Объектом изобретения являются пептиды, способные ингибировать высвобождение пепсина, их замещенные производные и их соли, а также фармацевтические композиции, содержащие эти пептиды. Пептиды могут использоваться при лечении болезней, связанных с высвобождением пепсина, и, в частности, при лечении язв или эзофагита.
В последнее время был выделен новый пептид из кишечника свиньи: этот пептид, названный сорбином, насчитывает 153 природные аминокислоты (WO 89/06241). Сорбин, также как и пептидные фрагменты C-концевой части сорбина (содержащие, кроме того, 40 аминокислот), способны вызывать увеличение поглощения слизистой оболочкой. Однако модификация этих пептидных фрагментов путем включения по меньшей мере одного радикала аминокислоты D-конфигурации, оказалось, обладает новой биологической активностью, способной замедлять высвобождение пепсина, что не было присуще немодифицированным пептидам.
Эта активность представляет особый интерес. Общие механизмы желудочной секреции у млекопитающих в настоящее время хорошо известны. Желудочное пищеварение осуществляется под действием ферментов, соляной кислоты и пепсина. Пепсин - это белок, который наряду с гастрином, является одним из основных компонентов желудочного сока. Его основной физиологической функцией является инициация разложения белков. Однако многочисленные исследования показали, что он способствует образованию язв. Следовательно, в определенных ситуациях желательно замедлить, по меньшей мере частично, высвобождение пепсина.
Объектом изобретения является пептид общей формулы I
A1-A2-A3-X
в которой A1 означает радикал L-Thr или D-Thr; или одну из следующих последовательностей, в которой, по меньшей мере, один радикал аминокислоты может иметь D-конфигурацию, представленную после формулы изобретения;
A2 означает последовательность Lys-Pro-Gln-Ala, в которой по меньшей мере один радикал аминокислоты может иметь D-конфигурацию;
A3 означает ковалентную связь или пептидную последовательность - Gly-A4-A5, в которой A4 и A5 означают каждый, независимо друг от друга, радикал основной аминокислоты; и
X означает гидрокси, амино, алкиламино группу, причем названный пептид формулы I отличается тем, что он содержит по меньшей мере один радикал аминокислоты D-конфигурации.
Изобретение также касается замещенных производных пептидов общей формулы I, в которой один или несколько радикалов аминокислоты замещены одной или несколькими защитными группами пептидов, которые широко используются в химии и биологии, в случае замещения несколькими защитными группами их идентичность не обязательна. Предпочтительно, защитные группы выбирают из групп низшего алкила, таких как метил или третичный бутил; фенил; бензил или замещенный бензил, такой как триметоксибензил; 2-хлорбензилоксикарбонил, 9-фторенилметилоксикарбонил, трет-бутилоксикарбонил; ацетил; сульфонил и фосфорил.
Объектом изобретения также являются пептиды, содержащие последовательность аминокислот A1-A2-A3, в которой A1, A2 и A3 являются такими, как они определены выше.
Изобретение касается также фармацевтически приемлемых солей пептидов, определенных выше. Эти соли можно получить с органическими кислотами, такими как уксусная кислота, молочная, малеиновая, лимонная, яблочная, аскорбиновая, бензойная, салициловая, янтарная, метансульфокислота, толуолсульфокислота, или с минеральными кислотами, такими как соляная кислота, серная кислота или фосфорная, или же с полимерными кислотами, такими как танин или карбоксиметил целлюлоза.
A4 и A5 означают, независимо друг от друга, когда они присутствуют в пептидах по изобретению, радикалы основной аминокислоты конфигурации D или L. Преимущественно A4 и A5 представляют, независимо, радикал аминовой кислоты Lys, D-Lys, Arg или D-Arg.
Вышеназванные пептиды по изобретению содержат один или несколько радикалов аминокислоты D-конфигурации. Объектом изобретения, в частности, являются пептиды, содержащие радикал аминокислоты D-конфигурации, при этом названные пептиды могут замещаться защитными группами, указанными в настоящем изобретении. Можно привести следующий пример пептидов, который представлен в конце описания.
Радикал аминокислоты D-конфигурации располагается преимущественно на C-концевом или N-концевом участке. Предпочитаемые пептиды, которые имеют радикал аминокислоты D-конфигурации на C-конце, являются пептидами, в которых A2 означает Lys-Pro-Gln-D-Ala и A3 означает ковалентную связь. Предпочтительно, чтобы пептиды, которые имеют радикал аминокислоты D-конфигурации на N-конце, представляли собой пептиды, в которых A1 означает D-Thr или вышеуказанную последовательность, и радикал аминокислоты на N-конце этой последовательности имеет D-конфигурацию.
Объектом изобретения также, в частности, являются пептиды, которые имеют два радикала аминокислоты D-конфигурации, причем названные пептиды могут замещаться защитными группами, определенными настоящим изобретением. Можно привести следующий пример пептидов.
D-Pro-Val-Thr-Lys-Pro-Gln-D-Ala-NH2,
D-Pro-Val-Thr-Lys-Pro-Gln-Ala-Gly-D-Lys-NH2,
Pro-D-Val-Thr-Lys-Pro-Gln-D-Ala-NH2 и
D-Pro-Val-Thr-(ацетил)Lys-Pro-Gln-D-Ala-NH2.
Преимущественно, пептиды, содержащие два радикала аминокислоты D-конфигурации, имеют один радикал на C-конце, а второй может располагаться в любом месте на пептидной цепи, но преимущественно он находится на N-конце пептида. Предпочтительными пептидами являются те, в которых A1 представляет собой D-Thr или вышеназванную последовательность, в которой радикал аминокислоты на N-конце этой последовательности имеет D-конфигурацию, A2 означает Lys-Pro-Gln-D-Ala, и A3 означает ковалентную связь. Такими пептидами, преимущественно, являются пептиды, имеющие одну или несколько защитных групп, и, в частности, ацетиловую защитную группу на радикалe Lys.
Пептиды по изобретению могут быть получены в соответствии с одним из известных классических методов в области пептидных синтезов, например, с помощью твердофазного синтеза, который осуществляется по следующей схеме: образование пептидной цепи начинается фиксацией первой аминокислоты с C-конца цепи, на смоле, посредством ее карбоксильной группы; ее аминогруппу защищают присоединением трет-бутилоксикарбонила (Boc). После присоединения COOH-группы первой аминокислоты снимают защиту с NH2-группы путем промывания смолы кислотой. В случае защитной группы Boc, снятие защиты может производиться с помощью трифторуксусной кислоты. Вторая аминокислота с защищенной аминогруппой присоединяется посредством своей карбоксильной группы по освободившейся аминогруппе первой аминокислоты. Связывание осуществляется в основном в присутствии агента связывания, такого как дициклогексилкарбодиимид или диизопропилкарбодиимид. Пептидная цепь, образованная таким образом, включает две аминокислоты с защищенной аминогруппой. Как указано выше, снимают защиту с концевой аминогруппы и затем осуществляют присоединение третьей аминокислоты. Таким образом, происходит последовательное присоединение аминокислот одна за другой и построение желаемой пептидной цепи. После удаления всех защитных групп пептид отщепляют от смолы.
Синтез пептида по изобретению, а именно Pro-Val-Thr-Lys-Pro-Gln-D-Ala-NH2, кратко описан далее. Остальные пептиды по изобретению могут быть получены соответствующими модификациями описанного ниже пептидного синтеза, которые хорошо знакомы специалистам в данной области.
Синтез осуществляется в твердой фазе при комнатной температуре. Используемый рабочий режим включает следующие этапы: снятие защиты, нейтрализация и связывание. Используется смола типа полистирола, структурированного на 1% дивинилбензолом (смола Мерифилда). Фиксация Boc-D-Ala на смоле Мерифилда производится в присутствии карбоната цезия в толуоле и диметилформамиде (ДМФ). Концевая аминогруппа используемых аминокислот защищается группой Boc.
Эти группы Boc замещаются трифторуксусной кислотой, затем производят несколько промываний метиленхлоридом и изопропанолом. Аминогруппы нейтрализуются триэтиламином, затем следует несколько промываний. Треонин и валин перед связыванием превращаются в сложный эфир гидроксибензотриазол в присутствии диизопропилкарбодиимида (DIPCDI), для глутамина сложный эфир гидроксибензотриазола производится прямо в реакторе. Лизин и оба пролина трансформируются в симметричный ангидрид перед связыванием. Во всех случаях связывание осуществляется в присутствии диизоопропилэтиламина. Боковая цепь лизина защищается группой Fmoc, в то время как боковая цепь треонина не защищается. Когда заканчивается последнее связывание, группу Fmoc снимают с помощью пиперидина в диметилформамиде перед замещением группы Boc N-концевой аминогруппы пролина. Пептид получают отщеплением от смолы после обработки нашатырным спиртом в смеси метанол/ДМФ. Полученный таким образом исходный продукт затем очищается.
Целью изобретения также являются фармацевтические композиции, содержащие в качестве активного ингредиента по меньшей мере один вышеназванный пептид формулы I, одно вышеназванное замещенное производное такого пептида или пептид, содержащий вышеназванную последовательность аминокислоты A1-A2-A3, вместе с фармацевтически приемлемым носителем или растворителем.
Пептиды настоящего изобретения могут вводиться орально, внутривенно, парентерально, подкожно, внутрибрюшинно или внутримышечно.
Фармацевтическая композиция может быть в виде желатиновой капсулы, таблетки, лиофилизата или жидкости в соответствии с выбранным способом введения. Фармацевтическая композиция также может быть в виде препарата с замедленным выделением.
Пептид настоящего изобретения можно назначать человеку для орального применения в дозе от 5 до 100 мкг/кг в день.
Внутривенно или подкожно соединение по изобретению можно назначать человеку в дозе от 1 до 12 мкг/кг от одного до трех раз в день. В организме животных соединения настоящего изобретения обнаружены в значительном количестве через несколько дней после глубокого применения, в частности, пептид Pro-Val-Thr-Lys-Pro-Gln-D-Ala-NH2 обнаружен в количестве, превышающем 10%.
Токсичность
Токсичность была изучена на крысах и собаках. Через четыре недели после введения доз, достигающих 4000 мкг/кг/день, не было обнаружено никаких признаков токсичности и никаких признаков, говорящих о мутагенной способности. Для человека подкожная или внутривенная инъекция дозы 200 мкг/кг не влечет за собой никакой биологической, клинической или патологической аномалии.
Фармакология
Терапевтический интерес соединений настоящего изобретения определяется следующим экспериментом.
Интенсивность желудочной реакции измеряется при определении объема вызванной желудочной секреции.
Котам была проведена операция под общей анестезией. Желудок был разделен на две части: на так называемый карман Хайденхейма и желудочную фистулу. Эти два кармана выводят наружу для сбора секреции соляной кислоты, пепсина и желудочного сока как во время основной фазы, так и после стимуляции. Поскольку эти животные имели хронические фистулы, они подвергались определенному количеству тестов каждую неделю и служили одновременно контрольными образцами. Стимулирование секреции пепсина достигалось путем перфузионного введения животным пентагастрина (PG) и VIP (вазоактивный желудочный пептид) в течение 2 часов из расчета 2 и 4 мкг/кг/ч.
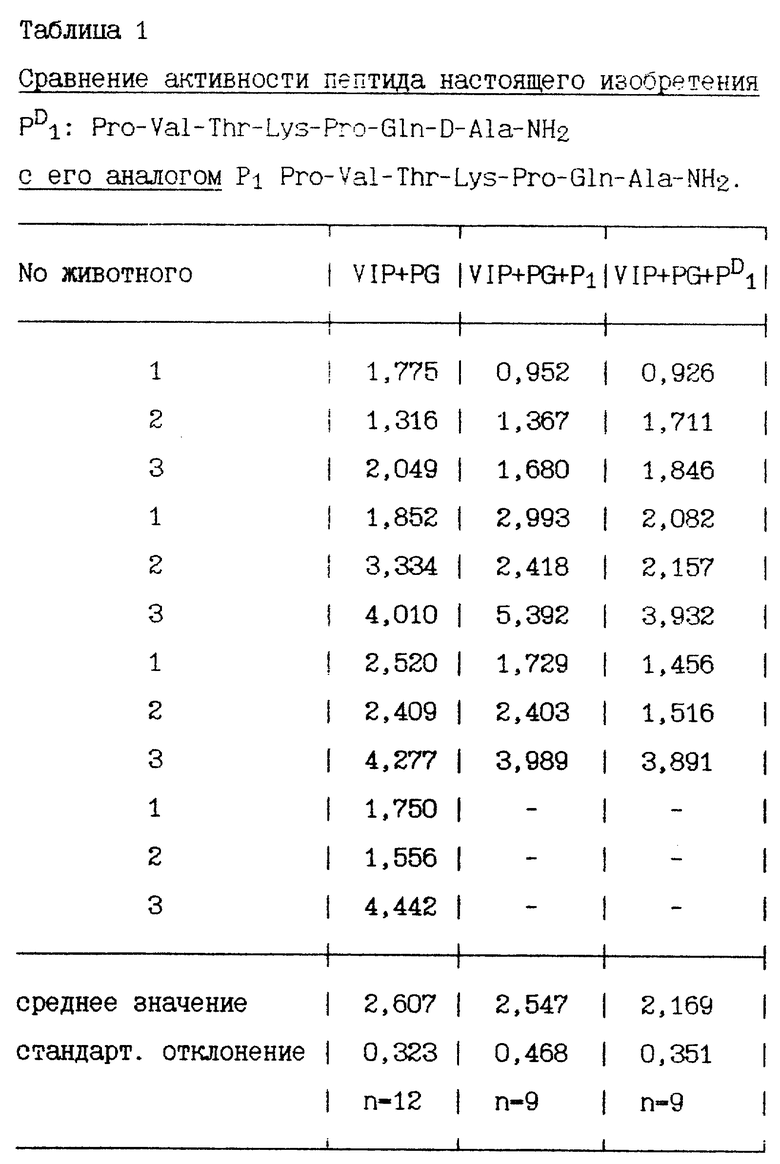
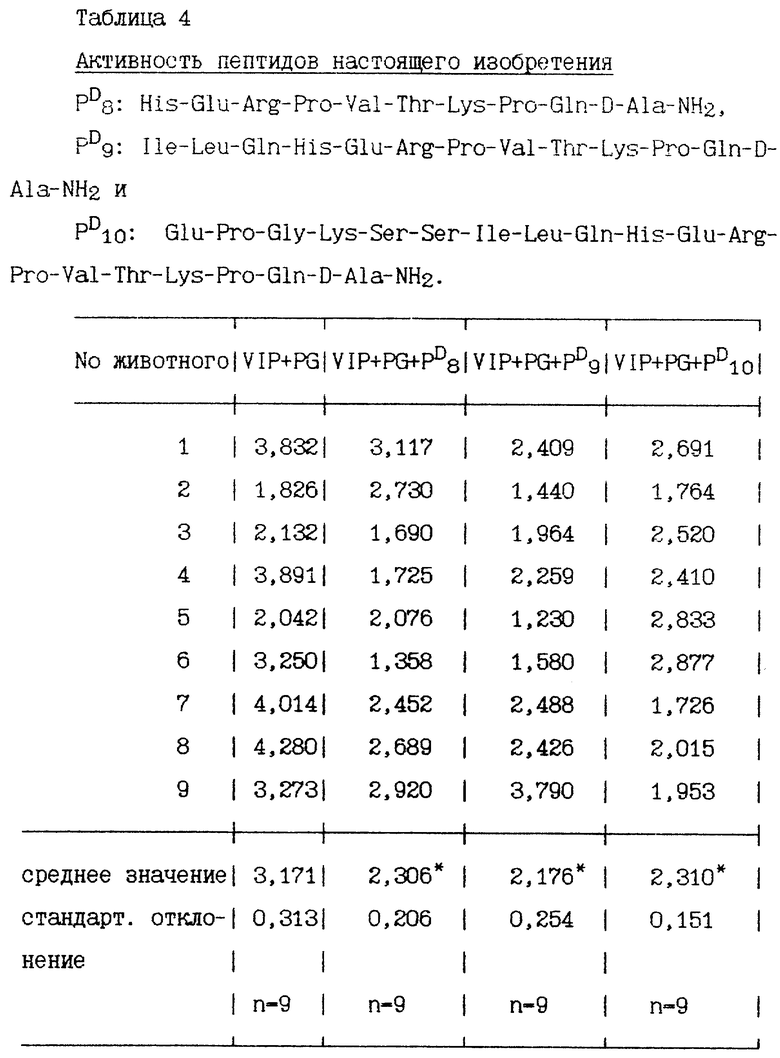
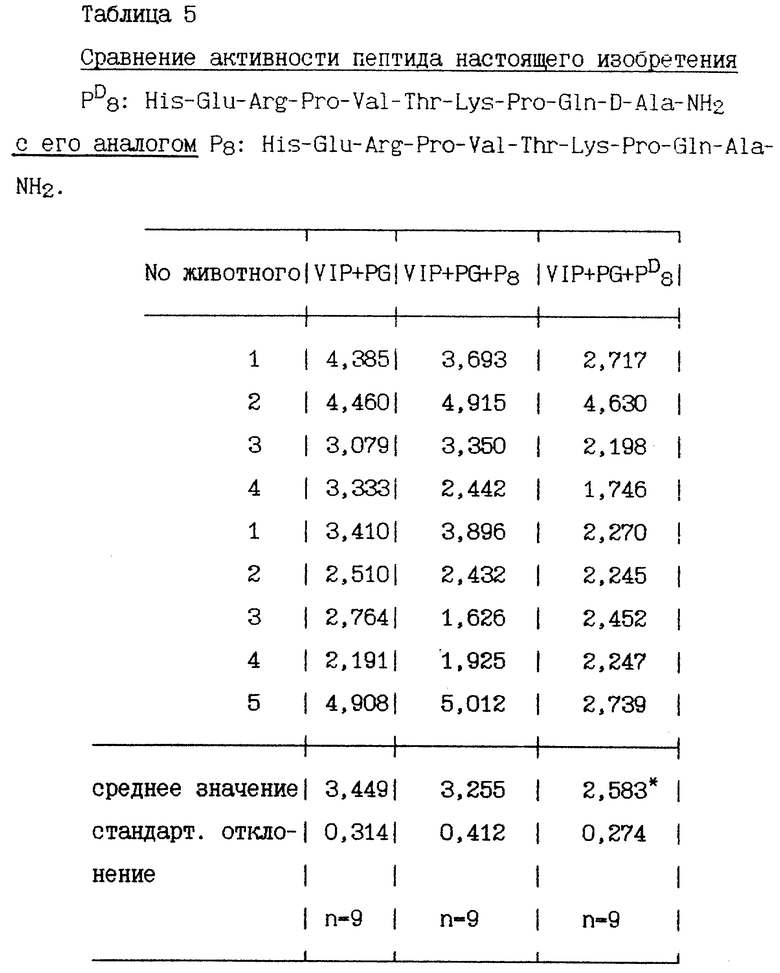
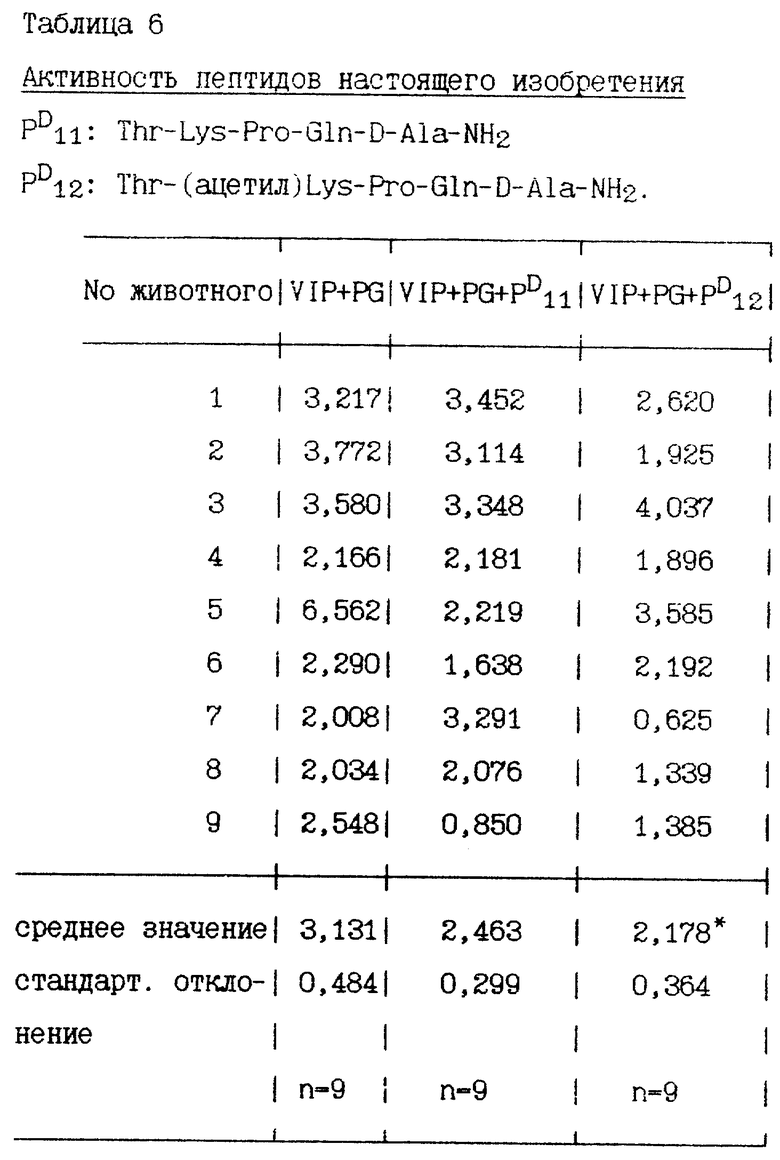
Через час после стимулирования пентагастрином и VIP перфузионно были добавлены пептиды в дозе 100 пмоль/кг/ч. Объем желудочного сока измеряют в течение 30 минут до и после перфузии. Количество пепсина в желудочном соке определяется протеолитическим спектрофотометрическим методом. Результаты, полученные в 9-12 экспериментах, представлены в нижеприведенных таблицах, секреция пепсина выражена в мг/15 минут, средняя величина двух периодов по 15 минут во время базальной секреции и средняя величина 6 периодов по 15 минут во время стимулированной секреции.
Некоторые пептиды изобретения, имеющие хотя бы один радикал D-аминокислоты, сравниваются со своими аналогами, у которых все радикалы аминокислот имеют L-конфигурацию.
Пример 1
Конкретные примеры приготовления фармкомпозиции.
В виде лиофилизата:
Пептид согласно изобретению - 0,10 мг
Маннит - 25,00 мг
Хлористый натрий - 4,50 мг
В виде раствора:
Пептид согласно изобретению - 0,30 мг
Спирт - 250,00 мг
Вода - 0,75 мг
В виде эмульсии:
Пептид согласно изобретению: - 0,30 мг
Полисорбат - 5,00 мг
Нейтральное масло - 500,00 мг
Вода - 0,50 мг
В виде таблеток:
Пептид согласно изобретению - 0,45 мг
Гидроксипролилцеллюлоза - 22,00 мг
Стеарат магния - 0,10 мг
Пример 2
Аминокислоты пептидов PD 2-PD 7 связывают друг с другом согласно указанному в описании классическому методу с использованием бутилоксикарбонила (Boc), который применяли для получения пептида PD 1, а также с использованием в качестве исходного смолы 4-метилбензгидриламина.
Связывание различных аминокислот производят в диметилформамиде (DMF) в присутствии бензотриазол-1-ил-окситрис(диметиламино)фосфоний-гексафторфосфата (BOP), 1-гидроксибензотриазола (HOBt) и N, N-диизопропилэтиламина (DIEA).
Процесс связывания и снятия защиты поэтапно приведен ниже:
1 - TFA - (1х1 мин)
2 - TFA - (1х3 мин)
3 - DCM - Быстрое отмывание
4 - изопропиловый спирт - (1х1 мин)
5 - DMF - (1х3 мин)
6 - связывание/DMF - (1х12 мин)
7 - DMF - (1х1 мин)
8 - связывание/DMF - (1х12 мин)
9 - DMF - (2х1 мин)
(TFA - трифторуксусная кислота, DCM - дихлорметан)
На каждом этапе использовали по 10 мл растворителя на 1 г смолы.
В процессе синтеза пептидов использовали следующие защитные группы боковых цепей:
- бензил для треонина
- 2-CIZ для лизина
- ацетил для лизина в пептидах PD 7 и PD 3.
Для прерывания процесса с помощью смолы к смеси добавляли по 10 мл фтористоводородной кислоты на 1 грамм пептидной смолы и инкубировали 45 минут при 0oC в присутствии паракрезола в качестве очистителя.
После выпаривания фтористоводородной кислоты продукт синтеза отмывали в эфире, растворяли в TFA, осаждали эфиром и высушивали.
Фракции, достаточно очищенные, разделяли и лиофилизировали. Конечные продукты проверяли методами аналитической высокоэффективной жидкостной хроматографии (HPLC), масс-спектроскопии и аминокислотного анализа.




Описываются новые синтетические пептиды общей формулы I A1-A2-A3-X где A1 = L- или D-Thr, или одну из следующих последовательностей, в которой по меньшей мере один радикал аминокислоты может иметь D-конфигурацию, приведенную в конце формулы изобретения; A2-Lys-Pro-Gln-Ala, в которой по меньшей мере один радикал аминокислоты может иметь D-конфигурацию; A3означает ковалентную связь; X = гидрокси, амино, алкиламино группу, причем названный пептид формулы I содержит по меньшей мере один радикал аминокислоты D-конфигурации и может содержать одну или множество одинаковых или различных защитных групп или их фармацевтически приемлемые соли. Описывается также фармацевтическая композиция, ингибирующая высвобождение пепсина, на основе соединений формулы I. 3 с. и 18 з.п.ф-лы, 6 табл.
A1-A2-A3-X,
где A1 означает радикал L-Thr или D-Thr; или одну из следующих последовательностей, в которой, по меньшей мере, один радикал аминокислоты может иметь D-конфигурацию, приведенную в графической части;
A2 представляет собой последовательность Lys-Pro-Gln-Ala, в которой по меньшей мере один радикал аминокислоты может иметь D-конфигурацию;
A3 означает ковалентную связь;
Х представляет собой гидрокси, амино, алкиламиногруппу, причем названный пептид формулы I содержит, по меньшей мере, 1 радикал D-конфигурации и может содержать одну или множество одинаковых или различных защитных групп, или их фармацевтически приемлемые соли.
| Способ получения калликреинтрипсин-ингибитора | 1972 |
|
SU503511A3 |
| Гептапептид,обладающий способностью ингибировать миотропное действие брадикинина | 1982 |
|
SU1083560A1 |
| US 3971736 A, 1976 | |||
| US 5276016 A, 04.01.94 | |||
| НОЖНИЦЫ С ПЕРЕМЕННЫМ НАКЛОНОМ | 2013 |
|
RU2601020C2 |
| СПОСОБ ПРОИЗВОДСТВА ЛИМОННОГО НАПОЛНИТЕЛЯ | 2005 |
|
RU2298334C1 |

