Изобретение относится к замещенным производным азолонов, которые являются мощными агентами против Helicobacter.
Патент США N 4791111 описывает азолоны, которые имеют строение, сходное со строением соединений настоящего изобретения, и которые являются промежуточными соединениями при получении [[4-[4-(4-фенил-1-пиперазинил)феноксиметил] -1,3-диоксолан-2- ил]метил]-1H-имидазолов и -1H-1,2,4-триазолов.
В патенте США N 4931444 описывают замещенные производные азолонов, обладающие ингибирующей 5-липоксигеназу активностью. Соединения настоящего изобретения отличаются от них полезной активностью против Helicobacter.
Для уничтожения Helicobacter в организме двойная терапия, включающая раздельное введение двух антибиотиков, не является удовлетворительной в силу одной или более следующих причин: малая скорость уничтожения, многочисленные побочные эффекты и развитие устойчивости у Helicobacter. Показано, что тройная терапия, включающая введение двух антибиотиков и соединения висмута, является эффективной, но предъявляет к пациенту повышенные требования и также скомпрометирована побочными эффектами. Настоящие соединения имеют то преимущество, что их можно применять в монотерапии для уничтожения Helicobacter pylori и родственных видов.
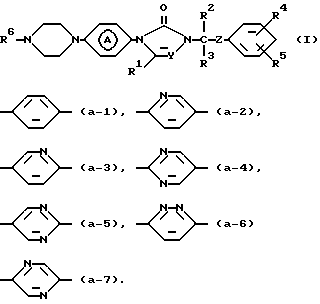
Настоящее изобретение относится к соединениям формулы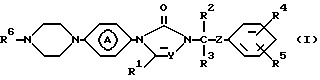
их фармацевтически приемлемым солям присоединения кислот и стереохимически изомерным формам,
где Y представляет CH или N;
R1, R2, и R3 каждый независимо представляет водород или С1-4алкил;
R4 и R5 каждый независимо представляет водород, галоген, С1-4алкил, С1-4алкилокси, гидрокси, трифторметил, трифторметилокси или дифторметилокси;
R6 представляет пиридинил, необязательно, замещенный вплоть до двух групп С1-4алкил; ди (С1-4алкил) гидроксипиридинил; ди (С1-4алкил) С1-4-алкилоксипиридинил; пиридазинил, необязательно, замещенный С1-4-алкилокси; пиримидинил, необязательно, замещенный гидрокси или С1-4алкилокси; тиазолил, необязательно, замещенный С1-4алкилом; тиадиазолил, необязательно, замещенный С1-4алкилом; бензоксазолил или бензотиазолил;
R6 представляет пиразинил или пиридазинил, замещенный С1-4алкилом;
Z представляет С=O или CHOH; и  является радикалом формулы
является радикалом формулы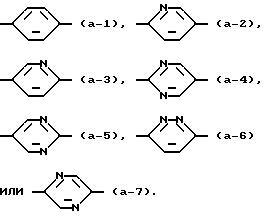
Употребляемые в настоящем документе определения галогены обозначает фтор, хлор, бром и йод; С1-4алкил обозначает насыщенные углеводородные радикалы с прямой и разветвленной цепью, содержащей от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил и 1,1- диметилэтил. C1-6алкил обозначает С1-4алкильные радикалы, как определено выше в настоящем документе, и их более высокие гомологи, содержащие от 5 до 6 атомов углерода, такие как, например, пентил или гексил.
Употребляемый в настоящей заявке термин фармацевтически приемлемая соль присоединения кислоты обозначает нетоксичные, терапевтически активные формы - соли присоединения кислот, которые могут образовываться соединениями формулы (I). Соединения формулы (I), обладающие основными свойствами, могут превращаться в соответствующие терапевтически активные, нетоксичные соли присоединения кислот путем действия на форму свободного основания подходящим количеством соответствующей кислоты согласно обычным методикам. Подходящие кислоты включают, например неорганические кислоты, такие как галогеноводородные кислоты, например хлористо-водородная или бромисто-водородная кислота; серная; азотная; фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, оксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламеновая, салициловая, п-аминосалициловая, памоевая и подобные кислоты. Термин соль присоединения, употребляемый в настоящем документе, также включает сольваты, которые соединения формулы (I), а также их соли, могут образовывать. Такими сольватами являются, например, гидраты, алкоголяты и т. п. Термин стереохимически изомерные формы, употребляемый в настоящем документе, обозначает различные изомерные, а также конформационные формы, которые могут иметь соединения формулы (I). Если не указывается или не упоминается иное, химическое обозначение соединений обозначает смесь всех возможных стереохимически или конформационно изомерных форм; причем указанная смесь содержит все диастереоизомеры, энантиомеры и/или конформеры основной молекулярной структуры. Все стереохимически изомерные формы соединений формулы (I), как в чистом виде, так и в виде смеси друг с другом, следует рассматривать как находящиеся в пределах объема настоящего изобретения.
Абсолютная конфигурация каждого хирального центра может обозначаться стереохимическими значками R и S. Для соединений, имеющих два хиральных центра, соответственно используются стереохимические обозначения R* и S*, в соответствии с правилами химических обозначений (Chemical Substance Name Selection Manual (CA), издание 1982, том III, глава 20).
Некоторые соединения по настоящему изобретению могут существовать в различных таутомерных формах и все такие таутомерные формы следует рассматривать как находящиеся в пределах объема настоящего изобретения.
Первую группу соединений, представляющих интерес, составляют соединения формулы (I), в которых R4 является галогеном, a R5 является водородом.
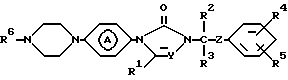
Вторую группу соединений, представляющих интерес, составляют соединения формулы (I), в которых является радикалом формулы (а-1) или (а-2).
является радикалом формулы (а-1) или (а-2).
Третью группу соединений, представляющих интерес, составляют со единения формулы (I), в которых Y является N, а R1 является водородом.
Четвертую группу соединений, представляющих интерес, составляют соединения формулы (I), в которых R2 является С1-4алкилом, а R3 является водородом.
Пятую группу соединений, представляющих интерес, составляют соединения формулы (I), в которых R6 является пиридинилом, тиазолилом или пиримидинилом.
Также шестую группу соединений, представляющих интерес, составляют соединения формулы (I), в которых R6 является пиразинилом.
Предпочтительными соединениями являются соединения формулы (I), в которых R1, R3 и R5 являются водородом; R2 является С1-4алкилом; R4 является галогеном и Y является N.
Более предпочтительными соединениями являются соединения формулы (I), в которых R1, R3 и R5 являются водородом; R2 является этилом; R4 является галогеном; Y является N; R6 является пиридинилом, тиазолилом или пиримидинилом и является радикалом формулы (а-1) или (а-2).
является радикалом формулы (а-1) или (а-2).
Наиболее предпочтительными соединениями являются
2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(2-пиримидинил)-1 -пиперазинил]-3-пиридинил]-3Н-1,2,4-триазол-3-он;
2-[1-(4-хлорбензоил)пропил-2,4-дигидро-4-[6-[4-(2-пиридинил)-1- пиперазинил]-3-пиридинил]-3Н-1,2,4-триазол-3-он;
2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4-[6- [4-(2-пиридинил)-1-пиперазинил]-3-пиридинил]-3Н-1,2,4- триазол-3-он; и
2-[1-[(4-хлорфенил)гидроксиметил] пропил]-2,4-дигидро-4-[6-[4- (2-тиазолил)-1-пиперазинил] -3-пиридинил] -3Н-1,2,4-триазол-3-он; их фармацевтически приемлемые соли присоединения кислот и стереохимически изомерные формы.
Аналогичные методики получения соединений, таких как соединения формулы (I) по настоящему изобретению, описаны в патенте США N 4791111 и патенте США N 4931444.
В частности, соединения формулы (I) могут быть получены путем N-алкилирования промежуточного соединения формулы (II) реагентом формулы (III).
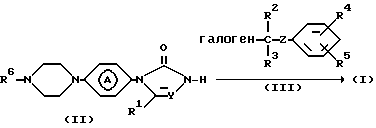
Реакцию N-алкилирования между (II) и (III) можно осуществить обычным путем посредством перемешивания и нагревания смеси реагентов в подходящем растворителе в присутствии подходящего основания. Подходящими растворителями являются, например, биполярные апротонные растворители, например N,N-диметилформамид, N, N-диметилацетамид, 1,3-диметил-2-имидазолидинон; ароматические растворители, например бензол, метилбензол, эфир, например 1,1'- оксибисэтан, тетрагидрофуран, 1-метокси-2-пропанол; галогенированные углеводороды, например дихлорметан, трихлорметан или смеси таких растворителей.
Подходящими основаниями являются, например, бис (триметилсилил) амид натрия, карбонаты или гидрокарбонаты щелочных и щелочно-земельных металлов, например карбонат натрия или калия; или органические основания, например триэтиламин и подобные основания.
Соединения формулы (I) могут также быть получены путем N-алкилирования промежуточного соединения формулы (IV) реагентом формулы (V).
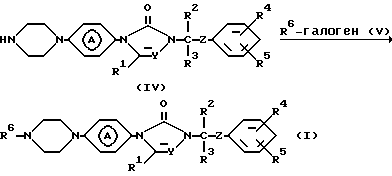
Вышеприведенную реакцию N-алкилирования можно удобно провести с использованием растворителей и оснований, описанных в настоящем документе выше для N-алкилирования промежуточного соединения (II).
Соединения формулы (I) могут также превращаться друг в друга с помощью следующих известных специалистам методик для трансформации функциональных групп.
Например, соединения формулы (I), в которых Z представляет C=O, могут превращаться в соединения формулы (I), в которых Z представляет CHOH, с помощью следующих известных специалистам методик восстановления. Например, указанное восстановление можно удобно провести с помощью взаимодействия с гидридом металла или комплексным гидридом металла, например боргидридом натрия, цианборгидридом натрия и т. п. в воде, 1-метилпирролидиноне, ацетонитриле, в спиртовой среде, например метаноле, этаноле или в эфире, например тетрагидрофуране, 1,4-диоксане или в смеси таких растворителей.
Альтернативно, указанное восстановление можно провести с помощью взаимодействия с гидроборатом трис (1- метилэтокси) калия, гидроборатом трис (1-метилпропил) натрия или гидроборатом (1-метилпропил) калия в реакционно инертном растворителе, например тетрагидрофуране или N,N-диметилформамиде.
Далее, соединения формулы (I), в которых R6 несет гидроксильный заместитель, могут быть получены из соответствующих производных С1-4алкилокси посредством подходящей реакции дезалкилирования, например с помощью трифторуксусной кислоты, неорганической кислоты, такой как концентрированная галогеноводородная кислота, например бромисто-водородная кислота, йодисто-водородная кислота, необязательно, в смеси с насыщенным раствором бромисто-водородной кислоты в ледяной уксусной кислоте; кислоты Льюиса, например трибромида бора в реакционно инертном растворителе, например дихлорметане или N, N-диметилацетамиде. В случае, когда используется бромистоводородная кислота, может быть выгодным проведение указанной реакции дезалкилирования в присутствии поглотителя брома, такого как, например, сульфит или гидросульфит натрия.
И наконец, чистые изомерные формы соединений формулы (I) можно разделить в смеси с помощью стандартных способов разделения. В частности, энантиомеры можно разделить с помощью колоночной хроматографии с использованием хиральной стационарной фазы, такой как подходящие производные целлюлозы, например три(диметилкарбамоил) целлюлоза (Chiralcel OD®) и подобных хиральных стационарных фаз.
Во всех ранее описанных и последующих реакциях продукты реакции можно выделять из реакционной среды и, если это необходимо, подвергать дальнейшей очистке согласно обычным методикам, хорошо известным специалистам.
Некоторые промежуточные и исходные материалы в описанных выше реакциях являются известными соединениями, которые можно получить с помощью известных специалистам методик получения указанных или подобных соединений. Другие промежуточные соединения являются новыми, такие как промежуточные соединения формулы (II).
Промежуточные соединения формулы (II) можно получить путем циклизации промежуточного соединения формулы (VI) с реагентом формулы (VII) или его производным.
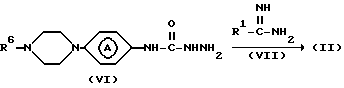
Подходящим реакционно инертным растворителем для вышеприведенной реакции циклизации является, например, биполярный апротонный растворитель, например N, N-диметилформамид, диметилсульфоксид и т.п., или спирт, например этанол, 1-бутанол и т.п.
Альтернативно, промежуточные соединения формулы (II) можно получить путем взаимодействия промежуточного соединения формулы (IX) с реагентом формулы (V) согласно известным методикам.
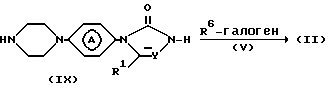
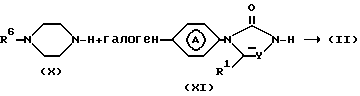
Промежуточные соединения формулы (II) можно также получить путем N-алкилирования промежуточного соединения формулы (Х) с реагентом формулы (XI) согласно известным методикам N-алкилирования.
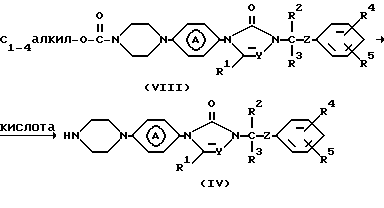
Промежуточные соединения формулы (IV) можно получить путем взаимодействия соединения формулы (VIII) с кислотой, например бромисто-водородной кислотой и т.п.
Соединения формулы (I) и их фармацевтически приемлемые соли присоединения кислот и стереохимически изомерные формы обладают полезной фармакологической активностью против видов Helicobacter, например Helicobacter pylori, Helicobacter mustelae, Helicobacter felis и т. п., особенно Helicobacter pylori.
Особенно важным в этом контексте является открытие того, что настоящие соединения демонстрируют ингибирующую активность в отношении роста Helicobacter, а также бактерицидной активностью в отношении указанной бактерии. Бактерицидный эффект в отношении Helicobacter определяли на суспензиях культур с помощью методики, описанной в Antimicrob. Agents Chemother., 1991, том 35, стр. 869-872.
Интересная отличительная особенность настоящих соединений связана с их высокоспецифичной активностью против Helicobacter. Соединения формулы (I), как оказалось, не обладают ингибирующей активностью в отношении любого из следующих видов: Campylobacter jejuni, Campylobacter coli, Campylobacter fetus, Campylobacter sputorum, Vibrio spp., Staphylococcus aureus и Escherichia coli в концентрациях до 10-5 M.
Важным ценным качеством настоящих соединений является их пролонгированная активность против Н.pylori при pH ниже нейтральной. Активность при низких значениях pH in vitro может указывать на то, что на соединение не оказывает неблагоприятного влияния кислая среда желудка in vivo.
Таким образом, настоящие соединения рассматриваются как ценные терапевтические лекарственные средства для лечения теплокровных животных, в частности людей, страдающих от заболеваний или состояний, связанных с Helicobacter. Примерами указанных заболеваний или состояний являются гастриты, язвы желудка и двенадцатиперстной кишки и рак желудка.
С точки зрения их полезных анти-Helicobacter свойств настоящие соединения могут быть помещены в различные фармацевтические формы для различных способов введения. Для изготовления фармацевтических композиций по настоящему изобретению эффективное количество конкретного соединения, в форме основания или соли присоединения, в качестве активного ингредиента комбинируют в виде гомогенной связи с фармацевтически приемлемым носителем, который может принимать самый разнообразный вид в зависимости от желательной для введения формы препарата. Желательно, чтобы эти фармацевтические композиции находились в виде стандартных доз, удобных, предпочтительно, для перорального, ректального или парентерального введения. Например, при изготовлении композиций в форме для перорального введения можно использовать любое из обычных фармацевтических веществ, такое как, например, вода, гликоли, масла, спирты и т. п., в случае жидких препаратов для перорального введения, таких как крахмалы сахара, каолин, смазывающие агенты, связывающие агенты, разрыхлители и т. п. , в случае порошков, пилюль, капсул и таблеток. Для парентеральных композиций носитель обычно включает стерильную воду, по меньшей мере в большей части, хотя можно включать и другие ингредиенты, например, для усиления растворимости. Например, можно изготавливать растворы для инъекций, в которых носитель включает солевой раствор, раствор глюкозы или смесь растворов соли и глюкозы. Можно изготавливать также суспензии для инъекций и в этом случае использовать подходящие жидкие носители, суспендирующие агенты и т.п.
Если фармацевтическую композицию изготавливают в форме водного раствора, те соединения формулы (I), которые обладают малой растворимостью, можно преобразовать в форму соли и добавить сорастворитель, который смешивается с водой и является физиологически приемлемым, например, диметилсульфоксид и т. п. , или соединения формулы (I) можно солюбилизировать подходящим носителем, например циклодекстрином (ЦД) или, в частности, производным циклодекстрина, таким как производные циклодекстрина, описанные в патенте США N 3459731, ЕР-А-149197 (24 июля 1985), EP-A-197571 (15 октября 1986), патенте США N 4535152 или WO 90/12035 (18 октября 1990). Подходящими производными циклодекстрина являются α-,β-,γ-циклодекстрины или их эфиры и смешанные эфиры, в которых одна или более гидроксильных групп в единицах кольцевидной формы глюкозы в циклодекстрине замещены следующими группами: С1-6алкил, особенно метил, этил или изопропил; гидроксиС1-6алкил, особенно гидроксиэтил, гидроксипропил или гидроксибутил; карбокси С1-6алкил, особенно карбоксиметил или корбоксиэтил; С1-6алкилкарбонил, особенно ацетил; С1-6алкилоксикарбонил С1-6алкил или карбокси С1-6алкилокси С1-6алкил, особенно карбоксиметоксипропил или карбоксиэтоксипропил; С1-6алкилкарбонилокси С1-6алкил, особенно 2-ацетилоксипропил. Особенно заслуживающими внимания в качестве комплексообразующих агентов и/или солюбилизирующих агентов являются β-ЦД, 2,6-диметил-β-ЦД, 2-гидроксиэтил-β-ЦД, 2-гидроксиэтил-γ-ЦД, 2-гидроксипропил-γ-ЦД и (2-карбоксиметокси)-пропил-β-ЦД, а особенно 2-гидроксипропил-β-ЦД.
Термин смешанный эфир означает производные циклодекстрина, в которых по меньшей мере две гидроксильные группы циклодекстрина этерифицированы различными группами, такими как, например, гидроксипропил или гидроксиэтил.
Среднее молярное замещение (МЗ) применяли в качестве измерения среднего количества молей алкокси единиц на моль глюкозы в виде кольца, образованного внутренним ангидридом. Величину МЗ можно определить с помощью различных аналитических методик, таких как ядерный магнитный резонанс (ЯМР), масс-спектрометрия (МС) и инфракрасная спектроскопия (ИК). В зависимости от используемого способа анализа для каждого конкретного производного циклодекстрина могут быть получены слегка отличающиеся друг от друга величины. У гидроксиалкильных производных циклодекстрина, используемых для композиций по настоящему изобретению, величина МЗ по результатам масс-спектрометрии находится в пределах от 0,125 до 10, в частности от 0,3 до 3 или от 0,3 до 1,5. Предпочтительно величина МЗ варьирует приблизительно от 0,3 до 0,8, в частности приблизительно от 0,35 до 0,5 и особенно предпочтительно приблизительно 0,4. Величины МЗ, определенные с помощью ЯМР или ИК, предпочтительно варьируют от 0,3 до 1, в частности от 0,55 до 0,75.
Средняя степень замещения (СЗ) относится к среднему количеству замещенных гидроксильных групп на единицу глюкозы в виде кольца, образованного внутренним ангидридом. Величину СЗ можно определить с помощью различных аналитических методик, таких как ядерный магнитный резонанс (ЯМР), масс-спектрометрия (МС) и инфракрасная спектроскопия (ИК). В зависимости от используемого способа анализа для каждого конкретного производного циклодекстрина могут быть получены слегка отличающиеся друг от друга величины. У производных циклодекстрина, используемых для композиций по настоящему изобретению, величина CЗ по результатам МС находится в пределах от 0,125 до 3, в частности, от 0,2 до 2 или от 0,2 до 1,5. Предпочтительно, величина CЗ варьирует приблизительно от 0,2 до 0,7, в частности, приблизительно от 0,35 до 0,5 и особенно предпочтительно, приблизительно 0,4. Величины CЗ, определенные с помощью ЯМР или ИК, предпочтительно варьируют от 0,3 до 1, в частности от 0,55 до 0,75.
Более конкретно, гидроксиалкильные производные β- и γ-циклодекстрина для использования в композициях по настоящему изобретению являются частично замещенными производными циклодекстрина, в которых средняя степень алкилирования при гидроксильных группах в различных положениях единиц глюкозы в виде кольца, образованного внутренним ангидридом, составляет приблизительно от 0 до 20% для 3 положения, от 2 до 70% для 2 положения и приблизительно от 5 до 90% для 6 положения. Предпочтительно количество незамещенных β- и γ-циклодекстринов составляет менее 5% от общего содержания циклодекстрина и, в частности, составляет менее 1,5%. Другим особенно интересным производным циклодекстрина является произвольно метилированный β-циклодекстрин.
Наиболее предпочтительными производными циклодекстрина для использования в композициях по настоящему изобретению являются частично замещенные эфиры или смешанные эфиры β-циклодекстрина, имеющие в качестве заместителей гидроксипропил, гидроксиэтил и в особенности 2-гидроксипропил и/или 2-(1-гидроксипропил).
Наиболее предпочтительным производным циклодекстрина для использования в композициях по настоящему изобретению является гидроксипропил-β-циклодекстрин, имеющий МЗ в пределах от 0,35 до 0,50 и содержащий менее 1,5% незамещенного β-циклодекстрина. Величины МЗ, определенные с помощью ЯМР или ИК, предпочтительно варьируют от 0,55 до 0,75.
Для удобства применения и стандартности дозировок особенно выгодно создавать вышеупомянутые фармацевтические композиции в форме стандартных дозированных единиц. Стандартная дозированная единица, упоминаемая в настоящем описании и формуле изобретения, относится к физически дискретным единицам, удобным в качестве стандартных дозировок; причем каждая единица содержит строго определенное количество активного ингредиента, рассчитанное для обеспечения желаемого терапевтического эффекта, в сочетании с требующимся фармацевтическим носителем. Примерами таких стандартных дозированных единиц являются таблетки (включая таблетки с засечками и таблетки в оболочке), капсулы, пилюли, пакетики с порошком, облатки, растворы или суспензии для инъекций и т.п., а также упаковки из множества этих изолированных единиц.
С точки зрения полезности настоящих соединений для лечения заболеваний, связанных с Helicobacter, очевидно, что настоящее изобретение обеспечивает способ лечения теплокровных животных, в частности людей, страдающих от заболеваний, связанных с Helicobacter, который включает в себя системное введение фармацевтически эффективного количества соединения формулы (I), его фармацевтически приемлемой соли присоединения или стереохимически изомерной формы, в смеси с фармацевтическим носителем. В еще одном аспекте настоящего изобретения, рассматриваемые соединения применяются в качестве лекарственного средства.
В общем, предполагается, что эффективное суточное количество составит от 0,05 до 50 мг/кг веса тела, предпочтительно от 0,1 до 30 мг/кг веса тела и наиболее предпочтительно от 0,5 до 10 мг/кг веса тела.
Очевидно, что указанное эффективное суточное количество может быть уменьшено или увеличено в зависимости от реакции пациента, подвергаемого лечению, и/или в зависимости от оценки терапевта, назначающего соединения настоящего изобретения. Таким образом, указанные выше эффективные пределы являются только руководством и не предназначены для ограничения в какой бы то ни было степени объема или применения настоящего изобретения.
Необязательно, в сочетании с соединениями по настоящему изобретению могут вводиться другие активные соединения, используемые для уничтожения Helicobacter. Введение можно осуществлять раздельно (т.е. одновременно или последовательно) или различные лекарственные средства могут объединяться в одну лекарственную форму. Подходящими соединениями для комбинированной терапии являются соединения висмута, например висмута субцитрат, висмута субсалицилат и т. п., антибиотики, например ампициллин, амоксициллин, кларитромицин и т. п., антагонисты H2 - рецепторов, например циметидин, ранитидин и т. п. и, в частности, ингибиторы протонного насоса, например омепразол, ланзопразол, пантопразол и т.п. Для перечисленных соединений, которые могут быть полезными для комбинированной терапии с соединениями формулы (I), эффективное суточное количество будет составлять от 0,05 до 50 мг/кг веса тела.
Экспериментальная часть
Далее в настоящей заявке "ДМФ" означает N,N-диметилформамид, "ДМСО" означает диметилсульфоксид и "КТ" означает комнатную температуру.
Пример 1
a) 2-(1-пиперазинил) пиримидин (5,4 г), 4-(6-хлор-3 - пиридинил)-2,4-дигидро-3Н-1,2,4-триазол-3 он (7 г) и карбонат натрия (4 г) перемешивали при 190oC в течение 1 часа. Смесь охлаждали и добавляли воду. Осадок отфильтровывали и перекристаллизовывали из 2-метоксиэтанола. Осадок отфильтровывали и высушивали, получая 6,5 г (61%) 2,4-дигидро -4-[6-[4-(2-пиримидинил)-1-пиперазинил] -3-пиридинил] -3Н-1,2,4-триазол-3-она (промежуточное соединение 1).
б) Смесь промежуточного соединения 1 (5,9 г), (±)-2-бром-1-(4-хлорфенил)-1-бутанон (5,75 г) и карбонат натрия (4,3 г) в 1-метокси-2 пропаноле (150 мл) перемешивали и нагревали с обратным холодильником в течение 8 часов. Растворитель выпаривали, добавляли воду и смесь экстрагировали CH2Cl2. Органический слой отделяли, высушивали, фильтровали и растворитель выпаривали. Остаток перекристаллизовывали из 2-пропанола. Осадок отфильтровывали и перекристаллизовывали из С2H5OH. Осадок отфильтровывали и высушивали, получая 7,6 г (84%) (±)-2-[1-(4- хлорбензоил) пропил]-2,4-дигидро -4-[6-[4-(2-пиримидинил)-1-пиперазинил] -3-пиридинил]-3Н-1,2,4-триазол-3-она (соединение 1).
Подобным образом были получены также (±)-2-[1 -(4-хлорбензоил)пропил]-2,4-дигидро-4-[6-[4-(5-метил -1,3,4-тиадиазол-2-ил)-1-пиперазинил] -3-пиридинил] -3H -1,2,4-триазол-3-он; т.пл. 200oC (соединение 2); (±) -2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(2 - тиазолил)-1-пиперазинил]-3-пиридинил] -3Н-1,2,4 - триазол-3 -он; (соединение 3); (±)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[6- [4-(6-метокси-3-пиридазинил)-1-пиперазинил]-3 - пиридинил] -3Н-1,2,4-триазол-3-он; т. пл. 133oC (соединение 4); (±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро -4-[6-[4- (2-пиридинил)-1-пиперазинил]-3-пиридинил] -3Н-1,2,4 - триазол-3-он; т.пл. 134,1oC (соединение 17); 2-[2-(4- хлорфенил)-2-оксоэтил] -2,4-дигидро-4-[6-[4-(5-метокси - 4,6-диметил-2-пиридинил)-1-пиперазинил] -3-пиридинил] -5 метил-3Н- 1,2,4-триазол-3-он; (соединение 18) и (±)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[6-[4- (3-пиридинил)-1-пиперазинил] -3-пиридинил]-3Н-1,2,4 - триазол-3-он; (соединение 21).
Пример 2
а) Смесь 5-метокси-2-(1-пиперазинил) пиримидин (8,8 г), 1-фтор-4-нитробензол (7 г) и карбонат натрия (7,5 г) в ДМСО (100 мл) перемешивали при КТ в течение 4 часов. Смесь выливали в воду. Осадок отфильтровывали и растворяли в CH2Cl2. Добавляли SiO2 (5 г) и смесь перемешивали, отфильтровывали, а растворитель выпаривали. Остаток доводили до кипения в 1-пропаноле, отфильтровывали и высушивали в вакууме при 75oC, получая 8,5 г (60%) 5-метокси-2-[4-(4-нитрофенил)-1-пиперазинил] пиримидина; т. пл. 212,3oC (промежуточное соединение 2).
б) Смесь промежуточного соединения 2 (68 г) в 4% растворе тиофена (2 мл) и метаноле (600 мл) гидрогенизировали при 50oC с палладием на активированном угле с содержанием палладия 10% (4 г) в качестве катализатора. После поглощения водорода (3 экв.) катализатор отфильтровывали, а фильтрат высушивали. Остаток размешивали в CH3ОН, отфильтровывали, высушивали в вакууме при 75oC и перекристаллизовывали из 1-пропанола обрабатывали норитом и фильтровали через декалит. Фильтрат кристаллизовали. Осадок отфильтровывали и высушивали в вакууме при 75oC, получая 36,7 г (59%) 4-[4-(5-метокси-2- пиримидинил)-1-пиперазинил]бензоламина; т.пл. 125,2oC (промежуточное соединение 3).
в) Промежуточное соединение 3 (47,7 г) в N,N-диметилацетамиде (500 мл) перемешивали на водяной бане со льдом. Добавляли по каплям фенилхлорформиат (23 мл) и смесь перемешивали в течение 3 часов. Смесь выливали в воду и отфильтровывали. Осадок растворяли в CH2Cl2. Водный слой отделяли и выпаривали. Остаток размешивали в диизопропиловом эфире, отфильтровывали и высушивали, получая 55,2 г (80%) фенил [4-[4-(5-метокси-2- пиримидинил)-1-пиперазинил]фенил]карбамата (промежуточное соединение 4).
г) Смесь промежуточного соединения 4 (55,2 г) в моногидрате гидразина (60 мл) и 1,4-диоксана (1000 мл) перемешивали при КТ в течение ночи. Смесь выливали в воду и отфильтровывали. Осадок высушивали в вакууме при 50oC, получая 42 г (87%) N-[4-[4-(5-метокси-2-пиримидинил)-1-пиперазинил] фенил] гидразинкарбоксамида (промежуточное соединение 5).
д) Смесь промежуточного соединения 5 (42 г), моногидрохлорида этанимидата (47 г) и ацетата натрия (49,2 г) в 1-бутаноле (750 мл) перемешивали и нагревали с обратным холодильником в течение 24 часов. Смесь охлаждали, добавляли воду и перемешивали. Осадок отфильтровывали, высушивали в вакууме при 70oC, кристаллизовали из ДМФ. Осадок отфильтровывали и высушивали в вакууме при 75oC, получая 19,6 г (45%) 2,4-дигидро-4-[4-[4-(5-метокси-2-пиримидинил)-1- пиперазинил] фенил]-5-метил-3Н-1,2,4-триазол-3-она; т.пл. > 300oC (промежуточное соединение 6).
е) Промежуточное соединение 6 (13 г) и ДМФ (400 мл) перемешивали при КТ. Добавляли по каплям 1 М NaN [Si (CH3)3]2 в тетрагидрофуране (38 мл) и смесь перемешивали в течение 1 часа при КТ. Добавляли 2-бром-1-(4- хлорфенил) этанон (9,4 г) и смесь перемешивали в течение 5 часов при КТ. Смесь выливали в воду и отфильтровывали. Осадок растворяли в CH2Cl2 и промывали водой. Органический слой высушивали, фильтровали и выпаривали. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: CH2Cl2/CH3ОН 99/1). Нужные фракции собирали и выпаривали. Остаток перекристаллизовывали из 1-пропанола, отфильтровывали и высушивали в вакууме при 70oC, получая 8,4 г (46%)2-[2-(4-хлорфенил)-2-оксоэтил] -2,4-дигидро-4-[4-[4- (5-метокси-2-пиримидинил)-1-пиперазинил] фенил] -5- метил-3Н-1,2,4-триазол-3-она; т.пл. 220oC (соединение 5).
Подобным образом были получены (±)-2-[1-(4- хлорбензоил)пропил]-2,4-дигидро-4-[6-[4-(5-метокси-2-пиримидинил)- 1-пиперазинил]-3-пиридинил]-3Н-1,2,4-триазол-3-он; т. пл. 152oC (соединение 6); (±)-2-[1-(4-хлорбензоил) пропил] -2,4-дигидро-4-[6-[4-(4-метокси-2-пиримидинил)- 1-пиперазинил]-3-пиридинил] -3Н-1,2,4-триазол-3-он; т. пл. 162,5oC (соединение 22) и (±)-2-[1-(4-хлорбензоил) пропил] -2,4-дигидро-4-[4-[4-(4-метокси-2-пиримидинил)- 1-пиперазинил] фенил]-3Н-1,2,4-триазол-3-он; т. пл. 149,6oC (соединение 34).
Пример 3
а) Смесь (±)- этил 4-[4-[2-[1-(4-хлорбензоил) пропил]-2,3-дигидро-3-оксо-4H-1,2,4-триазол-4-ил] фенил-1-пиперазинкарбоксилата (15 г) в 48% водном растворе бромисто-водородной кислоты (150 мл) перемешивали и нагревали с обратным холодильником в течение ночи. Растворитель выпаривали, остаток растворяли в CH2Cl2 и промывали NaHCO3/H2O. Органический слой высушивали, фильтровали и выпаривали. Остаток растворяли в 2-пропаноле и кристаллизовали соль хлористо-водородной кислоты (1:2) в 2-пропаноле. Осадок отфильтровывали и перекристаллизовывали из CH3CN, получая 7,9 г (2,2%) (±)-2-[1-(4-хлорбензоил)пропил] -2,4 - дигидро-4-[4-(1-пиперазинил]фенил]-3Н-1,2,4-триазол-3 - она дигидрохлорида моногидрата; т. пл. 175,9oC (промежуточное соединение 7).
б) Смесь 2-хлорбензотиазола (3,7 г), свободного основания промежуточного соединения 7 (7,5 г) и карбоната натрия (5 г) в ДМФ (100 мл) перемешивали при 70oC в течение ночи. Смесь охлаждали, выливали в смесь льда с водой и перемешивали в течение 1 часа. Осадок отфильтровывали и растворяли в CH2Cl2. Органический слой высушивали, фильтровали и выпаривали. Остаток кристаллизовали из С2H5ОН. Осадок отфильтровывали и высушивали, получая 6,5 г (78%) (±)-4-[4-[4-(2-бензотиазолил)-1-пиперазинил] фенил] -2-[1-(4-хлорбензоил) пропил]-2,4-дигидро-3Н-1,2,4 - триазол-3-он; т. пл. 162oC (соединение 7).
Подобным образом были получены (±)-4-[4-[4-(2- бензоксазолил)- 1-пиперазинил] фенил] -2-[1-(4-хлорбензоил) пропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он; т. пл. 150oC (соединение 8); (±)-2-[1-(4-хлорбензоил)пропил]-2,4- дигидро-4-[6-[4-(4-пиридинил)-1-пиперазинил] -3-пиридинил] -3H-1,2,4-триазол-3-он; т.пл. 173,8oC (соединение 23) и (±)-2-[1-(4-хлорбензоил)пропил]-2,4-дигидро-4-[6- [4-(6-метил-3-пиридазинил)-1-пиперазинил] -3- пиридинил]-3H-1,2,4-триазол-3-он; т. пл. 212,1oC (соединение 24).
Пример 4
Раствор трибромборана (150 мл) в CH2Cl2 (100 мл) перемешивали при КТ. Добавляли раствор соединения 5 (7,1 г) в CH2Cl2 (300 мл) и полученную реакционную смесь перемешивали в течение 3 часов при КТ. Эту смесь по каплям добавляли к смеси льда и аммиака (200 мл). Отделенный органический слой высушивали над MgSO4, фильтровали и растворитель выпаривали. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: CH2Cl2/CH3O 98/2). Чистые фракции собирали и растворитель выпаривали. Остаток кристаллизовали из 1-пропанола. Кристаллы отфильтровывали и высушивали (вакуум; 70oC), получая 4,3 г (65%) 2-[2-(4-хлорфенил)-2-оксоэтил]-2,4-дигидро-4-[4-[4-(5- гидрокси-2-пиримидинил)-1-пиперазинил] фенил]-5-метил-3H- 1,2,4-триазол-3-она; т.пл. 225oC (соединение 9).
Подобным образом были получены (±)-2-[1-(4-хлорбензоил) пропил]-2,4-дигидро-4-[6-[4-(5-гидрокси-2-пиримидинил)-1-пиперазинил] -3- пиридинил] -3Н-1,2,4-триазол-3-она 1-пропанолят (2: 1); т.пл. 109oC (соединение 10) и 2-[2-(4-хлорфенил)-2- оксоэтил] -2,4-дигидро-4-[6-[4-(5-гидрокси-4,6-диметил-2 -пиридинил)-1-пиперазинил]-3-пиридинил]-5-метил-3Н- 1,2,4-триазол-3-он; т. пл. 212,1oC (соединение 19).
Пример 5
Смесь соединения 9 (4,3 г) в CH3CN (150 мл) и CH3ОН (36 мл) перемешивали на ледяной бане при 0oC. Добавляли по каплям боргидрид натрия (1,6 г) в воде (12 мл) при 0-10oC и смесь перемешивали в течение 3 часов при КТ. Смесь выливали в воду и нейтрализовали CH3COOH. Осадок отфильтровывали и кристаллизовали из 1-пропанола. Смесь охлаждали, фильтровали и высушивали в вакууме при 75oC, получая 1,7 г (40%) (±)-2-[2-(4-хлорфенил)-2-оксоэтил]-2,4-дигидро-4-[6-[4-(5 -гидрокси-4,6-диметил-2- пиридинил)-1-пиперазинил]-3-пиридинил]-5-метил-3Н-1,2,4- триазол-3-он; т.пл.212,1oC (соединение 19).
Пример 5
Смесь соединения 9 (4,3 г) в CH3CN (150 мл) и CH3ОН (36 мл) перемешивали на ледяной бане при 0oC. Добавляли по каплям боргидрид натрия (1,6 г) в воде (12 мл) при 0-10oC и смесь перемешивали в течение 3 часов при КТ. Смесь выливали в воду и нейтрализовали CH3COOH. Осадок отфильтровывали и кристаллизовали из 1-пропанола. Смесь охлаждали, фильтровали и высушивали в вакууме при 75oC, получая 1,7 г (40%) (±)-2-[2-(4-хлорфенил)-2-гидроксиэтил]-2,4- дигидро-4-[4-[4-(5-гидрокси-2-пиримидинил)-1-пиперазинил] фенил] -5- метил-3Н-1,2,4-триазол-3-она; т. пл. 249oС (соединение 11).
Подобным образом были получены (±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил]- 2,4-дигидро-4-[6-[4-(6-метокси-3-пиридазинил)-1- пиперазинил]-3-пиридинил]-3Н-1,2,4-триазол-3-он; т.пл. 244oC (соединение 12) и (±)-(R*, R*)-2-[1-[(4-хлорфенил) гидроксиметил]пропил]-2,4-дигидро-4-[6-[4-(5-метил-1,3,4- тиадиазол-2-ил)-1-пиперазинил] -3-пиридинил] -3Н-1,2,4-триазол- 3-он; т. пл. 237,4oC (соединение 25).
Пример 6
Смесь соединения 8 (3,2 г) в ДМФ (80 мл) перемешивали при -20oC. Добавляли по каплям три (изобутил) боргидрид калия (20 мл) и смесь перемешивали при КТ в течение ночи. Смесь выливали в лед/воду с HCl и перемешивали при КТ в течение 1 часа. Осадок отфильтровывали и кристаллизовали из 2-метоксиэтанола. Осадок отфильтровывали и высушивали. Остаток очищали посредством ЖХВР. Чистые фракции собирали и выпаривали. Остаток кристаллизовали из 2-метоксиэтанола. Осадок от фильтровывали и высушивали, получая 0,79 г (26%) (±)-(R*, R*)-4-[4-[4-(2-бензоксазолил)-1- пиперазинил] -фенил] -2-[1-[(4-хлорфенил)гидроксиметил] пропил]-2,4-дигидро-3Н-1,2,4-триазол-3-он; т. пл. 221oС (соединение 13).
Подобным образом были получены (±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4- дигидро-4-[6-[4-(2-тиазолил)-1-пиперазинил] -3-пиридинил] -3Н-1,2,4-триазол-3-он; т.пл. 215oC (соединение 14); (±)-(R*, R*)-2-[1-[(4-хлорфенил) гидроксиметил] пропил]-2,4- дигидро-4-[6-[4-(5-метокси-2-пиримидинил)-1-пиперазинил] -3-пиридинил]-3Н-1,2,4-триазол-3-он; т. пл. 220oC (соединение 15); (±)-(R*, R*)-2-[1-[(4-хлорфенил) гидроксиметил] пропил] -2,4-дигидро-4-[6-[4-(2-пиримидинил)-1- пиперазинил]-3-пиридинил]-3Н-1,2,4-триазол-3-он; т. пл. 231oC (соединение 16); (±)-(R*, R*)-2-[1-[(4-хлорфенил) гидроксиметил] пропил] -2,4-дигидро-4-[6-[4-(2-пиридинил-1-пиперазинил] -3-пиридинил] -3Н-1,2,4-триазол-3-он; т. пл. 219,7oC (соединение 20); (±)-(R*, R*)-2-[1-[(4-хлорфенил) гидроксиметил]пропил]-4-[6-[4-(4,6-диметил-2-пиридинил)-1-пиперазинил] -3- пиридинил] -2,4-дигидро-3Н-1,2,4-триазол-3-он; т. пл. 171,4oC (соединение 26); (±)-(R*, R*)-2-[1-[(4-хлорфенил) гидроксиметил] пропил]-2,4-дигидро-4-[6-[6-(4-метокси-2- пиримидинил)-1-пиперазинил] -3-пиридинил] -3Н-1,2,4- триазол-3-он; т. пл. 215,3oC (соединение 27); (±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил]-2,4-дигидро-4-[4-[4-(4-метокси-2-пиримидинил)-1-пиперазинил] фенил] -3Н-1,2,4-триазол-3-он; т. пл. 161,2oC (соединение 28); (±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил] -2,4-дигидро-4-[6-[4-(3-пиридинил-1-пиперазинил] * -3-пиридинил] -1,2,4-триазол-3-он (соединение 29); и (±)-(R*, R*)-2-[1-[(4-хлорфенил)гидроксиметил] пропил]-2,4-дигидро-4-[6-[4-(2-пиразинил)-1 -пиперазинил]-3-пиридинил]-3Н-1,2,4-триазол-3-он; (соединение 30).
Пример 7
а) Смесь этил 4-[5-(2,3-дигидро-3-оксо-4H-1,2,4-триазол-4 ил)-2-пиридинил] -1-пиперазинкарбоксилата (15,7 г), полученного способом, описанным в примерах от 2б до 2д, в бромисто-водородной кислоте (48% водный раствор) (100 мл) перемешивали и нагревали с обратным холодильником в течение 2 часов. Смесь охлаждали до комнатной температуры и растворитель выпаривали, получая 17,2 г (85,3%) 2,4-дигидро-4-[6-(1-пиперазинил)-3-пиридинил]-3Н-1,2,4- триазол-3-она дигидробромида; т. пл. > 300oC (промежуточное соединение 8).
б) Смесь промежуточного соединения 8 (9,81 г) и 2- хлорпиразина (10,31 г) в 1-метил-2-пирролидиноне (100 мл) и триэтиламине (20 мл) перемешивали и нагревали с обратным холодильником в течение 15 часов.
На смесь воздействовали водой (200 мл) и отфильтровывали. Осадок растворяли в CH2Cl2 и слои разделяли. Органический слой высушивали над MgSO4, отфильтровывали и выпаривали. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: CH2Cl2/CH3OH от 100/0 до 93/7). Чистые фракции собирали и выпаривали, получая 7,4 г (95,1%) 2,4-дигидро-4-[6-[4-(2-пиразинил)-1-пиперазинил]-3-пиридинил]-3Н-1,2,4 -триазол-3-она (промежуточное соединение 9).
в) Смесь промежуточного соединения 9 (7,1 г) и (±)-2-бром-1-(4-хлорфенил)-1-бутанона (6,3 г) в ДМФ (200 мл) перемешивали в атмосфере N2 в течение 5 минут. Добавляли карбонат натрия (2,76 г) и смесь перемешивали и нагревали при 60oC в течение 15 минут. Смесь охлаждали в ледяной бане, медленно добавляли воду (350 мл) и отфильтровывали. Остаток перекристаллизовывали из CH3ОН, получая 7,9 г (71,4%) (±)-2-[1 -(4-хлорбензоил) пропил]-2,4-дигидро-4-[6-[4-(2-пиразинил)-1-пиперазинил] -3-пиридинил] -3Н- 1,2,4-триазол-3-она; т. пл. 190,0oC (соединение 31).
Пример 8
(±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-(1- пиперазинил)-3-пиридинил]-3Н-1,2,4-триазол-3-он; (6,02 г) и 2-бром-4,6-диметилпиридин (5,25 г) перемешивали и нагревали в атмосфере N2 при 140oC в течение 2 дней. Смесь растворяли в CH2Cl2 и очищали на стеклянном фильтре с силикагелем (элюент: CH2Cl2/CH3ОН от 100/0 до 97/3). Чистые фракции собирали и выпаривали, получая 3,6 г (48%) (±)-2-[1-(4-хлорбензоил) пропил]-4-[6-[4-(4,6-диметил-2-пиридинил)-1- пиперазинил]-3-пиридинил]-2,4-дигидро-3Н-1,2,4-триазол-3-она; т. пл. 113,2oC (соединение 32).
Пример 9
а) Смесь 2-хлор-4-метоксипиримидина (29 г) и 1-(5- нитро-2-пиридинил)-пиперазина (35 г) в 1,1-диоксиде тетрагидротиофена (50 мл) перемешивали при 130oC в течение 1 часа. Смесь выливали в карбонат натрия и воду, перемешивали 30 минут и экстрагировали CH2Cl2. Органический слой высушивали над MgSO4, фильтровали и выпаривали. Остаток растирали с диизопропиловым эфиром, получая 23 г (43%) 4-метокси-2[4-(5-нитро-2- пиридинил)-1-пиперазинил] пиримидина (промежуточное соединение 10).
б) Исходя из промежуточного соединения 10, был получен (±)-2-[1-(4-хлорбензоил)пропил] -2,4-дигидро-4-[6-[4-(4- метокси-2-пиримидинил)-1-пиперазинил] -3-пиридинил] - 3Н-1,2,4-триазол-3-он, согласно процедуре, аналогичной описанной в примерах от 2б до 2e; т.пл. 162,5oC (соединение 33).
Фармакологический пример
Активность испытуемых соединений против Helicobacter оценивали с помощью следующей тест-процедуры in vitro.
Пример 10
Активность испытуемых соединений против Helicobacter
Активность испытуемых соединений против Helicobacter pylori определяли в отношении стандартного набора из 5 штаммов Н.pylori, полученных из клинического материала. Минимальные ингибирующие концентрации (МИК) определяли путем измерения активности уреазы Н. pylori после обработки растущих культур бактерий антимикробными агентами.
Испытуемые соединения растворяли в ДМСО до концентрации 10-3 М. Также готовили разведение 10-4 в ДМСО. 10 мкл объемы этих растворов с помощью пипеток помещали в лунки планшетов Repli (® Sterilin). Лунки, содержавшие только ДМСО, включали в каждый планшет Repli в качестве контролей. В каждую серию тестов в качестве эталонов для сравнения включали ампициллин ((+)-6-[(2-амино-2 -фенилацетил)амино] 3,3-диметил-7-оксо-4-тиа-1- азабицикло [3.2.0] гептан-2-карбоксильная кислота тригидрат) и метронидазол (2-метил-5-нитро-1Н-имидазол- 1-этанол). (Эти соединения испытывали в конечных концентрациях 10-5, 10-6, 10-7 и 10-8 М). Тестовые планшеты хранили до использования при 4oC.
Пять штаммов Н.pylori поддерживали пересеиванием на 10% кровяном агаре каждые 2 - 3 дня. Бактерии выращивали при 37oC в атмосфере, содержащей 5% кислорода, 10% CO2 и 85% азота. Суспензии Helicobacter pylori для инокулятов получали на бульоне из настоя сердечной и мозговой ткани и доводили до поглощения 1,5 ± 0,3 на 530 нм.
Свежеприготовленный 10% кровяной агар с температурой 45oC в объеме 1 мл добавляли в лунки тестовых планшетов, разводя, таким образом, испытуемые соединения до 10-5 и 10-6. Среде позволяли остыть, затем пипеткой помещали 10 мкл объемы бактериальной суспензии на поверхность агара. Планшеты инкубировали в течение 48 часов при 37oC в микроаэрофильной атмосфере, описанной выше. Для облегчения чтения планшетов и гарантии того, что любой рост в среде действительно является Н.pylori, предпочтение отдавали высокой активности в отношении мочевины, уникальной для этих видов. После 48 часов инкубации 1 мл объемы бульона, содержавшего уреазу, осторожно добавляли в каждую лунку планшетов Repli и планшеты инкубировали при 37oC в течение 2 часов. Затем 100 мкл образцы жидкости из каждой лунки пипеткой переносили в лунки 96-луночных микротитрационных планшетов. Пурпурный цвет интерпретировали как рост, желто-оранжевый - как отсутствие роста Н.pylori. Таким способом достигалась точка, начиная с которой ингибирующие эффекты невозможно определить. Все соединения, показавшие активность при любой из двух концентраций, подвергали испытанию с дальнейшими разведениями для определения МИК и с более широким спектром бактериальных видов как организмов-мишеней. Таким способом было установлено, что величины МИК для соединений 1-4, 8, 10, 12, 14-16, 20, 23 и 26-29 равны или составляют менее 1 мкМ.
Примеры композиций
"Активный ингредиент" (АИ) в приведенных примерах относится к соединению формулы (I), его фармацевтически приемлемой соли присоединения кислоты или стереохимически изомерной форме.
Пример 11: КАПЛИ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ
500 граммов АИ растворяли в 0,5 л 2-оксипропановой кислоты и 1,5 л полиэтиленгликоля при ~ 60 - 80oC. После охлаждения до ~ 30 - 40oC добавляли 35 л полиэтиленгликоля и смесь тщательно перемешивали. Затем добавляли раствор 1750 граммов сахарин-натрия в 2,5 л очищенной воды. При перемешивании добавляли 2,5 л корригента со вкусом какао и полиэтиленгликоль q.s. до объема 50 л, в результате чего получали раствор для пероральных капель, содержавший 10 мг/мл АИ. Полученным раствором наполняли подходящие контейнеры.
Пример 12: КАПСУЛЫ
20 граммов АИ, 6 граммов лаурилсульфата натрия, 56 граммов крахмала, 56 граммов лактозы, 0,8 грамма коллоидного диоксида кремния и 1,2 грамма стеарата магния интенсивно смешивали друг с другом. Полученной смесью затем наполняли 1000 подходящих твердых желатиновых капсул, которые содержали по 20 мг активного ингредиента.
Пример 13: ТАБЛЕТКИ С ПЛЕНОЧНЫМ ПОКРЫТИЕМ
Изготовление ядра таблетки
Смесь 100 граммов АИ, 570 граммов лактозы и 200 граммов крахмала тщательно перемешивали, а затем увлажняли раствором 5 граммов додецилсульфата натрия и 10 граммов поливинилпирролидона в 200 мл воды. Влажную порошковую смесь просеивали, высушивали и вновь просеивали. Добавляли 100 граммов микрокристаллической целлюлозы и 15 граммов гидрогенизированного растительного масла. Смесь хорошо перемешивали и прессовали в таблетки, получая 10000 таблеток по 10 мг активного ингредиента каждая.
Покрытие
К раствору 10 граммов метилцеллюлозы в 75 мл денатурированного этанола добавляли раствор 5 граммов этилцеллюлозы в 150 мл дихлорметана. Затем добавляли 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. 10 граммов полиэтиленгликоля расплавляли и растворяли в 75 мл дихлорметана. Последний раствор добавляли к предыдущему, а затем добавляли 2,5 грамма октадеканоата магния, 5 граммов поливинилпирролидона и 30 мл концентрированной суспензии красителя и все вместе гомогенизировали. Ядра таблеток покрывали полученной таким способом смесью в машине для нанесения покрытия.
Пример 14: СУППОЗИТОРИИ
3 грамма АИ растворяли в растворе 3 граммов 2,3- дигидроксибутандикислоты в 25 мл полиэтиленгликоля 400. Совместно расплавляли 12 граммов поверхностно-активного вещества и триглицеридов q.s. до 300 граммов. Последнюю смесь хорошо перемешивали с предыдущим раствором. Полученную таким способом смесь выливали в формы при температуре 37 - 38oC, получая 100 суппозиториев с содержанием в каждом 30 мг/мл АИ.
Гетероциклические производные азолонов формулы I, где Y - СН, N; R1, R2, R3 - Н или С1-4алкил; R4, R5 - Н, галоген, С1-4алкил, С1-4алкокси, гидрокси, CF3, ОСF3, ОСНF2; R6 - пиридинил, возможно замещенный 1-2 алкилами; ди(С1-4алкил)гидроксипиридинил; ди(C1-4алкил)-алкилоксипиридинил, пиридазинил, необязательно замещенный C1-4алкилокси; пиримидинил, необязательно замещенный гидрокси или C1-4алкилокси; тиазолил, необязательно замещенный С1-4алкилом; бензоксазолил или бензотиазолил, или R6 - пиразинил или пиридазинил, замещенный С1-4алкилом; Z - С=O или СНОН; - радикал формулы (а-1) - (a-7), являются мощными агентами против Helicobacter. 4 с. и 5 з.п.ф-лы.
- радикал формулы (а-1) - (a-7), являются мощными агентами против Helicobacter. 4 с. и 5 з.п.ф-лы.
их фармацевтически приемлемые соли присоединения кислот и стереохимически изомерные формы,
где Y - CH или N;
R1 - R3 каждый независимо - водород или C1-6алкил;
R4 и R5 каждый независимо - водород, галоген, C1-4алкил, C1-4алкилокси, гидрокси, трифторметил, трифторметилокси или дифторметилокси;
R6 - пиридинил, необязательно замещенный вплоть до двух групп C1-4алкил; ди(C1-4алкил)гидроксипиридинил; ди(C1-4алкил)C1-4алкилоксипиридинил; пиридазинил, необязательно замещенный C1-4алкилокси; пиримидинил, необязательно замещенный гидрокси или C1-4алкилокси; тиазолил и тиадиазолил, необязательно замещенные C1-4алкилом; бензоксазолил или бензотиазолил или R6 - пиразинил или пиридазинил, замещенный C1-4алкилом;
Z - С = O или CHOH; - радикал формулы
- радикал формулы
2. Соединение по п. 1, отличающееся тем, что R6 - пиридинил, необязательно замещенный вплоть до двух групп C1-4алкил; ди(C1-4алкил)гидроксипиридинил; ди(C1-4алкил)C1-4алкилоксипиридинил; пиридазинил, необязательно замещенный C1-4алкилокси; пиримидинил, необязательно замещенный гидрокси или C1-4алкилокси; тиазолил и тиадиазолил, необязательно замещенные C1-4алкилом; бензоксазолил или бензотиазолил. - радикал формулы (а-1) или (а-2).
- радикал формулы (а-1) или (а-2).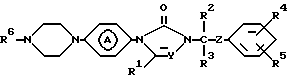
значения радикалов определены в п.1, отличающийся тем, что он включает N-алкилирование промежуточного соединения II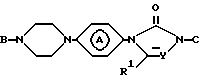
в котором В - водород или R6;
С - водород или радикал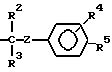
с реагентом формулы ГалR (III), в котором Гал - галоген, R - R6 или радикал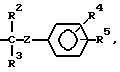
при условии, что, если в промежуточном соединении II В - водород и С - радикал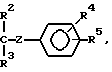
реагент III не может быть
и, если в промежуточном соединении II, В является R6 и С - водородом, реагент III не может быть ГалR6,
в подходящем растворителе и в присутствии основания, и далее, если это желательно, превращением соединений формулы I друг в друга известными способами, превращением соединений формулы I в соль присоединения кислоты посредством воздействия фармацевтически приемлемой кислотой, или, наоборот, превращением соли в свободное основание посредством воздействия щелочью, и/или получение их стереохимически изомерных форм.
| Способ получения производных пиперазина | 1973 |
|
SU503516A3 |
| US 4931444, 05.06.90 | |||
| Способ получения анионообменных мембран | 1972 |
|
SU464604A1 |
