й и 14-й дни, соответственно. С другой стороны, в группе ТРО отмечали снижение до приблизительно 38% от величины перед введением ACNU на 5-й день и затем ситуация изменялась до увеличения. Так, на 8-й день величина восстанавливалась приблизительно до 63% величины перед введением ACNU и на 10-й день и после него количества тромбоцитов были больше, чем перед введением АСNU. Так, на 12-й и 14-й дни увеличения были приблизительно до 300% и 400%, соответственно.
Как отмечалось выше, на 8-10 дни, когда отмечали снижение в контрольной группе, в группе ТРО уже наблюдали увеличение и на 10-й день количества тромбоцитов были выше количеств перед введением АСNU. Следовательно, было подтверждено, что ТРО человека ускоряет возвращение к норме при тромбоцитопении, вызываемой противораковым агентом.
Принимая во внимание также результаты Примера 74, можно ожидать, что ТРО человека ингибирует снижение количеств тромбоцитов и ускоряет возвращение к норме в случае тромбоцитопении, независимо от типа противоракового агента.
Пример 76. Терапевтическое действие ТРО на тромбоцитопению после пересадки костного мозга (ВМТ)
48 самцов мышей штамма С3Н/НеN семинедельного возраста облучали 10 Gy радиоактивного облучения (все тело) и сразу же после этого их внутривенно инъецировали 1•106 клетками костного мозга из мышей того же штамма. Затем мышей произвольно разделяли на две группы со следующего дня (1-й день) одну из групп (контрольную группу) инъецировали РВS, тогда как другую группу (группу ТРО) инъецировали ТРО-активной фракцией, применяемой в Примере 73 (относительная активность 44000 в М-07е-тесте) с дозой 100 мкл подкожно 1 раз в день в течение 20 последующих дней.
После начала инъекции каждые 6 мышей выбирали произвольно на 5-й, 10-й, 14-й и 21-й дни, кровь брали из их глазничных вен и количества тромбоцитов считали в микроцитометре (типа Е-2500, Toa Iyo Denshi).
Изменения в количествах тромбоцитов даны на фиг.28. Так, во всех группах количества тромбоцитов снижались с течением времени после введения клеток костного мозга. На 10-й день количества были самыми низкими (приблизительно 3% от величины перед применением ВМТ). После этого отмечали постепенное восстановление и на 4-й день количества тромбоцитов были 11 и 13% в контрольной группе и в группе ТРО, соответственно. На 21-й день отмечали только 37% восстановления в контрольной группе, тогда как в группе ТРО восстановление было приблизительно 65%, после чего наблюдали значимый эффект ускорения восстановления. Описанные результаты подтвердили, что ТРО человека, полученный, как в Примере 56, усиливает восстановление к норме количеств тромбоцитов после ВМТ.
Пример 77. Терапевтическое действие ТРО на тромбоцитопению после облучения радиоактивными лучами
36 самцов мышей штамма 1CR восьминедельного возраста облучали рентгеновскими лучами 5Gy на все тело и произвольно разделяли на две группы. Со следующего дня (1-й день) одну из этих групп (контрольную группу) инъецировали РВS, тогда как другую группу (группу ТРО) инъецировали ТРО-активной фракцией, применяемой в Примере 73 (относительная активность 1440000 в М-07е-тесте) один раз в день подкожно в течение 3 последующих дней и со следующего дня относительную активность 360000 (согласно тому же тесту) инъецировали подкожно один раз в день в течение 7 последующих дней. После начала инъекций каждую из групп разделяли на три группы: группу, из которой кровь брали на 4-й, 11-й и 21-й дни; другую группу, из которой кровь брали на 7-й и 13-й дни, и третью группу, из которой кровь брали на 9-й и 15-й дни. Кровь брали из глазничных вен и количества тромбоцитов считали при помощи микроцитометра (типа F-800, Тоа Iуо Denshi). Изменения в количествах тромбоцитов даны на фиг.29. Так, в контрольной группе количества тромбоцитов снижались с течением времени после облучения рентгеновскими лучами и на 9-й день количества были самыми низкими (приблизительно 25% от величины перед облучением рентгеновскими лучами). На 11-й и 13-й дни количества тромбоцитов восстанавливались приблизительно до 38 и 67%, соответственно, и на 15-й день они восстанавливались до величин перед облучением рентгеновскими лучами. В группе ТРО эти количества снижались приблизительно до 24% величин перед облучением рентгеновскими лучами на 7-й день. После этого отмечали увеличение и на 9-й день количества тромбоцитов восстанавливались приблизительно до 82%. На 11-й день и после него количества были больше величин перед облучением рентгеновскими лучами и до 21-го дня сохранялась эта тенденция. Как упоминалось выше, на 9-й день, когда в контрольной группе наблюдали еще снижение, количества тромбоцитов в группе ТРО уже изменялись в сторону увеличения и на 11-й день количества тромбоцитов были больше величин перед облучением рентгеновскими лучами, оставаясь высокими и после этого. С другой стороны, в контрольной группе требовались 15 дней для восстановления чисел тромбоцитов до величин перед облучением рентгеновскими лучами. Эти результаты показали, что ТРО человека, полученный в Примере 56, сокращает время, необходимое для восстановления до нормы после снижения количеств тромбоцитов после облучения рентгеновскими лучами, и что он проявляет терапевтическое действие на тромбоцитопению после облучения рентгеновскими лучами.
Пример 78. Действие hTРO163 на увеличение количества тромбоцитов
15 здоровых самцов мышей штамма С3Н/НеN, из которых уже брали кровь из глазничных вен и считали в ней число тромбоцитов при помощи микроцитометра (типа F-800, Тоа Iyо Denshi), разделяли произвольно на 4 группы: контрольную группу и группы А, В и С. Первую группу (контрольную группу)
инъецировали подкожно РВS, содержащим 0,1% сыворотки из мышей, 1 раз в день в течение 5 последовательных дней. Группы А, В и С инъецировали ТРО-активной фракцией SР Sepharose Fast Flow, полученной в Примере 65, с последующим разведением РВS, содержащим 0,1% мышиную сыворотку, при дозах приблизительно 40000000, приблизительно 8000000 и приблизительно 1600000/кг массы тела, соответственно, согласно относительной активности в М-07е-тесте HTРO163, подкожно в течение 5 последовательных дней.
На 6-й, 8-й, 10-й и 12-й дни после инъекции брали кровь из глазничной вены каждой мыши и количества тромбоцитов считали при помощи микроцитометра (типа F-800, производимого Toa Iyo Denshi). Изменения в количествах тромбоцитов представлены на фиг.30.
Так, в контрольной группе почти не было изменения в количествах тромбоцитов, тогда как в получаемых ТРО группах увеличения в числах тромбоцитов были замечены во всех случаях и наивысшие величины были на 8-й день после инъекций. Среди каждой из получаемых ТРО групп была отмечена зависимость ответной реакции от дозы. Так, в группе А увеличения приблизительно 88% и приблизительно 100% отмечали на 6-й и 8-й дни, соответственно, и даже на 10-й день сохранялось приблизительно 97% увеличение, после чего все результаты показали значимое отличие от контрольной группы (р<0,01 согласно Dunnet-тесту). Подобные увеличения отмечали также в группе В и, хотя степень увеличения была ниже, чем в группе А, увеличения примерно 65% и примерно 84% отмечали на 6-й и 8-й дни, соответственно, со значимой разницей по сравнению с контрольной группой (р<0,01, Dunnet-тест). Повышающее действие в группе С было ниже, чем в группе В. Все еще наблюдали приблизительно 31% увеличение на 8-й день, когда отмечали самые высокие количества тромбоцитов. Описанные результаты подтвердили, что hТРО163, полученный в Примере 65, увеличивает количества тромбоцитов и что его действие обнаруживает зависимость ответной реакции от дозы.
Пример 79. Терапевтическое действие hТРO163 на тромбоцитопению
100 самцов мышей штамма С3Н/НеN восьминедельного возраста инъецировали внутривенно 50 мг/кг гидрохлорида нимустина (ACANU) и затем произвольно разделили на 4 группы: контрольную группу и группы А, В и С, каждая из которых состояла из 25 мышей. Со следующего дня (1-й день) контрольную группу инъецировали подкожно 5 мл/кг РВS один раз в день в течение последующих дней, тогда как группы А, В и С инъецировали подкожно ТРО-активной фракцией SP Sepharose Fast Flow, полученной в Примере 65, разбавленной РВS, при дозах 16000000, приблизительно 48000000 и приблизительно 160000000/кг массы тела, соответственно, в значениях относительной активности hТРО163 в М-07е-тесте, один раз в день в течение последующих дней.
После начала инъекций мышей в каждой из групп разделяли произвольно на 5 групп и кровь брали на 5-й, 8-й, 10-й, 12-й и 14-й дни, соответственно. Кровь брали из глазничной вены и количества тромбоцитов считали при помощи микроцитометра (типа Е-2500, производимого Тоа Iyо Denshi). Изменения в количествах тромбоцитов представлены на фиг.31. Так, в контрольной группе количества тромбоцитов снижались с течением времени после введения АСNU, давая самые низкие величины (приблизительно 15-16% от величины перед введением ACNU) на 8-10-й дни, и степень восстановления была всего лишь приблизительно 28% и приблизительно 51% на 12-й и 14-й дни соответственно. С другой стороны, в группе А эти количества уменьшались приблизительно до 25% от величин перед введением АСNU на 8-й день. После этого, однако, они изменялись в сторону увеличения и восстанавливались приблизительно до 34% и приблизительно до 89% на 10-й и 12-й дни, соответственно. На 14-й день количество тромбоцитов было больше, чем перед введением АСNU.
Как упоминалось выше, самые низкие количества тромбоцитов наблюдали на 8-й день во всех группах, хотя в группах, получавших ТРО человека, уменьшение было более медленным, чем в контрольной группе, дающей самое низкое количество тромбоцитов. В контрольной группе почти не было восстановления количеств тромбоцитов даже на 10-й день, тогда как в группах, получавших ТРО человека, восстановление в количествах тромбоцитов отмечали во всех этих группах, хотя их уровни варьировали. В группе, которой вводили 50 мкг/кг, восстановление наблюдали на 12-й день, когда количества тромбоцитов были выше, чем величины перед введением ACNU. Если сравнить это с фактом, что в контрольной группе восстановление было всего лишь приблизительно 51%, по сравнению с величиной перед введением АСNU, даже на 14-й день, становится ясно, что hТРO163, полученный, как описано в Примере 65, усиливает восстановление количества тромбоцитов после их снижения. Тем самым было подтверждено, что hТРO163 имеет терапевтическое действие на тромбоцитопению, вызываемую противораковыми агентами.
Далее приводятся примеры фармацевтических препаратов.
Пример 80
Фракцию ТРО человека, полученную в Примере 56, концентрировали, подвергали асептической обработке и замораживали при -20oС, получая замороженный продукт для применения в качестве препарата для инъекций.
Пример 81
Фракцию ТРО человека, полученную в Примере 56, концентрировали, 5 мл ее помещали во флаконы на 10 мл при асептических условиях, высушивали лиофилизацией при -20oС и флакон закрывали резиновой пробкой, получая лиофилизированное вещество для применения в качестве инъецируемого препарата.
Пример 82
Фракцию ТРО человека, полученную в Примере 56, концентрировали, подвергали асептическому фильтрованию и помещали во флакон на 10 мл, получая продукт для применения в качестве инъецируемого препарата.
Пример 83
Фракцию hТРO163, полученную в Примере 65, концентрировали, подвергали асептической обработке и лиофилизировали при -20oС, получая замороженный продукт для применения в качестве препарата для инъекций.
Пример 84
Фракцию hТРO163, полученную в Примере 65, концентрировали, 5 мл ее помещали во флакон на 10 мл при помощи асептической операции, лиофилизировали при -20oС и флакон закрывали герметично резиновой пробкой, получая лиофилизированный продукт для применения в качестве препарата для инъекций.
Пример 85
Фракцию hТРO163, полученную в Примере 65, концентрировали, подвергали стерильному фильтрованию и помещали во флакон на 10 мл, получая продукт для применения в качестве препарата для инъекций.
Пример 86
Фракцию ТРО человека, полученную в Примере 57, концентрировали, подвергали асептической обработке и замораживали при -20oС, получая замороженный продукт для применения в качестве препарата для инъекций.
Пример 87
Фракцию ТРО человека, полученную в Примере 57, концентрировали, 5 мл ее помещали во флакон на 10 мл при стерильных условиях, лиофилизировали при -20oС и флакон закрывали герметично резиновой пробкой, получая лиофилизированный продукт для применения в качестве препарата для инъекций.
Пример 88
Фракцию ТРО человека, полученную в Примере 57, концентрировали, подвергали стерильному фильтрованию и помещали во флакон на 10 мл, получая продукт для применения в качестве препарата для инъекций.
Пример 89. Апластическая плазма собак
Гепаринизованную апластическую плазму собаки ("АРК9") или нормальную плазму собаки ("NК9") получали, как описано [Mazur, E. and South, K. Exp, Hematol. 13: 1164-1172 (1985); Arriaga, M., South K., Cohen. J.L. and Mazur, E.M. Blood 69: 486-492 (1987); Mazur, E., Basilico, D., Newton, J.L., Cohen, J. L. , Charland, C. , Sohl, P.A., and Narendran, A. Blood 76: 1771-1782 (1990)] , за исключением того, что 450 рад (4,5 Дж/кг) облучения всего тела применяли для реципиентов.
Пример 90. Тест для определения мегакариоцитов человека
Стандартные способы для многих процедур, описанных в следующих примерах, или других пригодных процедур даются в известных широко описаниях способов молекулярной биологии, таких как, например Sambrook, et al., (Eds), Molecular Cloning, Second. Edition, Cold Spring Harbor Laboratory Press (1987) ub Ausubel, et al., (Eds), Current Protocols in Molecular Biology, Green associates/Wiley Interscience, New York (1990).
APK9 и фракционированную АРК9 анализировали на способность стимулировать развитие мегакариоцитов человека из клеток-предшественников GD34+, СD34 - выбранные клетки получали из клеток периферической крови, как описано (Hokom, М. Н., Choi, Е., Nichol, Hornkohl, A., Arakawa, Т., and Hunt, P.Molecular Biology of Haematopoiesis 3:15-31, 1994), и инкубировали в следующей культуральной среде: среде Дульбекко, модифицированной по способу Исков (1MDM; G1ВСО, Grand Island, NY) с добавлением 1% Глутамин Pen-Strep (Irvine Scientific, Santa Ana, CA) и 10% гепаринизованной, с низким содержанием тромбоцитов АВ плазмы человека. Также в среду включали 2-меркаптоэтанол (10-4 М), пировиноградную кислоту (110 мкг/мл), холестерин (7,8 мкг/мл), аденозин, гуанин, цитидин, уридин, тимидин, 2-дезоксицитозин, 2-дезоксиаденозин, 2-дезоксигуанозин (10 мкг/мл каждого, Sigma); человеческий рекомбинантный инсулин (10 мкг/мл), человеческий трансферрин (300 мкг/мл), липиды сои (1%, Boehringer - Mannheim, Indianapolis, 1N); рекомбинантный основной фактор роста фибробластов человека (2 нг/мл), Genzyme, Cambridge, МА); рекомбинантный эпидермальный фактор роста человека (15 нг/мл), полученный из тромбоцитов фактор роста (10 нг/мл, Amgen, Inc., Thousand Oaks, CA). CD34-выбранные клетки засевали при 2•105 клеток/мл культуральной среды, с конечным объемом 15 мкл, в лунки микротитрационных планшетов Tepacaku (Vanguard, Inc., Neptune, NJ). Клетки инкубировали при 37oС в течение 8 дней в увлажняемых боксах в 5% СО2 в воздухе, фиксировали непосредственно в культуральных лунках 1% глутаральдегидом и инкубировали со смесью моноклональных антител (анти-GPIb, анти-GР11В, (Biodesign) и анти-GP1b (Dako, Carpinteria, CA). Иммунную реакцию проявляли системой детектирования со стрептавидин-β-галактозидазой (HistoMark, Kirgegaard and Perry). Мегакариоциты, идентифицированные по синей окраске, считали при помощи инвертационного микроскопа при 100Х увеличении. Результаты представляли в виде средних чисел мегакариоцитов на лунку ± стандартная ошибка среднего (SЕМ). В некоторых случаях данные представлены в "мегакариоцитных единицах/мл", где степень, до которой данная проба индуцировала развитие мегакариоцитов, нормализована до положительного АРК9 контроля для данного эксперимента. Одна единица определена как количество материала, которое приводит к такому же количеству мегакариоцитов, что и 1 мкл АРК9-стандартов. Активность считали связанной с MpL лигандом, если ее можно было блокировать 5-10 мкг/мл MPL-X (растворимым рецептором Мpl). Было показано, что АРК9 содержит фактор (факторы), которые стимулируют развитие мегакариоцитов человека в этой системе. СD34-выбранные клетки, инкубированные с 10% NK9 в течение 8 дней, обнаружили незначительное число окрашенных в синий цвет мегакариоцитов, тогда как CD34-выбранные клетки, инкубированные с 10% АРК9 в течение 8 дней, обнаружили очень большое число окрашенных в синий цвет мегакариоцитов.
Фиг.32 показывает, что увеличивающиеся концентрации Mpl-X, добавленные к системе культуры мегакариоцитов человека, увеличивающимся образом блокируют развитие мегакариоцитов. При концентрациях Мрl-Х более 5 мкг/мл ингибирование было полным. В этом эксперименте СD34-выбранные клетки стимулировались 5% АРК9. Это показывает, что активность, которая взаимодействует с Mpl-X (предпочтительно Мрl лиганда), необходима для развития мегакариоцитов человека, и подразумевает, что Mpl лиганд присутствует в самой АРК9.
Далее было показано, что активность Мрl лиганда, необходимая для развития мегакариоцитов человека, присутствует в АРК9. АРК9 (135 мл) разбавляли в 6 раз в среде Исков и наносили на аффинную колонку Мрl-Х. Несвязанный материал (проходящую через колонку фракцию) собирали и концентрировали до первоначального объема перед анализом. Связанный материал элюировали в 10 мл 1 М NаС1 и 20% пул диафильтровали и концентрировали в 4 раза перед анализом. СD34-выбранные клетки, инкубированные только в среде, не развивались в мегакариоциты. Клетки, инкубированные в 5% АРК9 (тот же пул, который нанесен на колонку), развивались в 48±8 мегакариоцитов на лунку. Клетки, инкубированные в 10% несвязанного материала, не развивались в мегакариоциты. Клетки, инкубированные в 10% элюированного пула, развивались в 120±44 мегакариоцитов на лунку. Как активность загрузки колонки, так и активность элюированного пула по существу полностью ингибировались 5 мкг/мл Mрl-X в этом тесте.
Пример 91. Трансфекция мышиного или человеческого рецептора Mpl в мышиную клеточную линию А. Мышиный рецептор Mpl
кДНК мышиного рецептора Mрl полной длины была субклонирована в экспрессирующий вектор, содержащий промотор транскрипции, полученный из LTR вируса Moloney саркомы мышей. 5 мкг этой конструкции и 1 мкг селектируемой маркерной плазмиды pWLneo (Stratagene) ко-электропорировали в 1L-3-зависимую мышиную клеточную линию (32D, clone 23; Greenberger et al., РNАS 80:2931-2936 (1983)). Клетки культивировали в течение 5 дней, извлекали и затем инкубировали в избирательных средах, содержащих 800 мкг/мл Geneticin (G418, Sigma) и 1 нг/мл мышиного 1L-3. Выжившие клетки затем делили на пулы по 2•105 клеток и культивировали до выращивания популяции клеток, которые затем анализировали. 6 популяций тестировали на поверхностную экспрессию рецептора Мрl при помощи FАСS анализа с применением поликлональной кроличьей антипептидной сыворотки. Одну популяцию выбирали для FАСS сортинга (с применением клеточного сортера с возбуждением флуоресценции) с применением той же самой антипептидной сыворотки. Одноклеточные клоны родительской клеточной линии отбирали по росту в 10% АРК9 и Генетицине. После отбора в АРК9 в течение 35 дней клетки выдерживали в 1 нг/мл мышиного 1L-3. Один из субклонов, 1А6.1, применяли для этой части работы.
В. Человеческий рецептор Мрl
Последовательность кДНК человеческого рецептора MPl полной длины (Vigon, 1. , et al., РNАS 89: 5640-5644 (1992)) субклонировали в экспрессирующий вектор, содержащий промотор транскрипции вируса Maloney саркомы мышей (тот же вектор, что и в случае мышиного рецептора). 6 мкг этой конструкции и 6 мкг амфотрофической ретровирусной упаковочной конструкции (Landau, N.R., Littman, D.R., J. Virology 66: 5110-5113 (1992)) трансфицировали в 3•106 клеток 293 с применением набора для трансфекции Stratagene с СаРO4. Те же самые клетки повторно трансфицировали после 2 дней и еще раз после 4 дней. На другой день после последней трансфекции 293 клетки культивировали совместно С 1L-3-зависимой мышиной клеточной линией (32D, клон 23; Greenberger et al., PNAS 80: 2931-2936 (1983). После 24 часов 32D клетки выделяли и получали в виде полосы в градиенте ВSА (Рath-о-сyte; mills 1nc.). Клетки увеличивали в объеме в 1 нг/мл мышиного 1L-3 и затем отбирали на рост в 20% АРК9. Клетки подвергали сортингу на экспрессию на их поверхности рецептора при помощи FACS с применением поликлональной кроличьей антипептидной сыворотки. Эти клетки затем применяли в тестах.
Пример 92. 1А6.1 анализ для определения Мрl лиганда
1А6.1 клетки промывали от 1L-3 культуры и помещали в микротитрационные планшеты Терасаки (1000 клеток/15 мкл общих кол./лунку) в α-MEM (Gibco) с добавлением 10% фетальной телячьей сыворотки (FCS), Генетицина (800 мкг/мл) и 10% пен/стреп (Gibco) в серийных разведениях 1:1 тест-проб. После 48 часов определяли под микроскопом число жизнеспособных клеток на лунку. За одну единицу активности принимали количество активности, которое приводило к 200 жизнеспособным клеткам на лунку. Активность считали связанной с Мрl лигандом, если ее можно было полностью блокировать добавлением в реакционную среду 5-10 мкг/мл Мрl-Х. Активность Мрl лиганда в АРК9 была в среднем 4400±539 единиц/мл апластической плазмы. Если нет других указаний, единицы активности Мрl лиганда определяли в 1А6.1-тесте.
Тесты с клетками, трансфицированными геном человеческого рецептора Mpl, проводили в основном так же, как с 1А6.1 клетками.
Пример 93. Демонстрация того, что Мрl-лиганд присутствует в апластической плазме или сыворотках таких источников, как мышь, собака, свинья и человек
MPL-лиганд присутствует в апластической плазме или сыворотках из мышей, собак, свиней и человека (таблица 9). Плазму собирали из BDF1 мышей с предоблучением и 12 днями пост-облучения (500 рад). Плазму тестировали в 1А6.1-тесте, в котором она показала 2000 единиц/мл активности, которая по существу полностью ингибировалась Mpl-X (10 мкг/мл). Облученная мышиная плазма была также положительна в тексте на мегакариоциты человека, в котором она обнаружила активность 1833 единиц/мл. Плазму собирали из собак с предоблучением и 10 днями пост-облучения (450 рад). Плазму тестировали как в 1А6.1 тесте, так и в тестах с развитием мегакариоцитов человека. Активность была обнаружена и полностью ингибировалась Mpl-X (10 мкг/мл) в обоих текстах. Плазму собирали из свиней, предоблученных с 10 днями пост-облучения (650 рад). Плазму тестировали как в 1А6.1 тесте, так и в тесте с развитием мегакариоцитов человека. В обоих тестах она обнаружила активность Mpl-лиганда (ингибируемую 10 мкг/мл Mpl-X), сравнимую с активностью, найденную в апластической плазме собак. Были получены сыворотки человека. Этот материал собирали из больных с пересадкой костного мозга. Сыворотки из 6 больных тестировали в 1А6.1 тесте, где они показали активность 903 единица/мл, 88% которой была связана с Mpl-лигандом (ингибировалась 10 мкг/мл Mpl-X). Сыворотки из 14 апластических больных (с аномалией развития) тестировали в тесте с развитием мегакариоцитов человека. Как группа, они обнаружили основную активность, 941 мегакариоцитных единиц/мл, которая полностью ингибировалась 10 мкг/мл Mpl-X. Приведены данные с мышиным 1L-3, чтобы показать специфичность 1А6.1 теста. Хотя этот рекомбинатный цитокин индуцирует рост этой клеточной линии, он не блокируется 10 мкг/мл Mpl-X.
Пример 94. Получение производных ТРО человека, экспрессируемых в системе Е.соli
С целью попытки улучшения биологической активности, стабильности и растворимости некоторые производные ТРО были сконструированы и экспрессированы в E. coli. Эти производные, генерированные с применением аминокислотной последовательности, представленной в SEQ 1D 11, включали: [Met-2, Lys-1, Аla1, Val3, Arg129] TPO(1-163), в котором аргинин заменяет лейцин в положении аминокислоты 129 (также называемый L129), [Met-2, Lys-1, Ala1, Val3, Arg133] TPO(1-163), в котором аргинин заменяет гистидин при положении аминокислоты 133 (также называемый Н133R), [Met-1, Lys-1, Ala1, Val3, Arg143] TPO(1-163), в котором аргинин заменяет метионин при положении аминокислоты 143 (также называемый M143R), [Met-2, Lys-1, Ala1, Val3, Leu82] TPO(1-163), в котором лейцин заменяет глицин при положении аминокислоты 82 (также называемый G82L), [Met-2, Lys-1, Ala1, Val3, Leu146] TPO(1-163), в котором лейцин заменяет глицин при положении аминокислоты 146 (также называемый G146L), [Met-2, Lys-1, Ala1, Val3, Pro148] TPO(1-163), в котором пролин заменяет серии при положении аминокислоты 148 (также называемый S148Р), [Меt-2, Lys-1, Ala1, Val3, Arg59] - TPO(1-163), в котором аргинин заменяет лизин при положении аминокислоты 59 (также называемый K59R), [Met-2, Lys-1, Аla1, Val3, Arg115] TPO(1-163), в котором аргинин заменяет глутаминовую кислоту при положении аминокислоты 115 (также называемый Q115R).
При получении этих производных ТРО в качестве матрицы использовали h6Т(1-163), описанный в примере 42, а в качестве праймеров применяли следующие синтезированные олигонуклеотиды:
Ll29R: 5'-АТСТТССGТТСТТТССАGСАССТ-3' (для получения Аrg-замещенного производного с Leu-129, замещенным Аrg в последовательности, представленной в SEQ 1D 11);
H133R: 5'-ТСТТТССАGСGТСТGСТGСGT-3' (для получения Аrg-замещенного производного с Нis-133, замещенным Arg в последовательности, представленной в SEQ 1D 11);
M143R: 5'-СGТТТССТGСGТСТGGТТGGС-3' (для получения Arg-замещенного производного с Met-143, замещенным Аrg в последовательности, представленной SEQ 1D 11);
G82L: 5'-GGССАGСТТСТGССGАССТGССТ-3' (для получения Lеu-замещенного производного Gly-82, замещенным Leu в последовательности, представленной в SEQ 1D 11);
G146L: 5'-ATGCTGGTTCTGGGTTCTACCCT-3' (для получения Leu-замещенного производного с Gly-146, замещенным Leu в последовательности, представленной в SЕQ 1D 11);
S148Р: 5'-GTTGGСGGТССGАСССТGТGСG-3' (для получения Рrо-замещенного производного с Sеr-148, замещенным Рrо в последовательности, представленной в SEQ 1ВD 11);
K59R: 5'-GААGАGАСССGСGСТСАGGАСАТСС-3' (для получения Arg-замещенного производного c Lys-59, замещенным Аrg в последовательности, представленной в SЕQ 1D 11) и
Q115R: 5'-CTGCCGCCACGTGGCCGTACCAC-3' (для получения Arg-замещенного производного с Gln-115, замещенным Аrg в последовательности, представленной в SЕQ 1D 11).
Мутантные плазмиды были сконструированы с применением набора для in vitro мутагенеза Sculptor (Amersham). Фрагмент Хbа 1 - Нind 111, полученный из pCFM536/h6Т(1-163), описанной в Примере 42, вводили в фаговый вектор Bluescript 11SK (-) (Stratagene, California, USA) после расщепления Xba 1 и Hind 111 для получения плазмиды рSКТО, которую затем трансформировали в E.coli JМ109. Одноцепочечную ДНК для мутагенеза получали из рSКТО с применением хелперного фага М13КO7 (Takara-Shuzo, Japan). Мутации вводили в ген ТРО в соответствии с протоколами поставщика с применением олигонуклеотидов, перечисленных выше. Секвенирование проводили с набором для секвенирования ДНК (Applied Biosystems, USA) в соответствии с протоколами поставщика и мутации ТРО были подтверждены.
Фрагменты Хbа 1 - Hind 111, полученные из мутированного рSКТРО, вводили в плазмиду рСЕМ536, расщепленную Хbа 1 и Hind 111. Плазмиды pCFM536, несущие мутированные гены, трансформировали в E.coli 261 для получения трансформанта, экспрессирующего гены производного ТРО.
Культивирование Е. coli 261, трансформированной мутированной рСFМ536, проводили в соответствии с Примером 43, как описано ранее. Осадок клеток трансформанта собирали и хранили в морозильнике не менее 1 дня. Замороженный клеточный осадок (3 г) суспендировали в 30 мл 20 мМ Трис-буфера (рН 8,5), содержащего 10 мМ ЭДТА, 10 мМ ДТТ и 1 мМ PMSF. Суспензию хранили на льду и обрабатывали ультразвуком 5 раз с интервалом 1 мин (по меньшей мере). Обработанные ультразвуком клетки собирали центрифугированием при 15000 об/мин, в течение 10 минут.
Осадок суспендировали в 30 мл 10 мМ Трис-буфера (рН 8,7), содержащего 8 М гуанидинхлорид, 5 мМ ЭДТА, 1 мМ PMSF и восстанавливали добавлением 50 мг ДТТ. После перемешивания в течение 1-1,5 ч рН раствора доводили до 5,0 разбавленной НС1 и затем охлаждали раствор до 4oС перед разведением.
Раствор для проб постепенно разбавляли в 1,5 л 10 мМ CAPS-буфера (рН 10,5), содержащего 30% глицерин, 3 М мочевину, 3 мМ цистамин и 1 мМ L-цистеин, в течение ночи. После перемешивания пробы не менее 2 дней осадок удаляли центрифугированием при 8000 об/мин в течение 45 минут. рН раствора доводили до 6,8 6M фосфорной кислотой и раствор разбавляли в 2 раза. Раствор фильтровали через 2 слоя фильтровальной бумаги 2 (диаметр 90 мм, Тоyо фильтровальная бумага, Japan) и затем наносили на колонку (2,6•10 см) СМ-Sepharose Fast Flow (Pharmacia). Колонку промывали 400 мл 10 мМ фосфатного буфера (рН 6,8), содержащего 15% глицерин и 1 М мочевину, и уравновешивали 10 мМ фосфатного буфера (рН 7,2), содержащего 15% глицерин. Белок элюировали из колонки при помощи системы линейного градиента от 10 мМ фосфатного буфера (рН 7,2), содержащего 15% глицерин, до того же буфера, содержащего 0,5 М NаС1, при скорости течения 1,0 мл/мин. Элюцию белка наблюдали по поглощению при 280 нм и фракцию, содержащую производное ТРО, подтверждали при помощи SDS-PAGE и собирали.
После концентрирования фракции ТРО (примерно 30 мл) до 2 мл при помощи Centriprep 10 (Omicon) мутантный ТРО очищали далее при помощи НРLС с обращенной фазой (Waters) с колонкой α Bondasphere, С4 (3,9•150 мм). Элюцию белка проводили с применением линейного градиента от 0,05% TFA в Н2О до 2 пропанола, содержащего 30% СН3СN и 0,02% TFA, при скорости течения 0,5 мл/мин в течение 40 минут. При этих условиях производные ТРО элюировались приблизительно после 30 минут.
Для биологического анализа очищенную таким образом пробу ТРО диализовали два раза против 1 л 10 мМ фосфатного буфера в течение 2 дней. После концентрирования при помощи Centriprep 10 (Omicon) приблизительно до 0,1 мг/мл биологическую активность оценивали в М-07е-тест-системе, как описано ранее. Было найдено, что все мутанты имеют почти одинаковую активность по сравнению с активностью h6Т(1-163), стандартной пробой ТРО (см. фиг.33 и 34).
Список депозитов
Escherichia coli (рHT1-231/DH5):
FERM BP-4564 и ССТСС-М95001
Escherichia coli (pEF18S-A2μ/DH5):
FERM BP-4565 и ССТСС-М95002
Escherichia coli (pHGT1/DH5):
FERM BP-4616 и CCTCC-M95003
Escherichia coli (pHTF1/DH5):
FERM BP-4617 и ССТСС-М95004
Мышь-Мышь-гибридома Р55:
FЕRМ ВР-4563 и ССТСС-С95001
СНO28/1/1/3-С6:
FERM ВР-4988 и ССТСС-С95004 СНO163Т-63-79-С1:
FERM BP-4989 и ССТСС-С95005.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРОМБОПОЭТИН | 1994 |
|
RU2245365C2 |
| ДЕГРАДИРОВАННОЕ АНТИТЕЛО, ЯВЛЯЮЩЕЕСЯ АГОНИСТОМ TPO | 2001 |
|
RU2287534C2 |
| ПОЛИНУКЛЕОТИДНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ (ВАРИАНТЫ), ПОЛИПЕПТИД (ВАРИАНТЫ), СЛИТЫЙ БЕЛОК, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, МОНОКЛОНАЛЬНОЕ АНТИТЕЛО, ШТАММ ГИБРИДНЫХ КУЛЬТИВИРУЕМЫХ КЛЕТОК | 1995 |
|
RU2143491C1 |
| ПРОИЗВОДНЫЕ ФАКТОРА ИНГИБИРОВАНИЯ ЛИПОГЕНЕЗА И ИХ ПОЛУЧЕНИЕ | 1993 |
|
RU2138554C1 |
| СПОСОБЫ И ПРЕПАРАТЫ ДЛЯ СТИМУЛИРОВАНИЯ РОСТА И ДИФФЕРЕНЦИРОВКИ МЕГАКАРИОЦИТОВ | 1995 |
|
RU2158603C2 |
| АНТИТЕЛО, НАПРАВЛЕННОЕ НА БЕЛОК SIGLEC-15, СВЯЗАННЫЙ С ОСТЕОКЛАСТАМИ | 2008 |
|
RU2475499C2 |
| АНТИТЕЛА, СОДЕРЖАЩИЕ ТЕРАПЕВТИЧЕСКИЕ ПЕПТИДЫ-МИМЕТИКИ ТРО/ЕРО | 2010 |
|
RU2559526C2 |
| НОВЫЕ БЕЛКИ И СПОСОБЫ ПОЛУЧЕНИЯ ТАКИХ БЕЛКОВ | 1996 |
|
RU2194714C2 |
| ПОЛИПЕПТИД, СПОСОБ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ДНК (ВАРИАНТЫ), ВЕКТОР (ВАРИАНТЫ), КЛЕТКА, ПРИМЕНЕНИЕ ПОЛИПЕПТИДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) | 1993 |
|
RU2177480C2 |
| АНТИТЕЛО ПРОТИВ Siglec-15 | 2010 |
|
RU2539790C2 |


































































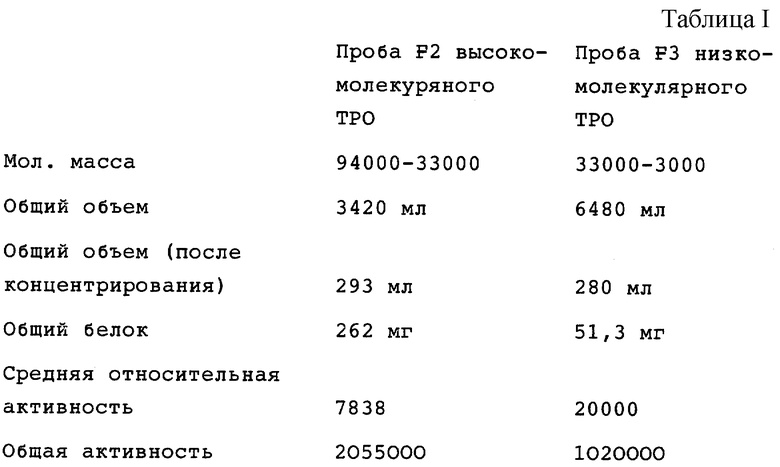

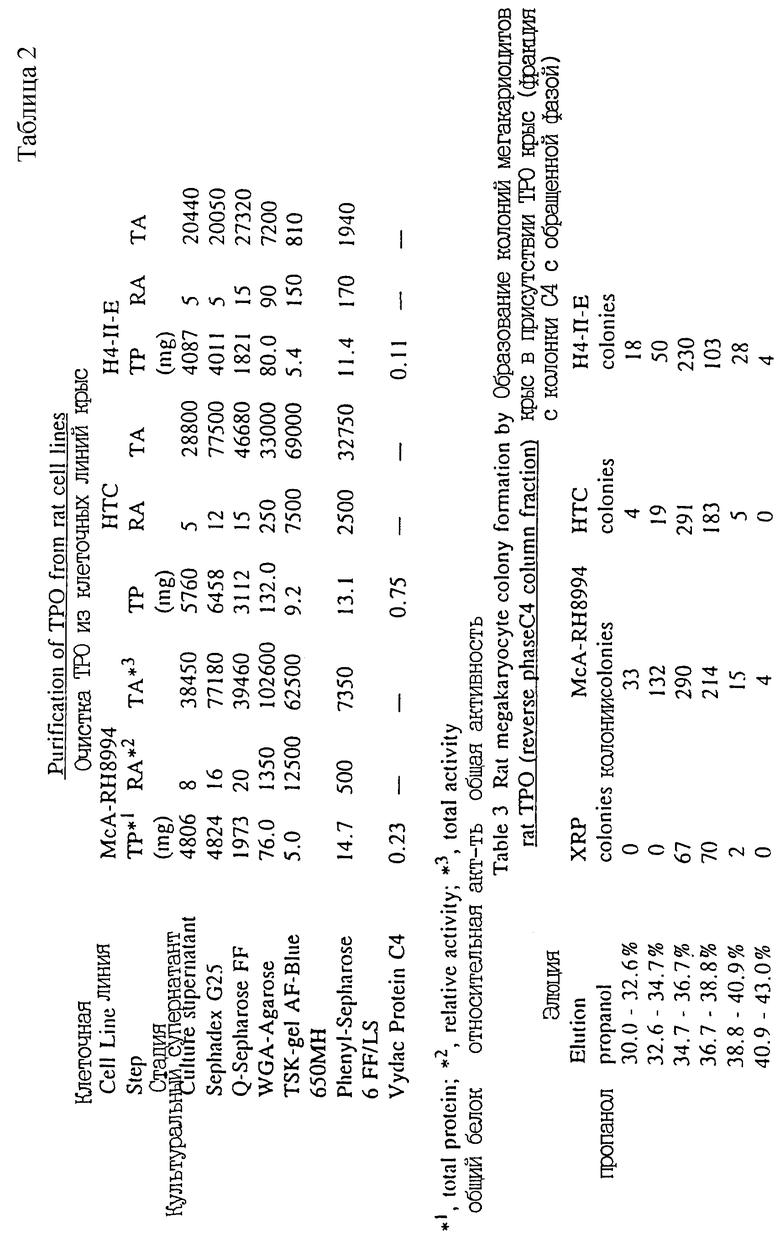










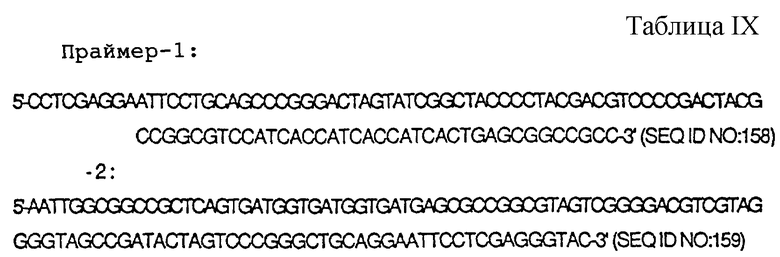


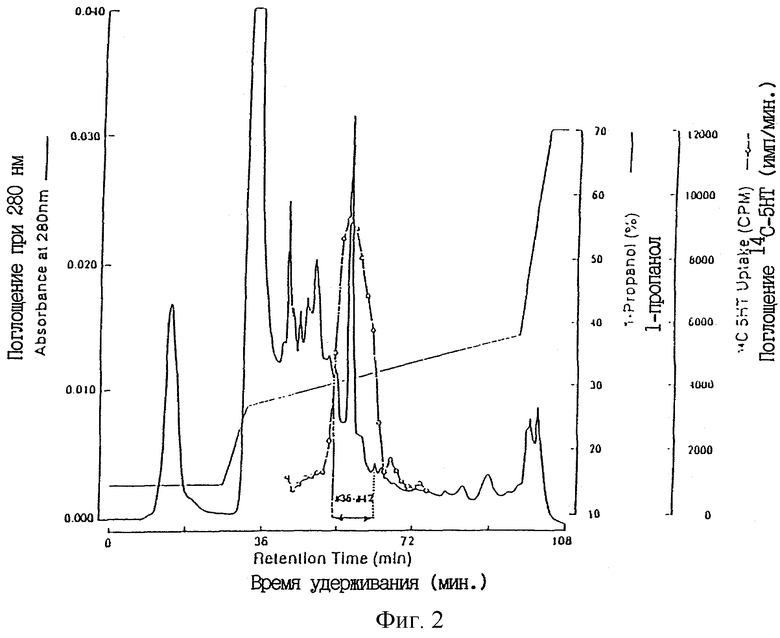
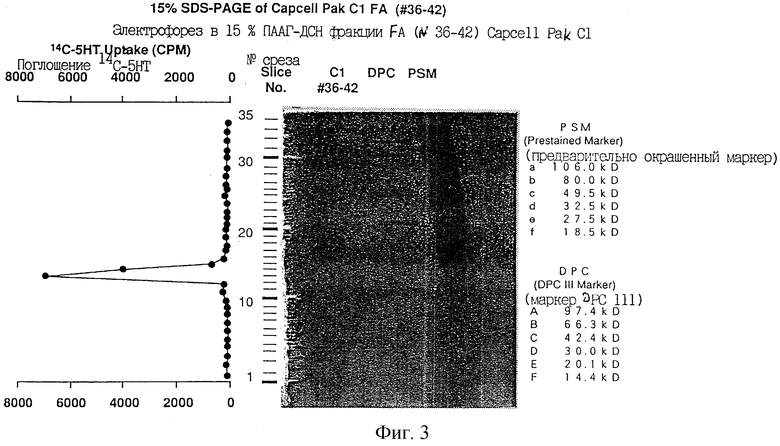
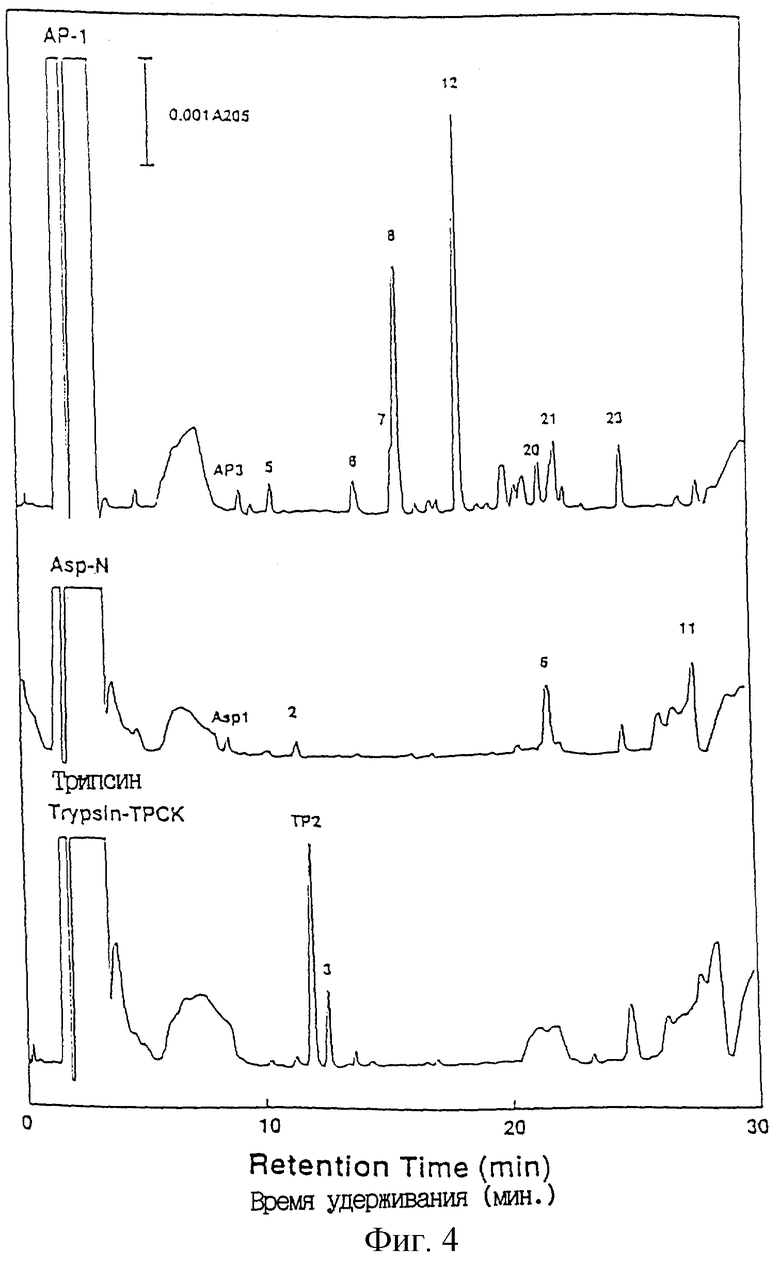
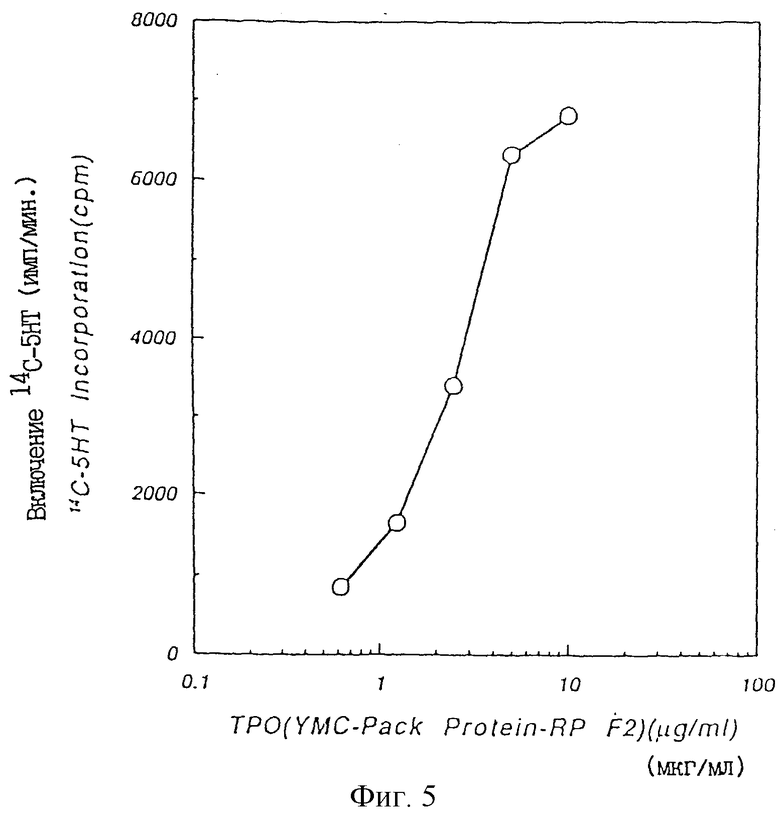
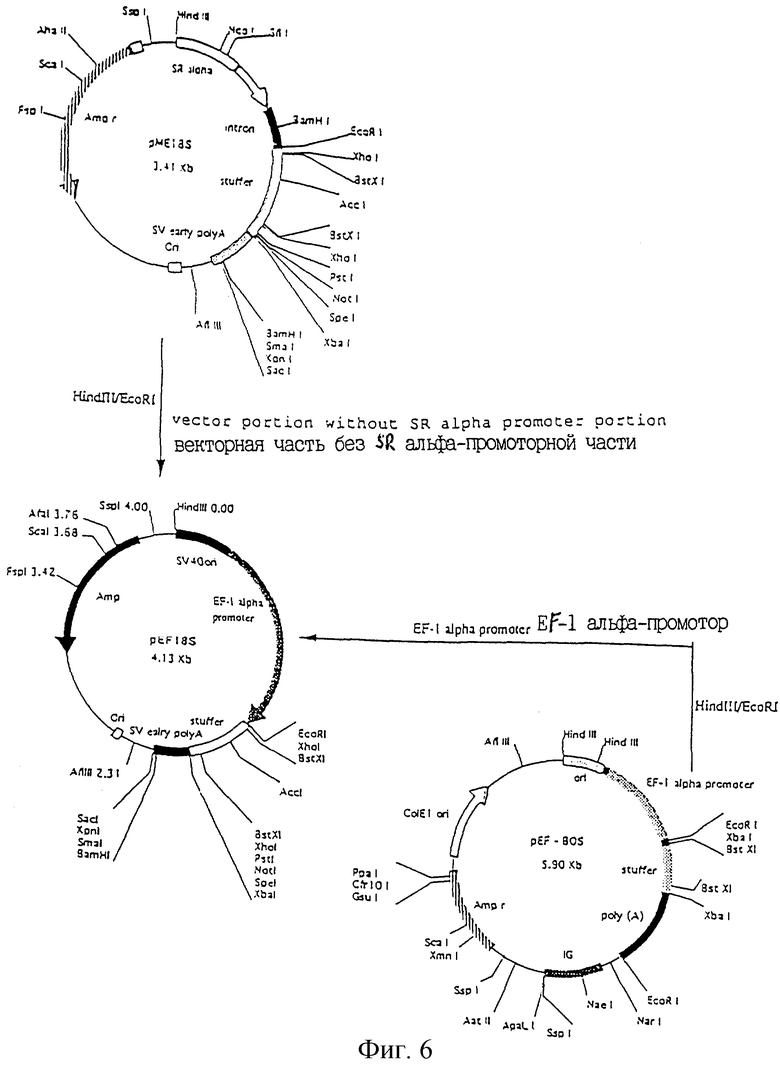
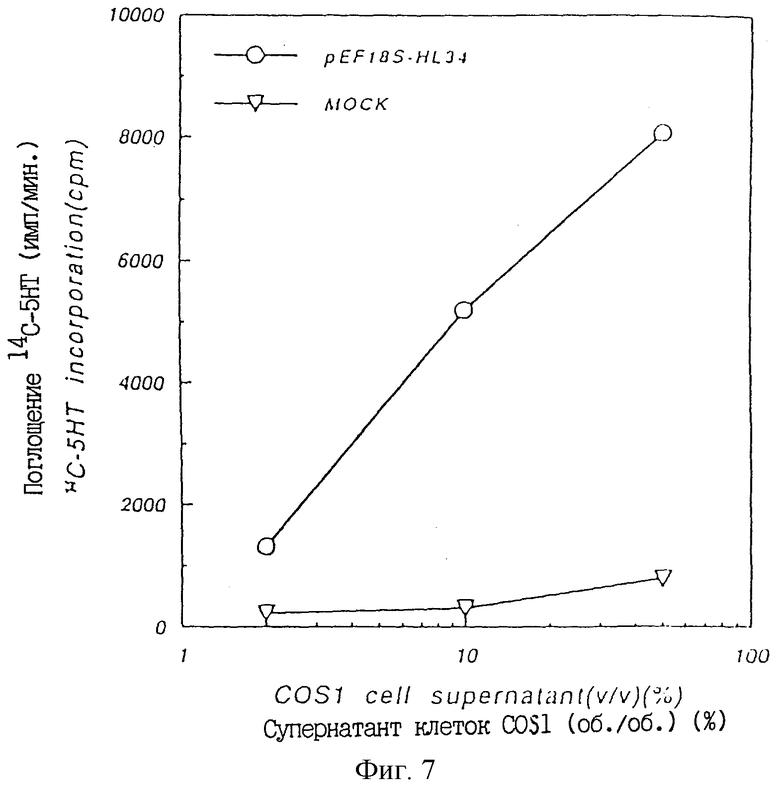
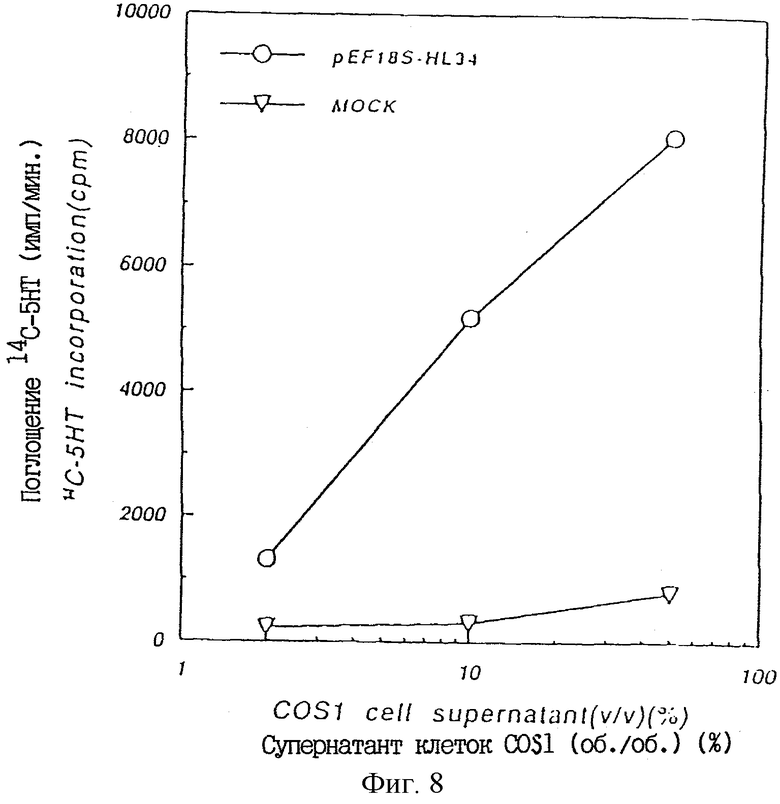
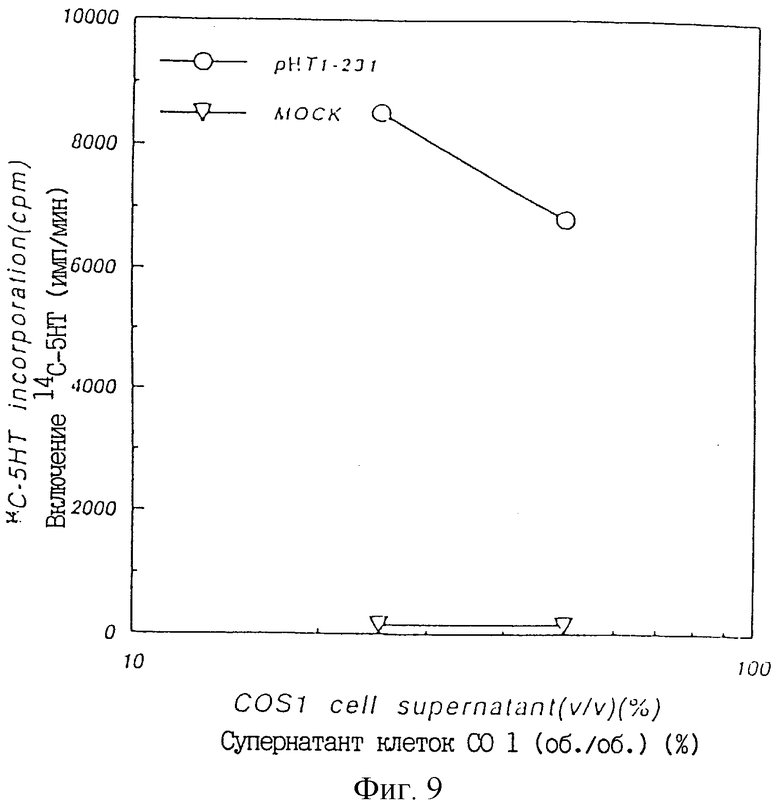
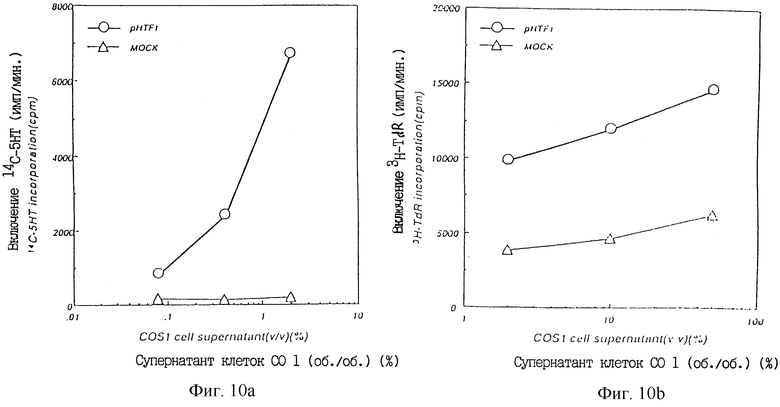
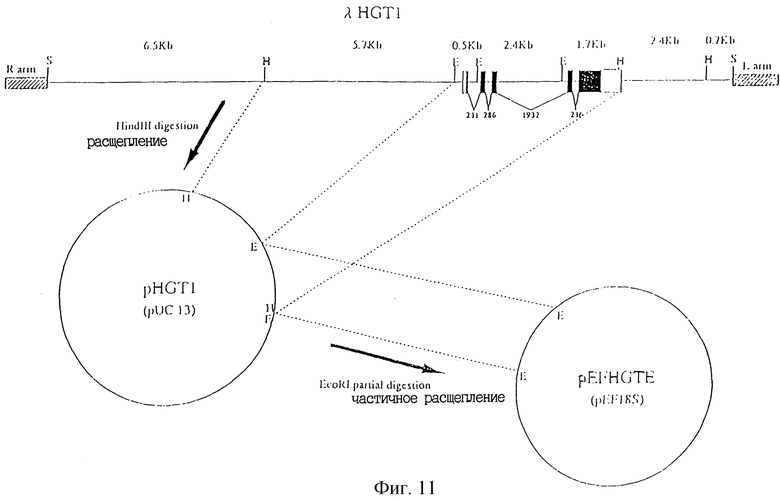
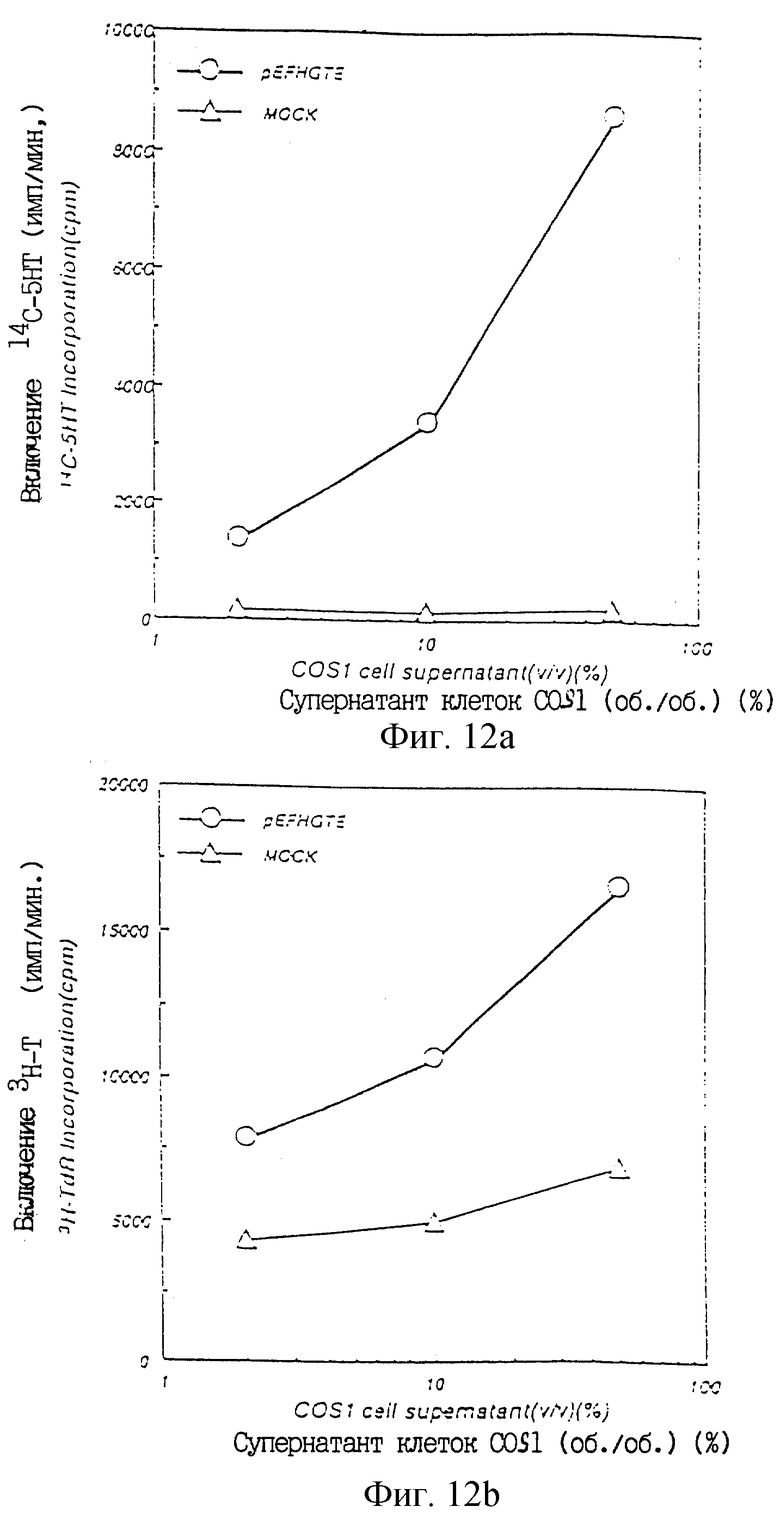
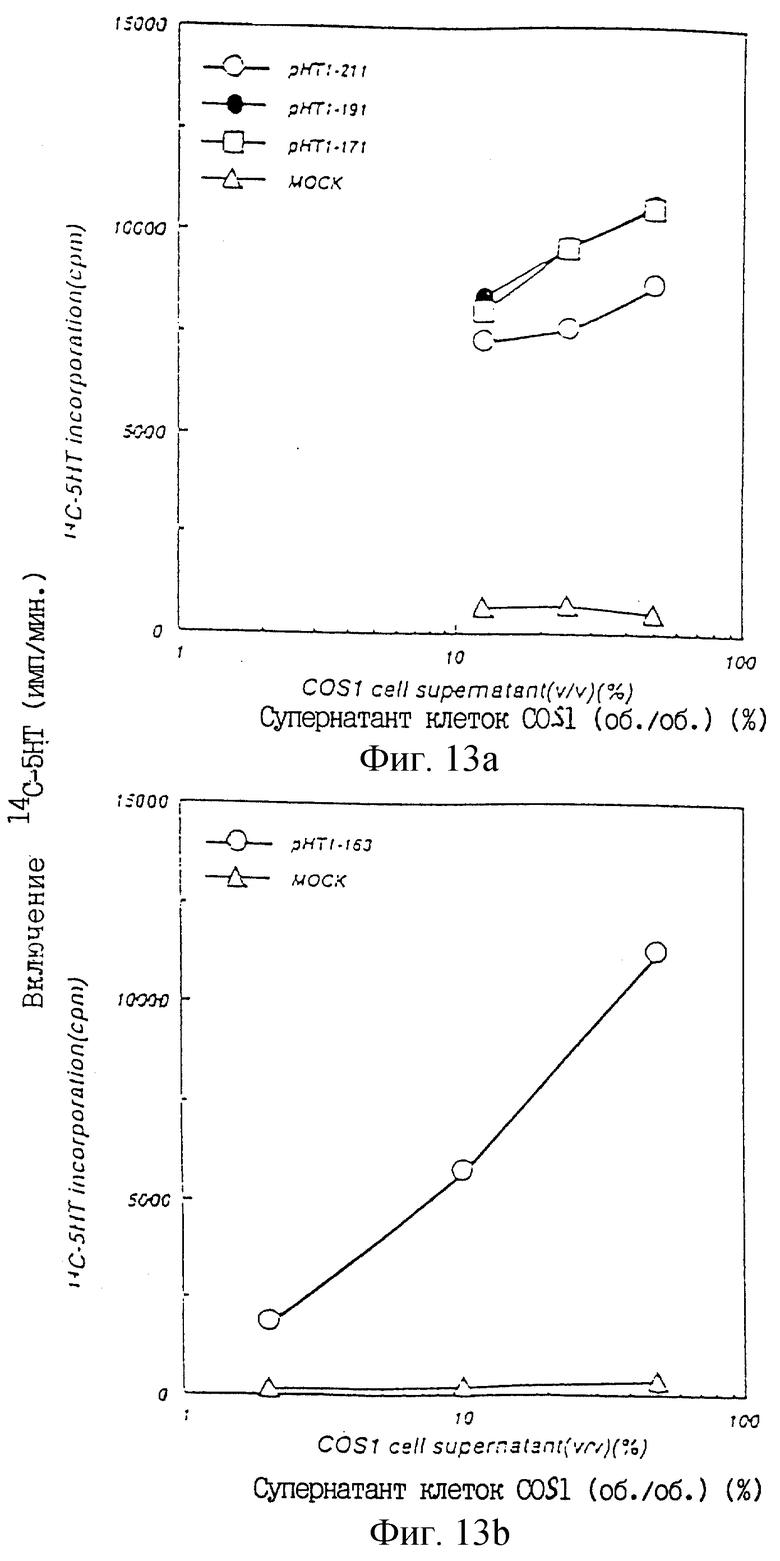
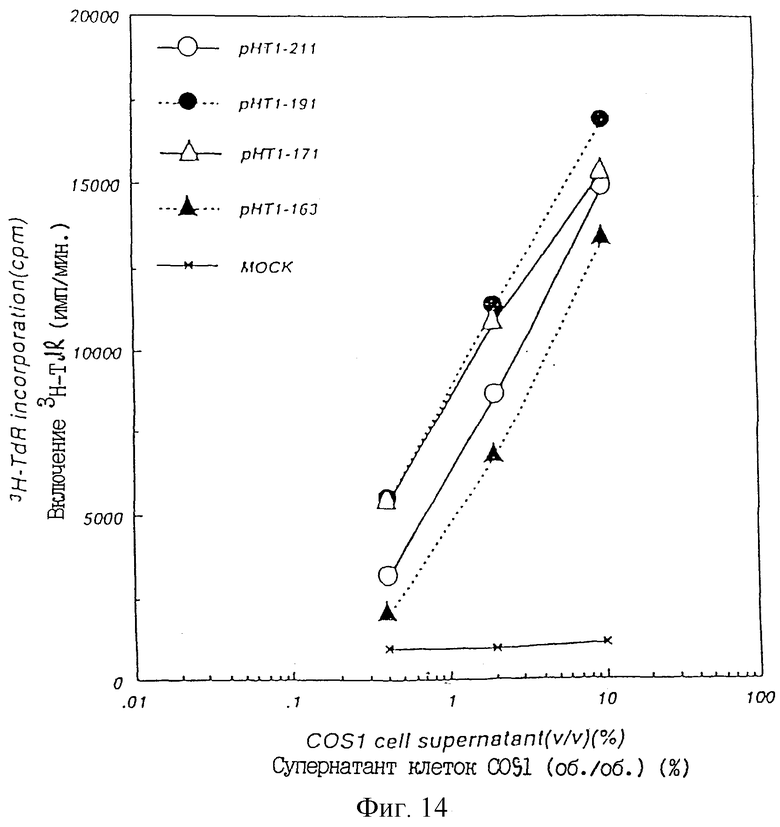
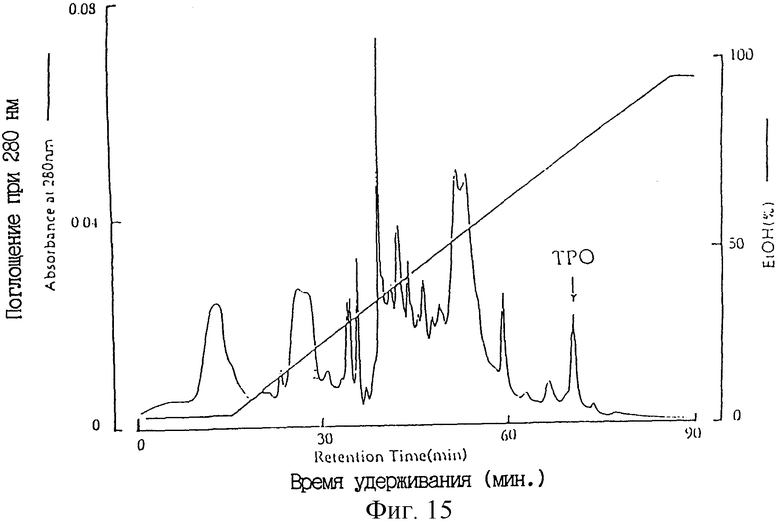
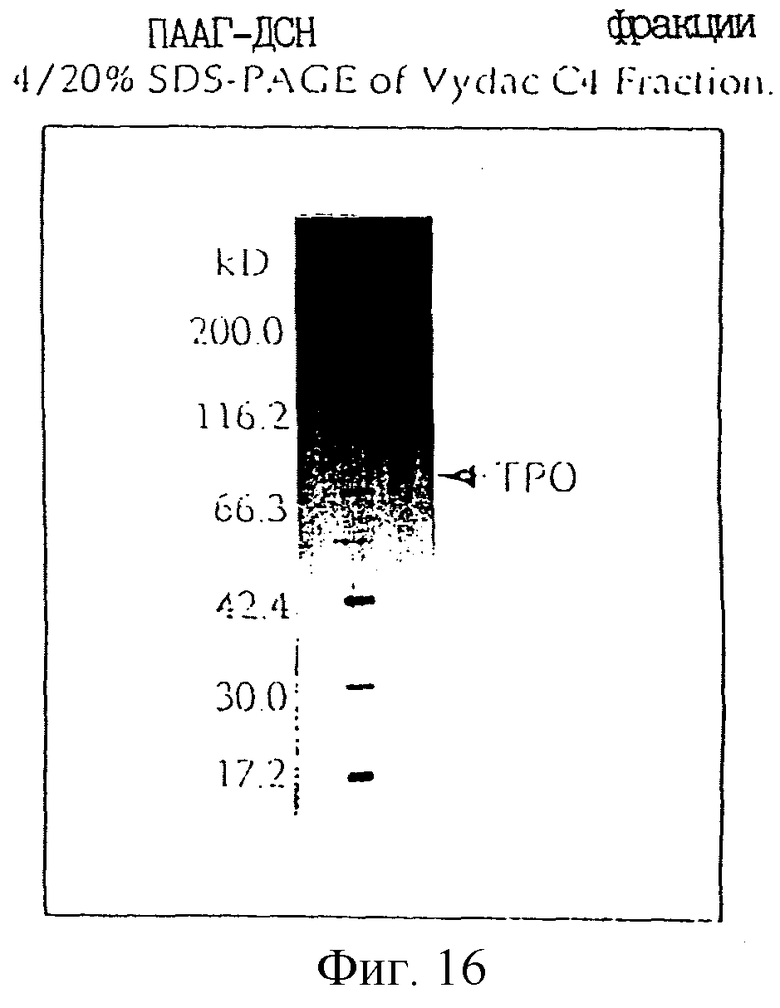
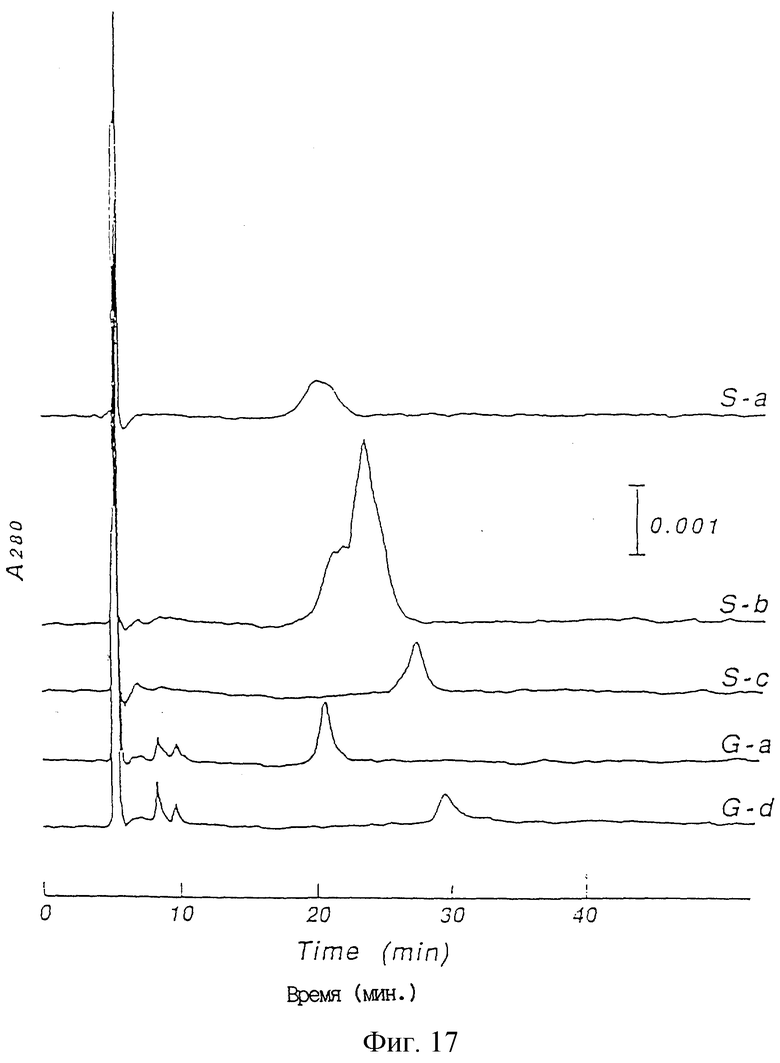

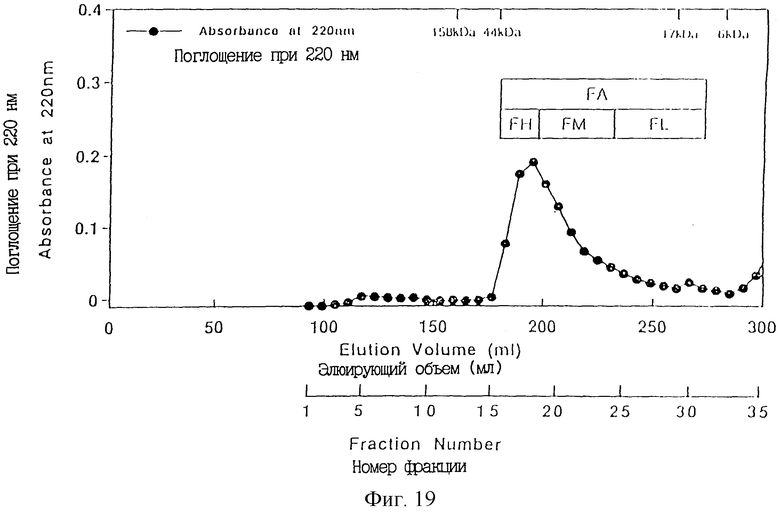
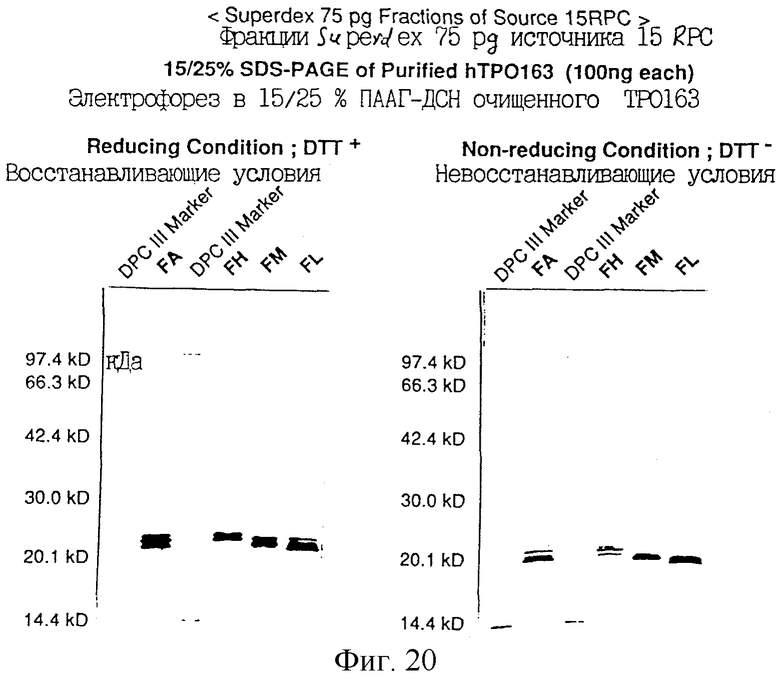
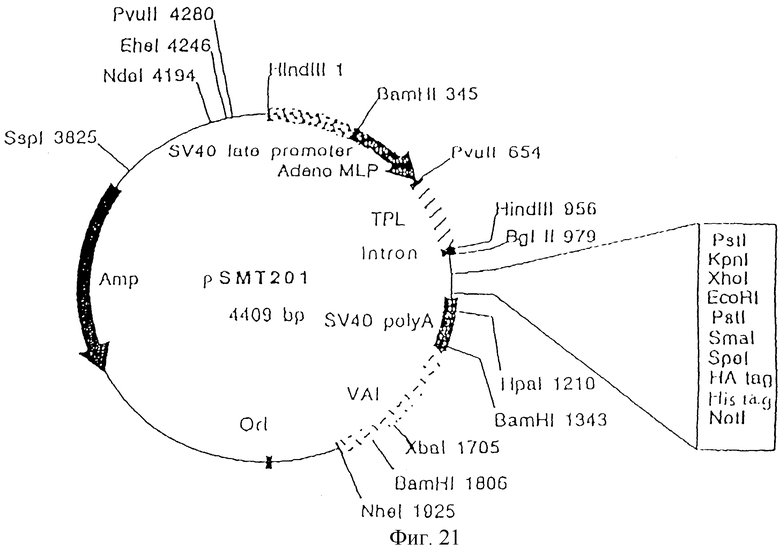
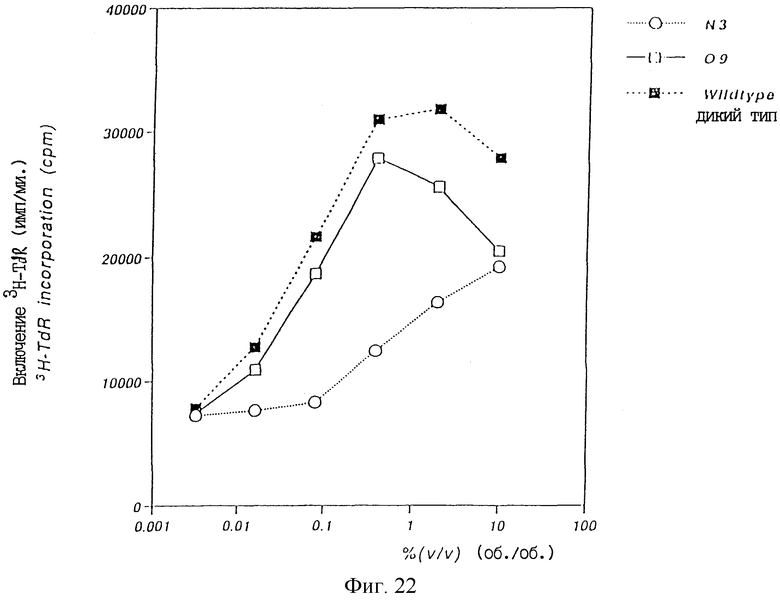
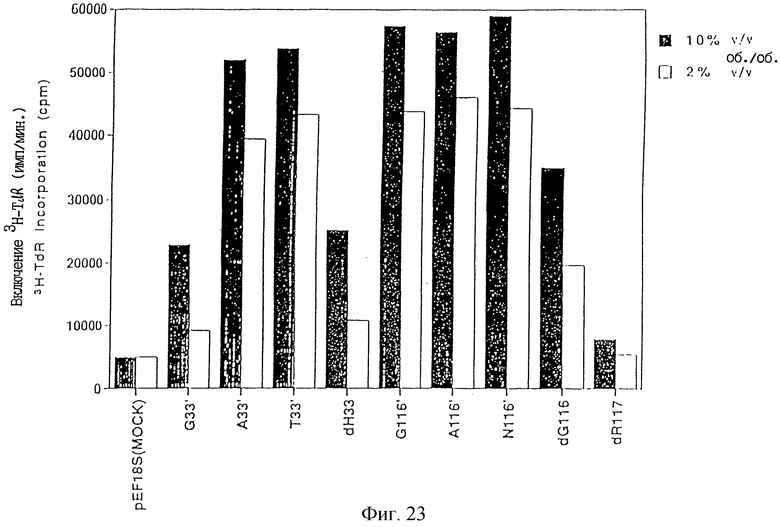
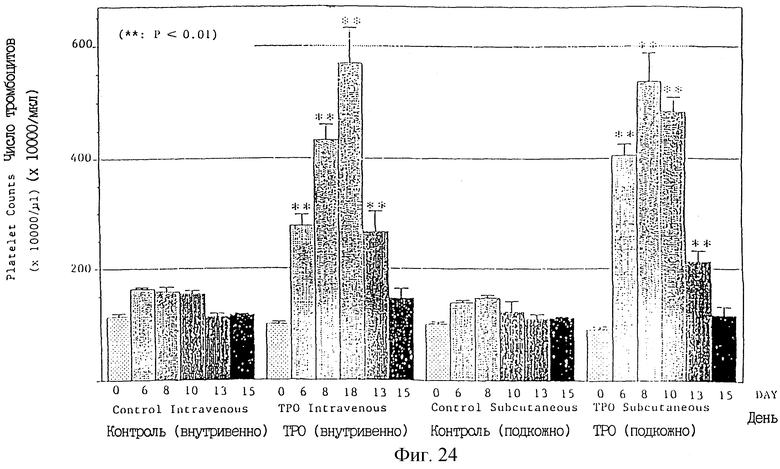

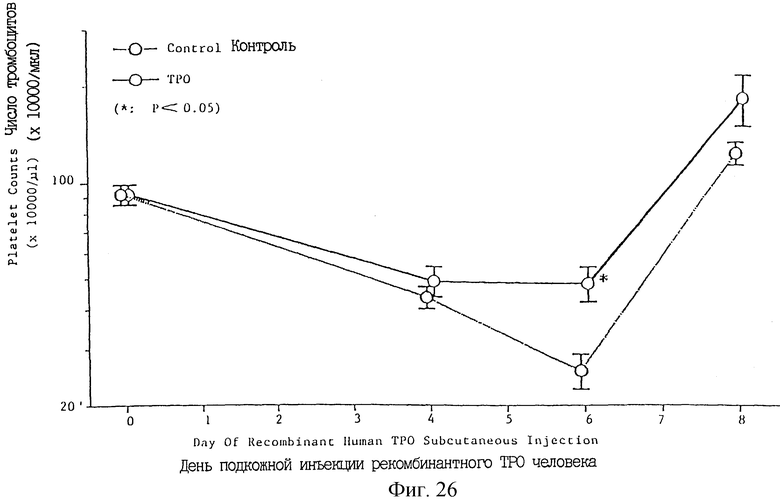

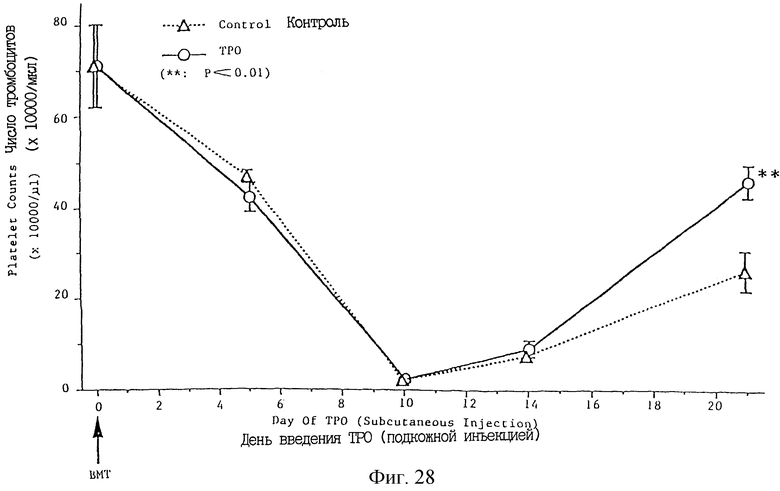
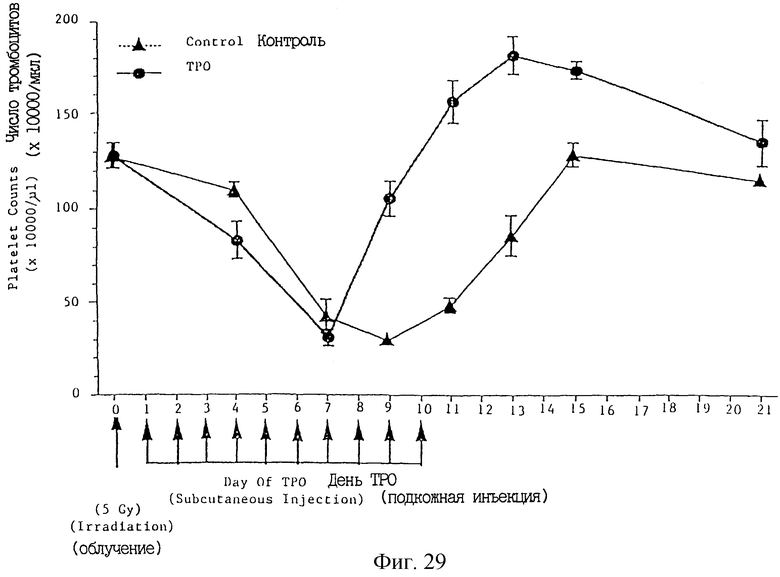
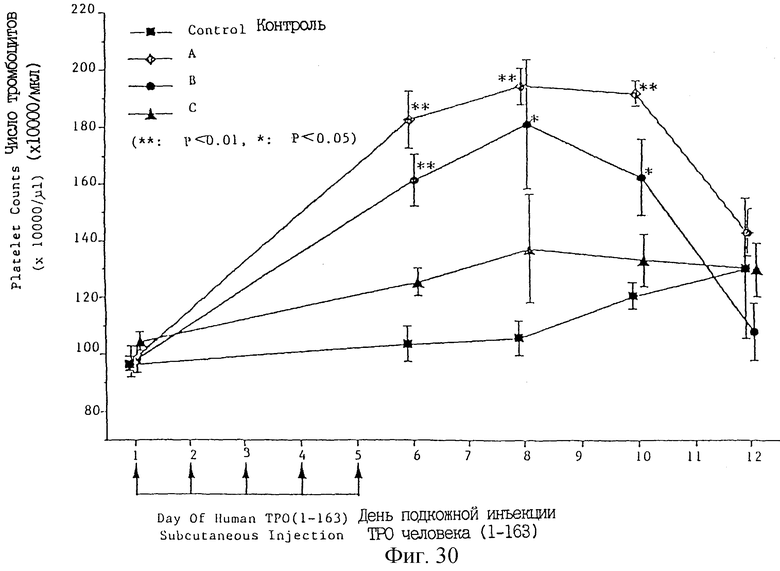
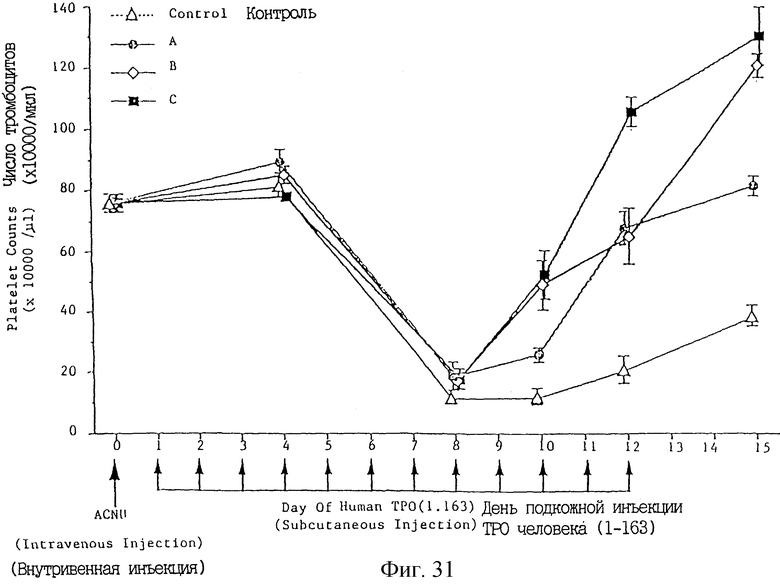
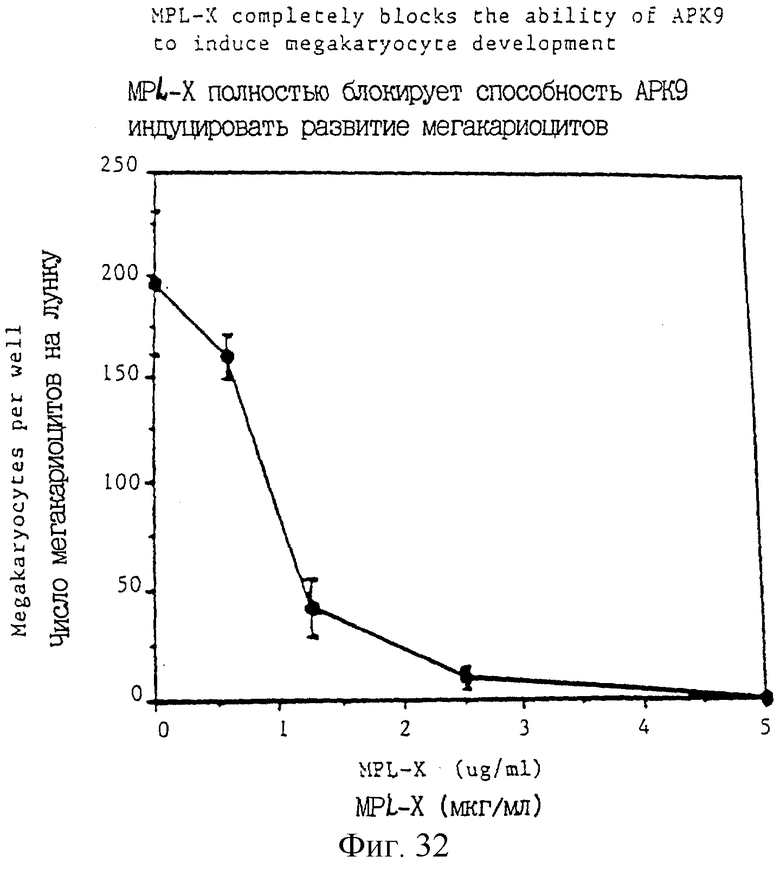
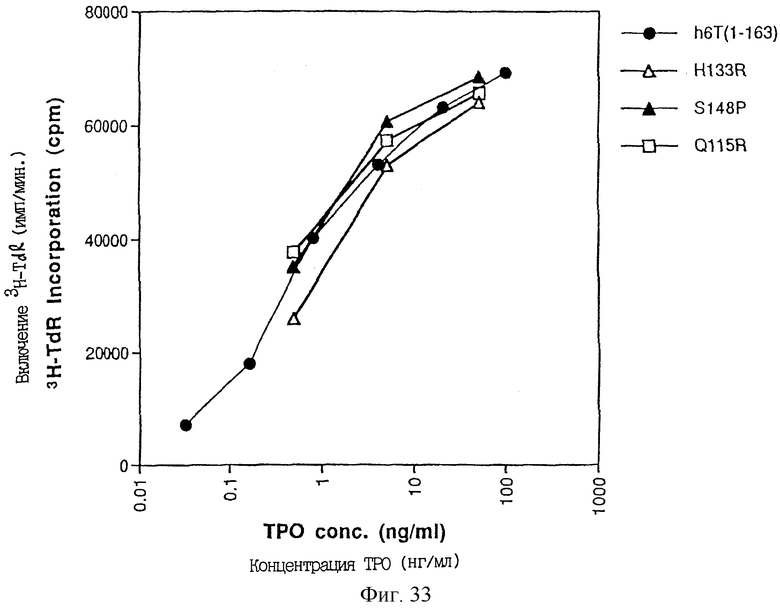
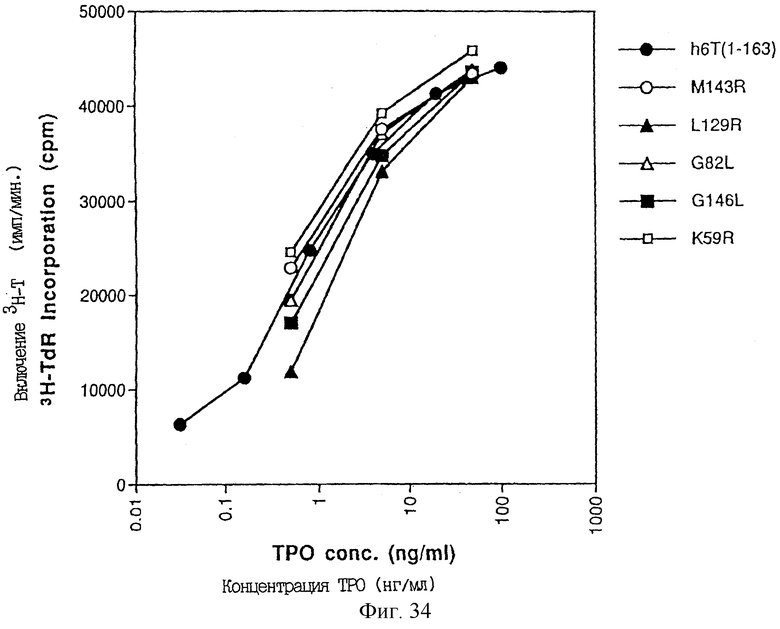
Изобретение касается генной инженерии. Полипептид тромбопоэтин (ТРО) имеет биологическую активность специфического стимулирования или увеличения образования тромбоцитов. Аминокислотная последовательность приведена в описании. ДНК, кодирующая полипептид ТРО с аминокислотной последовательностью, приведенной в описании. Полипептид ТРО получают экспрессией его в подходящем хозяине и последующим выделением. Фармкомпозиция для лечения связанных с тромбоцитами нарушений включает эффективное количество полипептида ТРО и приемлемый носитель. Фармкомпозицию вводят субъекту в эффективном количестве для лечения нарушений, связанных с тромбоцитами, например, тромбоцитопении. Проводят иммунизацию животного полипептидом ТРО и выделяют антитело к этому полипептиду. Изобретение позволяет осуществлять эффективное лечение нарушений, связанных с тромбоцитами. 12 с. и 22 з.п. ф-лы, 34 ил., 19 табл.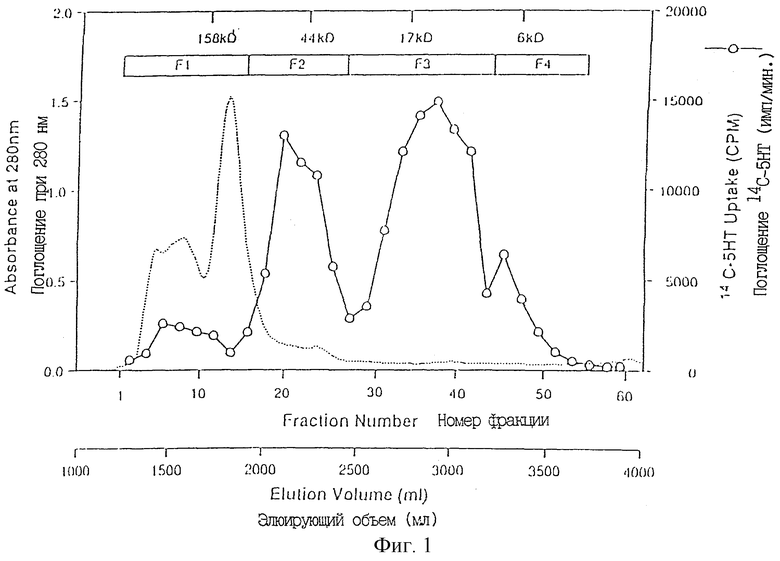
| EXPERIMENTAL HEMATOLOGY, vol 16, № 3, march 1998, р.201-205 | |||
| МАНИАТИС Г | |||
| и др | |||
| Методы генетической инженерии | |||
| - М.: Мир, 1984, с.9. |

