Настоящее изобретение относится к опиоидоподобным пептидным соединениям. Более конкретно, оно относится к опиоидоподобным пептидным соединениям, которые обладают анальгезирующей активностью периферического действия и избирательностью к подтипу μ-опиоидных рецепторов.
Предпосылки изобретения
Многие эндогенные пептиды млекопитающих и земноводных связываются со специфическими опиоидными рецепторами и вызывают анальгезирующую реакцию, подобную реакции, вызываемой классическими наркотическими опиатами. Было показано, что в высших животных одновременно присутствуют много различных типов опиоидных рецепторов. Например, см. W. Martin et al., J. Pharmacol. Exp. Ther., 197, p. 517 (1975) и J. Lord et al. Nature (London) 257, p. 495 (1977). Было идентифицировано три различных типа опиоидных рецепторов. Первый, δ, обнаруживает избирательное сродство к энкефалиноподобным пептидам. Второй, μ, обладает повышенной избирательностью к морфину и другим полицикличным алкалоидам. Третий, k, обладает равным сродством к любой группе из вышеуказанных лигандов и преимущественным сродством к динорфину. В анальгезирующих эффектах, по-видимому, в основном участвуют μ-рецепторы. Очевидно, что δ-рецепторы связаны с поведенческими эффектами, хотя δ- и k-рецепторы также могут опосредовать анальгезию.
Каждый опиоидный рецептор при взаимодействии с опиатом вызывает специфическую биологическую реакцию, уникальную для данного типа рецептора. Когда опиат активирует более одного рецептора, то затрачивается биологический отклик каждого рецептора, что дает побочные эффекты. Чем меньше специфичность и избирательность опиата, тем больше вероятность возникновения множественных побочных эффектов после введения опиата.
В предыдущих исследованиях опиаты, опиоидные пептиды и их аналоги не обнаруживали или обнаруживали ограниченную избирательность к типу рецептора или рецепторов, с которым они связываются.
Опиаты могут вызывать серьезные побочные эффекты, а иногда побочные эффекты, приводящие к летальному исходу. Побочные эффекты, такие как угнетение дыхания, привыкание к наркотическому средству, приобретение физической зависимости от наркотического средства и абстинентный синдром, вызываются неспецифическими взаимодействиями опиатов с рецепторами центральной нервной системы. См. К. Budd, в International Encyclopedia of Pharmacology and Therapeutics; N. E. Williams and H. Wilkinson, Eds., Pergammon: (Oxford), 112, p. 51 (1983). Исходя из этого следует ожидать, что опиоидные анальгетики, действующие только на опиоидные рецепторы периферической нервной системы, не будут вызывать такие же нежелательные побочные эффекты, какие вызывают опиоидные анальгетики центрального действия.
Одним из немногих известных в настоящее время классов (соединений), обладающих периодическим анальгезирующим действием, являются нестероидные противовоспалительные средства, такие как аспирин, ибупрофен и кеторолак. Эти вещества не взаимодействуют с опиоидными рецепторами, но известно, что они ингибируют циклооксигеназу и подавляют синтез простагландинов. Эти слабые анальгетики не оказывают побочного действия, связанного с рецепторами центральной нервной системы, однако могут вызывать другие побочные эффекты, такие как образование язв в желудочно- кишечном тракте.
Предполагалось, что неполярные пептиды более легко проникают в центральную нервную систему через гематоэнцефалический барьер, чем полярные пептиды. В опубликованных работах сообщалось, что пептид TAPP (H-Tyr-D-Ala-Phe-Phe-NH2) обладает антиноцицептивными (болеутоляющими) свойствами как периферического, так и центрального действия (P. Schiller et al. Proceedings of the 20th European Peptide Symposium, 1988). В противоположность этому, в настоящем изобретении было показано, что этот тетрапептид обладает каким-либо центральным действием даже при дозах в 100 мг/кг.
Целью настоящего изобретения является получение опиоидоподобных пептидных соединений периферического действия, которые не имеют нежелательных побочных эффектов, ассоциируемых с обычными анальгетиками периферического действия. Другой целью изобретения является получение пептидных соединений, которые избирательно связываются с μ-опиоидным рецептором.
Краткое описание изобретения
Настоящее изобретение относится к новым пептидным соединениям периферического действия, которые являются избирательными к μ-опиоидным рецепторам, которые могут быть представлены формулой I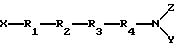
а также к их солям, производным и аналогам,
где R1 - Туг или 2'6'-диметилтирозин, или его производное, или аналог;
R2 - D-Ala или D-Arg;
R3 - Phe (p-F);
R4 - Phe или Phe(p-F);
X - H или C1-6-алкил;
Y и Z - независимо представляют H, аралкил или C1-6-алкил.
В другом аспекте настоящее изобретение относится к фармацевтическим композициям, включающим соединения формулы (I) в сочетании с фармацевтически приемлемым носителем и/или вторым терапевтически активным агентом.
В следующем аспекте, настоящее изобретение относится к способу устранения боли, предусматривающему введение млекопитающему, нуждающемуся в таком введении фармацевтически эффективного количества соединения формулы (I).
В еще одном аспекте, настоящее изобретение относится к использованию соединения формулы (I) для получения болеутоляющего лекарственного препарата.
Краткое описание чертежей
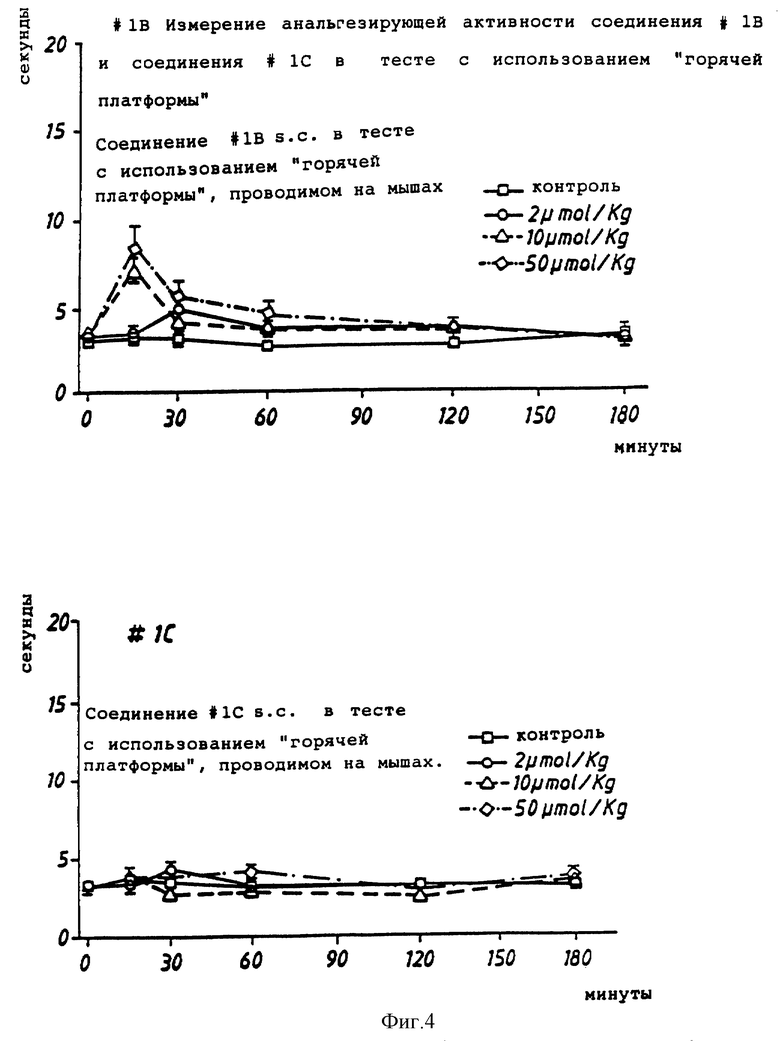
Фиг. 1, 2 и 4 иллюстрируют ингибирующее действие соединений настоящего изобретения в двух различных тестах, проводимых с использованием "горячей платформы".
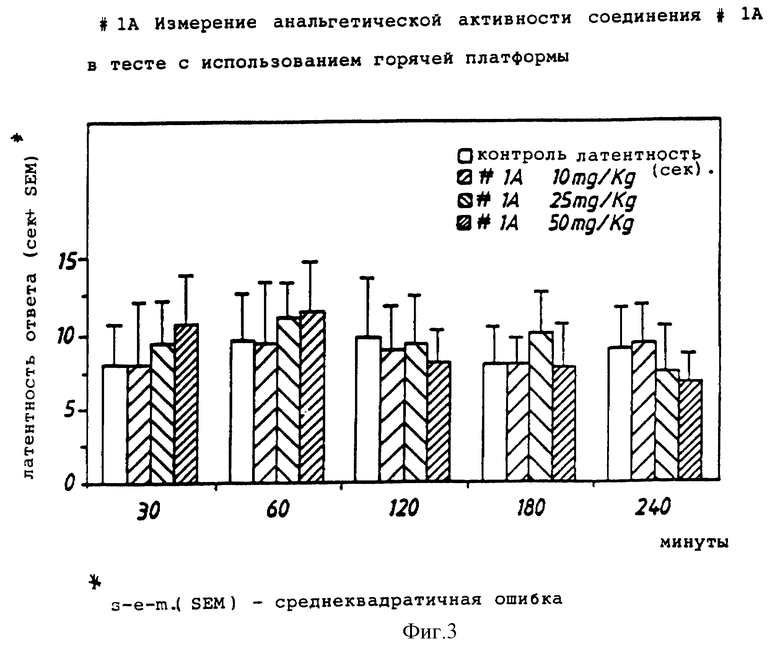
Фиг. 3 иллюстрирует ингибирующее действие H-Tyr-D-Ala-Phe-Phe-NH2 в тесте с использованием "горячей платформы".
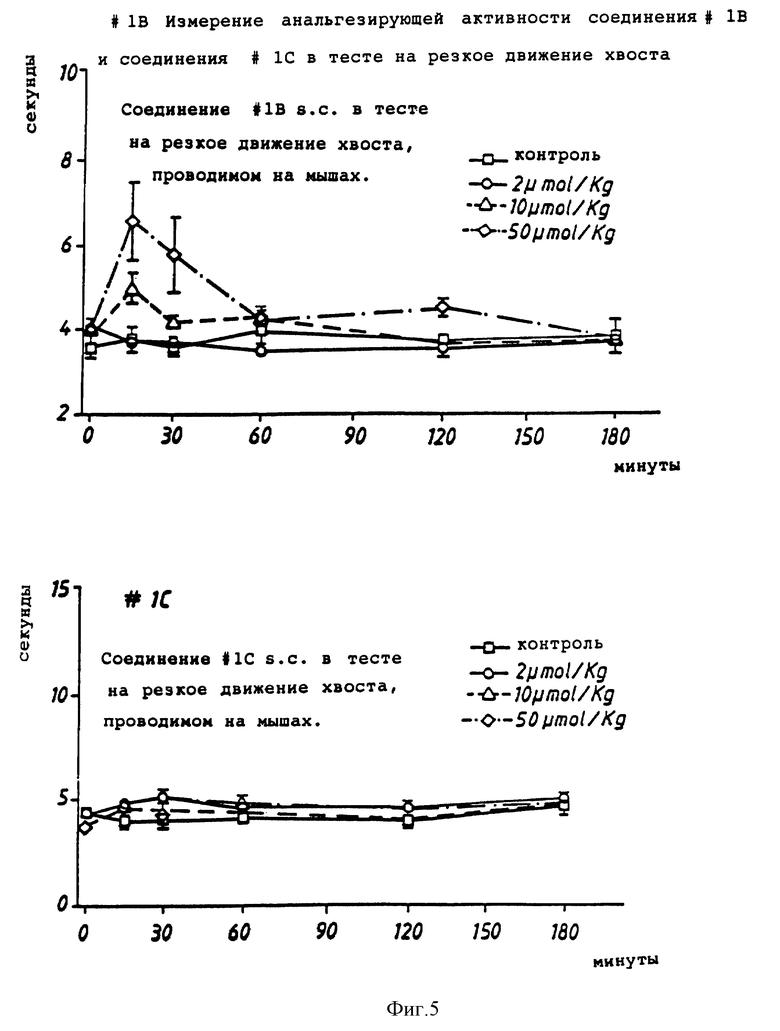
Фиг. 5 иллюстрирует ингибирующее действие соединений настоящего изобретения в тесте на резкие движения хвостом.
Описание изобретения
В описании и формуле изобретения используются следующие общепринятые сокращения:
Ala - аланин
Arg - аргинин
Phe - фенилаланин
Ser - серин
Tyr - тирозин
TAPP - H-Tyr-D-Ala-Phe-Phe-NH2
GPI - подвздошная кишка морской свинки
MVD - мышь vas deferens
Phe(p-F) - пара-фторфенилаланин
HOBT - N-гидроксибензотиазол
BOP - бензотриазолил-N-окси-трис(диметиламино) фосфонийгексафторфосфат
DMF - диметилформамид
TFA - трифторуксусная кислота
tBU - трет-бутил
Pmc - 2,2,5,7,8-пентаметилхроман-6-сульфонил
FMOC - 9-флуоренилметилоксикарбонил
PBQ - фенил-пара-бензохинон.
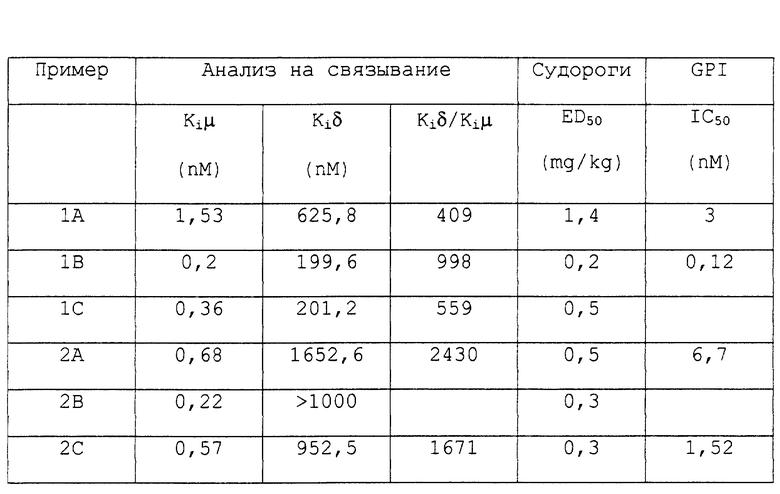
Величина "ED50", представленная в таблице для теста с PBQ-индуцированными судорогами, означает дозу лекарства, которая вызывает уменьшение количества судорог на 50% по сравнению с контролем. Величина "Ki", представленная в таблице 1 для анализа на связывание, означает константу ингибирования, полученную с использованием известного лиганда DAMGO для μ-рецептора и лиганда DADLE для δ-рецептора.
"K
Соединения настоящего изобретения представляют собой соединения формулы I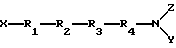
а также их соли, производные и аналоги.
X представляет H или метил, предпочтительно Н;
R1 представляет Tyr или 2'6'-диметилтирозин, предпочтительно Tyr, альфа-аминогруппа R1 замещена X с образованием аминогруппы, в случае если X представляет H, или алкиламиногруппы, если X представляет метил;
R2 представляет D-Ala или D-Arg, предпочтительно D-Ala;
R3 представляет Phe(p-F);
R4 представляет Phe или Phe(p-F), предпочтительно Phe;
Y и Z независимо представляют H, аралкил, такой как бензил, или C1-6-алкил, такой как метил, предпочтительно, чтобы Y и Z оба были Н.
Соединениями настоящего изобретения являются следующие соединения, но не ограничиваются только ими:
Соединение # 1B H-Tyr-D-Ala-Phe(p-F)-Phe(p-F)-NH2;
Соединение # 1C H-Tyr-D-Ala-Phe(p-F)-Phe-NH2;
Соединение # 2B H-Tyr-D-Arg-Phe(p-F)-Phe(p-F)-NH2;
Соединение # 2C H-Tyr-D-Arg-Phe(p-F)-Phe-NH2.
В предпочтительном варианте соединения настоящего изобретения выбираются из группы, включающей
Соединение # 1C H-Tyr-D-Ala-Phe(p-F)-Phe-NH2;
Соединение # 2C H-Tyr-D-Arg-Phe(p-F)-Phe-NH2.
В более предпочтительном варианте соединением настоящего изобретения является
Соединение # 1C H-Tyr-D-Ala-Phe(p-F)-Phe-NH2.
В опиоидных пептидных соединениях тирозин может быть заменен на аминокислотное производное 2'6'-диметилтирозин (Dmt). Эксперименты показали, что замена тирозина на Dmt в положении R1 первого аминокислотного остатка в общей формуле I увеличивает эффективность опиоидного пептида при связывании с μ-рецептором на 2 порядка величины. Избирательность к μ-рецепторам возрастает, если соединение содержит Dmt в положении R1. Эта замена приводит к соответствующему изменению отношения констант ингибирования связывания, что отражает увеличение избирательности к μ-рецепторам.
Опиоидную активность пептидов анализировали in vitro с использованием препарата продольной мышцы подвздошной кишки морской свинки, а их болеутоляющую активность определяли с использованием in vivo-модели PBQ-индуцированных судорог (периферическая активность) и двух тестах с использованием "горячей платформы" (центральная активность). Анальгезирующую активность соединения настоящего изобретения оценивали в тесте на резкие движения ("отдергивание") хвоста. Тест на резкие движения хвостом проводили для оценки центрального анальгезирующего действия испытуемого соединения. Сравнение активности соединений изобретения в экспериментах с индуцированными судорогами с использованием "горячей" платформы и в тестах на резкие движения хвостом продемонстрировало, что анальгезирующие эффекты были опосредованы преимущественно рецепторами периферической нервной системы. В тесте с индуцированными судорогами наблюдалась высокая степень периферической анальгезии, тогда как в тестах с использованием горячей платформы и в тесте на резкие движения хвостом отмечалась низкая степень анальгезии.
PBQ-индуцированные судороги у мыши (PBQ-фенилпарабензохинон) является критерием в оценке как периферической, так и центральной анальгезии. Схема эксперимента описана в работе Sigmund et al., Proc. Soc. Exp. Biol. Med., 95, p. 729 (1957), которая приводится здесь посредством ссылки. Центральную анальгезию определяли по ингибированию реакции у мыши в тесте с использованием "горячей платформы". Описание эксперимента смотри в работе G. Wolfe and A. MacDonald, J. Pharmacol. Exp. Ther., 80, p. 300 (1944), которое приводится здесь посредством ссылки. Тесты для определения аффинности связывания с μ- и δ-опиоидными рецепторами, а также тест с использованием GPI были описаны в работе в Schiller et al., Biophys. Res. Commun., 85, p. 1322 (1965), которая приводится здесь посредством ссылки.
Соединения настоящего изобретения могут быть получены методами, хорошо известными в практике пептидной химии. Например, смотри Principle of Peptide synthesis, Bodansky M., Springer-Verlag, Berlin, Heidelberg, New-York, Tokyo 1984 или The Peptides, Analysys, Synthesis, Biology, edited by Erhard Gross and Johaness Meienhofer, Academic Press 1979.
Соединения настоящего изобретения были получены путем твердофазного синтеза, как описано ниже, в соответствии с методами, обычно используемыми для синтеза пептидов. Коммерчески доступный пара-фторфенилаланин (Phe(p-F)) использовали на соответствующей стадии синтеза. 2'6'-диметилтирозин может быть использован в синтезе и получен известным методом химического синтеза.
Фармацевтически приемлемые соли пептидов данного изобретения могут быть получены обычным способом посредством реакции с соответствующей кислотой. Подходящие кислотно-аддитивные соли могут быть получены с использованием таких кислот, как соляная, бромистоводородная, фосфорная, уксусная, фумаровая, салициловая, лимонная, молочная, оксифенилуксусная, винная, щавелевая, метасульфоновая, а также других подходящих кислот, известных специалистам.
Настоящее изобретение также относится к фармацевтическим композициям. Подходящие композиции содержат фармацевтически эффективное количество соединений настоящего изобретения или их фармацевтически приемлемых солей в смеси с фармацевтически приемлемым носителем или добавками. Терапевтически эффективное количество пептида настоящего изобретения в сочетании с фармацевтически приемлемым носителем (т.е. карбонатом магния или лактозой) может быть использовано для получения терапевтического препарата, такого как
(i) пилюли, таблетки, капсулы или жидкость для перорального введения пациенту;
(ii) жидкость или мазь для введения путем ингаляции, чрескожно, интраназально, ректально или под язык;
(iii) раствор для внутривенного, парентерального, подкожного или внутрибрюшинного введения;
(iv) неоральный или парентеральный препарат пролонгированного действия.
Настоящее изобретение также относится к способу устранения боли у млекопитающих, включая человека. Этот способ предусматривает введение фармацевтически эффективного количества пептида формулы I или его фармацевтически пригодной соли или композиции одним из традиционных способов, т.е. орально, парентерально, чрескожно иди через слизистые. В виде препарата пролонгированного действия с использованием биологически совместимого полимера или посредством направленной доставки в нужные ткани-мишени с использованием мицелл, гелей и липосом. Пептиды могут быть введены человеку в дозе от 0,01 до 100 мг/кг, предпочтительно от 0,05 до 20 мг/кг и наиболее предпочтительно от 0,1 до 1 мкг.
Нижеследующие примеры приводятся для лучшего описания изобретения. Эти примеры представлены лишь в иллюстративных целях и не должны рассматриваться как некое ограничение изобретения.
Пример 1
Получение 1C H-Tyr-D-Ala-Phe(p-F)-Phe-NH2
Синтетический пептид был получен с использованием смолы (полимерный носитель) Knorr. Использованные аминокислоты имели Fmoc-замещенные альфа-аминогруппы и tBU-защищенную боковую цепь тирозина. Диметилформамид, использованный на стадии присоединения, не содержал диметиламина. DMF, использованный на стадии отмывки, и TFA были биологически чистыми (Biograde). Для стадии очистки использовались очищенная H2O (USP) и ацетонитрил, очищенный с помощью ВЭЖХ. Все оставшиеся растворители были ACS (спектрофотометрически) чистыми и использовались без дополнительной очистки.
Твердофазный синтез был проведен вручную на геле с нагрузкой в 0,84 мМоль/г. Конденсацию пептидов осуществляли с использованием по 1,5-2 эквивалента (каждого) Fmoc-аминокислоты, HOBT и BOP в DMF в течение 3-24 часов при комнатной температуре. Стадии по снятию Fmoc-защиты с альфа-аминогрупп были проведены с использованием 20% (v/v) пиперидина в DMF в течение 25 минут. Расщепление пептида и снятие защиты с боковых цепей осуществляли путем обработки TFA/CH2Cl2/анизолем. Пептидный полимерный носитель обрабатывали TFA в течение двух периодов по 90 минут при комнатной температуре в атмосфере азота. После отмывания CH2Cl2 и высушивания остаток обрабатывали этиловым эфиром, осадок фильтровали и осушали в условиях вакуума.
Полученный неочищенный пептид был очищен с помощью ВЭЖХ на колонке C1810μ-15μ 300A с обращенной фазой, с использованием градиента элюции 0,06% TFA/H2O и 0,06% TFA/ацетонитрила. Мониторинг был проведен при 220 нм. Чистые фракции объединяли и лиофилизировали. Очищенный материал превращали в его гидрохлоридную соль с получением чистого целевого соединения:
Таким же методом были синтезированы следующие пептиды:
1A H-Tyr-D-Ala-Phe-Phe-NH2
1B H-Tyr-D-Ala-Phe(p-F)-Phe(p-F)-NH2.
Пример 2
Получение 2C H-Tyr-D-Arg-Phe(p-F)-Phe-NH2.
Синтетический пептид был получен с использованием Knorr. Использованные аминокислоты имели Fmoc-защищенную альфа-аминогруппу, и Pmc-защищенную боковую цепь для D-аргинина, и tBU-защищенную боковую цепь для тирозина. Диметилформамид, использованный на стадии присоединения, не содержал диметиламина. DMF, использованный на стадии промывания, и TFA были биологически чистыми (Biograde). Для стадии очистки использовались USP-очищенная H2O и ацетонитрил, очищенный с помощью ВЭЖХ. Все оставшиеся растворители были ACS-чистыми и использовались как таковые без какой-либо очистки.
Твердофазный синтез был проведен вручную на полимерном носителе с нагрузкой в 0,84 мМоль/г. Конденсацию пептидов осуществляли с использованием по 2 эквивалента (каждого): Fmoc-аминокислоты HOBT и BOP в DMF в течение 2 - 5 часов при комнатной температуре. Стадии снятия Fmoc-защиты с альфа-аминогрупп были проведены с использованием 20% (v/v) (объемные проценты) пиперидина в DMF в течение 25 минут. Отщепление пептида и снятие защиты с боковых цепей осуществляли путем обработки TFA/CH2Cl2/анизолем. Пептидный полимер обрабатывали TFA в течение двух периодов по 90 минут при комнатной температуре в атмосфере азота. После промывания CH2Cl2 и высушивания остаток обрабатывали этиловым эфиром, а осадок фильтровали и осушали в условиях вакуума.
Полученный неочищенный пептид очищали с помощью ВЭЖХ на колонке C1810μ-15μ 300A с обращенной фазой с использованием градиента элюции 0,06% TFA/H2O и 0,06% TFA/ацетонитрила. Мониторинг был проведен при 220 нм. Чистые фракции объединяли и лиофилизировали.
Таким же образом были синтезированы следующие пептиды:
2A H-Tyr-D-Arg-Phe-Phe-NH2
2B H-Tyr-D-Arg-Phe(p-F)-Phe(p-F)-NH2.
Пример 3
Анализ на связывание радиоактивного меченого лиганда
Подготовка мембраны
Самцов крыс Sprague-Dawley весом от 350 до 450 г умерщвляли путем ингаляции CO2. Крыс обезглавливали, и головной мозг без мозжечка извлекали, помещали в ледяной (охлажденной до температуры льда) солевой раствор, а затем гомогенизировали в 50 mM холодного (температуры льда) буфера трис (pH 7,4) (10 мл/мозга). Мембраны были центрифугированы при 14000 об/мин в течение 30 минут при 4oC. Осадки после центрифугирования ресуспендировали в 50 mM ледяного буфера Трис (pH 7,4), приблизительно в соотношении 6 мл/мозг, и помещали на хранение при -78oC вплоть до использования. Количественную оценку белка в гомогенате головного мозга проводили с использованием имеющегося в продаже набора для анализа белков (Bio-Rad).
Ингибирование радиолиганда
(3H)-DAMGO и (3H)-DAGLE были использованы в качестве радиоактивных лигандов для μ- и δ-рецепторов соответственно. 50 мкл радиолиганда, 100 мкл мембраны и серийно разведенное контрольное соединение инкубировали в течение 1 часа при 22oC. Неспецифическое связывание определяли с использованием 500-кратного избытка немеченного лиганда в присутствии радиоактивной метки и мембран. Свободный лиганд отделяли от связанного путем фильтрации через бумагу Whatman GF/B (предварительно пропитанную в 1% водном растворе полиэтиленамина) и промывали в 500 mM ледяном буфере, pH 7,4, с использованием клеточного харвестера клеток Brandel. Фильтры осушали, а радиоактивность подсчитывали в 24-луночном планшете для микротитрования в присутствии 500 мл сцинциллятора на лунку. Радиоактивность измеряли при помощи счетчика Wallac 1450 Microbeta.
Ki (константы ингибирования) для различных соединений были определены исходя из IC50 по уравнению Cheng and Prusoff. Результаты анализа на связывание систематизированы в таблице.
Активность пептидных соединений в отношении μ-рецепторов определяли в тесте с использованием подвздошной кишки морской свинки (GPI) (препарат продольной мышцы) согласно методу, описанному Schiller et al., Biophys. Res, Communn., 85, p. 1322 (1975). Результаты активности систематизированы в таблице.
Пример 4
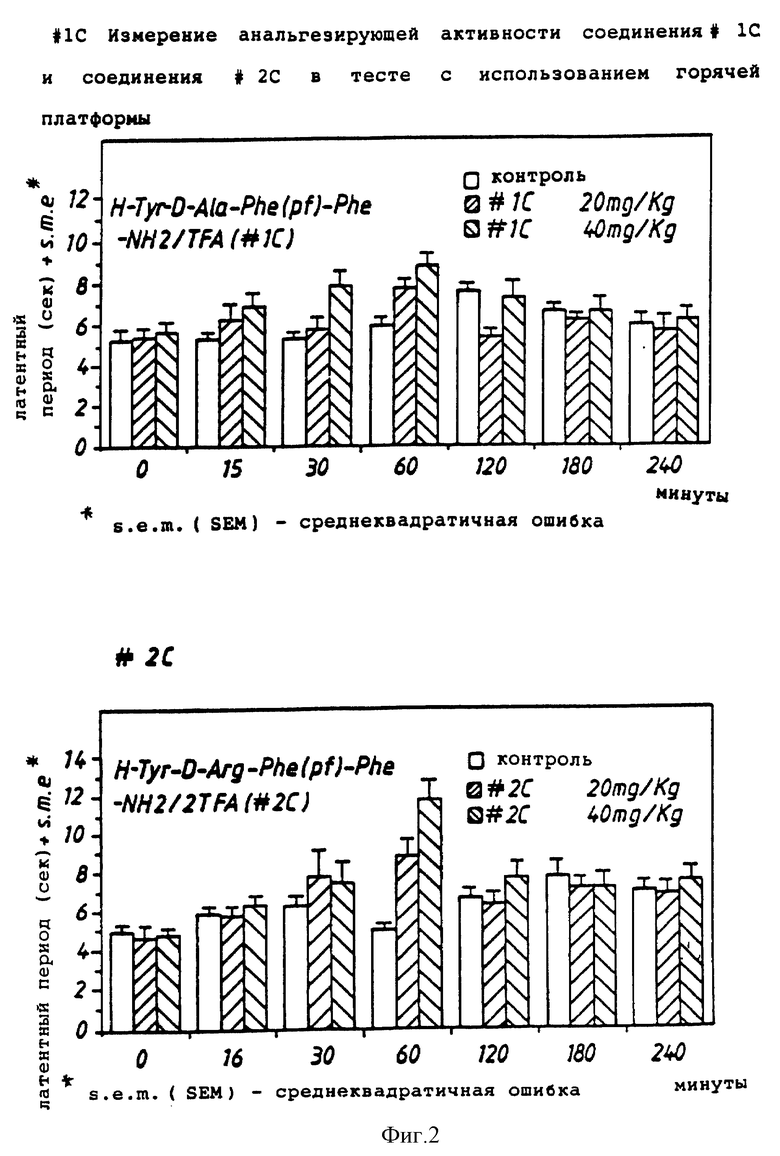
Тест с использованием "горячей платформы" (измерение анальгезирующей активности) проводили при 55oC.
Для этого теста использовали самцов мыши CD # 1 весом от 20 до 25 г. Мыши были взвешены, помечены и разделены на группы по 10.
Мышам была сделана подкожная инъекция соединения (или стандартного соединения, или среды) с объемом инъекции, эквивалентным 0,1 мл/10 г. р.с. (10 мл/кг).
Мышей индивидуально оценивали по времени реакции на "горячей платформе". Температура горячей платформы (Sorel, модель DS37) составляла 55oC. У мыши наблюдались признаки дискомфорта, такие как облизывание или подергивание лап, попытка ухода (спрыгнуть с платформы) или дрожание. Время реакции регистрировали, когда появлялся одни из этих признаков, и выражали в "секундах". Каждую мышь наблюдали максимум в течение 30 секунд, чтобы предотвратить поражение ткани лапы.
В каждом случае регистрации времени среднее время реакции в контрольной группе умножали на 1,5. Время реакции каждой тестируемой мыши сравнивали со "средним контрольным х 1,5". Если время реакции было меньше, чем "среднее контрольное х 1,5", то считали, что у мыши анальгезирующий эффект отсутствует. Если время реакции было выше, чем "среднее контрольное х 1,5", считали, что анальгезирующий эффект есть. Процент анальгезирующего действия соединения для данного времени теста определяли по числу анальгезированных (не чувствительных к боли) мышей в группе. Если процент анальгезирования был ниже 30%, то соединение считалось неактивным.
Результаты показаны на фиг. 1-3.
Пример 5
Тест с индуцированием судорог
Тест был проведен на самцах мыши CD # 1 весом от 18 до 22 г. Мыши были взвешены и помечены. Этим мышам инъецировали внутрибрюшинно (интраперитониально) 0,02% раствор фенилхинона в количестве 0,3 мл/20 г веса. Судороги, которые появлялись в течение 15 минутного периода времени после инъецирования, были подсчитаны. Фенилхинон подкожно инъецировали с интервалами времени в 5, 20 или 60 минут после введения соединения (или среды, или стандартного соединения).
0,02% раствор фенилхинона (2-фенил-1,4-бензохинон (Sigma)) был приготовлен следующим образом. 20 мг фенилхинона растворяли в 5 мл 90% этанола (Sigma, реактив, спирт). Растворенный фенилхинон медленно добавляли к 95 мл подогретой (не кипяченой) дистиллированной воды при постоянном встряхивании. Раствор фенилхинона выдерживали в течение 2 часов перед использованием и во всех случаях в условиях, защищенных от света. Каждый день для теста готовили новый раствор.
Результаты опытов систематизированы в таблице. Можно видеть, что пептидные соединения настоящего изобретения, в том случае, если один или оба R3 или R4 представляют Phe (p-F), показывают более высокую селективность к μ-опиоидным рецепторам по сравнению с соответствующими соединениями, не содержащим Phe (p-F), а также более сильную трансдукцию рецептора, как было определено в тесте с использованием GPI. Кроме того, соединения настоящего изобретения показывают более высокую периферическую анальгезирующую активность, как было показано в тесте с индуцированными судорогами.
Пример 6
Тест с использованием горячей платформы (измерение анальгезирующей активности) проводили при 58oC.
Для этого теста использовали самцов мыши NMRI весом от 20 до 25 г. Мыши были взвешены, помечены и разделены на группы по 6.
Мышам была сделана подкожная инъекция соединения (или стандартного соединения, или среды) при объеме инъекции эквивалентом 0,1 мл/10 г. р.с. (10 мл/кг).
Оценка времени реакции у мышей на горячей платформе была сделана индивидуально для каждой мыши. Температура горячей платформы (ПТС, Inc; модель 35-0) составляла 58oC. У мышей наблюдались признаки дискомфорта, такие как облизывание или подергивание лапы, попытка ухода (спрыгивания с платформы) или дрожание. Время реакции регистрировали, когда появлялся один из этих признаков, и выражали в "секундах". Каждую мышь наблюдали максимум в течение 20 секунд, чтобы предотвратить поражение ткани лапы.
Считалось, что соединение обладает анальгезирующим действием, если время реакции имело статистически значимое (p<0,05; двухфакторный ANOVA (дисперсионный анализ), sigma slot) отличие от времени реакции контрольной группы.
Результаты показаны на фиг. 4.
Пример 7
Тест на резкие движения хвоста
Для осуществления этого теста использовали мышей-самцов NMRI весом от 20 до 25 г. Мышей взвешивали, помечали и разделяли на группы по 6.
Мышам вводили подкожную инъекцию соединений (или стандартной среды при объеме инъекции, эквивалентном 0,1/10 г. р.с. (10 мл/кг)). Мышей индивидуально оценивали на время реакции в тесте на резкие движения хвоста. Латентный период времени до резкого движения хвостом измеряли в тот момент, когда регулируемый реостатом световой луч был направлен на кончик хвоста (ПТС Inc. Модель 33). Каждая мышь наблюдалась в течение максимум 10 секунд, чтобы предотвратить поражение тканей.
Соединение считалось анальгетиком, если время реакции значительно отличалось (p<05, двухфакторный ANOVA (дисперсионный анализ), Sigma Stat) от времени реакции для контрольной группы.
Результаты представлены на фиг. 5.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОПИОИДНЫЕ ПЕПТИДЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2146681C1 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ УНДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2541127C1 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ ТРИДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2538727C1 |
| ЛИНЕЙНЫЕ ИЛИ ЦИКЛИЧЕСКИЕ ОЛИГОПЕПТИДЫ, ОБЛАДАЮЩИЕ СРОДСТВОМ К ОПИАТНЫМ РЕЦЕПТОРАМ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2131438C1 |
| СРЕДСТВО ДЛЯ ЭФФЕКТИВНОГО КУПИРОВАНИЯ ОСТРОГО И/ИЛИ ХРОНИЧЕСКОГО БОЛЕВОГО СИНДРОМА И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2016 |
|
RU2622980C1 |
| ПЕПТИДНЫЕ КОМПОЗИЦИИ | 2014 |
|
RU2725150C2 |
| ПРОЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ С ВЫСОКОЙ СТЕПЕНЬЮ ПРОНИКНОВЕНИЯ НА ОСНОВЕ ПЕПТИДОВ И РОДСТВЕННЫХ ПЕПТИДАМ СОЕДИНЕНИЙ | 2010 |
|
RU2627065C2 |
| ЛЕЧЕНИЕ НЕВРОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2755997C2 |
| СИНТЕТИЧЕСКОЕ АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ ПРИРОДЫ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2017 |
|
RU2656188C1 |
| ДЕКАПЕПТИД, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2084458C1 |
Описываются новые опиоидоподобные пептиды формулы I Х-R1-R2-R3-R4-N(Z)-Y, а также их соли, производные и аналоги, где Х - водород; R1 - Tyr; R2 - D-Ala или D-Arg; R3 - Phe (p-F); R4 - Phe или Phe (p-F); Y и Z представляют H. Новые соединения обладают анальгезирующей активностью периферического действия и избирательностью к подтипу μ-опиоидных рецепторов. Описываются также способ их получения, фармацевтическая композиция. 3 с. и 10 з.п. ф-лы, 5 ил., 1 табл.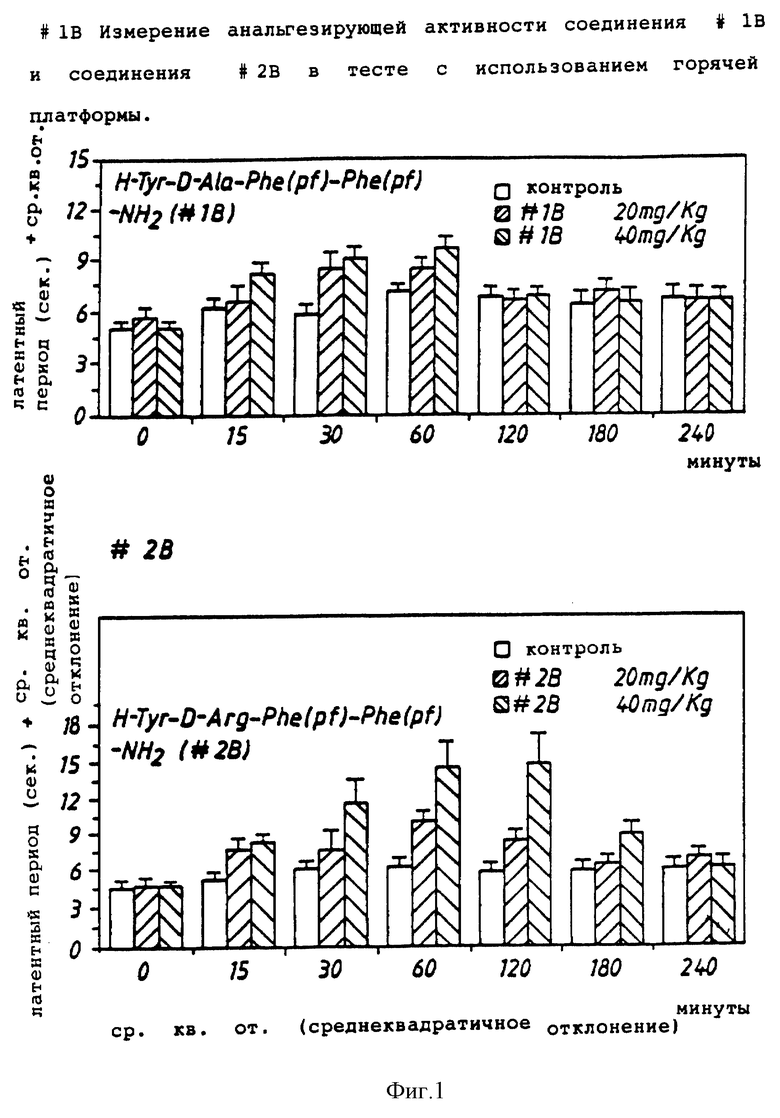
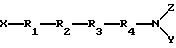
а также его соли, производные и аналоги,
где X - водород;
R1 - Tyr;
R2 - D-Ala или D-Arg;
R3 - Phe (p-F);
R4 - Phe или Phe (p-F);
Y и Z независимо представляют H.
H - Tyr - D - Ala - Phe (p-F) - Phe (p-F) - NH2.
H - Tyr - D - Ala - Phe (p-F) - Phe - NH2.
H - Tyr - D - Arg - Phe (p-F) - Phe (p-F) - NH2.
H - Tyr - D - Arg - Phe (p-F) - Phe - NH2.
Приоритет по пунктам:
18.08.95 по пп.7 - 11;
07.11.95 по пп.1 - 6, 12 и 13.
| Аналог энкефалина,обладающий анальгетическим действием | 1981 |
|
SU1048703A1 |
| Способ получения тетрапептидов или их кислотно-аддитивных солей | 1981 |
|
SU1082319A3 |
| RU 94035760 A1, 10.08.96 | |||
| СПОСОБ ПЕРЕВОЗБУЖДЕНИЯ СИНХРОННОГО ГИСТЕРЕЗИСНОГО ДВИГАТЕЛЯ РЕАКЦИЕЙ ЯКОРЯ | 2011 |
|
RU2465713C2 |
| US 5312899 A, 17.05.94 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Экономайзер | 0 |
|
SU94A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
