Настоящее изобретение относится к новым водорастворимым азольным противогрибковым средствам широкого спектра действия, а также к их предшественникам, обладающим противогрибковой активностью.
Системные микозы (грибковые инфекции) у людей в странах с умеренным климатом являются относительно редкими, и многие грибы, которые могут стать патогенными, нормально сосуществуют с организмом или являются обычными в окружающей среде. Однако за несколько последних десятилетий имеются свидетельства о возрастающей частоте ряда опасных для жизни микозов во всем мире, и в настоящее время они представляют главную опасность для многих чувствительных к ним пациентов, в особенности для тех, кто уже госпитализирован. Большая часть причин такого возрастания может быть приписана улучшенной выживаемости пациентов с ослабленным иммунитетом и постоянному применению противомикробных средств. Кроме того, флора, типичная для многих обычных микозов, также изменяется, и это обстоятельство является эпидемиологическим признаком возрастающего значения. К пациентам с наибольшим риском относятся пациенты с иммунными функциями, ослабленными либо непосредственно в результате иммуносупрессии от цитотоксических лекарственных препаратов или ВИЧ-инфекции, либо косвенно вследствие других истощающих заболеваний, таких как рак, острый лейкоз, инвазивная оперативная техника или длительное воздействие противомикробных средств. Наиболее распространенными системными микозами у людей являются кандидоз, аспергиллез, гистоплазмоз, кокцидиоидомикоз, паракокцидиоидомикоз, бластомикоз и криптококкоз.
Все в большей степени для лечения и профилактики системных микозов у пациентов с ослабленными иммунными функциями применяют такие противогрибковые средства, как кетоконазол, итраконазол и флуконазол. Однако, что касается резистентности грибов к некоторым из этих средств, в особенности к средствам с более узким спектром действия, например к флуконазолу, то она растет. Что еще хуже, в мировой медицине признают, что около 40% людей, страдающих от тяжелых системных микозов, могут с большим трудом или вовсе не в состоянии получать медикаментозное лечение через пероральное введение. Такая неспособность является следствием того факта, что такие пациенты находятся в коме или страдают от тяжелого гастропареза. Следовательно, применение нерастворимых или плохо растворимых противогрибковых средств, таких как итраконазол или саперконазол, которые тяжело вводить внутривенно, является весьма затруднительным.
Поэтому имеется потребность в новых противогрибковых средствах, предпочтительно в противогрибковых средствах широкого спектра действия, к которым не существует резистентности, и которые можно вводить внутривенно. Противогрибковые средства, предпочтительно, должны быть также доступны в фармацевтических композициях, подходящих для перорального введения. Это дает возможность лечащему врачу продолжать лечение тем же лекарственным препаратом после того, как пациент выведен из состояния, когда ему требуется внутривенное введение упомянутого лекарственного препарата.
Патент США 4267179 описывает гетероциклические производные (4-фенилпиперазин-1-ил-арилоксиметил-1,3-диоксолан-2-ил)-метил  имидазолов и
имидазолов и 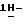 1,2,4-триазолов, пригодных для применения в качестве противогрибковых и противомикробных средств. Упомянутый патент включает итраконазол, который в настоящее время доступен как противогрибковое средство широкого спектра действия во всем мире.
1,2,4-триазолов, пригодных для применения в качестве противогрибковых и противомикробных средств. Упомянутый патент включает итраконазол, который в настоящее время доступен как противогрибковое средство широкого спектра действия во всем мире.
Патент США 4916134 описывает новые 4-[4-[4-[[2-(2,4-дифторфенил)-2-( 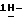 азолилметил)-1,3-диоксолан-4- ил] метокси] фенил]-1-пиперазинил]фенил]триазолоны, обладающие улучшенными противомикробными свойствами. Упомянутый патент включает в себя саперконазол.
азолилметил)-1,3-диоксолан-4- ил] метокси] фенил]-1-пиперазинил]фенил]триазолоны, обладающие улучшенными противомикробными свойствами. Упомянутый патент включает в себя саперконазол.
Патент США 4791111 описывает производные [[4-[[4-(4-фенил-1-пиперазинил)феноксиметил] -1,3-диоксолан-2-ил] метил] - 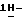 имидазолов и
имидазолов и 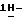 1,2,4-триазолов, по структуре родственные некоторым из соединений настоящего изобретения, которые описываются как соединения, обладающие подходящими противомикробными свойствами.
1,2,4-триазолов, по структуре родственные некоторым из соединений настоящего изобретения, которые описываются как соединения, обладающие подходящими противомикробными свойствами.
Патент США 5039676 описывает азолметил-замещенные тетрагидрофураны, родственные по строению некоторым соединениям настоящего изобретения, о которых говорится, что они обладают противогрибковой активностью, и европейский патент EP 0539938 описывает аналогичные тризамещенные тетрагидрофурановые противогрибковые средства.
Настоящее изобретение относится к новым соединениям формулы (I)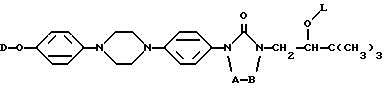 (I)
(I)
их фармацевтически премлемым солям присоединения кислот или оснований, и к их стереохимически изомерным формам, при этом в формуле (I)
A и B вместе образуют двухвалентный радикал формулы
-N=CH- (a),
-CH=N- (b),
-CH2-CH2- (c),
-CH=CH- (d),
-C(=O)-CH2- (e),
-CH2-C(=O)- (f),
при этом один атом водорода в радикалах (a) и (b) может быть замещен C1-6-алкильным радикалом, и до двух атомов водорода в радикалах (c), (d), (e) или (f) могут быть замещены C1-6-алкил-радикалом;
D представляет собой радикал формулы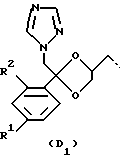
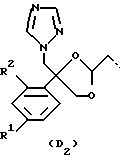 или
или 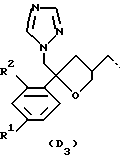
L представляет собой радикал формулы или
или 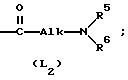
где Alk представляет собой радикал C1-4-алкандиил;
R1 представляет собой галоген;
R2 представляет собой водород или галоген;
R3 представляет собой водород, C1-6-алкил, фенил или галоидфенил;
R4 представляет собой водород, C1-6-алкил, фенил или галоидфенил;
R5 представляет собой водород или C1-6-алкил;
R6 представляет собой водород, C1-6-алкил, C1-6-алкилоксикарбонил, или
R5 и R6, вместе с атомом азота, к которому они присоединены, образуют пирролидиновое, пиперидиновое, морфолинное, пиперазиновое или замещенное пиперазиновое кольцо, причем упомянутое замещенное пиперазиновое кольцо представляет собой пиперазиновое кольцо, замещенное в положении 4 пиперазинового кольца C1-6-алкилом, гидрокси-C1-6-алкилом, амино-C1-6-алкилом, моно- или ди-(C1-6-алкил)амино-C1-6-алкилом.
В приведенных выше определениях и далее термин "галоген" определяет фтор, хлор, бром и йод; "C1-6-алкил" является общим названием линейных или разветвленных углеводородных радикалов, содержащих от 1 до 6 атомов углерода, таких как, например, метил, этил, пропил, бутил, пентил, или гексил, и их возможные разветвленные изомеры; термин "C1-6-алкил" в C1-6-алкилоксигруппе, амино-C1-6-алкиле, моно- или ди-(C1-6-алкил)амино-C1-6-алкиле имеет вышеустановленное значение.
Фармацевтически приемлемые соли присоединения кислот, которые упоминаются выше, включают терапевтически активные нетоксичные формы солей присоединения кислот, которые могут образовывать соединения формулы (I). Их можно удобно получать путем обработки соединения в форме свободного основания такими подходящими кислотами, как неорганические кислоты, например галогенводородные кислоты, например хлористоводородная, бромистоводородная кислота и т. п. ; серная кислота; азотная кислота; фосфорная кислота и т.п., или органические кислоты, например уксусная, пропановая, оксиуксусная, 2-оксипропановая, 2-оксопропановая, этандиовая, пропандиовая, бутандиовая, (Z)-2-бутендиовая, (E)-2-бутендиовая, 2-оксибутандиовая, 2,3-диоксибутандиовая, 2-окси-1,2,3-пропантрикарбоновая, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-оксибензойная, 4-амино-2-оксибензойная кислота и подобные кислоты. Напротив, солевая форма может быть превращена в форму свободного основания путем обработки щелочью. Соединения формулы (I), содержащие кислотные протоны, могут быть также превращены в формы их терапевтически активных нетоксичных солей металлов или присоединения аминов путем обработки соответствующими органическими и неорганическими основаниями. Подходящие формы солей присоединения основания включают, например, аммониевые соли, соли щелочных и щелочноземельных металлов, например соли лития, натрия, калия, магния, кальция и подобные соли, соли присоединения органических оснований, например бензатина,  метил-D-глюкамина, 2-амино-(гидроксиметил)-1,3-пропандиола, гидрабаминовые соли, и соли с аминокислотами, такими как, например, аргинин, лизин и подобные кислоты. И наоборот, путем обработки кислотой солевая форма может быть превращена в форму свободной кислоты.
метил-D-глюкамина, 2-амино-(гидроксиметил)-1,3-пропандиола, гидрабаминовые соли, и соли с аминокислотами, такими как, например, аргинин, лизин и подобные кислоты. И наоборот, путем обработки кислотой солевая форма может быть превращена в форму свободной кислоты.
Термин "соль присоединения кислоты" включает также гидраты и формы с присоединением растворителя, которые способны образовывать соединения формулы (I). Примерами таких форм являются, например, гидраты, алкоголяты и подобные сольваты.
Термин "стереохимически изомерные формы", который здесь используется, относится ко всем возможным изомерным формам, которыми могут обладать соединения формулы (I). Если не оговаривается или не указывается что-либо иное, химическое название соединений означает смесь всех стереохимически возможных изомерных форм, причем упомянутые смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. Точнее, стереогенные центры могут иметь R- или S-конфигурацию; заместители двухвалентных насыщенных циклических углеводородных радикалов, в частности заместители в диоксолановом или тетрагидрофурановом кольце, могут иметь либо цис-, либо транс-конфигурацию. Имеется в виду, очевидно, что стереохимически изомерные формы соединений формулы (I) входят в объем настоящего изобретения.
Соответственно, Alk представляет собой метилен или этандиил;
подходящим значением R1 является фтор, хлор или бром, предпочтительно фтор;
подходящим значением R2 является водород, фтор, хлор или бром, предпочтителен фтор;
подходящим значением R3 является водород или фенил, предпочтителен водород;
подходящим значением R4 является водород или фенил, предпочтителен водород;
подходящим значением R5 является водород, метил или этил;
подходящим значением R6 является водород, метил или этил; или
подходящим является случай, когда R5 и R6, вместе с атомом азота, к которому они присоединены, образуют пирролидиновое кольцо или замещенное пиперазиновое кольцо.
Подходящим является вариант, когда A и B вместе образуют радикалы формул (a), (b) или (c).
Соединения, представляющие интерес, являются такими соединениями формулы (I), в которых D представляет собой радикал формулы (D1) или (D2).
Соединениями, заслуживающими особого внимания, являются те соединения формулы (I), в которых L представляет собой радикал формулы (L2), при этом R5 и R6, каждый и независимо, представляет собой водород или C1-6-алкил; или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое кольцо или пиперазиновое кольцо, замещенное в своем положении 4 C1-6-алкилом или C1-6-алкилоксигруппой.
Более интересными соединениями являются те представляющие интерес соединения, в которых L представляет собой радикал формулы (L1).
Предпочтительными соединениями формулы (I) являются те соединения формулы (I), в которых D представляет собой радикал формулы (D1), при этом R1 представляет собой хлор или фтор, и R2 представляет собой водород, хлор или фтор; L представляет собой радикал формулы (L1), при этом R3 и R4, каждый и независимо, представляет собой фенил или водород.
Соединения формулы (I), в которых заместители в диоксолановом или тетрагидрофурановом кольце имеют цис-конфигурацию, т.е. триазолметиленовый заместитель и замещенный фенилоксиметиленовый заместитель располагаются по одну сторону плоскости диоксоланового или тетрагидрофуранового кольца, являются предпочтительными соединениями.
Более предпочтительные соединения выбирают из группы, в которую входят
(±)-аммоний  1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(  1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил]фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил]фенил] -4,5-дигидро-5-оксо  1,2,4-триазол-1-ил]метил]- 2,2-диметилпропилфосфат;
1,2,4-триазол-1-ил]метил]- 2,2-диметилпропилфосфат;
(±)-аммоний 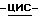 1-[[4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(  1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил]фенил] -4,5-дигидро-5-оксо-
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил]фенил] -4,5-дигидро-5-оксо-  1,2,4-триазол-1-ил] -метил] - 2,2-диметилпропилфосфат;
1,2,4-триазол-1-ил] -метил] - 2,2-диметилпропилфосфат;
моногидрат (±)-аммоний 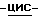 1-[[4-[4-[4-[4-[[2-(2-хлорфенил)- 2-(
1-[[4-[4-[4-[4-[[2-(2-хлорфенил)- 2-( 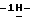 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо 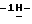 1,2,4-триазол-1-ил]-метил]- 2,2-диметилпропилфосфата (сложного эфира);
1,2,4-триазол-1-ил]-метил]- 2,2-диметилпропилфосфата (сложного эфира);
(±)-аммоний 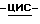 1-[[4-[4-[4-[4-[[2-(4-фторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(4-фторфенил)-2-( 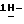 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-4,5-дигидро-5-оксо 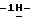 1,2,4-триазол-1-ил]метил]- 2,2-диметилпропилфосфат;
1,2,4-триазол-1-ил]метил]- 2,2-диметилпропилфосфат;
моногидрохлорид (±) 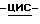 1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)- 2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)- 2-( 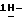 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо 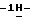 1,2,4-триазол-1-ил]метил]- 2,2-диметилпропил-4-метил-1-пиперазинацетата; и
1,2,4-триазол-1-ил]метил]- 2,2-диметилпропил-4-метил-1-пиперазинацетата; и
полугидрат (±) 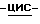 4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 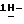 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1- пиперазинил]фенил]-2-[3,3-диметил-2-(фосфонокси)бутил]-2,4-дигидро- 3H-1,2,4-триазол-3-она.
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1- пиперазинил]фенил]-2-[3,3-диметил-2-(фосфонокси)бутил]-2,4-дигидро- 3H-1,2,4-триазол-3-она.
Наиболее предпочтительными являются
полугидрат (±)  4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 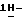 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1- пиперазинил]фенил]-2-[3,3-диметил-2-(фосфонокси)бутил] -2,4-дигидро- 3H-1,2,4-триазол-3-она, его изомерные формы и соли присоединения основания.
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1- пиперазинил]фенил]-2-[3,3-диметил-2-(фосфонокси)бутил] -2,4-дигидро- 3H-1,2,4-триазол-3-она, его изомерные формы и соли присоединения основания.
Соединения формулы (I) можно получить, главным образом, посредством 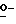 ацилирования или
ацилирования или 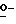 фосфорилирования промежуточного спирта формулы (II) ацилирующим или фосфорилирующим реагентом формулы (III), при этом W1 представляет собой реакционноспособную отщепляющуюся группу, такую как гидроксильная группа или галоген. Упомянутое взаимодействие можно осуществить с помощью известных в технике процедур алкилирования или фосфорилирования, например, путем перемешивания реагентов в инертном для реакции растворителе, необязательно с примесью основания для захвата кислоты, которая образуется во время реакции.
фосфорилирования промежуточного спирта формулы (II) ацилирующим или фосфорилирующим реагентом формулы (III), при этом W1 представляет собой реакционноспособную отщепляющуюся группу, такую как гидроксильная группа или галоген. Упомянутое взаимодействие можно осуществить с помощью известных в технике процедур алкилирования или фосфорилирования, например, путем перемешивания реагентов в инертном для реакции растворителе, необязательно с примесью основания для захвата кислоты, которая образуется во время реакции.

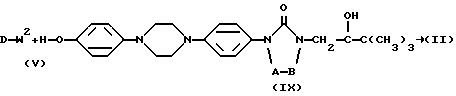
Соединения формулы (I) также можно получить посредством 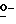 -алкилирования фенола формулы (IV) алкилирующим реагентом формулы (V), при этом W2 представляет собой реакционноспособную отщепляющуюся группу, такую как галоген или сульфонилоксигруппа. Упомянутое взаимодействие осуществляют путем перемешивания реагентов в инертном растворителе, необязательно с добавлением подходящего основания для связывания кислоты, которая образуется во время реакции. В упоминаемых здесь соединениях и в промежуточных соединениях заместители имеют установленные выше значения, если нет других указаний.
-алкилирования фенола формулы (IV) алкилирующим реагентом формулы (V), при этом W2 представляет собой реакционноспособную отщепляющуюся группу, такую как галоген или сульфонилоксигруппа. Упомянутое взаимодействие осуществляют путем перемешивания реагентов в инертном растворителе, необязательно с добавлением подходящего основания для связывания кислоты, которая образуется во время реакции. В упоминаемых здесь соединениях и в промежуточных соединениях заместители имеют установленные выше значения, если нет других указаний.
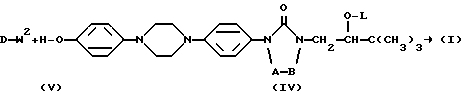
Получение промежуточных соединений формулы (V), в которых D представляет собой радикал формулы D1, описывается в патенте США N 4267179. Получение промежуточных соединений формулы (V), в которых D представляет собой радикал формулы D3, описывается в EP 0539938.
Соединения формулы (I), в которых L представляет собой радикал формулы L2, причем упомянутые соединения изображаются формулой (I-b), также можно получить 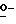 ацилированием промежуточного соединения формулы (II) реагентом формулы (VI), и последующим взаимодействием полученного таким образом промежуточного соединения формулы (VII) с амином формулы (VIII), получая таким образом соединение формулы (I-b).
ацилированием промежуточного соединения формулы (II) реагентом формулы (VI), и последующим взаимодействием полученного таким образом промежуточного соединения формулы (VII) с амином формулы (VIII), получая таким образом соединение формулы (I-b).
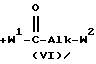

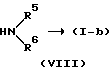
Соединения формулы (I) также могут быть превращены в каждое другое такое соединение посредством трансформаций, известных в технике. Например, соединения, в которых L представляет собой радикал формулы L1, причем упомянутые соединения изображаются формулой (I-a), могут участвовать в следующих взаимопревращениях. Соединения формулы (I-a), в которых R3 и/или R4 представляют собой C1-6-алкил, фенил или галоидфенил, можно превратить в соединение формулы (I-a), в которых R3 и/или R4 представляют собой водород, используя известные в технике способы гидролиза, например, посредством взаимодействия с гидроксидом натрия в подходящем растворителе, например в воде или в 1,4-диоксане.
Соединения формулы (I-b) могут быть превращены в другие соединения такой формулы следующим образом. Соединения формулы (I-b), в которых R5 и/или R6 представляют собой водород, могут быть превращены в соединения формулы (I-b), в которых R5 и/или R6 представляют собой C1-6-алкил, известными в технике реакциями  алкилирования. Соединения формулы (I-b), в которых R6 представляет собой водород, могут быть превращены в соединения формулы (I-b), в которых R6 представляет собой C1-6-алкилоксикарбонил, посредством известных в технике реакций
алкилирования. Соединения формулы (I-b), в которых R6 представляет собой водород, могут быть превращены в соединения формулы (I-b), в которых R6 представляет собой C1-6-алкилоксикарбонил, посредством известных в технике реакций  ацилирования. Напротив, соединения формулы (I-b), в которых R6 представляет собой C1-6-алкилоксикарбонил, могут быть превращены в соединения формулы (I-b), в которых R6 представляет собой водород, известными в технике реакциями гидролиза.
ацилирования. Напротив, соединения формулы (I-b), в которых R6 представляет собой C1-6-алкилоксикарбонил, могут быть превращены в соединения формулы (I-b), в которых R6 представляет собой водород, известными в технике реакциями гидролиза.
Промежуточные соединения формулы (II) могут быть получены  алкилированием реагента формулы (IX) алкилирующим реагентом формулы (V) в соответствии со способами
алкилированием реагента формулы (IX) алкилирующим реагентом формулы (V) в соответствии со способами  алкилирования, описанными выше для получения соединений формулы (I).
алкилирования, описанными выше для получения соединений формулы (I).
Промежуточные соединения формулы (II) также можно получить посредством  алкилирования реагента формулы (X) алкилирующим реагентом формулы (V) в соответствии с процедурами
алкилирования реагента формулы (X) алкилирующим реагентом формулы (V) в соответствии с процедурами 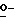 алкилирования, описанными выше для получения соединений формулы (I), и последующим восстановлением образованных таким образом промежуточных соединений формулы (XI). Упомянутое восстановление может быть осуществлено при перемешивании промежуточного соединения формулы (XI) с восстановителем, таким как, например, боргидрид натрия, в инертном растворителе, таком как, например, галоидированный углеводород, например дихлорметан, спирт, например метанол, и их смеси.
алкилирования, описанными выше для получения соединений формулы (I), и последующим восстановлением образованных таким образом промежуточных соединений формулы (XI). Упомянутое восстановление может быть осуществлено при перемешивании промежуточного соединения формулы (XI) с восстановителем, таким как, например, боргидрид натрия, в инертном растворителе, таком как, например, галоидированный углеводород, например дихлорметан, спирт, например метанол, и их смеси.
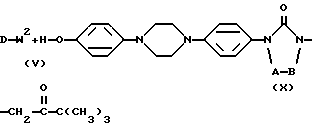
Получение промежуточных соединений формулы (X) описывается в патенте США 4931444.
Промежуточные соединения формулы (XI) также могут быть получены посредством  алкилирования промежуточных соединений формулы (XII), в соответствии с известными в технике способами
алкилирования промежуточных соединений формулы (XII), в соответствии с известными в технике способами 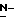 алкилирования, алкилирующим реагентом формулы (XIII), при этом W3 представляет собой подходящую отщепляющуюся группу, например, галоген.
алкилирования, алкилирующим реагентом формулы (XIII), при этом W3 представляет собой подходящую отщепляющуюся группу, например, галоген.
Чистые стереохимически изомерные формы соединений и промежуточных соединений по настоящему изобретению могут быть получены посредством применения способов, известных в технике. Диастереоизомеры можно разделить способами физического разделения, такими как селективная кристаллизация, и техническими приемами хроматографии, например жидкостной хроматографией. Энантиомеры можно отделить друг от друга селективной кристаллизацией их диастереомерных солей с активными кислотами. С другой стороны, энантиомеры можно разделить хроматографическими способами, используя хиральные неподвижные фазы. Упомянутые чистые стереохимически изомерные формы также можно получить из соответствующих чистых стереохимически изомерных форм подходящих исходных веществ, при условии, что реакция происходит стереоспецифически. Если нужен специфический стереоизомер, упомянутое соединение предпочтительно будет синтезировано стереоспецифическими способами получения. В этих способах преимущественно будут использоваться энантиомерно чистые исходные вещества. Предполагается, очевидно, что стереохимически изомерные формы соединений формулы (I) включаются в объем настоящего изобретения.
Соединения формулы (I), их фармацевтически приемлемые соли присоединения кислот или оснований и их стереохимически изомерные формы являются подходящими средствами для борьбы с грибами и бактериями in vivo. Кроме того, соединения формулы (I) растворяются в водных растворах, что делает их пригодными для внутривенного введения. Обнаружено, что упомянутые соединения являются активными против широкого ряда грибов, таких как Candida albicans, Aspergillus fumigatus, Cryptococcus neoformans, Coccidioides immitis, Histoplasma capsulatum, Blastomyces dermatitidis, Sporothrix schenkii, вид Fonsecaea, Microsporum canis, Paracoccidioldes immitis, вид Trichophyton, Cladosporium carrionii, и против бактерий, таких как, например, Erysipelotrix insidiosa, стафилококка, такого как Staphylococcus haemolyticus, и стрептококка, такого как Streptococcus pyogenes.
Промежуточные соединения формулы (II), их фармацевтически приемлемые соли присоединения кислот и их стереохимически изомерные формы также пригодны для лечения или предупреждения заболеваний, ассоциируемых с микозами, и, таким образом, составляют другой аспект настоящего изобретения. Представляющую интерес группу соединений формулы (II) составляют  4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 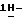 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)  1,2,4-триазол-3-он, его фармацевтически приемлемые соли присоединения кислот и стереохимически изомерные формы.
1,2,4-триазол-3-он, его фармацевтически приемлемые соли присоединения кислот и стереохимически изомерные формы.
Настоящее изобретение также относится к композициям для лечения или предупреждения микозов, содержащим противогрибково эффективное количество соединения формулы (I) или промежуточного соединения формулы (II) и фармацевтически приемлемый носитель или разбавитель.
Принимая во внимание их полезные фармакологические свойства, соединения, являющиеся предметом настоящего изобретения, могут быть включены в состав различных фармацевтических форм с целью введения. Чтобы получить фармацевтические композиции настоящего изобретения, эффективное количество определенного соединения, в форме соли присоединения основания или кислоты, как активного ингредиента, соединяют при тщательном перемешивании с фармацевтически приемлемым носителем, и этот носитель может иметь самые разные формы в зависимости от формы препарата, нужного для введения. Такие фармацевтические композиции являются пригодными в виде стандартных лекарственных форм, подходящих, предпочтительно, для перорального введения, ректального и подкожного введения, или для введения посредством парентеральной инъекции. Например, при получении композиции для пероральной лекарственной формы можно использовать любую из обычных фармацевтических сред, такую, как, например, воду, гликоли, масла, спирты и подобное - в случае жидких препаратов для перорального введения, таких как суспензии, сиропы, эликсиры и растворы; или применять твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие, дезинтегрирующие средства и подобные вещества - в случае порошков, пилюль, капсул и таблеток. Вследствие простоты введения наибольший интерес из единиц лекарственной формы для перорального введения представляют таблетки и капсулы, в которых, очевидно, используются твердые фармацевтические носители. Носителем для парентеральных композиций будет, как правило, стерильная вода, по крайней мере, в большинстве случаев, хотя могут быть включены другие ингредиенты, улучшающие растворимость, например циклодекстрины. Например, могут быть приготовлены растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Могут быть также приготовлены суспензии для инъекций, в которых могут быть использованы подходящие жидкие носители, суспендирующие агенты и подобные вещества. В композициях, подходящих для подкожного введения, носитель необязательно включает средство, усиливающее проникновение, и/или подходящий смачиватель, необязательно, в сочетании с подходящими добавками любой природы, которые берутся в небольшом количестве и которые не оказывают заметного вредного действия на кожу. Упомянутые добавки могут облегчать введение под кожу и/или могут способствовать получению нужных композиций. Такие композиции могут вводиться различными способами, например, в виде трансдермального пэтча, наноситься в виде пятнышка на пораженное место, применяться в виде мази. Особенно выгодно составлять вышеупомянутые фармацевтические композиции в виде стандартной лекарственной формы для облегчения введения и равномерности дозировки. Стандартная лекарственная форма, о которой идет речь в описании и формуле изобретения, имеет в виду физически раздельные единицы, подходящие для единичных доз, причем каждая единица содержит заранее определенное количество активного ингредиента, вычисленное с таким расчетом, чтобы получить желаемое терапевтическое действие, в сочетании с требуемым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая рифленые таблетки и таблетки с покрытием), капсулы, пилюли, пакетики с порошком, облатки, растворы или суспензии для инъекций, чайная ложка, столовая ложка и подобное, и их множество, разделенное по-своему.
Подходящими производными циклодекстрина (CD) являются α-, β-, γ- циклодекстрины или их простые эфиры и смешанные простые эфиры, в которых одна или несколько гидроксильных групп безводных глюкозных звеньев циклодекстрина замещается C1-6-алкилом, в частности этилом, метилом или изопропилом; гидрокси-C1-6-алкилом, в частности гидроксиэтилом, гидроксипропилом или гидроксибутилом; карбокси-C1-6-алкилом, в частности карбоксиметилом или карбоксиэтилом; C1-6-алкилкарбонилом, в частности ацетилом; C1-6-алкилоксикарбонил-C1-6-алкилом или карбокси-C1-6-алкилокси-C1-6-алкилом, в частности карбоксиметоксипропилом или карбоксиэтоксипропилом; C1-6-алкилкарбонилокси-C1-6-алкилом, в частности 2-ацетилоксипропилом. В качестве комплексантов и/или солюбилизаторов особого внимания заслуживают β-CD, 2,6-диметил-β-CD, 2-гидроксиэтил-β-CD, 2-гидроксиэтил-γ-CD, 2-гидроксипропил-γ-CD и (2-карбоксиметокси)-пропил-β-CD, в особенности 2-гидроксипропил-β-CD.
Термин "простой смешанный эфир" обозначает производные циклодекстрина, в которых по крайней мере две циклодекстриновые гидроксильные группы этерифицированы различными группами, такими как, например, гидроксипропильная и гидроксиэтильная.
Специалисты в области лечения теплокровных животных, страдающих от заболеваний, вызванных грибами и/или бактериями, могут легко определить эффективное количество по результатам приведенных здесь экспериментов. Вообще, ожидается, что эффективное количество должно составлять от 0,01 мг/кг до 50 мг/кг массы тела, предпочтительнее от 0,05 мг/кг до 20 мг/кг массы тела.
Экспериментальная часть
Стереохимическая конфигурация некоторых соединений формулы (I) экспериментально не определяется. В таких случаях стереохимически изомерную форму, которую выделяют первой, обозначают "A", а вторую, которую выделяют, обозначают "B".
A. Получение промежуточных соединений
Пример 1
К смеси 2-(2,4-дифторфенил)-3-( 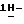 1,2,4-триазол-1-ил)-1,2-пропандиола (30 г), метансульфоновой кислоты (50 мл) и дихлорметана (500 мл) при перемешивании и охлаждении (ледяная баня) добавляют по каплям 1-бром-2,2-диэтоксиэтан (17 мл). После перемешивания в течение 3 часов при 0oC реакционную смесь выливают в гидрокарбонат натрия (водный раствор). Продукт реакции экстрагируют дихлорметаном, и экстракт сушат, фильтруют и упаривают. Остаток дважды очищают колоночной хроматографией (силикагель; CHCl3/CH3OH 99:1; CHCl3/CH3OH/гексан/CH3COOC2H5 49:1:20:30). Элюат нужной фракции упаривают, и получают 8 г (19,0)
1,2,4-триазол-1-ил)-1,2-пропандиола (30 г), метансульфоновой кислоты (50 мл) и дихлорметана (500 мл) при перемешивании и охлаждении (ледяная баня) добавляют по каплям 1-бром-2,2-диэтоксиэтан (17 мл). После перемешивания в течение 3 часов при 0oC реакционную смесь выливают в гидрокарбонат натрия (водный раствор). Продукт реакции экстрагируют дихлорметаном, и экстракт сушат, фильтруют и упаривают. Остаток дважды очищают колоночной хроматографией (силикагель; CHCl3/CH3OH 99:1; CHCl3/CH3OH/гексан/CH3COOC2H5 49:1:20:30). Элюат нужной фракции упаривают, и получают 8 г (19,0) 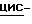 1-[[2-(бромметил)-4-(2,4-дифторфенил)-1,3-диоксолан-4-ил] метил]
1-[[2-(бромметил)-4-(2,4-дифторфенил)-1,3-диоксолан-4-ил] метил] 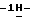 1,2,4-триазола; т.пл. 76,3oC (промежуточное соединение 1).
1,2,4-триазола; т.пл. 76,3oC (промежуточное соединение 1).
Пример 2
Смесь 2-(3,3-диметил-2-оксобутил)-2,4-дигидро-4-[4-[4-(4- гидроксифенил)-1-пиперазинил]фенил] 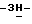 1,2,4-триазол-3-она (0,01 моль) и гидрида натрия (0,012 моль) в
1,2,4-триазол-3-она (0,01 моль) и гидрида натрия (0,012 моль) в 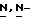 диметилформамиде (100 мл) перемешивают в атмосфере азота при 70oC. Добавляют промежуточное соединение (1) (0,012 моль), и смесь перемешивают еще в течение ночи. Снова добавляют промежуточное соединение (1) (2 г), и смесь перемешивают при 70oC в течение 6 часов, а затем в течение ночи при комнатной температуре. Смесь упаривают, остаток растворяют в дихлорметане и промывают. Органический слой сушат, фильтруют и упаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент CH2Cl2/гексан/этилацетат, 50/20/30). Очищенные фракции собирают и упаривают. Остаток дополнительно очищают на стеклянном фильтре на силикагеле/NH2 (элюент CH2Cl2). Чистые фракции собирают и упаривают. Остаток кристаллизуют из этилацетата и получают 2,2 г (31%) (±)
диметилформамиде (100 мл) перемешивают в атмосфере азота при 70oC. Добавляют промежуточное соединение (1) (0,012 моль), и смесь перемешивают еще в течение ночи. Снова добавляют промежуточное соединение (1) (2 г), и смесь перемешивают при 70oC в течение 6 часов, а затем в течение ночи при комнатной температуре. Смесь упаривают, остаток растворяют в дихлорметане и промывают. Органический слой сушат, фильтруют и упаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент CH2Cl2/гексан/этилацетат, 50/20/30). Очищенные фракции собирают и упаривают. Остаток дополнительно очищают на стеклянном фильтре на силикагеле/NH2 (элюент CH2Cl2). Чистые фракции собирают и упаривают. Остаток кристаллизуют из этилацетата и получают 2,2 г (31%) (±) 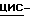 4-[4-[4-[4-[[4-(2,4-дифторфенил)-4-(
4-[4-[4-[4-[[4-(2,4-дифторфенил)-4-( 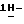 1,2,4-триазол-1-илметил)- 1,3-диоксолан-2-ил]-метокси]фенил] -1-пиперазинил] фенил]-2-[3,3- диметил-2-оксобутил]-2,4-дигидро
1,2,4-триазол-1-илметил)- 1,3-диоксолан-2-ил]-метокси]фенил] -1-пиперазинил] фенил]-2-[3,3- диметил-2-оксобутил]-2,4-дигидро 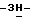 1,2,4-триазол-3-она; т.пл. 197,1oC (промежуточное соединение 2).
1,2,4-триазол-3-она; т.пл. 197,1oC (промежуточное соединение 2).
Пример 3
a) Смесь 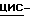 2-(2,4-дифторфенил)-2-(
2-(2,4-дифторфенил)-2-( 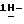 1,2,4-триазол-1- илметил)-1,3-диоксолан-4-метанола (0,2 моль) с пиридином (400 мл) и дихлорметаном (250 мл) перемешивают при комнатной температуре. Добавляют по каплям раствор 4-цианобензоилхлорида (0,22 моль) в дихлорметане (150 мл). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре. Разбавляют реакционную смесь водой. Отделяют органический слой, сушат его (MgSO4), фильтруют и испаряют растворитель. Остаток кристаллизуют из метилбензола. Кристаллы отфильтровывают и сушат. Эту фракцию очищают колоночной хроматографией на колонке Chiracell OD (элюент C2H5OH). Фракции первого пика объединяют и испаряют растворитель. Остаток кристаллизуют из 4-метил-2-пентанона. Кристаллы отфильтровывают и сушат, получают 21,2 г (24,9%) (+)
1,2,4-триазол-1- илметил)-1,3-диоксолан-4-метанола (0,2 моль) с пиридином (400 мл) и дихлорметаном (250 мл) перемешивают при комнатной температуре. Добавляют по каплям раствор 4-цианобензоилхлорида (0,22 моль) в дихлорметане (150 мл). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре. Разбавляют реакционную смесь водой. Отделяют органический слой, сушат его (MgSO4), фильтруют и испаряют растворитель. Остаток кристаллизуют из метилбензола. Кристаллы отфильтровывают и сушат. Эту фракцию очищают колоночной хроматографией на колонке Chiracell OD (элюент C2H5OH). Фракции первого пика объединяют и испаряют растворитель. Остаток кристаллизуют из 4-метил-2-пентанона. Кристаллы отфильтровывают и сушат, получают 21,2 г (24,9%) (+) 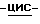 [2-(2,4-дифторфенил)-2-(
[2-(2,4-дифторфенил)-2-( 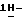 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метил-4-цианобензоата; т. пл. 146,3oC; [α]
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метил-4-цианобензоата; т. пл. 146,3oC; [α]
Объединяют фракции второго пика и испаряют растворитель. Остаток кристаллизуют из 4-метил-2-пентанона. Кристаллы отфильтровывают и сушат, получают 21,4 г (25,1%) (-) 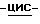 [2-(2,4-дифторфенил)-2-(
[2-(2,4-дифторфенил)-2-( 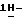 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метил-4-цианобензоата; т. пл. 144,0oC; [α]
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метил-4-цианобензоата; т. пл. 144,0oC; [α]
b) Смесь промежуточного соединения (3) (0,049 моль) и 50% раствора гидроксида натрия (0,059 моль) в воде (300 мл) и 1,4-диоксане (300 мл) перемешивают в течение 24 часов при комнатной температуре. Растворитель испаряют. Остаток обрабатывают водой и дихлорметаном. Органический слой отделяют, сушат (MgSO4), фильтруют, и испаряют растворитель. Остаток кристаллизуют из 4-метил-2-пентанона. Кристаллы отфильтровывают и сушат, получают 10,1 г (70%) (+) 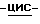 2-(2,4-дифторфенил)-2-(
2-(2,4-дифторфенил)-2-( 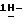 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола; т. пл. 123,0oC; [α]
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола; т. пл. 123,0oC; [α]
Подобным способом получают также
(-) 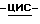 2-(2,4-дифторфенил)-2-(
2-(2,4-дифторфенил)-2-( 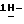 1,2,4-триазол-1-илметил)-1,3- диоксолан-4-метанол; т. пл. 123,2oC; [α]
1,2,4-триазол-1-илметил)-1,3- диоксолан-4-метанол; т. пл. 123,2oC; [α]
c) Смесь промежуточного соединения (5) (0,02 моль) и 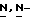 диэтилэтанамина (0,03 моль) в дихлорметане (50 мл) перемешивают при комнатной температуре. Добавляют по каплям метансульфонилхлорид (0,03 моль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель испаряют. Остаток обрабатывают метилбензолом и водой. Образуется осадок, который отфильтровывают, сушат и перекристаллизовывают из 2,2'-оксибиспропана (диизопропиловый эфир) и 4-метил-2-пентанона. Осадок отфильтровывают и сушат (вакуум; 40oC), получают две фракции. Эти фракции объединяют и перекристаллизовывают из 2,2'-оксибиспропана и 4-метил-2-пентанона. Осадок отфильтровывают и сушат (вакуум; 40oC), получают 5,88 г (78,3%) метансульфоната (+)
диэтилэтанамина (0,03 моль) в дихлорметане (50 мл) перемешивают при комнатной температуре. Добавляют по каплям метансульфонилхлорид (0,03 моль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель испаряют. Остаток обрабатывают метилбензолом и водой. Образуется осадок, который отфильтровывают, сушат и перекристаллизовывают из 2,2'-оксибиспропана (диизопропиловый эфир) и 4-метил-2-пентанона. Осадок отфильтровывают и сушат (вакуум; 40oC), получают две фракции. Эти фракции объединяют и перекристаллизовывают из 2,2'-оксибиспропана и 4-метил-2-пентанона. Осадок отфильтровывают и сушат (вакуум; 40oC), получают 5,88 г (78,3%) метансульфоната (+) 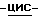 2-(2,4-дифторфенил)-2-(
2-(2,4-дифторфенил)-2-(  1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (сложный эфир); [α]
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (сложный эфир); [α]
Подобным способом получают также
метансульфонат (-)  2-(2,4-дифторфенил)-2-(
2-(2,4-дифторфенил)-2-(  1,2,4-триазол-1- илметил)-1,3-диоксолан-4-метанола (сложный эфир); [α]
1,2,4-триазол-1- илметил)-1,3-диоксолан-4-метанола (сложный эфир); [α]
Пример 4
а) Смесь 2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)-4-[4-[4-(4- гидроксифенил)-1-пиперазинил] фенил]  1,2,4-триазол-3-она (0,046 моль), 2-(хлордиметилсилил)-2-метилпропана (0,063 моль) и
1,2,4-триазол-3-она (0,046 моль), 2-(хлордиметилсилил)-2-метилпропана (0,063 моль) и  имидазола (0,19 моль) в
имидазола (0,19 моль) в  диметилформамиде (300 мл) перемешивают в течение 4 часов при 50oC. Реакционную смесь выливают в воду. Получающийся в результате осадок отфильтровывают и сушат, получают 21 г (83%) продукта реакции. Образец (1 г) обрабатывают 2,2'-оксибиспропаном, отфильтровывают и сушат, получают 0,7 г (±)-4-[4-[4-[4-[[(1,1-диметилэтил)диметилсилил] окси]фенил]-1- пиперазинил]фенил]-2-(2-гидрокси-3,3-диметилбутил)-2,4-дигидро
диметилформамиде (300 мл) перемешивают в течение 4 часов при 50oC. Реакционную смесь выливают в воду. Получающийся в результате осадок отфильтровывают и сушат, получают 21 г (83%) продукта реакции. Образец (1 г) обрабатывают 2,2'-оксибиспропаном, отфильтровывают и сушат, получают 0,7 г (±)-4-[4-[4-[4-[[(1,1-диметилэтил)диметилсилил] окси]фенил]-1- пиперазинил]фенил]-2-(2-гидрокси-3,3-диметилбутил)-2,4-дигидро 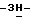 1,2,4-триазол-3-она; т.пл. 196,1oC (промежуточное соединение 9).
1,2,4-триазол-3-она; т.пл. 196,1oC (промежуточное соединение 9).
b) Промежуточное соединение (9) (0,036 моль, смесь энантиомеров) разделяют на составляющие энантиомеры колоночной хроматографией на колонке ChiracellR OD (элюент н-гексан/2-пропанол, 65/35). Фракцию, соответствующую первому хроматографическому пику, собирают, и испаряют растворитель. Остаток кристаллизуют из ацетонитрила. Осадок отфильтровывают и сушат (вакуум; 50oC); получают 1,56 г (7,8%) (-)-4-[4-[4-[4-[[(1,1-диметилэтил)диметилсилил] окси] фенил]-1- пиперазинил]фенил]-2-(2-гидрокси-3,3-диметилбутил)-2,4-дигидро 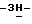 1,2,4-триазол-3-она (промежуточное соединение 10). Собирают фракцию, соответствующую второму хроматографическому пику, и испаряют растворитель. Остаток кристаллизуют из ацетонитрила. Осадок отфильтровывают и сушат (вакуум; 50oC); получают 2,28 г (11,4%) (+)-4-[4-[4-[4-[[(1,1-диметилэтил)диметилсилил] окси] фенил] -1- пиперазинил]фенил]-2-(2-гидрокси-3,3-диметил-бутил)-2,4-дигидро
1,2,4-триазол-3-она (промежуточное соединение 10). Собирают фракцию, соответствующую второму хроматографическому пику, и испаряют растворитель. Остаток кристаллизуют из ацетонитрила. Осадок отфильтровывают и сушат (вакуум; 50oC); получают 2,28 г (11,4%) (+)-4-[4-[4-[4-[[(1,1-диметилэтил)диметилсилил] окси] фенил] -1- пиперазинил]фенил]-2-(2-гидрокси-3,3-диметил-бутил)-2,4-дигидро 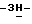 1,2,4-триазол-3-она (промежуточное соединение 11).
1,2,4-триазол-3-она (промежуточное соединение 11).
c) Смесь промежуточного соединения (10) (0,0135 моль) с дихлорметаном (150 мл) перемешивают до полного растворения. Добавляют в один прием раствор тетрабутиламмонийфторида в тетрагидрофуране (0,015 моль), и реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Смесь разбавляют водой (150 мл) и перемешивают в течение 1 часа. Осадок отфильтровывают и перекристаллизовывают из 2-метоксиэтанола. Продукт отфильтровывают и сушат (вакуум; 60oC), получают 4,7 г (79,6%) (-)-2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)- 4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] 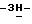 1,2,4-триазол-3-она; [α]
1,2,4-триазол-3-она; [α]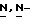 диметилформамиде) (промежуточное соединение 12).
диметилформамиде) (промежуточное соединение 12).
Подобным способом получают также (+)-2,4-дигидро-2-(2-гидрокси- 3,3-диметилбутил)-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] 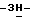 1,2,4-триазол-3-он; [α]
1,2,4-триазол-3-он; [α]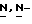 диметилформамиде) (промежуточное соединение 13).
диметилформамиде) (промежуточное соединение 13).
Пример 5
Смесь 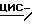 -4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
-4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 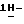 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил]фенил]-2,4-дигидро
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил]фенил]-2,4-дигидро 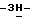 1,2,4-триазол-3-она (9,3 г), 1-бром-3,3-диметил-2-бутанона (2,8 г), карбоната натрия (6,4 г) и 1,3-диметил-2-имидазолидинона (52,2 г) перемешивают в течение 5 часов при 100oC. После охлаждения реакционную смесь выливают в воду. Образовавшийся осадок отфильтровывают и растворяют в дихлорметане. Раствор сушат, фильтруют и упаривают. Остаток очищают колоночной хроматографией (силикагель; CH2Cl2/CH3OH, 98:2). Испаряют элюент из нужной фракции, и остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и сушат, получают 5,5 г (51,3%)
1,2,4-триазол-3-она (9,3 г), 1-бром-3,3-диметил-2-бутанона (2,8 г), карбоната натрия (6,4 г) и 1,3-диметил-2-имидазолидинона (52,2 г) перемешивают в течение 5 часов при 100oC. После охлаждения реакционную смесь выливают в воду. Образовавшийся осадок отфильтровывают и растворяют в дихлорметане. Раствор сушат, фильтруют и упаривают. Остаток очищают колоночной хроматографией (силикагель; CH2Cl2/CH3OH, 98:2). Испаряют элюент из нужной фракции, и остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и сушат, получают 5,5 г (51,3%) 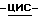 4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 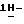 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил]фенил]-2-(3,3-диметил-2-оксобутил)-2,4-дигидро
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил]фенил]-2-(3,3-диметил-2-оксобутил)-2,4-дигидро 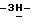 1,2,4-триазол-3-она; т.пл. 176,2oC (промежуточное соединение 14).
1,2,4-триазол-3-она; т.пл. 176,2oC (промежуточное соединение 14).
Пример 6
К смеси промежуточного соединения (14) (4,5 г), 1,4-диоксана (40 мл) и метанола (3 мл) добавляют по каплям раствор тетрагидробората натрия (0,3 г) в небольшом количестве воды. После перемешивания в течение ночи реакционную смесь выливают в воду и подкисляют уксусной кислотой до ~ pH 5. Осадок отфильтровывают, промывают водой, сушат и очищают колоночной хроматографией (силикагель; CH2Cl2/CH3OH, 98:2). Испаряют элюент из нужной фракции, и остаток кристаллизуют из 2-пропанола, получают 2,2 г (48,7%) 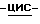 4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 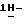 1,2,4-триазол-1-илметил)- 1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] -2,4-дигидро- 2-(2-гидрокси-3,3-диметилбутил)
1,2,4-триазол-1-илметил)- 1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] -2,4-дигидро- 2-(2-гидрокси-3,3-диметилбутил) 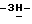 1,2,4-триазол-3-она; т.пл. 196,4oC (промежуточное соединение 15).
1,2,4-триазол-3-она; т.пл. 196,4oC (промежуточное соединение 15).
Пример 7
К смеси промежуточного соединения (12) (0,00595 моль) с 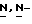 -диметилформамидом (50 мл) при перемешивании при комнатной температуре добавляют частями гидрид натрия (0,00675 моль). Смесь перемешивают в течение 90 минут при комнатной температуре. Добавляют промежуточное соединение (7) (0,0054 моль), и реакционную смесь перемешивают в течение 4 часов при 60oC. Смесь охлаждают, и испаряют растворитель. Остаток обрабатывают дихлорметаном и водой. Отделяют органический слой, сушат, фильтруют, и испаряют растворитель. Остаток очищают колоночной хроматографией на аминопропиловой колонке (элюент CH2Cl2/CH3OH, 96/4). Собирают чистые фракции и испаряют растворитель. Остаток кристаллизуют из 4-метил-2-пентанона. Осадок отфильтровывают и сушат (вакуум; 50oC), получают 1,67 г (43,1%) (+)-[
-диметилформамидом (50 мл) при перемешивании при комнатной температуре добавляют частями гидрид натрия (0,00675 моль). Смесь перемешивают в течение 90 минут при комнатной температуре. Добавляют промежуточное соединение (7) (0,0054 моль), и реакционную смесь перемешивают в течение 4 часов при 60oC. Смесь охлаждают, и испаряют растворитель. Остаток обрабатывают дихлорметаном и водой. Отделяют органический слой, сушат, фильтруют, и испаряют растворитель. Остаток очищают колоночной хроматографией на аминопропиловой колонке (элюент CH2Cl2/CH3OH, 96/4). Собирают чистые фракции и испаряют растворитель. Остаток кристаллизуют из 4-метил-2-пентанона. Осадок отфильтровывают и сушат (вакуум; 50oC), получают 1,67 г (43,1%) (+)-[ 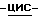 (+)(B)]-4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
(+)(B)]-4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 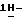 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил) 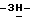 1,2,4-триазол-3-она; т. пл. 191,9oC; [α]
1,2,4-триазол-3-она; т. пл. 191,9oC; [α]
Пример 8
Смесь промежуточного соединения (2) (0,0025 моль) с дихлорметаном (100 мл) и метанолом (100 мл) перемешивают при комнатной температуре. Добавляют боргидрид натрия (0,005 моль), и смесь перемешивают в течение 4 часов. Добавляют воду (100 мл), смесь перемешивают в течение ночи и разделяют. Органический слой промывают, сушат, фильтруют и упаривают. Остаток кристаллизуют из этилацетата, и получают 1,6 г (89,3%) (±) 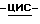 4-[4-[4-[4-[[4-(2,4-дифторфенил)- 4-(
4-[4-[4-[4-[[4-(2,4-дифторфенил)- 4-( 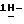 1,2,4-триазол-1-илметил)-1,3-диоксолан-2-ил]метокси] фенил] -1- пиперазинил] фенил] -2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)
1,2,4-триазол-1-илметил)-1,3-диоксолан-2-ил]метокси] фенил] -1- пиперазинил] фенил] -2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил) 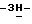 1,2,4-триазол-3-она; т.пл. 184,1oC (промежуточное соединение 24).
1,2,4-триазол-3-она; т.пл. 184,1oC (промежуточное соединение 24).
Пример 9
Смесь 2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)-4-[4-[4-(4- гидроксифенил)-1-пиперазинил] фенил] 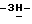 1,2,4-триазол-3-она (0,0035 моль), 4-метилбензолсульфоната
1,2,4-триазол-3-она (0,0035 моль), 4-метилбензолсульфоната 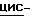 [5-(2,4-дифторфенил)-тетрагидро-5-(
[5-(2,4-дифторфенил)-тетрагидро-5-( 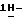 1,2,4-триазол-1-илметил)-3-фуранметанола (получение которого описывается в заявке на европейский патент N 0539938) (0,0033 моль) и гидроксида натрия (0,01 моль) в
1,2,4-триазол-1-илметил)-3-фуранметанола (получение которого описывается в заявке на европейский патент N 0539938) (0,0033 моль) и гидроксида натрия (0,01 моль) в 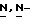 диметилформамиде (50 мл) перемешивают в атмосфере азота при 50oC в течение 4 часов, и затем смесь перемешивают в атмосфере азота при 60oC в течение 2 часов. Смесь охлаждают, и добавляют воду. Продукт выкристаллизовывают, фильтруют и сушат. Остаток очищают на стеклянном фильтре на силикагеле (элюент CH2Cl2/CH3OH, 99/1). Чистые фракции собирают и упаривают. Остаток перекристаллизовывают из диоксана/2,2'-оксибиспропана и получают 1,7 г (72%) (±)
диметилформамиде (50 мл) перемешивают в атмосфере азота при 50oC в течение 4 часов, и затем смесь перемешивают в атмосфере азота при 60oC в течение 2 часов. Смесь охлаждают, и добавляют воду. Продукт выкристаллизовывают, фильтруют и сушат. Остаток очищают на стеклянном фильтре на силикагеле (элюент CH2Cl2/CH3OH, 99/1). Чистые фракции собирают и упаривают. Остаток перекристаллизовывают из диоксана/2,2'-оксибиспропана и получают 1,7 г (72%) (±)  4-[4-[4-[4-[[5-(2,4-дифторфенил)тетрагидро-5-(
4-[4-[4-[4-[[5-(2,4-дифторфенил)тетрагидро-5-( 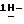 1,2,4-триазол-1-илметил)-3-фуранил] метокси] фенил] -1- пиперазинил]фенил]-2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил)
1,2,4-триазол-1-илметил)-3-фуранил] метокси] фенил] -1- пиперазинил]фенил]-2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил) 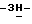 1,2,4-триазол-3-она; т.пл. 210,8oC (промежуточное соединение 25).
1,2,4-триазол-3-она; т.пл. 210,8oC (промежуточное соединение 25).
Пример 10
К смеси промежуточного соединения (15) (0,0082 моль) с дихлорметаном (100 мл) при перемешивании добавляют хлорацетилхлорид (0,02 моль). Добавляют по каплям пиридин (0,037 моль), и смесь перемешивают в течение 2 часов. Добавляют 1N соляную кислоту (50 мл), смесь перемешивают в течение 2 часов и разделяют. Органический слой промывают раствором гидрокарбоната натрия, сушат, фильтруют и упаривают. Остаток кристаллизуют и обрабатывают 4-метил-2-пентаноном/2,2'-оксибиспропаном, получают 6,3 г (96,8%) продукта. Образец (1 г) перекристаллизовывают из 4-метил-2-пентанона, и получают 0,6 г (±) 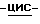 1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 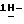 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-4,5-дигидро-5-оксо 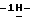 1,2,4-триазол-1-ил] метил] - 2,2-диметилпропилхлорацетата (промежуточное соединение 26).
1,2,4-триазол-1-ил] метил] - 2,2-диметилпропилхлорацетата (промежуточное соединение 26).
Пример 11
Смесь (±)-2,4-дигидро-4-(2-гидрокси-3,3-диметилбутил)- 2-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] -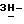 -1,2,4-триазол-3-она (0,0068 моль), метансульфоната (±)
-1,2,4-триазол-3-она (0,0068 моль), метансульфоната (±)  (2-(2,4-дифторфенил)-2-(
(2-(2,4-дифторфенил)-2-( -1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (сложный эфир) (0,0082 моль) и гидроксида натрия (0,025 моль) в
-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (сложный эфир) (0,0082 моль) и гидроксида натрия (0,025 моль) в 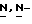 диметилформамиде (100 мл) перемешивают в атмосфере азота при 60oC в течение 4 часов. Смесь охлаждают, добавляют воду, и смесь перемешивают. Осадок отфильтровывают и сушат. Остаток очищают на стеклянном фильтре на силикагеле (элюент CH2Cl2/CH3OH/этилацетат/н-гексан, 48/2/30/20). Чистые фракции собирают и упаривают. Остаток перекристаллизовывают из 4-метил-2-пентанона, получают 1,1 г (22%) (±)
диметилформамиде (100 мл) перемешивают в атмосфере азота при 60oC в течение 4 часов. Смесь охлаждают, добавляют воду, и смесь перемешивают. Осадок отфильтровывают и сушат. Остаток очищают на стеклянном фильтре на силикагеле (элюент CH2Cl2/CH3OH/этилацетат/н-гексан, 48/2/30/20). Чистые фракции собирают и упаривают. Остаток перекристаллизовывают из 4-метил-2-пентанона, получают 1,1 г (22%) (±)  2-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
2-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 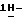 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил] фенил]-2,4-дигидро-4-(2-гидрокси-3,3-диметилбутил)
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил] фенил]-2,4-дигидро-4-(2-гидрокси-3,3-диметилбутил) 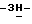 1,2,4-триазол-3-она; т.пл. 201,2oC (промежуточное соединение 27).
1,2,4-триазол-3-она; т.пл. 201,2oC (промежуточное соединение 27).
Пример 12
Перемешивают смесь (±) 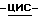 1-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 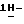 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -3-(3,3-диметил-2-оксобутил)-1,3-дигидро
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -3-(3,3-диметил-2-оксобутил)-1,3-дигидро 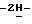 имидазол-2-она (0,0036 моль) с метанолом (50 мл) и дихлорметаном (50 мл). Добавляют боргидрид натрия (0,01 моль), и смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют воду (100 мл), и смесь перемешивают в течение 1 часа. Смесь разделяют, и водный слой экстрагируют дихлорметаном. Органический слой сушат, фильтруют и упаривают. Остаток очищают на стеклянном фильтре с силикагелем (элюент CH2Cl2/CH3OH, 99/1). Чистые фракции собирают и упаривают. Остаток перекристаллизовывают из 4-метил-2-пентанона, получают 1,9 г (73%) (±)
имидазол-2-она (0,0036 моль) с метанолом (50 мл) и дихлорметаном (50 мл). Добавляют боргидрид натрия (0,01 моль), и смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют воду (100 мл), и смесь перемешивают в течение 1 часа. Смесь разделяют, и водный слой экстрагируют дихлорметаном. Органический слой сушат, фильтруют и упаривают. Остаток очищают на стеклянном фильтре с силикагелем (элюент CH2Cl2/CH3OH, 99/1). Чистые фракции собирают и упаривают. Остаток перекристаллизовывают из 4-метил-2-пентанона, получают 1,9 г (73%) (±) 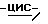 1-[4-[4-[4-[[2-(2,4-дифторфенил)- 2-(
1-[4-[4-[4-[[2-(2,4-дифторфенил)- 2-( 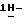 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-3-(2-гидрокси-3,3-диметилбутил)-2-имидазолидинона; т. пл. 196,8oC (промежуточное соединение 29).
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-3-(2-гидрокси-3,3-диметилбутил)-2-имидазолидинона; т. пл. 196,8oC (промежуточное соединение 29).
B. Получение конечных соединений
Пример 13
Смесь промежуточного соединения (15) (0,0014 моль), дифенилхлорфосфата (0,003 моль) и 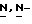 диметил-4-пиридинамина (1 г) в дихлорметане (30 мл) перемешивают при комнатной температуре в течение 2 часов. Смесь очищают на стеклянном фильтре с силикагелем (элюент CH2Cl2/CH3OH, 98/2). Чистые фракции собирают и упаривают.
диметил-4-пиридинамина (1 г) в дихлорметане (30 мл) перемешивают при комнатной температуре в течение 2 часов. Смесь очищают на стеклянном фильтре с силикагелем (элюент CH2Cl2/CH3OH, 98/2). Чистые фракции собирают и упаривают.
Остаток перекристаллизовывают из 4-метил-2-пентанона/2,2'-оксибиспропана и получают 1,1 г (83%) (±) 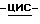 1-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 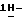 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо 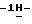 1,2,4-триазол-1-ил]метил]- 2,2-диметилпропилдифенилфосфата; т.пл. 170,8oC (соединение 1).
1,2,4-триазол-1-ил]метил]- 2,2-диметилпропилдифенилфосфата; т.пл. 170,8oC (соединение 1).
Пример 14
Смесь соединения (1) (0,0029 моль) и дисперсии 50% раствора гидроксида натрия (5 г) в 1,4-диоксане (50 мл) перемешивают при комнатной температуре в течение 6 часов. Добавляют воду (200 мл), смесь фильтруют через дикалит и фильтрат подкисляют соляной кислотой до pH 2-3. Смесь экстрагируют три раза дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Остаток растворяют в насыщенном растворе гидрокарбоната натрия (100 мл), раствор промывают 2,2'-оксибиспропаном и экстрагируют дважды дихлорметаном (500 мл) и метанолом (100 мл). Объединенные органические слои сушат, фильтруют и упаривают. Остаток кристаллизуют из 2-пропанола и небольшого количества воды, получают 1,2 г (46%) (±) 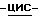 1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 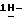 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо 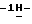 1,2,4-триазол-1-ил]метил]-2,2-диметилпропилфенилфосфата натрия; т. пл. 167,0oC (соединение 2).
1,2,4-триазол-1-ил]метил]-2,2-диметилпропилфенилфосфата натрия; т. пл. 167,0oC (соединение 2).
Пример 15
Смесь соединения (1) (0,0038 моль) и 50% раствора гидроксида натрия (5 г) в 1,4-диоксане (100 мл) перемешивают при комнатной температуре в течение ночи. Добавляют воду (600 мл), смесь фильтруют через дикалит и фильтрат подкисляют соляной кислотой. Осадок отфильтровывают (*) и фильтрат экстрагируют дихлорметаном. Смесь упаривают. Остаток, осадок (*) и 50% гидроксид натрия (5 г) перемешивают при 60oC в течение 24 часов. Снова добавляют 50% гидроксид натрия (3 г) и смесь перемешивают при 60oC в течение 48 часов. Смесь охлаждают, подкисляют 1N соляной кислотой до pH 4 и экстрагируют дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Остаток доводят до кипения в метаноле (70 мл), фильтруют и к фильтрату добавляют метанол с аммиаком (20 мл). Смесь доводят до кипения в 2-пропаноле (20 мл) и охлаждают. Смесь фильтруют, осадок растворяют в воде (200 мл) и раствор дважды промывают этилацетатом. Водный слой подкисляют 1N соляной кислотой и экстрагируют дихлорметаном три раза. Объединенные органические слои сушат, фильтруют и упаривают. Остаток растворяют в метаноле (70 мл) и добавляют метанол с аммиаком (10 мл). Осадок отфильтровывают и сушат, получают 1,6 г (51%) (±)  1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 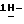 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил]фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил]фенил] -4,5-дигидро-5-оксо 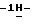 1,2,4-триазол-1-ил]метил]-2,2- диметилпропилфосфата аммония; т. пл. 189,6oC (соединение 3).
1,2,4-триазол-1-ил]метил]-2,2- диметилпропилфосфата аммония; т. пл. 189,6oC (соединение 3).
Пример 16
К суспензии (±) 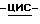 1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 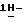 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил] -4,5-дигидро-5-оксо 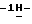 1,2,4-триазол-1-ил]метил]-2,2- диметилпропилдифенилфосфата (0,07 моль) в 1,4-диоксане (1400 мл) в потоке N2 добавляют 50% гидроксид натрия (1,4 моль). Получающуюся в результате суспензию нагревают до 60oC. Реакционный раствор перемешивают при 60oC в течение 92 часов. Смеси дают возможность охладиться до 25oC. Смесь выливают в дистиллированную воду (5,25 л), и эту смесь энергично перемешивают в течение 1 часа. Смесь фильтруют. Фильтрат подкисляют (pH 2,7) соляной кислотой, и в результате выпадает осадок. Водный слой экстрагируют CH2Cl2 (1х2 л; 1х1,5 л). Объединенные экстракты сушат (Na2SO4), фильтруют, и испаряют растворитель. Остаток (71,02 г; выход 124,7%) перемешивают с 2-пропанолом (1050 мл), нагревают до кипения, перемешивают и кипятят с обратным холодильником в течение 5 минут, охлаждают на ледяной бане при энергичном перемешивании, охлаждают до 20oC, и перемешивание продолжают в течение ночи. Осадок отфильтровывают, промывают 2-пропанолом (1х35 мл), диизопропиловым эфиром (2х35 мл) и сушат (вакуум; 50oC), получают 48,10 г полугидрата (±)
1,2,4-триазол-1-ил]метил]-2,2- диметилпропилдифенилфосфата (0,07 моль) в 1,4-диоксане (1400 мл) в потоке N2 добавляют 50% гидроксид натрия (1,4 моль). Получающуюся в результате суспензию нагревают до 60oC. Реакционный раствор перемешивают при 60oC в течение 92 часов. Смеси дают возможность охладиться до 25oC. Смесь выливают в дистиллированную воду (5,25 л), и эту смесь энергично перемешивают в течение 1 часа. Смесь фильтруют. Фильтрат подкисляют (pH 2,7) соляной кислотой, и в результате выпадает осадок. Водный слой экстрагируют CH2Cl2 (1х2 л; 1х1,5 л). Объединенные экстракты сушат (Na2SO4), фильтруют, и испаряют растворитель. Остаток (71,02 г; выход 124,7%) перемешивают с 2-пропанолом (1050 мл), нагревают до кипения, перемешивают и кипятят с обратным холодильником в течение 5 минут, охлаждают на ледяной бане при энергичном перемешивании, охлаждают до 20oC, и перемешивание продолжают в течение ночи. Осадок отфильтровывают, промывают 2-пропанолом (1х35 мл), диизопропиловым эфиром (2х35 мл) и сушат (вакуум; 50oC), получают 48,10 г полугидрата (±) 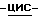 4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 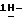 1,2,4-триазол-1-илметил)-1,3- диоксолан-4-ил]метокси] фенил] -1-пиперазинил] фенил]-2-[3,3-диметил- 2-(фосфоноокси)бутил]-2,4-дигидро-3H-1,2,4-триазол-3-она; т.пл. 156,2oC (соединение 17).
1,2,4-триазол-1-илметил)-1,3- диоксолан-4-ил]метокси] фенил] -1-пиперазинил] фенил]-2-[3,3-диметил- 2-(фосфоноокси)бутил]-2,4-дигидро-3H-1,2,4-триазол-3-она; т.пл. 156,2oC (соединение 17).
Пример 17
К смеси соединения (17) (0,005 моль) с водой (70 мл) добавляют 1-деокси-1-(метиламино)-D-глюцит (0,02 моль), и смесь перемешивают до полного растворения (30 минут). Испаряют растворитель. Добавляют толуол, и смесь азеотропируют на роторном испарителе. Добавляют этанол (250 мл), и смесь энергично перемешивают. Охлаждают смесь на ледяной бане и перемешивают в течение 1 часа, в результате выпадает осадок. Смеси дают возможность нагреться до комнатной температуры (20oC). Смесь перемешивают в течение 18 часов при комнатной температуре, и получающийся в результате осадок отфильтровывают, промывают этанолом, диизопропиловым эфиром (2х10 мл) и сушат (вакуум; 50oC). Получают 6,14 г (1). Фильтрат упаривают. Остаток сушат (вакуум; 50oC), получают 1,98 г (2). Фракцию (1) измельчают, энергично перемешивают в течение 5 часов в этаноле (200 мл), и получающийся в результате осадок отфильтровывают, промывают этанолом (4х5 мл) и сушат (вакуум; 45-50oC; 64 часа). Получают 4,98 г моногидрата (±) 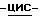 4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(  1,2,4-триазол-1-илметил)-1,3- диоксолан-4-ил]метокси]фенил]-1-пиперазинил] фенил]-2-[3,3-диметил- 2-(фосфоноокси)бутил]-2,4-дигидро-3H-1,2,4-триазол-3-она-1-деокси- 1-(метиламин)-D-глюцита (1:2) (соединение 18).
1,2,4-триазол-1-илметил)-1,3- диоксолан-4-ил]метокси]фенил]-1-пиперазинил] фенил]-2-[3,3-диметил- 2-(фосфоноокси)бутил]-2,4-дигидро-3H-1,2,4-триазол-3-она-1-деокси- 1-(метиламин)-D-глюцита (1:2) (соединение 18).
Пример 18
Смесь промежуточного соединения (26) (0,0025 моль) и пирролидина (0,014 моль) в  диметилформамиде (50 мл) перемешивают при комнатной температуре в течение 4 часов. Добавляют воду, и смесь перемешивают. Отфильтровывают осадок, промывают его водой и очищают на стеклянном фильтре с силикагелем (элюент 1 - CH2Cl2/CH3OH, 98/2, и элюент 2 - CH2Cl2/CH3OH, 95/5). Собирают подходящие фракции и упаривают. Остаток растворяют в дихлорметане (100 мл), и перемешивают раствор с 0,4N соляной кислотой (50 мл). Смесь разделяют, и водный слой экстрагируют четыре раза дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Маслянистый остаток растворяют в дихлорметане, промывают раствором гидрокарбоната натрия, сушат, фильтруют и упаривают. Остаток кристаллизуют из 4-метил-2-пентанона, и получают 1,1 г (53%) (±)
диметилформамиде (50 мл) перемешивают при комнатной температуре в течение 4 часов. Добавляют воду, и смесь перемешивают. Отфильтровывают осадок, промывают его водой и очищают на стеклянном фильтре с силикагелем (элюент 1 - CH2Cl2/CH3OH, 98/2, и элюент 2 - CH2Cl2/CH3OH, 95/5). Собирают подходящие фракции и упаривают. Остаток растворяют в дихлорметане (100 мл), и перемешивают раствор с 0,4N соляной кислотой (50 мл). Смесь разделяют, и водный слой экстрагируют четыре раза дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Маслянистый остаток растворяют в дихлорметане, промывают раствором гидрокарбоната натрия, сушат, фильтруют и упаривают. Остаток кристаллизуют из 4-метил-2-пентанона, и получают 1,1 г (53%) (±)  1-[[4-[4-[4-[4-[[2-(2,4- дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4- дифторфенил)-2-(  1,2,4-триазол-1-илметил)-1,3-диоксолан-4- ил] метокси] фенил] -1-пиперазинил]фенил]-4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4- ил] метокси] фенил] -1-пиперазинил]фенил]-4,5-дигидро-5-оксо 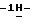 1,2,4-триазол-1-ил]метил]-2,2-диметилпропил-1-пирролидинацетата; т. пл. 156,7oC (соединение 28).
1,2,4-триазол-1-ил]метил]-2,2-диметилпропил-1-пирролидинацетата; т. пл. 156,7oC (соединение 28).
Пример 19
Смесь промежуточного соединения (16) (0,0066 моль), дигидрохлорида 4-метил-1-пиперазинуксусной кислоты (0,013 моль), 1,3-дициклогексилкарбодиимида (0,026 моль) и 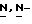 диметил-4-пиридинамина (0,026 моль) в дихлорметане (100 мл) перемешивают при комнатной температуре в течение 4 часов. Добавляют 1N соляную кислоту (200 мл), и смесь перемешивают в течение 1 часа. Отфильтровывают осадок, добавляют воду (600 мл) и смесь разделяют (*). Водный слой промывают дихлорметаном (100 мл) и отделяют. Водный слой нейтрализуют пиридином и экстрагируют пять раз дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают, получают 4,5 г фракции 1. Органический слой (*) промывают 1N соляной кислотой (100 мл) и отделяют. Водный слой нейтрализуют пиридином и экстрагируют дважды дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают, получают 2 г фракции 2. Фракции 1 и 2 соединяют и перекристаллизовывают из смеси 2% ацетонитрила, вода/2,2'-оксибиспропан, и получают 3,8 г (61%) полугидрата моногидрохлорида (±)
диметил-4-пиридинамина (0,026 моль) в дихлорметане (100 мл) перемешивают при комнатной температуре в течение 4 часов. Добавляют 1N соляную кислоту (200 мл), и смесь перемешивают в течение 1 часа. Отфильтровывают осадок, добавляют воду (600 мл) и смесь разделяют (*). Водный слой промывают дихлорметаном (100 мл) и отделяют. Водный слой нейтрализуют пиридином и экстрагируют пять раз дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают, получают 4,5 г фракции 1. Органический слой (*) промывают 1N соляной кислотой (100 мл) и отделяют. Водный слой нейтрализуют пиридином и экстрагируют дважды дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают, получают 2 г фракции 2. Фракции 1 и 2 соединяют и перекристаллизовывают из смеси 2% ацетонитрила, вода/2,2'-оксибиспропан, и получают 3,8 г (61%) полугидрата моногидрохлорида (±) 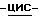 1-[[4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-( 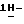 1,2,4-триазол-1-илметил)- 1,3-диоксолан-4-ил] метокси] фенил]-1-пиперазинил]фенил]-4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)- 1,3-диоксолан-4-ил] метокси] фенил]-1-пиперазинил]фенил]-4,5-дигидро-5-оксо  1,2,4-триазол-1-ил]метил]-2,2-диметилпропил-4-метил-1-пиперазинацетата; т. пл. 156,0oC (соединение 32).
1,2,4-триазол-1-ил]метил]-2,2-диметилпропил-4-метил-1-пиперазинацетата; т. пл. 156,0oC (соединение 32).
Пример 20
Смесь соединения (17) (0,0034 моль) с раствором хлористоводородной кислоты в 2-пропаноле (10 мл) и дихлорметаном (60 мл) перемешивают и кипятят с обратным холодильником в течение 30 минут. Смесь упаривают. Остаток растворяют в воде и фильтруют. Фильтрат нейтрализуют раствором гидрокарбоната натрия и экстрагируют дихлорметаном. Органический слой сушат, фильтруют и упаривают. Остаток растворяют в 0,5N соляной кислоте (50 мл), и раствор три раза промывают этилацетатом (100 мл). Водный слой нейтрализуют пиридином и экстрагируют три раза дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Остаток кристаллизуют из ацетонитрила и небольшого количества  диметилформамида, и получают 1,3 г (46%) моногидрохлорида (±)
диметилформамида, и получают 1,3 г (46%) моногидрохлорида (±)  1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(  1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1- пиперазинил]фенил]-4,5-дигидро-5-оксо 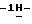 1,2,4-триазол-1-ил]метил]-2,2-диметилпропил -β- аланина; т. пл. 217,9oC (соединение 36).
1,2,4-триазол-1-ил]метил]-2,2-диметилпропил -β- аланина; т. пл. 217,9oC (соединение 36).
Пример 21
Смесь моногидрата (±) 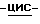 N-[1-[3-[[4-[4-[4-[4-[[2-(2,4- дифторфенил)-2-(
N-[1-[3-[[4-[4-[4-[4-[[2-(2,4- дифторфенил)-2-( 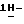 1,2,4-триазол-1-илметил)-1,3-диоксолан-4- ил]метокси]фенил]-1-пиперазинил] фенил] -4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4- ил]метокси]фенил]-1-пиперазинил] фенил] -4,5-дигидро-5-оксо 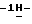 1,2,4-триазол-1-ил]метил]-2,2-диметилпропокси]-3-оксопропил]-1,4- дигидро-4-пиридинилиден]
1,2,4-триазол-1-ил]метил]-2,2-диметилпропокси]-3-оксопропил]-1,4- дигидро-4-пиридинилиден] 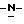 метилметанаминохлорида (0,0024 моль) и пирролидина (0,01 моль) в
метилметанаминохлорида (0,0024 моль) и пирролидина (0,01 моль) в 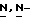 диметилформамиде (50 мл) перемешивают при комнатной температуре в течение 1 часа. Смесь выливают в воду и экстрагируют три раза дихлорметаном. Объединенные органические слои промывают водой, сушат, фильтруют и упаривают. Остаток растворяют в 0,5N соляной кислоте (500 мл), и раствор три раза промывают этилацетатом (100 мл). Водный слой нейтрализуют пиридином и экстрагируют три раза дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Остаток кристаллизуют из 4-метил-2-пентанона и воды (0,5 мл). Остаток очищают на стеклянном фильтре с силикагелем (элюент CH2Cl2/CH3OH, 96/4). Подходящие фракции собирают и упаривают. Остаток растворяют в 1N соляной кислоте (50 мл), нейтрализуют пиридином и экстрагируют пять раз дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Остаток кристаллизуют из 4-метил-2-пентанона и воды (5 капель), получают 1,1 г (51%) моногидрата моногидрохлорида (±)
диметилформамиде (50 мл) перемешивают при комнатной температуре в течение 1 часа. Смесь выливают в воду и экстрагируют три раза дихлорметаном. Объединенные органические слои промывают водой, сушат, фильтруют и упаривают. Остаток растворяют в 0,5N соляной кислоте (500 мл), и раствор три раза промывают этилацетатом (100 мл). Водный слой нейтрализуют пиридином и экстрагируют три раза дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Остаток кристаллизуют из 4-метил-2-пентанона и воды (0,5 мл). Остаток очищают на стеклянном фильтре с силикагелем (элюент CH2Cl2/CH3OH, 96/4). Подходящие фракции собирают и упаривают. Остаток растворяют в 1N соляной кислоте (50 мл), нейтрализуют пиридином и экстрагируют пять раз дихлорметаном. Объединенные органические слои сушат, фильтруют и упаривают. Остаток кристаллизуют из 4-метил-2-пентанона и воды (5 капель), получают 1,1 г (51%) моногидрата моногидрохлорида (±) 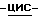 1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
1-[[4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 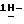 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил] фенил]-4,5-дигидро-5-оксо
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил] фенил]-4,5-дигидро-5-оксо 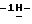 1,2,4-триазол-1-ил]метил]- 2,2-диметилпропил-1-пирролидинпропаноата; т. пл. 154,4oC (соединение 37).
1,2,4-триазол-1-ил]метил]- 2,2-диметилпропил-1-пирролидинпропаноата; т. пл. 154,4oC (соединение 37).
C. Пример определения физико-химических свойств
Пример 22
Растворимость
Избыточное количество соединения добавляют к 5 мл растворителя (тип растворителя указывается в таблице). Смесь встряхивают в течение 1 часа при комнатной температуре. Отфильтровывают осадок. Измеряют pH оставшегося растворителя и указывают в таблице 5. Концентрацию соединения измеряют УФ-спектроскопией и указывают в колонке "растворимость".
D. Фармакологические примеры
Пример 23
Мышиная модель тройного микоза
Испытуемые соединения оценивают на их активность в мышиной модели микоза, в которой одновременно устраивают три микоза - вагинальный кандидоз, кожный трихофитоз и диссеминированный аспергиллез. Мышей, в группах по 10 особей, которым предварительно дают подкожные инъекции эстрадиолвалерата (500 мкг), затем инокулируют, в 0 сутки, следующим образом: 100000 колониеобразующих единиц, (КОЕ)/г Aspergillus fumigatus B19119, внутривенно; суспензия, содержащая 108 клеток Candida albicans, интравагинально; и водная суспензия Trichophyton quinckeanum, на слегка надсеченную кожу спины. Обработку испытуемыми соединениями (перорально или внутривенно) начинают в день заражения и продолжают в течение 5 суток. У всех животных, которые погибают сами, и которые выживают, и их умерщвляют на 6 сутки, отбирают образцы на число КОЕ/г Aspergillus fumigatus в почках и селезенке, для оценки поражений кожи (0 = отсутствуют видимые поражения; 1 = несколько точечных поражений; 2 = умеренные поражения; 3 = тяжелые поражения) и для оценки КОЕ Candida albicans из влагалищного мазка.
Мышиная модель диссеминированного Candida
Мышей, в группах по 10 особей, инфицируют внутривенно 8•105 КОЕ Candida albicans. Лечение начинают в день заражения, и повторяют в течение 9 суток ежесуточно. КОЕ/г Candida albicans в почках определяют для всех мышей, которые погибли сами или были умерщвлены на 10 сутки.
В таблице 6 приводятся наименьшие концентрации указанных соединений, которые дают уменьшение среднего числа Candida в 1 log (т.е., 10-кратное снижение) или больше, либо на модели тройного микоза, либо на модели заражения диссеминированным Candida; а также наименьшие концентрации, которые снижают средний показатель поражений кожи от кожного дерматомикозного компонента ниже 1,0 на этой модели (NT = не испытывались, IV = внутривенный).
Пример 24
Определение чувствительности грибов
Для оценки активности проверяемых соединений in vitro используют набор культур Candida и отделение культуры дерматофитов Microsporum canis, Trichophyton rubrum и T. Mentagrophytes; Aspergillus fumigates и Cryptococcus neoformans. Инокулянты получают в виде бульонных культур (дрожжи) или в виде суспензий грибного вещества, приготовленного из культур скошенного агара (плесени). Проверяемые соединения пипетируют из основного ДМСО-раствора в воду, чтобы получить серии 10-кратных разведений. Грибной инокулят суспендируют в питательной среде CYG (F.C. Odds, Journal of Clinical Microbiology, 29, (2735-S740, 1991) приблизительно при 50000 колониеобразующих единиц (КОЕ) на 1 мл и добавляют к водным проверяемым лекарственным средствам.
Культуры высевают в 96-луночные планшеты, и инкубируют их в течение 2 суток при 37oC (виды Candida) или в течение 5 суток при 30oC (другие грибы). Развитие микрокультур измеряют по их оптической плотности (OD), измеряемой при длине волны 405 нм. OD для культур с проверяемыми соединениями вычисляют как процент от OD контрольных культур без лекарственного средства. Ингибирование роста до 35% или меньше от контрольных образцов регистрируют как существенное ингибирование.
Минимальная ингибирующая концентрация (MIC) промежуточных соединений 15, 16, 17,18,24 находится в интервале от ≤0,01 до 10 мкМ для Candida glabrata, Candida krusei, Candida parapsilosis, азолонеризистентного Candida albicans, Candida kefyr, Microsporum canis, Trichophton rubrum, Trichophton mentagrophytes, Cryptococcus neoformans, Aspergillus fumigatus.
E. Примеры композиций
Используемый в настоящих примерах термин "активный ингредиент" (A.I.) относится к соединению формулы I, его фармацевтически приемлемой соли присоединения кислоты или его стереохимически изомерной форме.
Пример 25. Капли для перорального введения
Растворяют 500 г A.I. в 0,5 л раствора гидроксида натрия и 1,5 л полиэтиленгликоля при 60-80oC. После охлаждения до 30-40oC добавляют 35 л полиэтиленгликоля, и смесь тщательно перемешивают. Затем добавляют раствор 1750 г натрийсахарина в 2,5 л очищенной воды, и при перемешивании добавляют 2,5 л какао-одушки и полиэтиленгликоль q.s. до объема 50 л, и получают раствор капель для перорального введения, содержащий 10 мг/мл A.I. Получающийся в результате раствор разливают в подходящие емкости.
Пример 26. Капсулы
При энергичном перемешивании смешивают 20 г A.I., 6 г лаурилсульфата натрия, 56 г крахмала, 56 г лактозы, 0,8 г коллоидального диоксида кремния и 1,2 г стеарата магния. Получающейся в результате смесью затем наполняют 1000 подходящих твердых желатиновых капсул, причем каждая капсула содержит 20 мг активного ингредиента.
Пример 27. Таблетки с пленочным покрытием
Получение сердцевинной составляющей
Смесь 100 г A.I., 570 г лактозы и 200 г крахмала тщательно перемешивают, и затем увлажняют раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона в 200 мл воды. Смесь в виде влажного порошка просеивают, сушат и снова просеивают. Затем к ней добавляют 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Все тщательно перемешивают и прессуют в таблетки, получая 10000 таблеток, каждая из которых содержит 10 мг активного ингредиента.
Покрытие
К раствору 10 г метилцеллюлозы в 75 мл денатурированного этанола добавляют раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола (глицерин). Расплавляют 10 г полиэтиленгликоля и растворяют в 75 мл дихлорметана. Последний раствор добавляют к первому раствору, и затем добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной красящей суспензии, и все гомогенизируют. На сердцевины таблеток наносят покрытие из полученной таким образом смеси в аппарате для нанесения покрытий.
Пример 28. Раствор для инъекций
Растворяют 1,8 г метил-4-гидроксибензоата и 0,2 г гидроксида натрия в 0,5 л кипящей воды для инъекций. После охлаждения до 50oC добавляют при перемешивании 0,05 г пропиленгликоля и 4 г A.I. Раствор охлаждают до комнатной температуры и добавляют воду для инъекций q.s. до 1 л, получая раствор, содержащий 4 мг/мл A.I. Раствор стерилизуют посредством фильтрации и разливают в стерильные емкости.
Пример 29. Суппозитории
Растворяют 3 г A.I. в растворе 3 г 2,3-диоксибутандиовой кислоты в 25 мл полиэтиленгликоля 400. Сплавляют вместе 12 г поверхностно-активного вещества (SPAN®) и триглицериды (Witepsol 555® ) q.s. до 300 г. Последнюю смесь смешивают с первым раствором. Полученную таким образом смесь выливают в формы при температуре 37-38oC, и формуют 100 суппозиториев, каждый из которых содержит 30 мг/мл A.I.
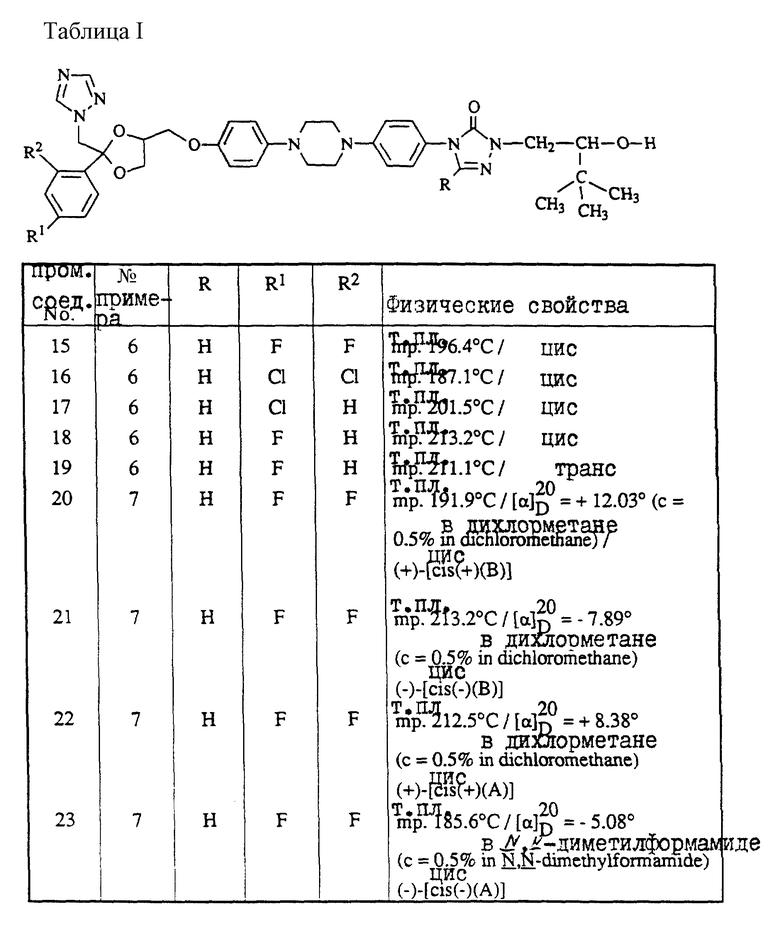

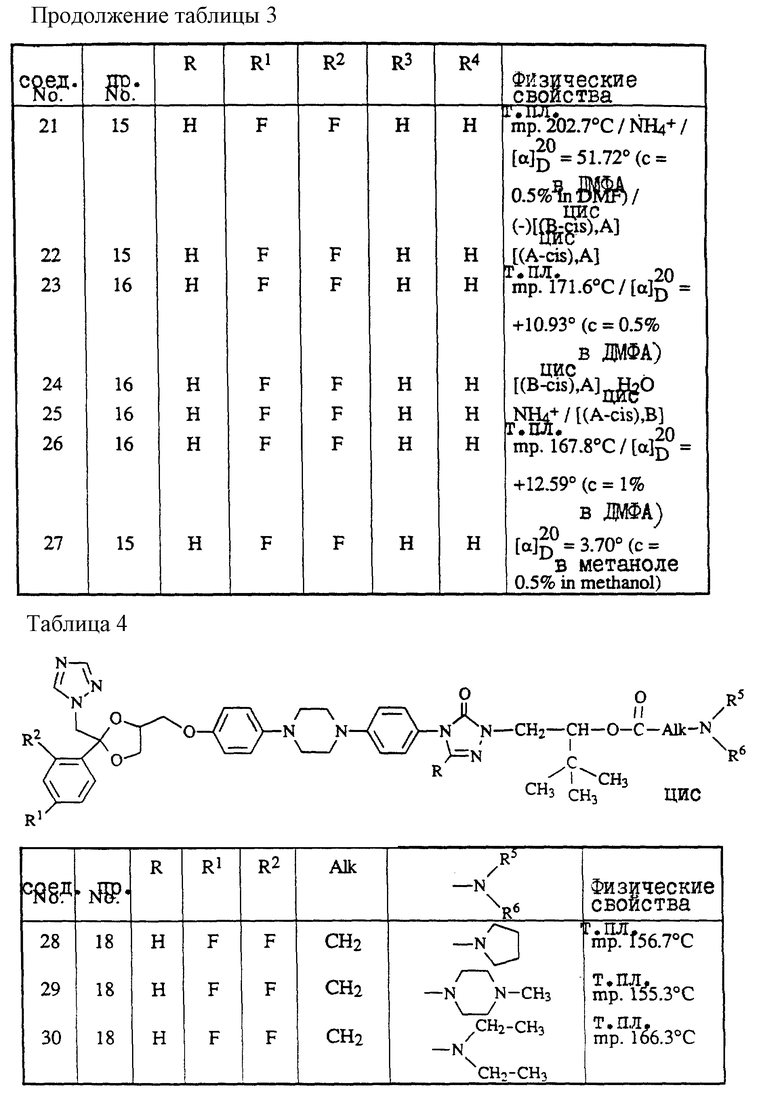
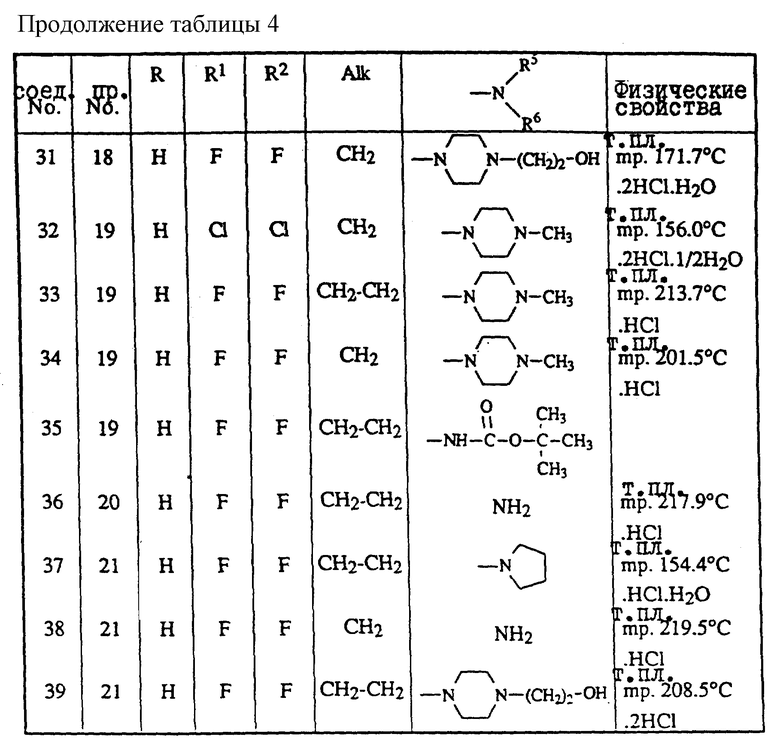
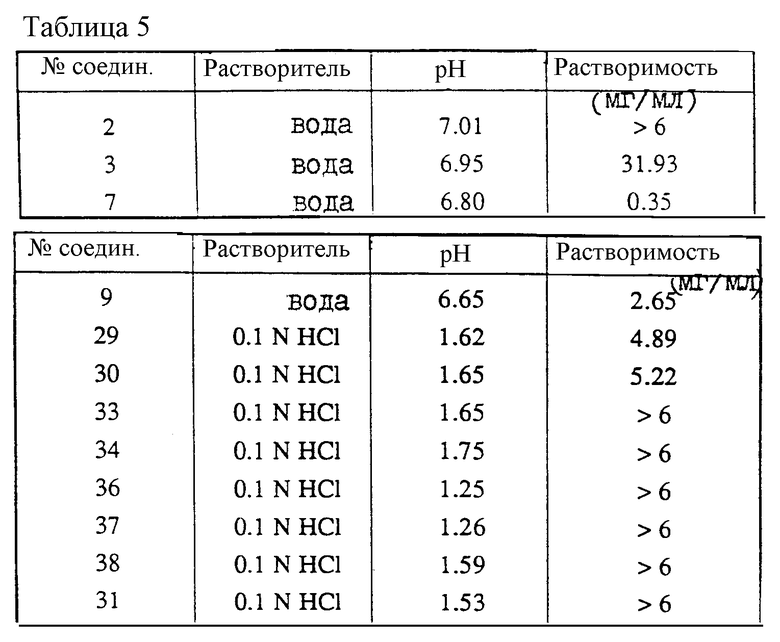
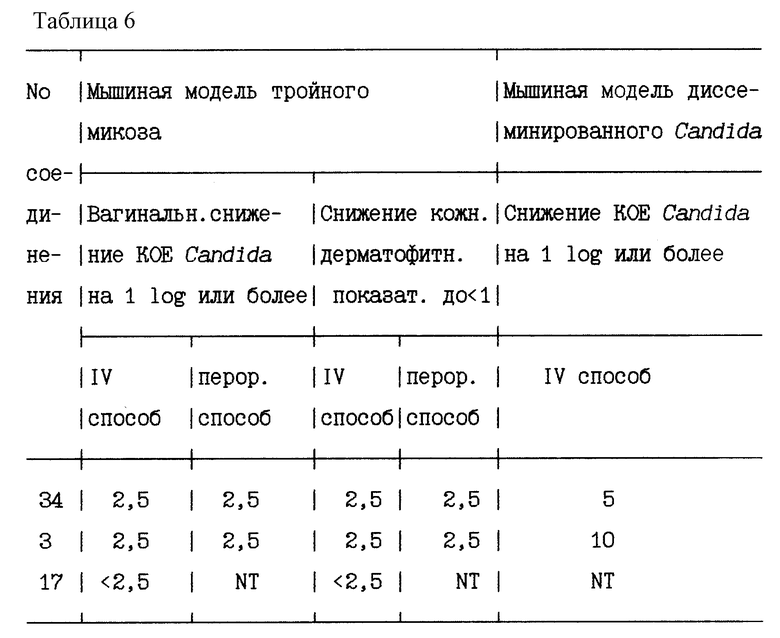
Описываются новые азольные соединения общей формулы (I), его фармацевтически приемлемая соль или основание, или стереохимически изомерная форма, где А и В вместе образуют двухвалентный радикал формулы: - СН=В, (в), D - радикал (D1) или (D3), L - радикал (L1) или (L2), Alk является С1-4алкандиил, R1 является галогеном; R2 - водородом или галогеном, R3 - водородом или фенилом, R4 - водородом или фенилом, R5 - водородом или С1-6алкилом, R6 - водородом или С1-6алкилом, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое или замещенное пиперазиновое кольцо, причем замещенное пиперазиновое кольцо замещено в 4-положении пиперазинового кольца С1-6алкилом, гидроксиС1-6алкилом. Соединение формулы (I) является новым водорастворимым азольным противогрибковым средством широкого спектра действия. Описываются также способ их получения, противогрибковая композиция, способ получения композиции, способ ингибирования развития и/или разрастания грибов у теплокровных животных, промежуточные соединения. 7 с. и 4 з.п. ф-лы, 6 табл.
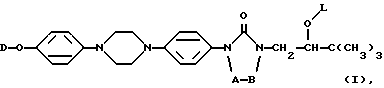
D является радикалом формулы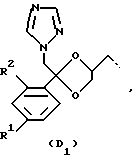
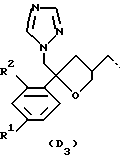
L является радикалом формулы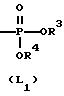
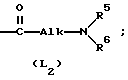
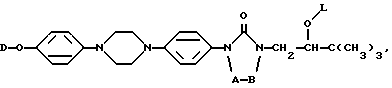
их фармацевтически приемлемые соли присоединения кислоты или основания, или стереохимически изомерные формы,
где А и В вместе образуют двухвалентный радикал формулы -CH=N-;
D - радикал формулы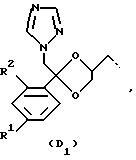
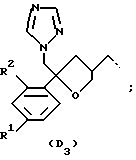
L - радикал формулы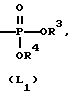
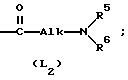
Alk - радикал C1-4алкандиил;
R1 - галоген;
R2 - водород или галоген;
R3 - водород или фенил;
R4 - водород или фенил;
R5 - водород или C1-6алкил;
R6 - водород или C1-6алкил, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют пирролидиновое или замещенное пиперазиновое кольцо, причем заместитель находится в положении 4 пиперазинового кольца и представляет собой C1-6алкил или гидроксиC1-6алкил.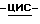 4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-( 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1-пиперазинил]фенил]-2-[3,3-диметил-2-(фосфоноокси)-бутил] -2,4-дигидро-3Н-1,2,4-триазол-3-он, его стереохимически изомерную форму или его соль присоединения основания.
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил]метокси]фенил]-1-пиперазинил]фенил]-2-[3,3-диметил-2-(фосфоноокси)-бутил] -2,4-дигидро-3Н-1,2,4-триазол-3-он, его стереохимически изомерную форму или его соль присоединения основания.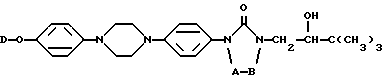
его фармацевтически приемлемая соль присоединения кислоты или стереохимически изомерная форма, где радикалы -А-В- и D имеют указанные в п.1 значения.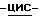 4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(
4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(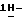 1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] -2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил]
1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил] -1-пиперазинил] фенил] -2,4-дигидро-2-(2-гидрокси-3,3-диметилбутил] 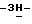 1,2,4-триазол-3-он, его фармацевтически приемлемую соль присоединения кислоты или его стереохимически изомерную форму.
1,2,4-триазол-3-он, его фармацевтически приемлемую соль присоединения кислоты или его стереохимически изомерную форму.
W1-L,
где W1 - реакционноспособная отщепляемая группа, такая, как гидроксильная или галоген, а L имеет значения, указанные в п.1, с последующим, в случае необходимости, превращением целевого продукта в форму соли посредством обработки фармацевтически приемлемыми кислотой или основанием и/или получают их стереохимически изомерные формы.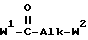
где W1 представляет собой реакционноспособную отщепляемую группу, такую, как гидроксильная группа или галоген, а W2 - реакционноспособная отщепляемая группа, такая, как галоген или сульфонилоксигруппа, и затем полученное соединение формулы VII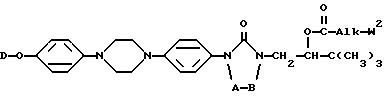
подвергают взаимодействию с амином формулы VIII
где R5 и R6 имеют указанные в п.1 значения, с последующим, в случае необходимости, превращением целевого продукта в форму соли посредством обработки фармацевтически приемлемыми кислотой или основанием и/или получают их стереохимически изомерные формы.
| EP, 0283992 A, 1988 | |||
| US, 5039676 A, 1991 | |||
| EP, 0539988 A, 1993 | |||
| SU, 1069625 A, 1984 | |||
| SU, 1635900 A3, 1991 | |||
| US, 4267179, 1980. |
